
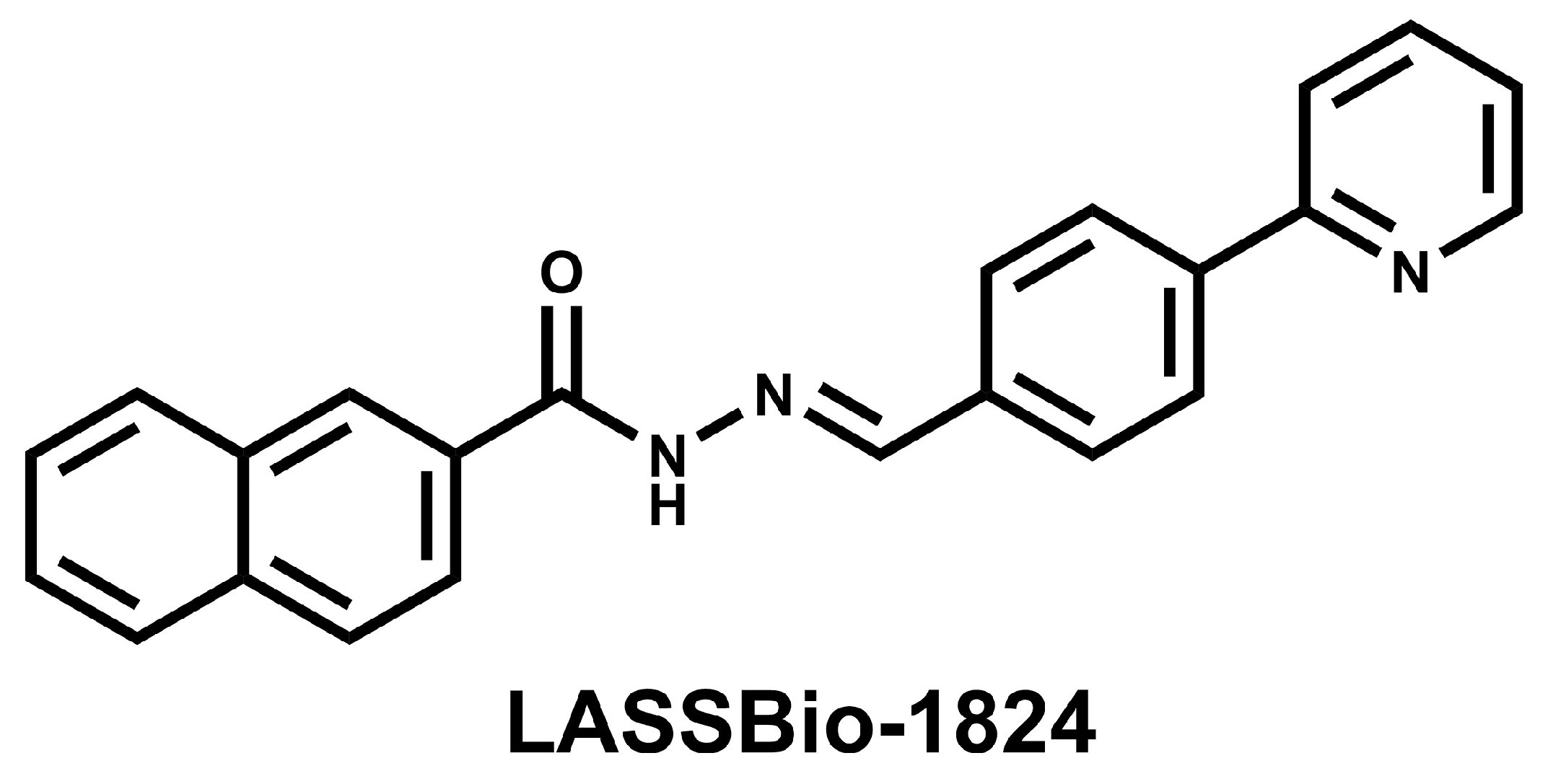
Novel p38 Mitogen-Activated Protein Kinase Inhibitor Reverses Hypoxia-Induced Pulmonary Arterial Hypertension in Rats

 , , ,
, , ,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Results
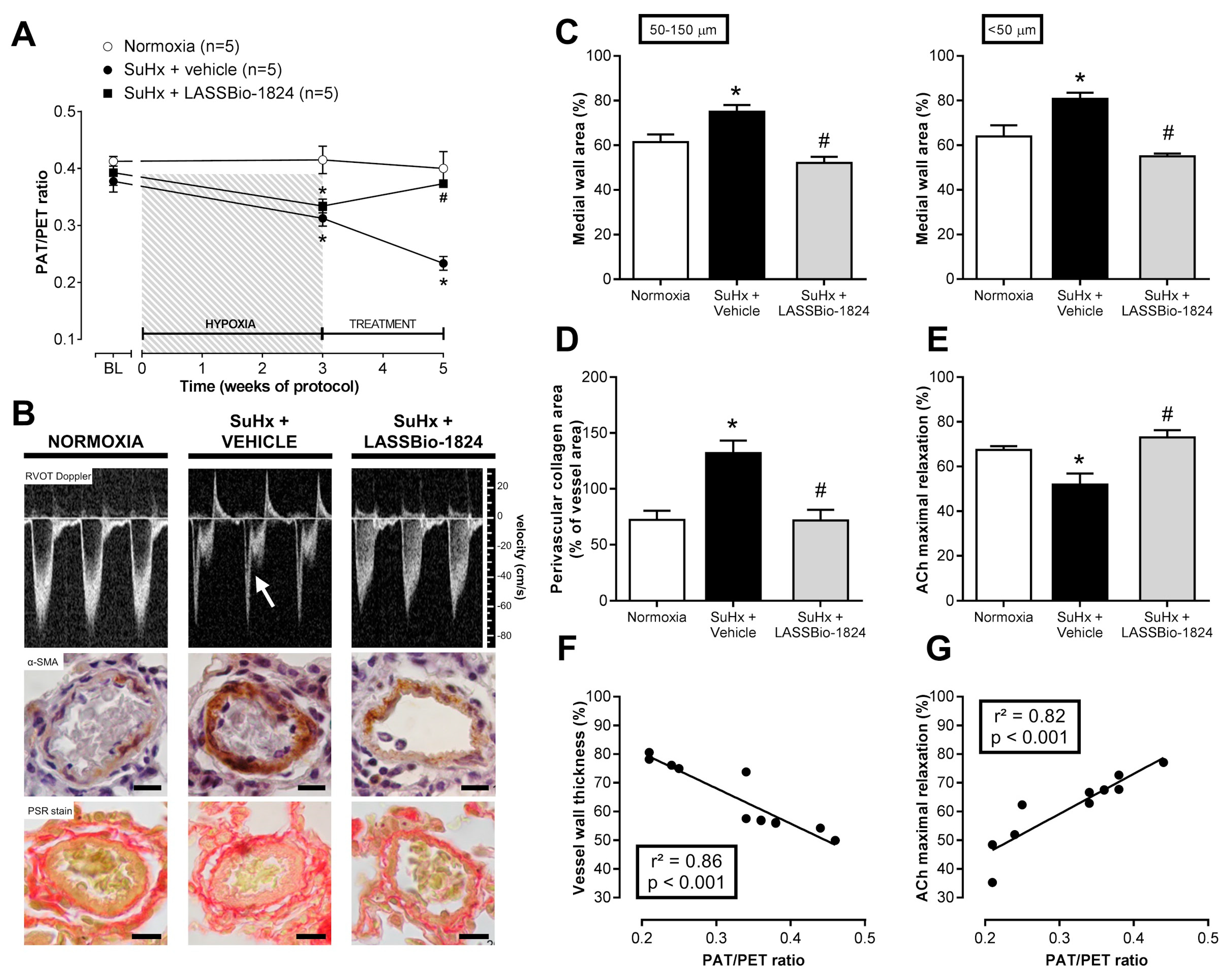
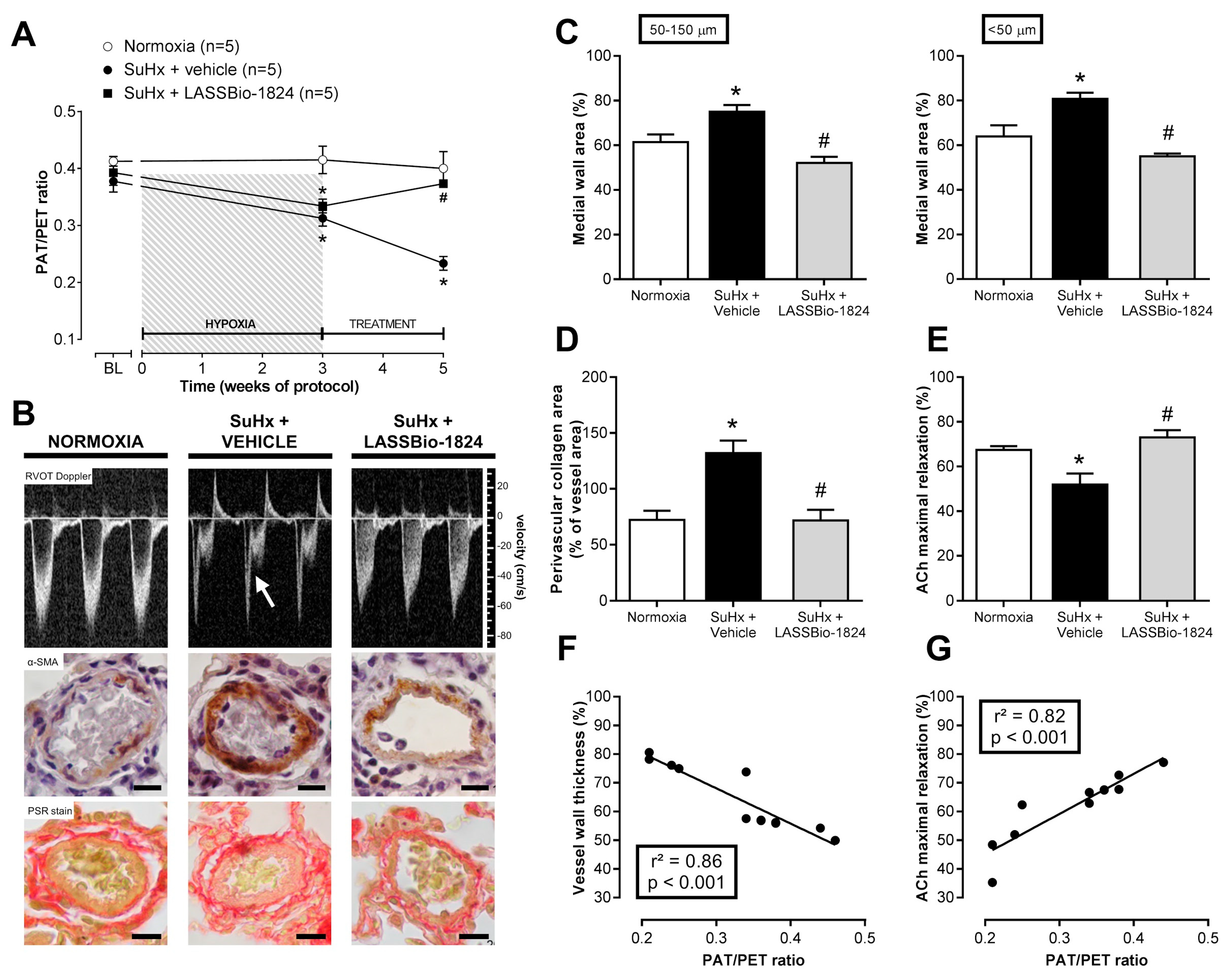
2.1. LASSBio-1824 Selectively Decreases Pulmonary Vascular Resistance
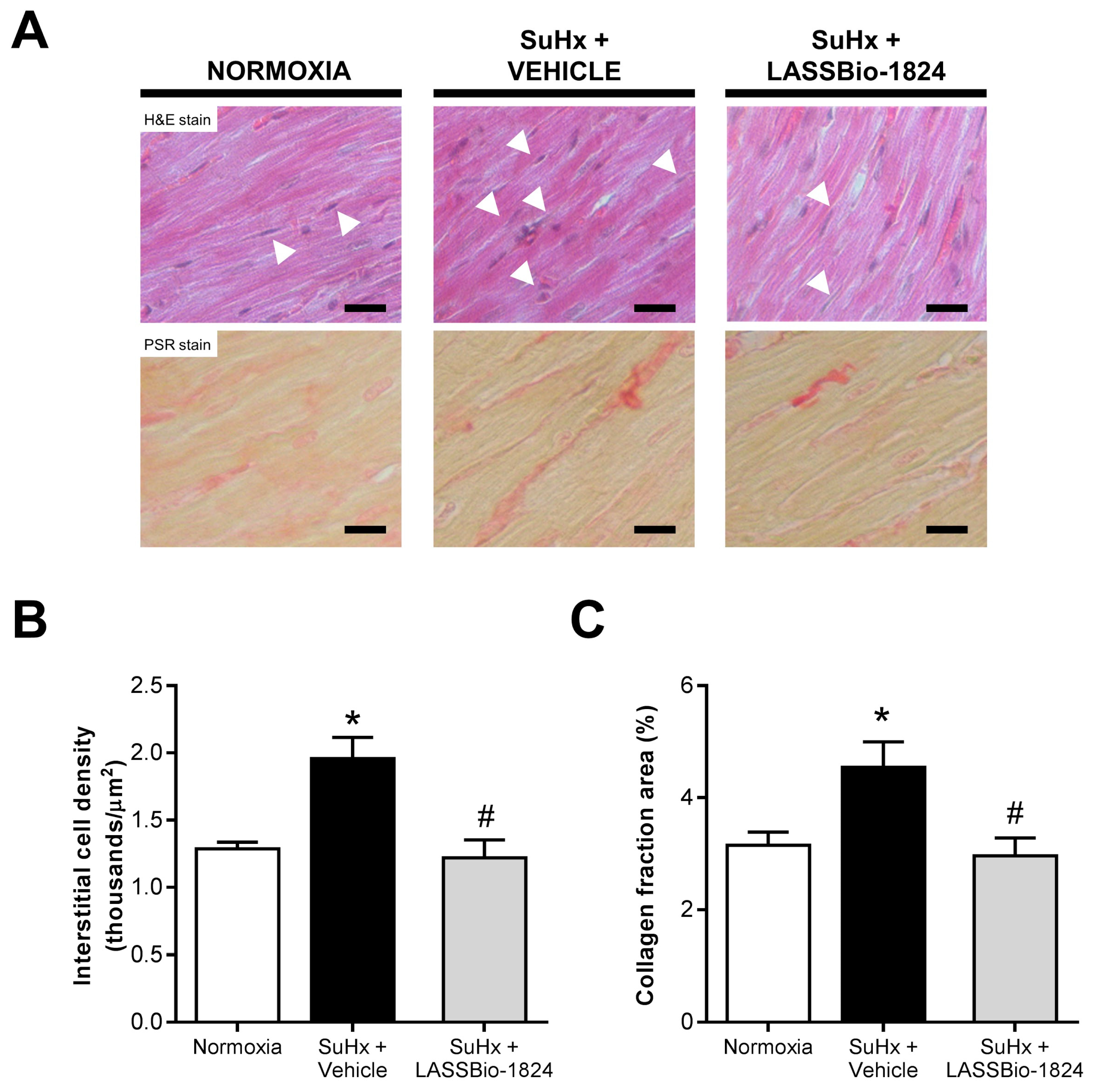
2.2. LASSBio-1824 Reduces Medial Hypertrophy and Endothelial Dysfunction in Pulmonary Arteries
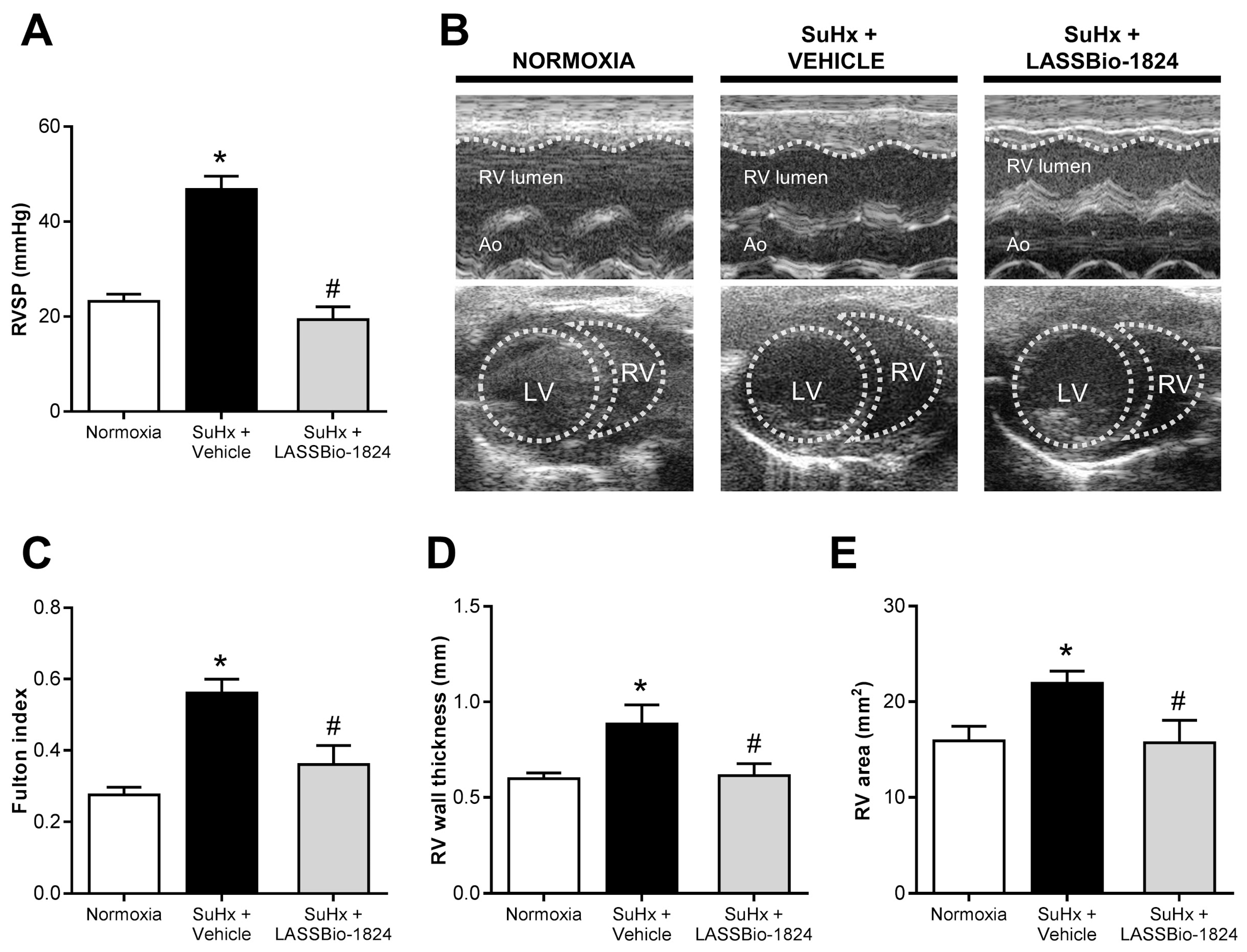
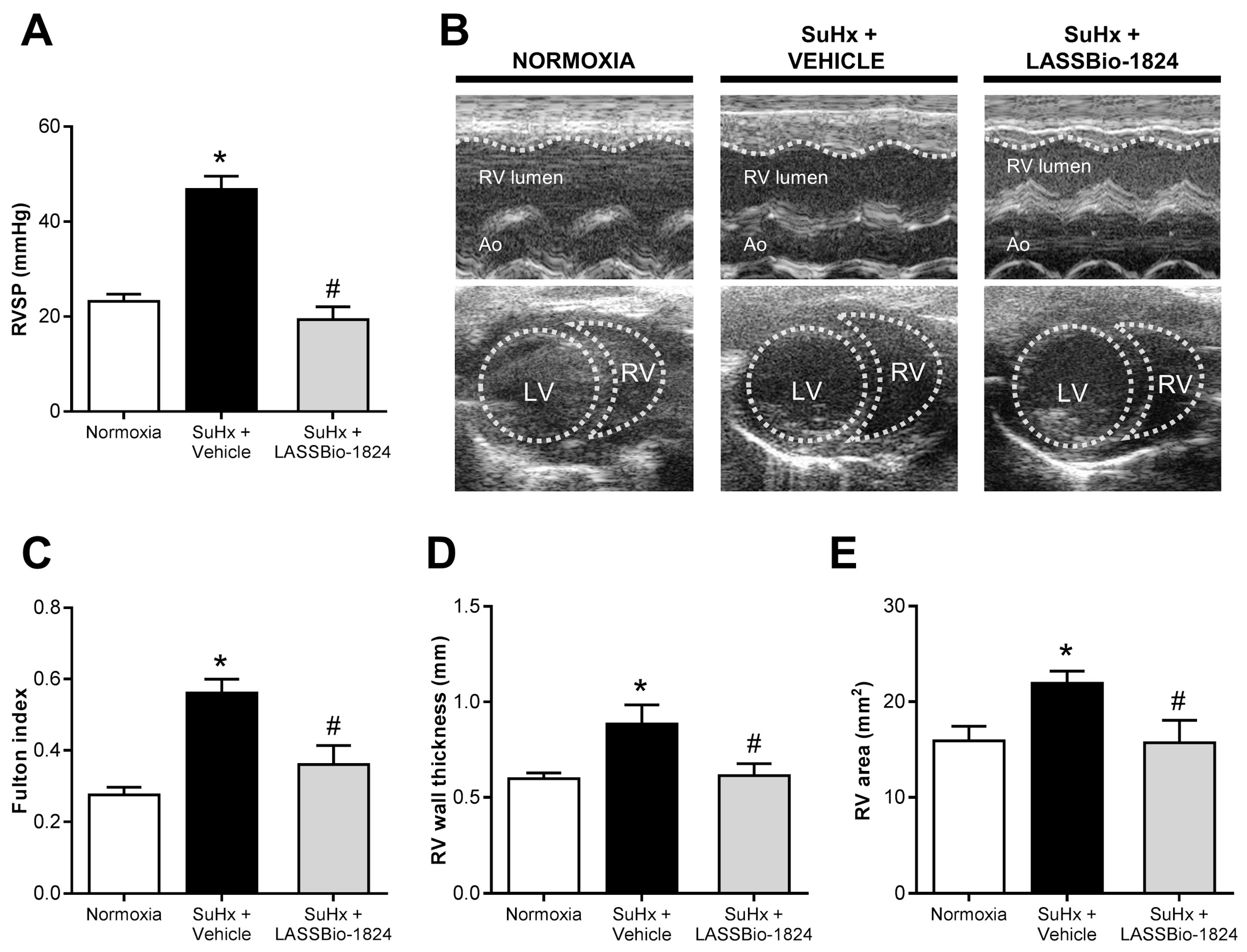
2.3. LASSBio-1824 Alleviates RV Overload and Remodeling and Preserves RV Function
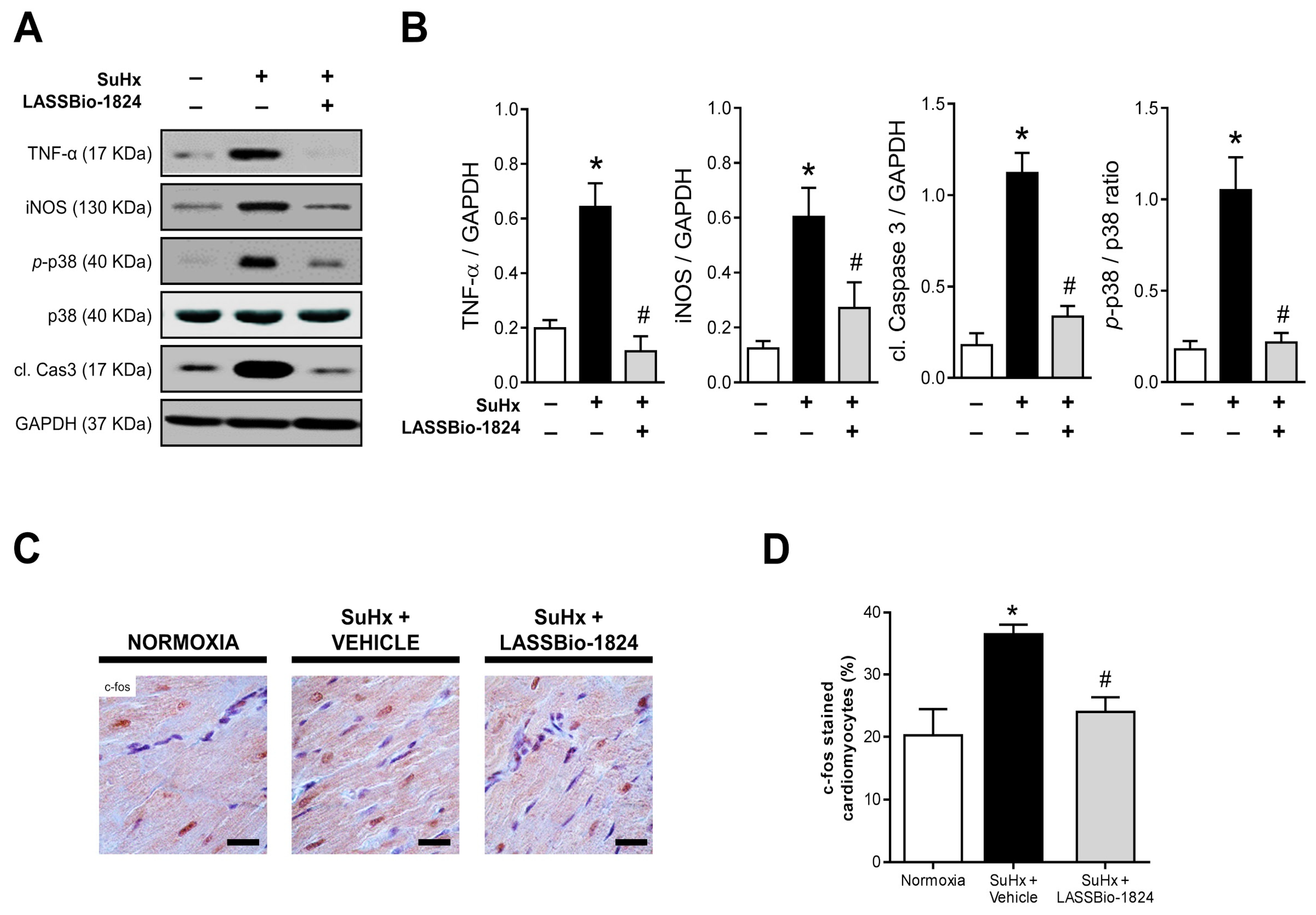
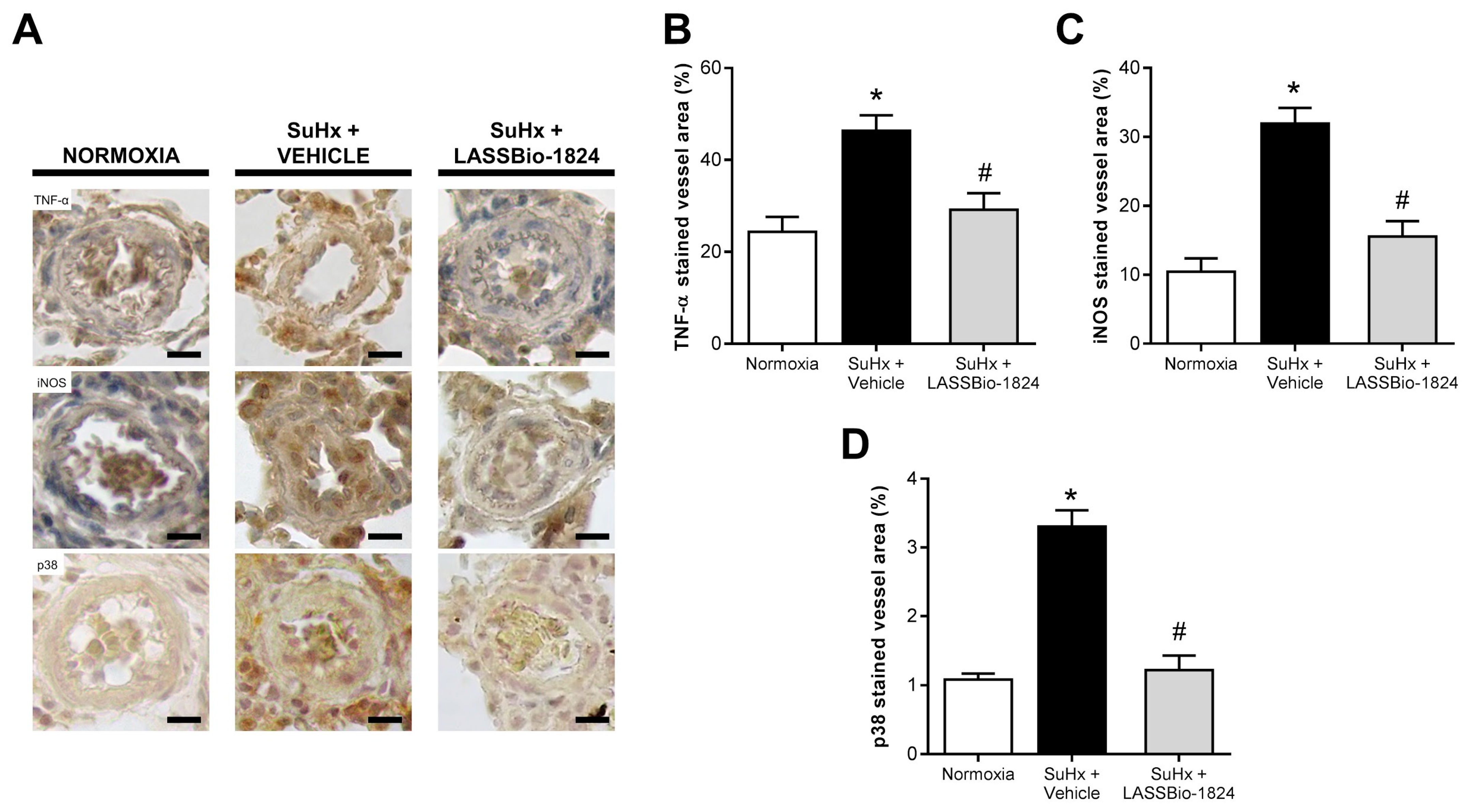
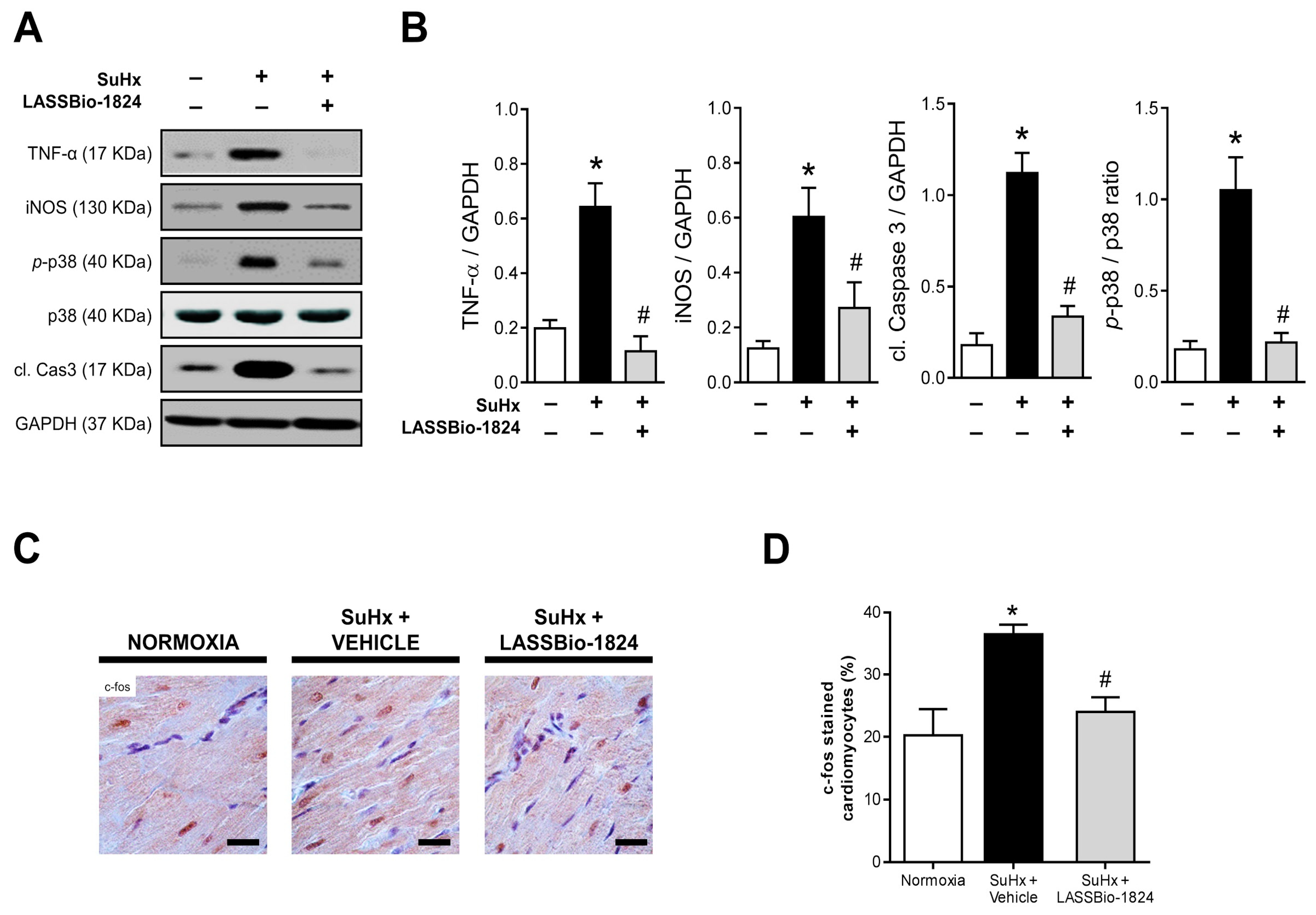
2.4. Administration of LASSBio-1824 Normalized Inflammation in Lung and RV
3. Discussion
4. Materials and Methods
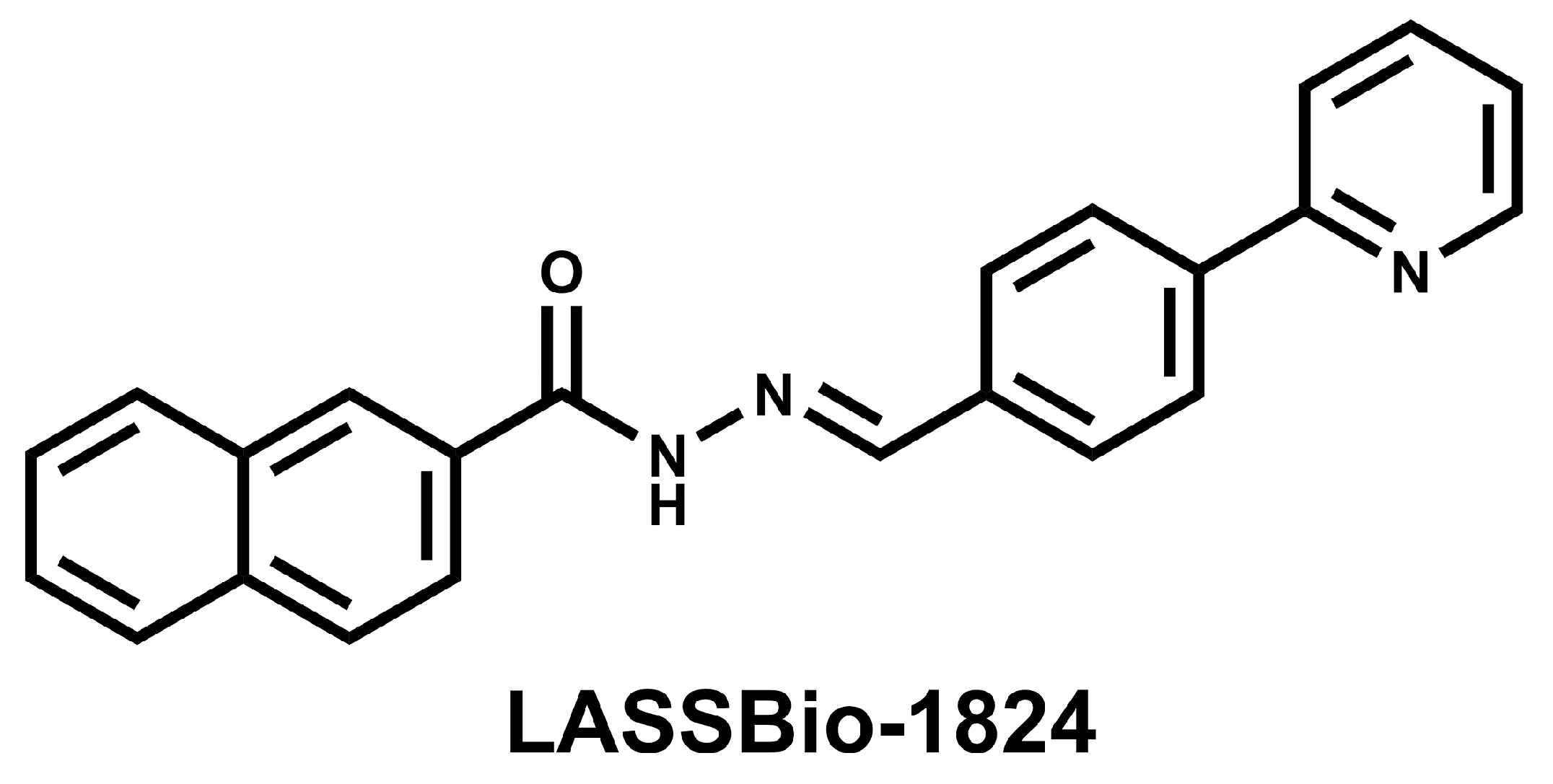
4.1. Drugs and Reagents
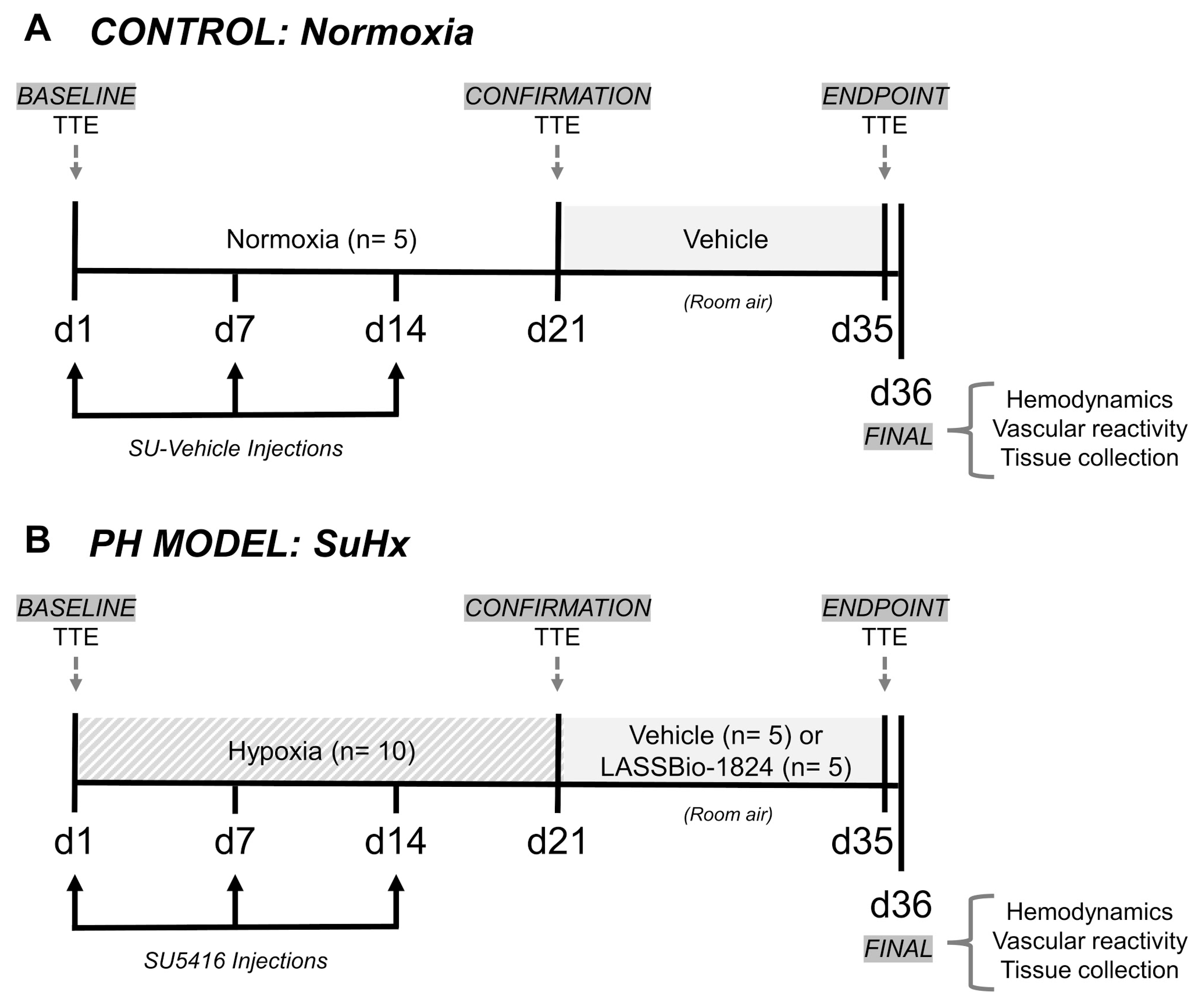
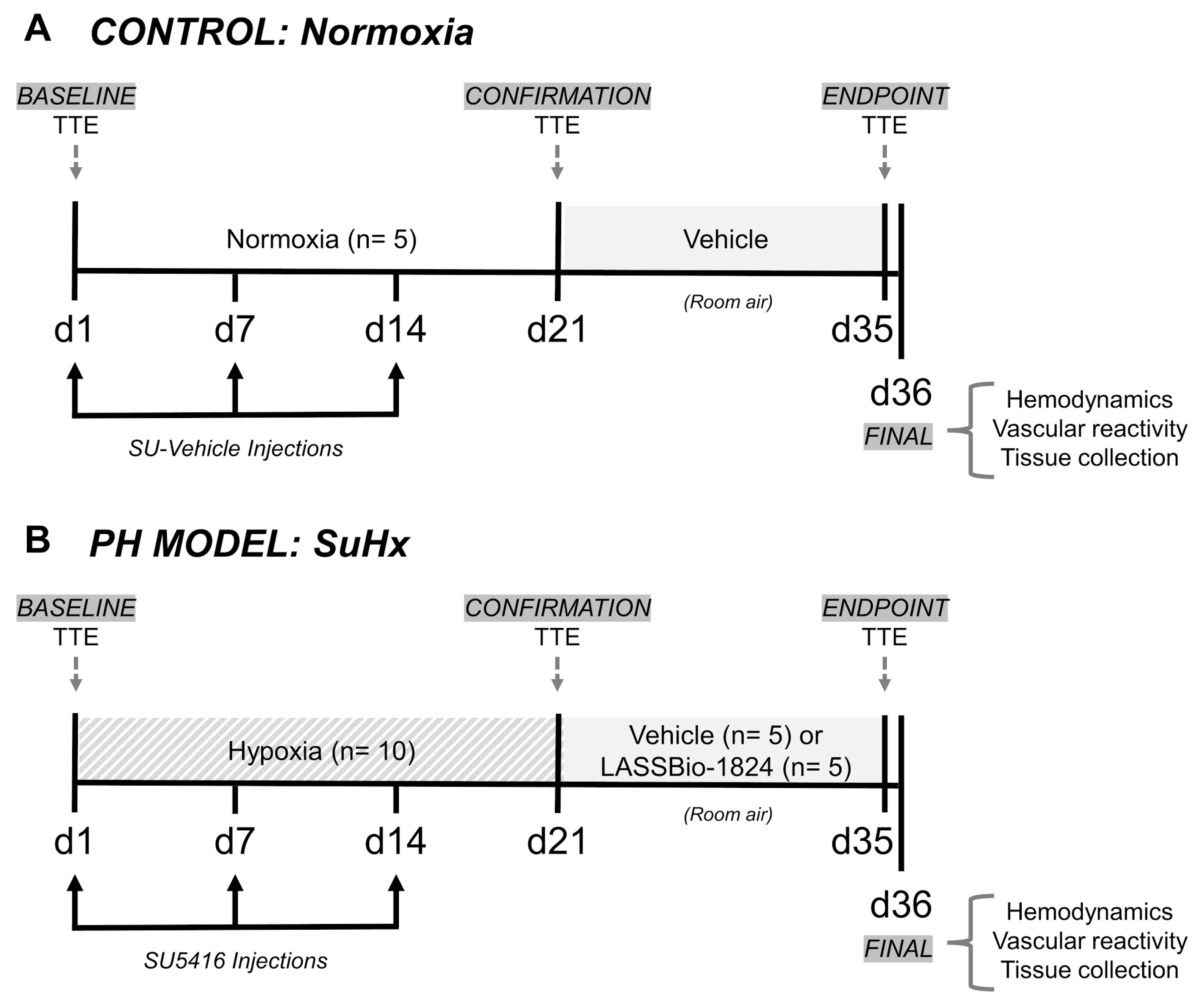
4.2. Animals and Experimental Design
4.3. Transthoracic Echocardiography (TTE)
4.4. Vascular and Intraventricular Hemodynamic Measurements
4.5. Vascular Reactivity of Pulmonary Artery Rings
4.6. Morphometric and Histochemical Analysis
4.7. Membrane Preparation and Western Blot
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmüller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019, 53, 1801887. [Google Scholar] [CrossRef] [Green Version]
- Hoeper, M.M.; Humbert, M.; Souza, R.; Idrees, M.; Kawut, S.M.; Sliwa-Hahnle, K.; Jing, Z.-C.; Gibbs, J.S.R. A global view of pulmonary hypertension. Lancet Respir. Med. 2016, 4, 306–322. [Google Scholar] [CrossRef]
- Hoeper, M.M.; Ghofrani, H.-A.; Grünig, E.; Klose, H.; Olschewski, H.; Rosenkranz, S. Pulmonary Hypertension. Dtsch. Arztebl. Int. 2017, 114, 73–84. [Google Scholar] [CrossRef]
- Wood, C.; Balciunas, M.; Lordan, J.; Mellor, A. Perioperative Management of Pulmonary Hypertension: A Review. J. Crit. Care Med. 2021, 7, 83–96. [Google Scholar] [CrossRef]
- Galiè, N.; McLaughlin, V.V.; Rubin, L.J.; Simonneau, G. An overview of the 6th World Symposium on Pulmonary Hypertension. Eur. Respir. J. 2019, 53, 1802148. [Google Scholar] [CrossRef] [Green Version]
- Rajagopal, S.; Yu, Y.-R.A. The Pathobiology of Pulmonary Arterial Hypertension. Cardiol. Clin. 2022, 40, 1–12. [Google Scholar] [CrossRef]
- Lechartier, B.; Berrebeh, N.; Huertas, A.; Humbert, M.; Guignabert, C.; Tu, L. Phenotypic Diversity of Vascular Smooth Muscle Cells in Pulmonary Arterial Hypertension. Chest 2022, 161, 219–231. [Google Scholar] [CrossRef]
- Wang, R.; Yuan, T.; Wang, J.; Chen, Y.; Zhao, J.; Li, M.; Fang, L.; Du, G. Immunity and inflammation in pulmonary arterial hypertension: From pathophysiology mechanisms to treatment perspective. Pharmacol. Res. 2022, 180, 106238. [Google Scholar] [CrossRef]
- Shafiq, M.; Lone, Z.R.; Bharati, P.; Singh, H.; Jagavelu, K.; Verma, N.K.; Ghosh, J.K.; Gaestel, M.; Hanif, K. Involvement of mitogen-activated protein kinase (MAPK)-activated protein kinase 2 (MK2) in endothelial dysfunction associated with pulmonary hypertension. Life Sci. 2021, 286, 120075. [Google Scholar] [CrossRef]
- Huertas, A.; Tu, L.; Humbert, M.; Guignabert, C. Chronic inflammation within the vascular wall in pulmonary arterial hypertension: More than a spectator. Cardiovasc. Res. 2020, 116, 885–893. [Google Scholar] [CrossRef]
- Huertas, A.; Guignabert, C.; Barberà, J.A.; Bärtsch, P.; Bhattacharya, J.; Bhattacharya, S.; Bonsignore, M.R.; Dewachter, L.; Dinh-Xuan, A.T.; Dorfmüller, P.; et al. Pulmonary vascular endothelium: The orchestra conductor in respiratory diseases. Eur. Respir. J. 2018, 51, 1700745. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.; Boehm, M.; Egemnazarov, B.; Grimminger, F.; Savai Pullamsetti, S.; Kwapiszewska, G.; Schermuly, R.T. Kinases as potential targets for treatment of pulmonary hypertension and right ventricular dysfunction. Br. J. Pharmacol. 2021, 178, 31–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humbert, M.; McLaughlin, V.; Gibbs, J.S.R.; Gomberg-Maitland, M.; Hoeper, M.M.; Preston, I.R.; Souza, R.; Waxman, A.; Escribano Subias, P.; Feldman, J.; et al. Sotatercept for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2021, 384, 1204–1215. [Google Scholar] [CrossRef] [PubMed]
- Burton, J.C.; Antoniades, W.; Okalova, J.; Roos, M.M.; Grimsey, N.J. Atypical p38 Signaling, Activation, and Implications for Disease. Int. J. Mol. Sci. 2021, 22, 4183. [Google Scholar] [CrossRef]
- Canovas, B.; Nebreda, A.R. Diversity and versatility of p38 kinase signalling in health and disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 346–366. [Google Scholar] [CrossRef]
- Whitaker, R.H.; Cook, J.G. Stress Relief Techniques: p38 MAPK Determines the Balance of Cell Cycle and Apoptosis Pathways. Biomolecules 2021, 11, 1444. [Google Scholar] [CrossRef]
- Kim, E.K.; Choi, E.-J. Compromised MAPK signaling in human diseases: An update. Arch. Toxicol. 2015, 89, 867–882. [Google Scholar] [CrossRef]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [Green Version]
- Kojonazarov, B.; Novoyatleva, T.; Boehm, M.; Happe, C.; Sibinska, Z.; Tian, X.; Sajjad, A.; Luitel, H.; Kriechling, P.; Posern, G.; et al. p38 MAPK Inhibition Improves Heart Function in Pressure-Loaded Right Ventricular Hypertrophy. Am. J. Respir. Cell Mol. Biol. 2017, 57, 603–614. [Google Scholar] [CrossRef]
- Church, A.C.; Martin, D.H.; Wadsworth, R.; Bryson, G.; Fisher, A.J.; Welsh, D.J.; Peacock, A.J. The reversal of pulmonary vascular remodeling through inhibition of p38 MAPK-alpha: A potential novel anti-inflammatory strategy in pulmonary hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2015, 309, L333–L347. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Shimpo, H.; Shimamoto, A.; Chong, A.J.; Hampton, C.R.; Spring, D.J.; Yada, M.; Takao, M.; Onoda, K.; Yada, I.; et al. Specific inhibition of p38 mitogen-activated protein kinase with FR167653 attenuates vascular proliferation in monocrotaline-induced pulmonary hypertension in rats. J. Thorac. Cardiovasc. Surg. 2004, 128, 850–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, C.; Guo, J.; Qiu, X.; Ma, K.; Xiang, M.; Zhu, X.; Guo, J. IGF-1 induces iNOS expression via the p38 MAPK signal pathway in the anti-apoptotic process in pulmonary artery smooth muscle cells during PAH. J. Recept. Signal Transduct. 2014, 34, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Predescu, D.; Bardita, C.; Chen, J.; Jeganathan, N.; Pritchard, M.; DiBartolo, S.; Machado, R.; Predescu, S. Modulation of Intersectin-1s Lung Expression Induces Obliterative Remodeling and Severe Plexiform Arteriopathy in the Murine Pulmonary Vascular Bed. Am. J. Pathol. 2017, 187, 528–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Y.; Yang, Y.; Wu, Q.; Gao, G.; Liu, Y.; Xiong, Y.; Huang, C.; Wu, S. Activation of LXRα improves cardiac remodeling induced by pulmonary artery hypertension in rats. Sci. Rep. 2017, 7, 6169. [Google Scholar] [CrossRef] [Green Version]
- Freitas, R.H.C.N.; Cordeiro, N.M.; Carvalho, P.R.; Alves, M.A.; Guedes, I.A.; Valerio, T.S.; Dardenne, L.E.; Lima, L.M.; Barreiro, E.J.; Fernandes, P.D.; et al. Discovery of naphthyl- N -acylhydrazone p38α MAPK inhibitors with in vivo anti-inflammatory and anti-TNF-α activity. Chem. Biol. Drug Des. 2018, 91, 391–397. [Google Scholar] [CrossRef]
- Mortimer, H.J.; Peacock, A.J.; Kirk, A.; Welsh, D.J. p38 MAP kinase: Essential role in hypoxia-mediated human pulmonary artery fibroblast proliferation. Pulm. Pharmacol. Ther. 2007, 20, 718–725. [Google Scholar] [CrossRef]
- Costa, G.C.; Montagnoli, T.L.; Da Silva, J.S.; de Alencar, A.K.N.; Reina Gamba, L.E.; Alves, B.E.O.; da Silva, M.M.C.; Trachez, M.M.; do Nascimento, J.H.M.; Pimentel-Coelho, P.M.; et al. New Benzofuran N-Acylhydrazone Reduces Cardiovascular Dysfunction in Obese Rats by Blocking TNF-Alpha Synthesis. Drug Des. Devel. Ther. 2020, 14, 3337–3350. [Google Scholar] [CrossRef]
- Alencar, A.K.N.; Pereira, S.L.; Montagnoli, T.L.; Maia, R.C.; Kümmerle, A.E.; Landgraf, S.S.; Caruso-Neves, C.; Ferraz, E.B.; Tesch, R.; Nascimento, J.H.M.; et al. Beneficial effects of a novel agonist of the adenosine A 2A receptor on monocrotaline-induced pulmonary hypertension in rats. Br. J. Pharmacol. 2013, 169, 953–962. [Google Scholar] [CrossRef] [Green Version]
- Toba, M.; Alzoubi, A.; O’Neill, K.D.; Gairhe, S.; Matsumoto, Y.; Oshima, K.; Abe, K.; Oka, M.; McMurtry, I.F. Temporal hemodynamic and histological progression in Sugen5416/hypoxia/normoxia-exposed pulmonary arterial hypertensive rats. Am. J. Physiol. Circ. Physiol. 2014, 306, H243–H250. [Google Scholar] [CrossRef] [Green Version]
- Abe, K.; Toba, M.; Alzoubi, A.; Ito, M.; Fagan, K.A.; Cool, C.D.; Voelkel, N.F.; McMurtry, I.F.; Oka, M. Formation of Plexiform Lesions in Experimental Severe Pulmonary Arterial Hypertension. Circulation 2010, 121, 2747–2754. [Google Scholar] [CrossRef] [Green Version]
- Silva, M.D.M.C.D.; Alencar, A.K.N.D.; Silva, J.S.D.; Montagnoli, T.L.; Silva, G.F.D.; Rocha, B.D.S.; Montes, G.C.; Mendez-Otero, R.; Pimentel-Coelho, P.M.; Vasques, J.F.; et al. Therapeutic Benefit of the Association of Lodenafil with Mesenchymal Stem Cells on Hypoxia-induced Pulmonary Hypertension in Rats. Cells 2020, 9, 2120. [Google Scholar] [CrossRef] [PubMed]
- Alencar, A.K.; Montes, G.C.; Montagnoli, T.; Silva, A.M.; Martinez, S.T.; Fraga, A.G.; Wang, H.; Groban, L.; Sudo, R.T.; Zapata-Sudo, G. Activation of GPER ameliorates experimental pulmonary hypertension in male rats. Eur. J. Pharm. Sci. 2017, 97, 208–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, J.E.; Mendes, L.; Rudd, M.A.; Russo, G.; Loscalzo, J.; Zhang, Y.-Y. Serial noninvasive assessment of progressive pulmonary hypertension in a rat model. Am. J. Physiol. Circ. Physiol. 2002, 283, H364–H371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augustine, D.X.; Coates-Bradshaw, L.D.; Willis, J.; Harkness, A.; Ring, L.; Grapsa, J.; Coghlan, G.; Kaye, N.; Oxborough, D.; Robinson, S.; et al. Echocardiographic assessment of pulmonary hypertension: A guideline protocol from the British Society of Echocardiography. Echo Res. Pract. 2018, 5, G11–G24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goncharov, D.A.; Kudryashova, T.V.; Ziai, H.; Ihida-Stansbury, K.; DeLisser, H.; Krymskaya, V.P.; Tuder, R.M.; Kawut, S.M.; Goncharova, E.A. Mammalian Target of Rapamycin Complex 2 (mTORC2) Coordinates Pulmonary Artery Smooth Muscle Cell Metabolism, Proliferation, and Survival in Pulmonary Arterial Hypertension. Circulation 2014, 129, 864–874. [Google Scholar] [CrossRef] [Green Version]
- Rajkumar, R.; Konishi, K.; Richards, T.J.; Ishizawar, D.C.; Wiechert, A.C.; Kaminski, N.; Ahmad, F. Genomewide RNA expression profiling in lung identifies distinct signatures in idiopathic pulmonary arterial hypertension and secondary pulmonary hypertension. Am. J. Physiol. Circ. Physiol. 2010, 298, H1235–H1248. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Vinasco, L.; Gomberg-Maitland, M.; Maitland, M.L.; Desai, A.A.; Singleton, P.A.; Sammani, S.; Sam, L.; Liu, Y.; Husain, A.N.; Lang, R.M.; et al. Genomic assessment of a multikinase inhibitor, sorafenib, in a rodent model of pulmonary hypertension. Physiol. Genomics 2008, 33, 278–291. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Fantozzi, I.; Tigno, D.D.; Yi, E.S.; Platoshyn, O.; Thistlethwaite, P.A.; Kriett, J.M.; Yung, G.; Rubin, L.J.; Yuan, J.X.-J. Bone morphogenetic proteins induce apoptosis in human pulmonary vascular smooth muscle cells. Am. J. Physiol. Cell. Mol. Physiol. 2003, 285, L740–L754. [Google Scholar] [CrossRef] [Green Version]
- Bull, T.M.; Coldren, C.D.; Moore, M.; Sotto-Santiago, S.M.; Pham, D.V.; Nana-Sinkam, S.P.; Voelkel, N.F.; Geraci, M.W. Gene Microarray Analysis of Peripheral Blood Cells in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2004, 170, 911–919. [Google Scholar] [CrossRef] [Green Version]
- Hurst, L.A.; Dunmore, B.J.; Long, L.; Crosby, A.; Al-Lamki, R.; Deighton, J.; Southwood, M.; Yang, X.; Nikolic, M.Z.; Herrera, B.; et al. TNFα drives pulmonary arterial hypertension by suppressing the BMP type-II receptor and altering NOTCH signalling. Nat. Commun. 2017, 8, 14079. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Lee, P.J.; Long, L.; Trembath, R.C.; Morrell, N.W. BMP4 Induces HO-1 via a Smad-Independent, p38 MAPK-Dependent Pathway in Pulmonary Artery Myocytes. Am. J. Respir. Cell Mol. Biol. 2007, 37, 598–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.-W.; Yang, L.-H.; Ren, Z.-C.; Mu, D.-G.; Li, Y.-Q.; Yan, J.-P.; Wang, L.-X.; Chen, C. Resveratrol downregulates TNF-α-induced monocyte chemoattractant protein-1 in primary rat pulmonary artery endothelial cells by P38 mitogen-activated protein kinase signaling. Drug Des. Devel. Ther. 2019, 13, 1843–1853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, T.; Nagaya, N.; Ishibashi-Ueda, H.; Kyotani, S.; Oya, H.; Sakamaki, F.; Kimura, H.; Nakanishi, N. Increased plasma monocyte chemoattractant protein-1 level in idiopathic pulmonary arterial hypertension. Respirology 2006, 11, 158–163. [Google Scholar] [CrossRef]
- Ferrero, E.; Zocchi, M.R.; Magni, E.; Panzeri, M.C.; Curnis, F.; Rugarli, C.; Ferrero, M.E.; Corti, A. Roles of tumor necrosis factor p55 and p75 receptors in TNF-α-induced vascular permeability. Am. J. Physiol. Physiol. 2001, 281, C1173–C1179. [Google Scholar] [CrossRef]
- Yan, S.; Wang, Y.; Liu, P.; Chen, A.; Chen, M.; Yao, D.; Xu, X.; Wang, L.; Huang, X. Baicalin Attenuates Hypoxia-Induced Pulmonary Arterial Hypertension to Improve Hypoxic Cor Pulmonale by Reducing the Activity of the p38 MAPK Signaling Pathway and MMP-9. Evid. Based Complement. Altern. Med. 2016, 2016, 2546402-9. [Google Scholar] [CrossRef] [Green Version]
- See, F.; Thomas, W.; Way, K.; Tzanidis, A.; Kompa, A.; Lewis, D.; Itescu, S.; Krum, H. p38 mitogen-activated protein kinase inhibition improves cardiac function and attenuates left ventricular remodeling following myocardial infarction in the rat. J. Am. Coll. Cardiol. 2004, 44, 1679–1689. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Kuri, M.; Meka, S.H.; Salamanca-Buentello, F.; Hajjar, R.J.; Lipskaia, L.; Chemaly, E.R. Molecules linked to Ras signaling as therapeutic targets in cardiac pathologies. Biol. Res. 2021, 54, 23. [Google Scholar] [CrossRef]
- Mosele, F.; Tavares, A.M.V.; Colombo, R.; Caron-Lienert, R.; Araujo, A.S.R.; Ribeiro, M.F.; Belló-Klein, A. Effects of Purple Grape Juice in the Redox-sensitive Modulation of Right Ventricular Remodeling in a Pulmonary Arterial Hypertension Model. J. Cardiovasc. Pharmacol. 2012, 60, 15–22. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, M.; Goldstein, S.; Li, Y.; Ge, J.; He, B.; Ruiz, G. The Effect of c-fos on Acute Myocardial Infarction and the Significance of Metoprolol Intervention in a Rat Model. Cell Biochem. Biophys. 2013, 65, 249–255. [Google Scholar] [CrossRef]
- Min, W.; Bin, Z.W.; Quan, Z.B.; Hui, Z.J.; Sheng, F.G. The signal transduction pathway of PKC/NF-κB/c-fos may be involved in the influence of high glucose on the cardiomyocytes of neonatal rats. Cardiovasc. Diabetol. 2009, 8, 8. [Google Scholar] [CrossRef] [Green Version]
- Imoto, K.; Okada, M.; Yamawaki, H. Periostin Mediates Right Ventricular Failure through Induction of Inducible Nitric Oxide Synthase Expression in Right Ventricular Fibroblasts from Monocrotaline-Induced Pulmonary Arterial Hypertensive Rats. Int. J. Mol. Sci. 2018, 20, 62. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Fernandez, R.A.; Yuan, J.X.-J. miRNA208/Mef2 and TNF-α in Right Ventricular Dysfunction. Circ. Res. 2015, 116, 6–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jobe, L.J.; Meléndez, G.C.; Levick, S.P.; Du, Y.; Brower, G.L.; Janicki, J.S. TNF-α inhibition attenuates adverse myocardial remodeling in a rat model of volume overload. Am. J. Physiol. Circ. Physiol. 2009, 297, H1462–H1468. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.M.; McCarthy, J.; Sack, M.N. TNF alpha is required for hypoxia-mediated right ventricular hypertrophy. Mol. Cell. Biochem. 2001, 219, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; McTiernan, C.F.; Frye, C.S.; Slawson, S.E.; Lemster, B.H.; Koretsky, A.P.; Demetris, A.J.; Feldman, A.M. Dilated Cardiomyopathy in Transgenic Mice With Cardiac-Specific Overexpression of Tumor Necrosis Factor-α. Circ. Res. 1997, 81, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Westermann, D.; Linthout, S.; Dhayat, S.; Dhayat, N.; Schmidt, A.; Noutsias, M.; Song, X.-Y.; Spillmann, F.; Riad, A.; Schultheiss, H.-P.; et al. Tumor necrosis factor-alpha antagonism protects from myocardial inflammation and fibrosis in experimental diabetic cardiomyopathy. Basic Res. Cardiol. 2007, 102, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.L. Stress-Activated Cytokines and The Heart: From Adaptation to Maladaptation. Annu. Rev. Physiol. 2003, 65, 81–101. [Google Scholar] [CrossRef]
- Stenmark, K.R.; Meyrick, B.; Galie, N.; Mooi, W.J.; McMurtry, I.F. Animal models of pulmonary arterial hypertension: The hope for etiological discovery and pharmacological cure. Am. J. Physiol. Cell. Mol. Physiol. 2009, 297, L1013–L1032. [Google Scholar] [CrossRef]
- Bordenave, J.; Thuillet, R.; Tu, L.; Phan, C.; Cumont, A.; Marsol, C.; Huertas, A.; Savale, L.; Hibert, M.; Galzi, J.-L.; et al. Neutralization of CXCL12 attenuates established pulmonary hypertension in rats. Cardiovasc. Res. 2020, 116, 686–697. [Google Scholar] [CrossRef]
- Alencar, A.K.N.; Pereira, S.L.; da Silva, F.E.; Mendes, L.V.P.; Cunha, V.D.M.N.; Lima, L.M.; Montagnoli, T.L.; Caruso-Neves, C.; Ferraz, E.B.; Tesch, R.; et al. N-acylhydrazone derivative ameliorates monocrotaline-induced pulmonary hypertension through the modulation of adenosine AA2R activity. Int. J. Cardiol. 2014, 173, 154–162. [Google Scholar] [CrossRef]
- Urboniene, D.; Haber, I.; Fang, Y.-H.; Thenappan, T.; Archer, S.L. Validation of high-resolution echocardiography and magnetic resonance imaging vs. high-fidelity catheterization in experimental pulmonary hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2010, 299, L401–L412. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Normoxia | SuHx + Vehicle | SuHx + LASSBio-1824 |
|---|---|---|---|
| Body weight (g) | 236.4 ± 5.5 | 220.8 ± 9.1 | 206.5 ± 16.5 |
| Systolic BP (mmHg) | 106.9 ± 6.8 | 104.7 ± 3.8 | 97.2 ± 8.6 |
| Diastolic BP (mmHg) | 79.5 ± 7.1 | 81.6 ± 4.5 | 71.3 ± 5.4 |
| Mean BP (mmHg) | 93.3 ± 6.9 | 93.4 ± 4.1 | 84.2 ± 2.8 |
| Parameter | Normoxia | SuHx + Vehicle | SuHx + LASSBio-1824 |
|---|---|---|---|
| Phenylephrine EC50 (nM) | 62.6 ± 5.9 | 82.7 ± 22.8 | 91.1 ± 2.6 |
| Phenylephrine Emax (%) | 100.0 | 100.0 | 100.0 |
| Acetylcholine EC50 (nM) | 66.1 ± 18.6 | 72.2 ± 26.2 | 136.0 ± 49.2 |
| Acetylcholine Emax (%) | 67.5 ± 2.1 | 51.9 ± 5.0 * | 74.1 ± 3.0 # |
| Parameter | Normoxia | SuHx + Vehicle | SuHx + LASSBio-1824 |
|---|---|---|---|
| Heart rate (bpm) | 283.0 ± 110.2 | 302.5 ± 25.56 | 285.1 ± 13.67 |
| LV cardiac output (mL·min−1) | 86.18 ± 7.55 | 73.23 ± 3.91 | 81.36 ± 4.35 |
| LV systolic pressure (mmHg) | 98.1 ± 4.1 | 83.3 ± 5.2 | 89.6 ± 2.4 |
| LV area (mm2) | 35.3 ± 3.0 | 34.2 ± 3.6 | 32.4 ± 4.3 |
| RV cardiac output (mL·min−1) | 90.25 ± 3.9 | 67.45 ± 5.4 * | 91.21 ± 6.15 # |
| RV end diastolic pressure (mmHg) | 6.77 ± 1.23 | 7.01 ± 3.62 | 4.81 ± 1.66 |
| RV + dP/dt (mmHg·s−1) | 1162 ± 193 | 1608 ± 317 | 1114 ± 209 |
| RV − dP/dt (mmHg·s−1) | −1117 ± 171 | −1753 ± 426 | −1074 ± 165 |
| Target | Host | Clonality | Conjugate | Cat. No. | Vendor |
|---|---|---|---|---|---|
| p38 | Mouse | Monoclonal | – | ab1793 | AbCam (Cambridge, MA, USA) |
| phosphorylated-p38 | Rabbit | Polyclonal | – | ab13847 | AbCam (Cambridge, MA, USA) |
| TNF-α | Rabbit | Polyclonal | – | ab7952 | AbCam (Cambridge, MA, USA) |
| iNOS | Rabbit | Polyclonal | – | 4511 | Cell Signaling (Danvers, MA, USA) |
| cleaved caspase 3 | Rabbit | Polyclonal | – | 2982 | Cell Signaling (Danvers, MA, USA) |
| α-SMA | Mouse | Monoclonal | – | 5174 | Cell Signaling (Danvers, MA, USA) |
| GAPDH | Mouse | Monoclonal | – | A2547 | Sigma-Aldrich (St. Louis, MO, USA) |
| Rabbit IgG | Goat | Polyclonal | HRP | 1706515 | Bio-Rad (Hercules, CA, USA) |
| Mouse IgG | Goat | Polyclonal | HRP | 1706516 | Bio-Rad (Hercules, CA, USA) |
| Mouse/Rabbit IgG | Goat | Polyclonal | HRP | 414191F | Nichirei Biosciences (Tokyo, Japan) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, G.F.; da Silva, J.S.; de Alencar, A.K.N.; de Moraes Carvalho da Silva, M.; Montagnoli, T.L.; de Souza Rocha, B.; de Freitas, R.H.C.N.; Sudo, R.T.; Fraga, C.A.M.; Zapata-Sudo, G. Novel p38 Mitogen-Activated Protein Kinase Inhibitor Reverses Hypoxia-Induced Pulmonary Arterial Hypertension in Rats. Pharmaceuticals 2022, 15, 900. https://doi.org/10.3390/ph15070900
Silva GF, da Silva JS, de Alencar AKN, de Moraes Carvalho da Silva M, Montagnoli TL, de Souza Rocha B, de Freitas RHCN, Sudo RT, Fraga CAM, Zapata-Sudo G. Novel p38 Mitogen-Activated Protein Kinase Inhibitor Reverses Hypoxia-Induced Pulmonary Arterial Hypertension in Rats. Pharmaceuticals. 2022; 15(7):900. https://doi.org/10.3390/ph15070900
Chicago/Turabian StyleSilva, Grazielle Fernandes, Jaqueline Soares da Silva, Allan Kardec Nogueira de Alencar, Marina de Moraes Carvalho da Silva, Tadeu Lima Montagnoli, Bruna de Souza Rocha, Rosana Helena Coimbra Nogueira de Freitas, Roberto Takashi Sudo, Carlos Alberto Manssour Fraga, and Gisele Zapata-Sudo. 2022. "Novel p38 Mitogen-Activated Protein Kinase Inhibitor Reverses Hypoxia-Induced Pulmonary Arterial Hypertension in Rats" Pharmaceuticals 15, no. 7: 900. https://doi.org/10.3390/ph15070900
APA StyleSilva, G. F., da Silva, J. S., de Alencar, A. K. N., de Moraes Carvalho da Silva, M., Montagnoli, T. L., de Souza Rocha, B., de Freitas, R. H. C. N., Sudo, R. T., Fraga, C. A. M., & Zapata-Sudo, G. (2022). Novel p38 Mitogen-Activated Protein Kinase Inhibitor Reverses Hypoxia-Induced Pulmonary Arterial Hypertension in Rats. Pharmaceuticals, 15(7), 900. https://doi.org/10.3390/ph15070900