
Sensitive Detection of Pharmaceutical Drugs and Metabolites in Serum Using Data-Independent Acquisition Mass Spectrometry and Open-Access Data Acquisition Tools †

 , , and
, , and
Abstract
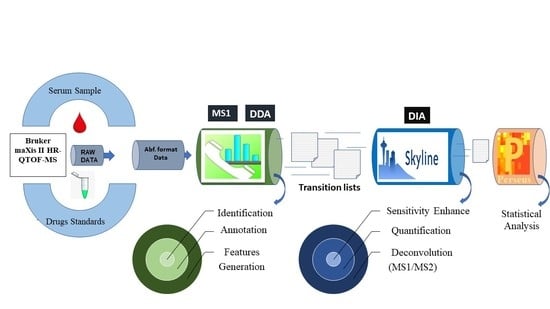
:
1. Introduction
2. Results
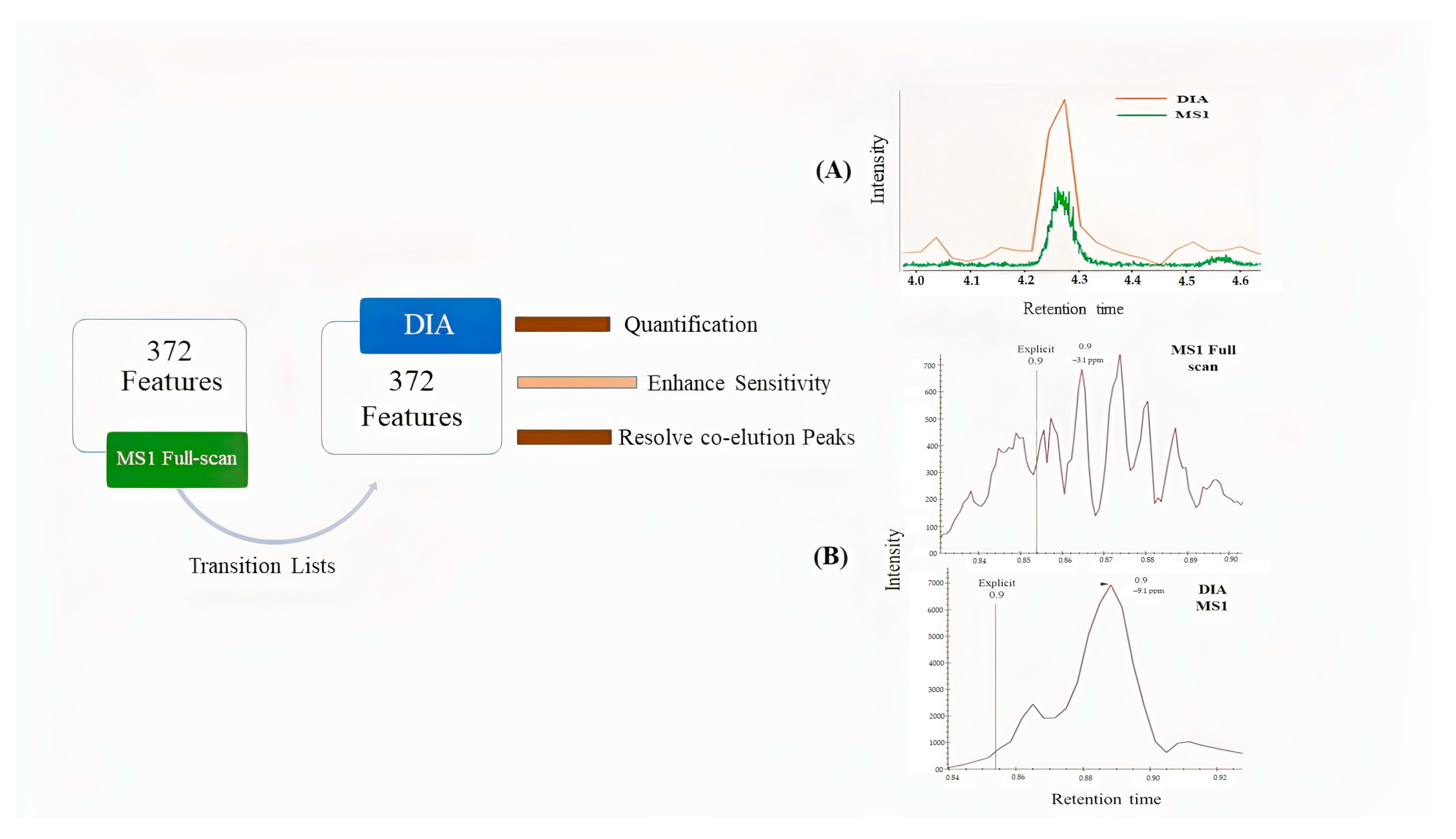
2.1. Identification by Using MS-DIAL
2.2. Deconvolution of Peak Areas by Using Skyline
2.3. Comparison of Quantitative Results
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Sample Preparation
4.3. LC-MS Analysis
4.4. Software and Data Processing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Klont, F.; Jahn, S.; Grivet, C.; König, S.; Bonner, R.; Hopfgartner, G. SWATH data independent acquisition mass spectrometry for screening of xenobiotics in biological fluids: Opportunities and challenges for data processing. Talanta 2020, 211, 120747. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Wang, R.; Cai, Y.; Wang, Z.; Zhu, Z.-J. DecoMetDIA: Deconvolution of multiplexed MS/MS spectra for metabolite identification in SWATH-MS-based untargeted metabolomics. Anal. Chem. 2019, 91, 11897–11904. [Google Scholar] [CrossRef]
- Kenar, E.; Franken, H.; Forcisi, S.; Wörmann, K.; Häring, H.-U.; Lehmann, R.; Schmitt-Kopplin, P.; Zell, A.; Kohlbacher, O. Automated label-free quantification of metabolites from liquid chromatography–mass spectrometry data. Mol. Cell. Proteom. 2014, 13, 348–359. [Google Scholar] [CrossRef] [PubMed]
- Zha, H.; Cai, Y.; Yin, Y.; Wang, Z.; Li, K.; Zhu, Z.-J. SWATHtoMRM: Development of high-coverage targeted metabolomics method using SWATH technology for biomarker discovery. Anal. Chem. 2018, 90, 4062–4070. [Google Scholar] [CrossRef]
- Li, H.; Cai, Y.; Guo, Y.; Chen, F.; Zhu, Z.-J. MetDIA: Targeted metabolite extraction of multiplexed MS/MS spectra generated by data-independent acquisition. Anal. Chem. 2016, 88, 8757–8764. [Google Scholar] [CrossRef]
- Pappireddi, N.; Martin, L.; Wühr, M. A review on quantitative multiplexed proteomics. ChemBioChem 2019, 20, 1210–1224. [Google Scholar] [CrossRef] [PubMed]
- Bilbao, A.; Varesio, E.; Luban, J.; Strambio-De-Castillia, C.; Hopfgartner, G.; Müller, M.; Lisacek, F. Processing strategies and software solutions for data-independent acquisition in mass spectrometry. Proteomics 2015, 15, 964–980. [Google Scholar] [CrossRef]
- Malipatil, N.; Fachim, H.A.; Siddals, K.; Geary, B.; Wark, G.; Porter, N.; Anderson, S.; Donn, R.; Harvie, M.; Whetton, A.D. Data independent acquisition mass spectrometry can identify circulating proteins that predict future weight loss with a diet and exercise programme. J. Clin. Med. 2019, 8, 141. [Google Scholar] [CrossRef]
- Zhou, J.; Li, Y.; Chen, X.; Zhong, L.; Yin, Y. Development of data-independent acquisition workflows for metabolomic analysis on a quadrupole-orbitrap platform. Talanta 2017, 164, 128–136. [Google Scholar] [CrossRef]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef]
- Röst, H.L.; Rosenberger, G.; Navarro, P.; Gillet, L.; Miladinović, S.M.; Schubert, O.T.; Wolski, W.; Collins, B.C.; Malmström, J.; Malmström, L. OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat. Biotech. 2014, 32, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, Y.; Chen, W.; Cao, Q.; Guo, Y.; Wan, N.; Jiang, X.; Tang, Y.J.; Wang, Q.; Shui, W. Integrating MS1 and MS2 scans in high-resolution parallel reaction monitoring assays for targeted metabolite quantification and dynamic 13C-labeling metabolism analysis. Anal. Chem. 2017, 89, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Barbier Saint Hilaire, P.; Rousseau, K.; Seyer, A.; Dechaumet, S.; Damont, A.; Junot, C.; Fenaille, F. Comparative evaluation of data dependent and data independent acquisition workflows implemented on an orbitrap fusion for untargeted metabolomics. Metabolites 2020, 10, 158. [Google Scholar] [CrossRef]
- Guo, J.; Huan, T. Comparison of Full-Scan, Data-Dependent, and Data-Independent Acquisition Modes in Liquid Chromatography–Mass Spectrometry Based Untargeted Metabolomics. Anal. Chem. 2020, 92, 8072–8080. [Google Scholar] [CrossRef]
- Van der Laan, T.; Boom, I.; Maliepaard, J.; Dubbelman, A.C.; Harms, A.C.; Hankemeier, T. Data-independent acquisition for the quantification and identification of metabolites in plasma. Metabolites 2020, 10, 514. [Google Scholar] [CrossRef] [PubMed]
- Pezzatti, J.; González-Ruiz, V.; Boccard, J.; Guillarme, D.; Rudaz, S. Evaluation of different tandem MS acquisition modes to support metabolite annotation in human plasma using ultra high-performance liquid chromatography high-resolution mass spectrometry for untargeted metabolomics. Metabolites 2020, 10, 464. [Google Scholar] [CrossRef]
- Ten-Doménech, I.; Martínez-Sena, T.; Moreno-Torres, M.; Sanjuan-Herráez, J.D.; Castell, J.V.; Parra-Llorca, A.; Vento, M.; Quintás, G.; Kuligowski, J. Comparing targeted vs. untargeted ms2 data-dependent acquisition for peak annotation in LC–MS metabolomics. Metabolites 2020, 10, 126. [Google Scholar] [CrossRef]
- Kumari, S.; Ali, A.; Roome, T.; Razzak, A.; Iqbal, A.; Siddiqui, A.J.; Azam, S.M.Z.; Shaikh, H.; El-Seedi, H.R.; Musharraf, S.G. Metabolomics Approach to Understand the Hepatitis C Virus Induced Hepatocellular Carcinoma using LC-ESI-MS/MS. Arab. J. Chem. 2020, 14, 102907. [Google Scholar] [CrossRef]
- Pino, L.K.; Searle, B.C.; Bollinger, J.G.; Nunn, B.; MacLean, B.; MacCoss, M.J. The Skyline ecosystem: Informatics for quantitative mass spectrometry proteomics. Mass Spectrom. Rev. 2020, 39, 229–244. [Google Scholar] [CrossRef]
- Treutler, H.; Tsugawa, H.; Porzel, A.; Gorzolka, K.; Tissier, A.; Neumann, S.; Balcke, G.U. Discovering regulated metabolite families in untargeted metabolomics studies. Anal. Chem. 2016, 88, 8082–8090. [Google Scholar] [CrossRef]
- Adams, K.J.; Pratt, B.; Bose, N.; Dubois, L.G.; John-Williams, L.S.; Perrott, K.M.; Ky, K.; Kapahi, P.; Sharma, V.; MacCoss, M.J. Skyline for Small Molecules: A Unifying Software Package for Quantitative Metabolomics. J. Proteome Res. 2020, 19, 1447–1458. [Google Scholar] [CrossRef] [PubMed]
- Rardin, M.J. Rapid assessment of contaminants and interferences in mass spectrometry data using skyline. J. Am. Soc. Mass Spectrom. 2018, 29, 1327–1330. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Cox, J. Perseus: A bioinformatics platform for integrative analysis of proteomics data in cancer research. In Cancer Systems Biology; Humana Press: New York, NY, USA, 2018; pp. 133–148. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drugs | Linear Calibration Range (ng/mL) | Acquisitions | Regression Equation | R2 | LOD (ng/mL) | LOQ (ng/mL) |
|---|---|---|---|---|---|---|
| Ranitidine | 0.3–100 10–100 0.3–100 3–100 | MS SCAN DDA DIA DIA (MS2) | y = 1094.9x + 686.35 y = 2438.7x + 31,531 y = 3475.3x + 8773 y = 39.904x + 610.2 | 0.9998 0.9988 0.9999 0.9993 | 1.23 6.9 0.50 1.4 | 3.75 21.01 1.51 4.25 |
| Ranolazine | 0.3–100 10–100 0.3–100 0.3–100 | MS SCAN DDA DIA DIA (MS2) | y = 1704.3x + 1808.7 y = 5460x + 60,586 y = 5792.1x + 29,669 y = 2409.6x + 8245.7 | 0.9998 0.9975 0.9999 0.9999 | 1.39 8.9 1.00 0.27 | 4.23 27.02 3.03 3.66 |
| Diphenhydramine | 0.3–100 10–100 0.3–100 0.3–100 | MS SCAN DDA DIA DIA (MS2) | y = 682.45x + 426.99 y = 1857.6x + 25,226 y = 2538x + 14,939 y = 4992.9x + 11,820 | 0.9998 0.9994 0.9999 0.9999 | 1.18 4.7 1.0 0.55 | 3.58 14.3 2.94 11.04 |
| Phenylbutazone | 0.3–100 10–100 0.3–100 0.3–100 | MS SCAN DDA DIA DIA (MS2) | y = 1297.7x + 508.63 y = 4897.3x − 2071.5 y = 4780x − 2760.8 y = 12.595x + 354.43 | 0.9998 0.9997 0.9999 0.9988 | 2.72 3.63 1.0 3.10 | 8.25 11.08 3.19 9.33 |
| Oxytetracycline | 3–100 10–100 0.3–100 0.3–100 | MS SCAN DDA DIA DIA (MS2) | y = 73.192x + 260.82 y = 382.84x + 2845.8 y = 383.1x + 2744.1 y = 265.17x + 307.1 | 0.9998 0.9997 0.9999 0.9999 | 1.85 3.32 0.65 0.29 | 5.60 10.21 1.97 0.90 |
| Duloxetine | 10–100 10–100 0.3–100 3–100 | MS SCAN DDA DIA DIA (MS2) | y = 21.694x + 427.04 y = 285.82x + 9676.9 y = 291.66x + 9101.4 y = 17.12x + 402.22 | 0.9989 0.9988 0.9999 0.9918 | 6.75 6.84 1.0 11.15 | 20.48 20.73 3.19 19.24 |
| Haloperidol | 3–100 10–100 0.3–100 3–100 | MS SCAN DDA DIA DIA (MS2) | y = 227.85x + 169.38 y = 900.54x + 9838.5 y = 972.34x + 3121.6 y = 34.018x + 151.12 | 0.9997 0.9996 0.9999 0.9999 | 2.06 4.02 0.3 0.68 | 6.2 12.20 1.01 2.07 |
| Finasteride | 0.3–100 10–100 0.3–100 0.3–100 | MS SCAN DDA DIA DIA (MS2) | y = 865.32x − 89.72 y = 3613.3x + 7669 y = 3469.7x + 19,199 y = 1366.3x + 553.75 | 0.9997 0.9995 0.9999 0.9999 | 1.55 3.65 0.92 0.39 | 4.69 10.11 2.77 1.44 |
| Dipyridamole | 0.3–100 10–100 0.3–100 0.3–100 | MS SCAN DDA DIA DIA (MS2) | y = 3700.3x + 1016.8 y = 1346.4x − 13,657 y = 11171x + 42,330 y = 449.73x + 2689.4 | 0.9999 0.9988 0.9999 0.9999 | 1.07 4.06 0.83 0.30 | 3.25 12.4 2.51 0.98 |
| Atropine | 0.3–100 10–100 0.3–100 3–100 | MS SCAN DDA DIA DIA (MS2) | y = 2458.2x + 3144 y = 7855.3x + 32,350 y = 8014.1x + 21,661 y = 17.495x + 111.04 | 0.9999 0.9996 0.9999 0.9993 | 1.01 4.02 0.9 3.32 | 3.06 12.18 2.98 10.08 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shah, S.M.Z.; Ali, A.; Khan, M.N.; Khadim, A.; Asmari, M.; Uddin, J.; Musharraf, S.G. Sensitive Detection of Pharmaceutical Drugs and Metabolites in Serum Using Data-Independent Acquisition Mass Spectrometry and Open-Access Data Acquisition Tools. Pharmaceuticals 2022, 15, 901. https://doi.org/10.3390/ph15070901
Shah SMZ, Ali A, Khan MN, Khadim A, Asmari M, Uddin J, Musharraf SG. Sensitive Detection of Pharmaceutical Drugs and Metabolites in Serum Using Data-Independent Acquisition Mass Spectrometry and Open-Access Data Acquisition Tools. Pharmaceuticals. 2022; 15(7):901. https://doi.org/10.3390/ph15070901
Chicago/Turabian StyleShah, Syed Muhammad Zaki, Arslan Ali, Muhammad Noman Khan, Adeeba Khadim, Mufarreh Asmari, Jalal Uddin, and Syed Ghulam Musharraf. 2022. "Sensitive Detection of Pharmaceutical Drugs and Metabolites in Serum Using Data-Independent Acquisition Mass Spectrometry and Open-Access Data Acquisition Tools" Pharmaceuticals 15, no. 7: 901. https://doi.org/10.3390/ph15070901
APA StyleShah, S. M. Z., Ali, A., Khan, M. N., Khadim, A., Asmari, M., Uddin, J., & Musharraf, S. G. (2022). Sensitive Detection of Pharmaceutical Drugs and Metabolites in Serum Using Data-Independent Acquisition Mass Spectrometry and Open-Access Data Acquisition Tools. Pharmaceuticals, 15(7), 901. https://doi.org/10.3390/ph15070901