
Neuronal Hyperexcitability and Free Radical Toxicity in Amyotrophic Lateral Sclerosis: Established and Future Targets

 ,
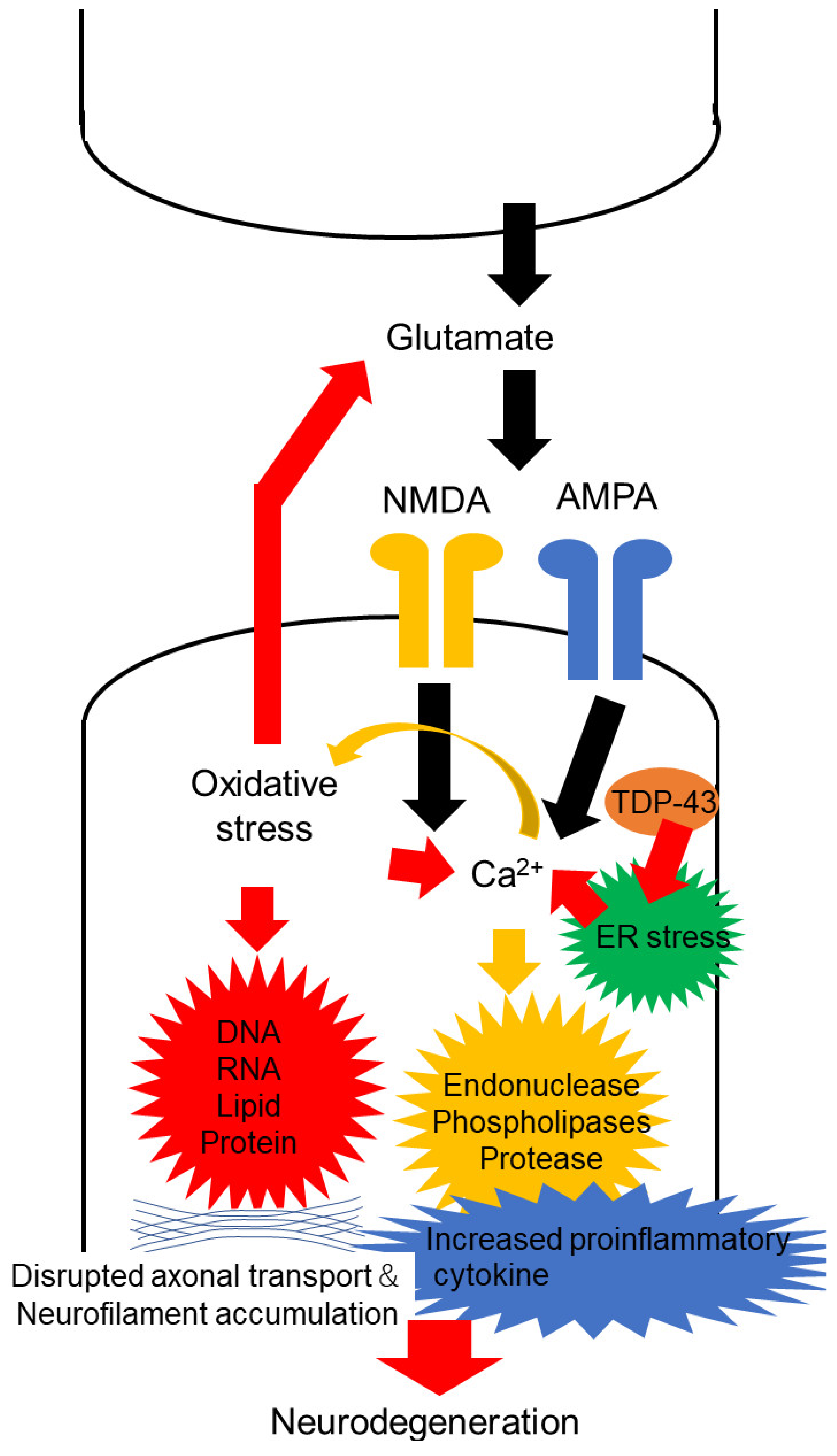
, 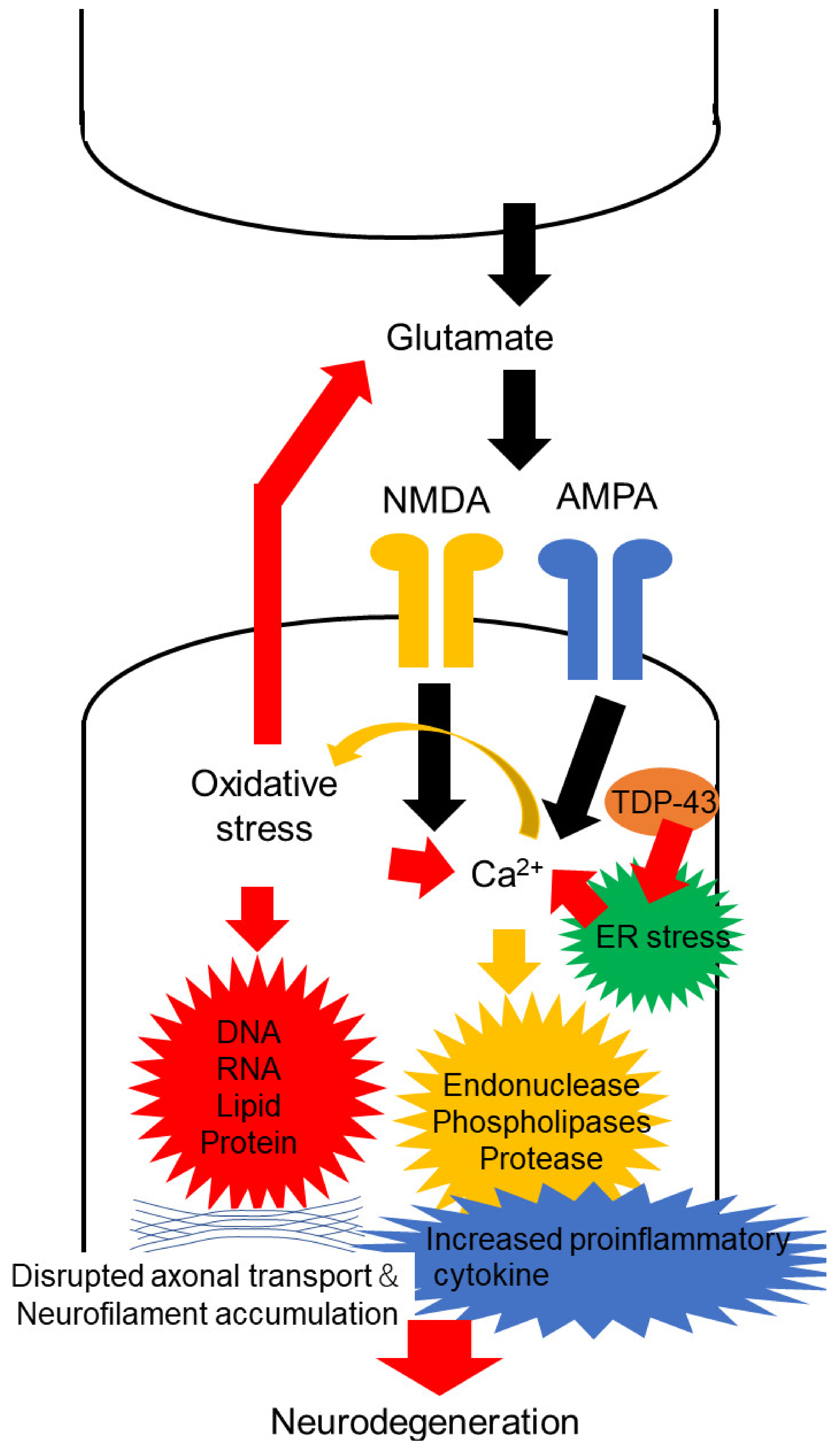{kind=link}
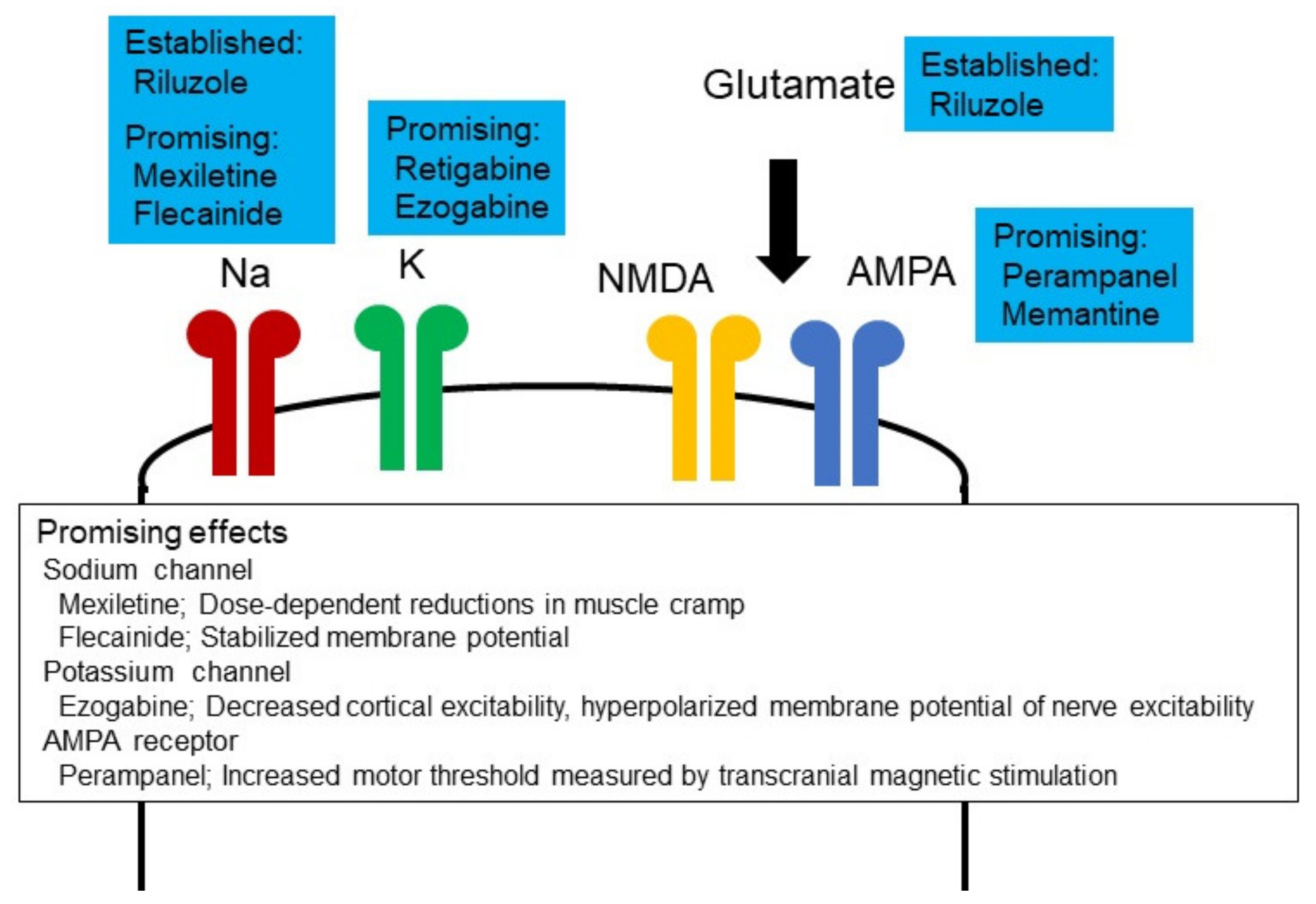
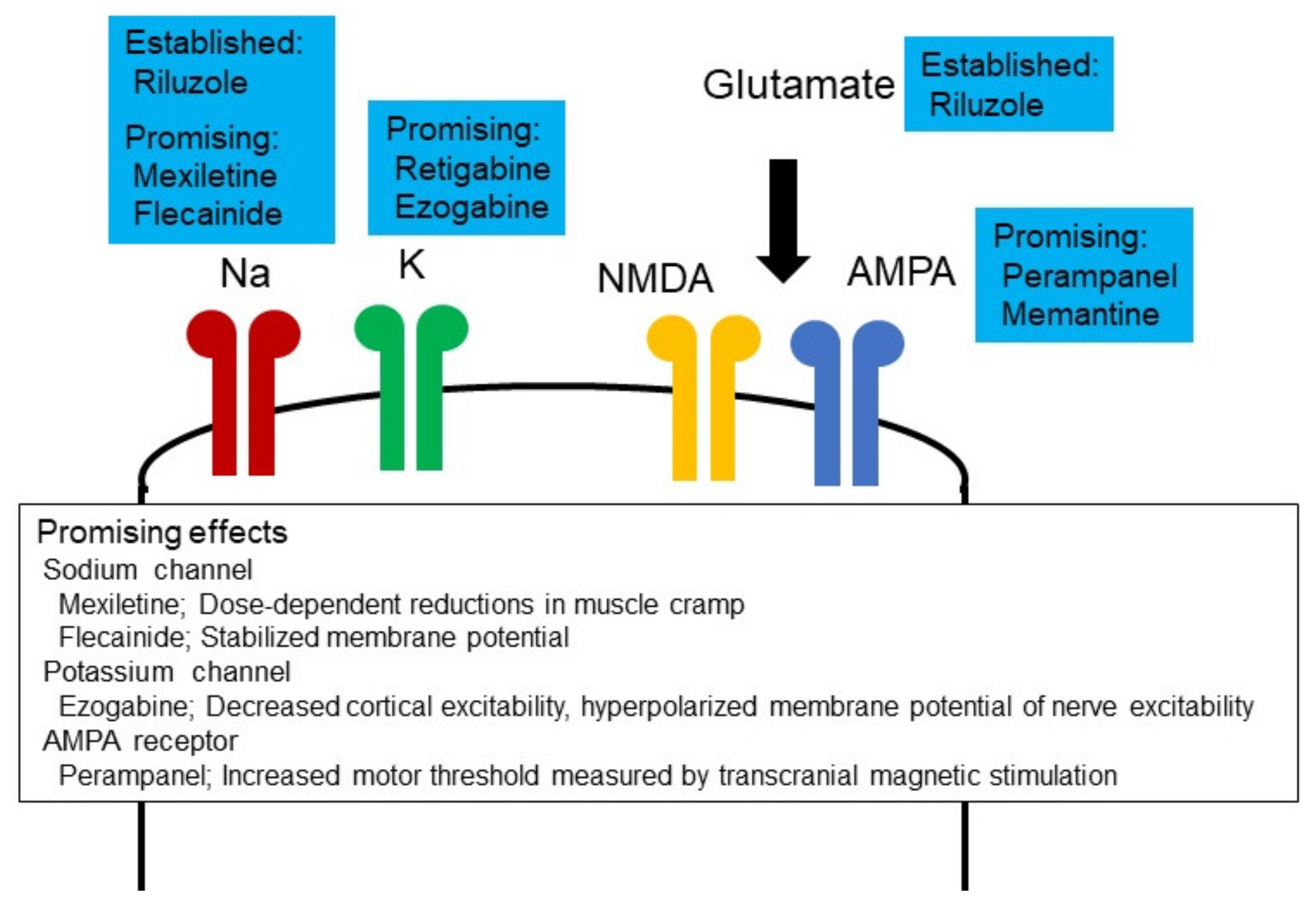{kind=link}
Abstract
:1. Introduction
2. Oxidative Stress in Motor Neuron Degeneration of ALS
3. Increased Cortical Excitability
3.1. Neurophysiological Evidence
3.2. Imaging Evidence
3.3. Pathological and Animal Model Evidence
3.4. Fluid Biomarkers
4. Increased Excitability of the Peripheral Nerve
4.1. Neurophysiological Evidence
4.2. Pathological Evidence
5. Therapeutic Potential of Altered Neuronal Excitability in ALS
5.1. Ion Channel Modulators
5.2. Non-Pharmacological Approaches
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mehta, P.; Raymond, J.; Punjani, R.; Larson, T.; Han, M.; Bove, F.; Horton, D.K. Incidence of amyotrophic lateral sclerosis in the united states, 2014–2016. Amyotroph. Lateral Scler. Front. Degener. 2022, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Hulisz, D. Amyotrophic lateral sclerosis: Disease state overview. Am. J. Manag. Care 2018, 24, S320–S326. [Google Scholar] [PubMed]
- Takeda, T.; Kitagawa, K.; Arai, K. Phenotypic variability and its pathological basis in amyotrophic lateral sclerosis. Neuropathol. Off. J. Jpn. Soc. Neuropathol. 2020, 40, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Giordano, A.; De Panfilis, L.; Perin, M.; Servidio, L.; Cascioli, M.; Grasso, M.G.; Lugaresi, A.; Pucci, E.; Veronese, S.; Solari, A. Advance care planning in neurodegenerative disorders: A scoping review. Int. J. Environ. Res. Public Health 2022, 19, 803. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.A.; Lally, C.; Kupelian, V.; Flanders, W.D. Estimated prevalence and incidence of amyotrophic lateral sclerosis and sod1 and c9orf72 genetic variants. Neuroepidemiology 2021, 55, 342–353. [Google Scholar] [CrossRef]
- De Boer, E.M.J.; Orie, V.K.; Williams, T.; Baker, M.R.; De Oliveira, H.M.; Polvikoski, T.; Silsby, M.; Menon, P.; van den Bos, M.; Halliday, G.M.; et al. Tdp-43 proteinopathies: A new wave of neurodegenerative diseases. J. Neurol. Neurosurg. Psychiatry 2020, 92, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Ji, Y.; Wang, W.; Zhang, L.; Chen, Z.; Yu, M.; Shen, Y.; Ding, F.; Gu, X.; Sun, H. Amyotrophic lateral sclerosis: Molecular mechanisms, biomarkers, and therapeutic strategies. Antioxidants 2021, 10, 1012. [Google Scholar] [CrossRef]
- Van Harten, A.C.M.; Phatnani, H.; Przedborski, S. Non-cell-autonomous pathogenic mechanisms in amyotrophic lateral sclerosis. Trends Neurosci. 2021, 44, 658–668. [Google Scholar] [CrossRef]
- Shah, S.; Dooms, M.M.; Amaral-Garcia, S.; Igoillo-Esteve, M. Current drug repurposing strategies for rare neurodegenerative disorders. Front. Pharmacol. 2021, 12, 768023. [Google Scholar] [CrossRef]
- Benson, B.C.; Shaw, P.J.; Azzouz, M.; Highley, J.R.; Hautbergue, G.M. Proteinopathies as hallmarks of impaired gene expression, proteostasis and mitochondrial function in amyotrophic lateral sclerosis. Front. Neurosci. 2021, 15, 783624. [Google Scholar] [CrossRef]
- Rodríguez, L.R.; Lapeña-Luzón, T.; Benetó, N.; Beltran-Beltran, V.; Pallardó, F.V.; Gonzalez-Cabo, P.; Navarro, J.A. Therapeutic strategies targeting mitochondrial calcium signaling: A new hope for neurological diseases? Antioxidants 2022, 11, 165. [Google Scholar] [CrossRef]
- Fourier, A.; Quadrio, I. Proteinopathies associated to repeat expansion disorders. J. Neural Transm. 2022, 129, 173–185. [Google Scholar] [CrossRef]
- Staats, K.A.; Borchelt, D.R.; Tansey, M.G.; Wymer, J. Blood-based biomarkers of inflammation in amyotrophic lateral sclerosis. Mol. Neurodegener. 2022, 17, 11. [Google Scholar] [CrossRef]
- Wong, C.; Stavrou, M.; Elliott, E.; Gregory, J.M.; Leigh, N.; Pinto, A.A.; Williams, T.L.; Chataway, J.; Swingler, R.; Parmar, M.K.B.; et al. Clinical trials in amyotrophic lateral sclerosis: A systematic review and perspective. Brain Commun. 2021, 3, fcab242. [Google Scholar] [CrossRef]
- Ortiz, J.F.; Khan, S.A.; Salem, A.; Lin, Z.; Iqbal, Z.; Jahan, N. Post-marketing experience of edaravone in amyotrophic lateral sclerosis: A clinical perspective and comparison with the clinical trials of the drug. Cureus 2020, 12, e10818. [Google Scholar] [CrossRef]
- Miller, R.G.; Mitchell, J.D.; Lyon, M.; Moore, D.H. Riluzole for amyotrophic lateral sclerosis (als)/motor neuron disease (mnd). Cochrane Database Syst. Rev. 2007, 1, Cd001447. [Google Scholar]
- Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017, 16, 505–512. [CrossRef]
- Houzen, H.; Kano, T.; Horiuchi, K.; Wakita, M.; Nagai, A.; Yabe, I. Improved long-term survival with edaravone therapy in patients with amyotrophic lateral sclerosis: A retrospective single-center study in japan. Pharmaceuticals 2021, 14, 705. [Google Scholar] [CrossRef]
- Kumar, V.; Islam, A.; Hassan, M.I.; Ahmad, F. Therapeutic progress in amyotrophic lateral sclerosis-beginning to learning. Eur. J. Med. Chem. 2016, 121, 903–917. [Google Scholar] [CrossRef]
- Lau, A.; Tymianski, M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflug. Arch. Eur. J. Physiol. 2010, 460, 525–542. [Google Scholar] [CrossRef]
- Sen, I.; Nalini, A.; Joshi, N.B.; Joshi, P.G. Cerebrospinal fluid from amyotrophic lateral sclerosis patients preferentially elevates intracellular calcium and toxicity in motor neurons via ampa/kainate receptor. J. Neurol. Sci. 2005, 235, 45–54. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative stress: A key modulator in neurodegenerative diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [Green Version]
- Chiurchiù, V.; Orlacchio, A.; Maccarrone, M. Is modulation of oxidative stress an answer? The state of the art of redox therapeutic actions in neurodegenerative diseases. Oxid. Med. Cell. Longev. 2016, 2016, 7909380. [Google Scholar] [CrossRef] [Green Version]
- Chiurchiù, V.; Maccarrone, M. Chronic inflammatory disorders and their redox control: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011, 15, 2605–2641. [Google Scholar] [CrossRef]
- Halliwell, B. Reactive species and antioxidants. Redox biology is a fundamental theme of aerobic life. Plant Physiol. 2006, 141, 312–322. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.D.; Weiss, J.H. Excitotoxic and oxidative cross-talk between motor neurons and glia in als pathogenesis. Trends Neurosci. 2004, 27, 17–23. [Google Scholar] [CrossRef]
- Zhang, B.; Tu, P.; Abtahian, F.; Trojanowski, J.Q.; Lee, V.M. Neurofilaments and orthograde transport are reduced in ventral root axons of transgenic mice that express human sod1 with a g93a mutation. J. Cell Biol. 1997, 139, 1307–1315. [Google Scholar] [CrossRef]
- Hemerková, P.; Vališ, M. Role of oxidative stress in the pathogenesis of amyotrophic lateral sclerosis: Antioxidant metalloenzymes and therapeutic strategies. Biomolecules 2021, 11, 437. [Google Scholar] [CrossRef]
- Genova, M.L.; Pich, M.M.; Bernacchia, A.; Bianchi, C.; Biondi, A.; Bovina, C.; Falasca, A.I.; Formiggini, G.; Castelli, G.P.; Lenaz, G. The mitochondrial production of reactive oxygen species in relation to aging and pathology. Ann. N. Y. Acad. Sci. 2004, 1011, 86–100. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, B. Oxidative stress and neurodegeneration: Where are we now? J. Neurochem. 2006, 97, 1634–1658. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, R.J.; Browne, S.E.; Shinobu, L.A.; Bowling, A.C.; Baik, M.J.; MacGarvey, U.; Kowall, N.W.; Brown, R.H., Jr.; Beal, M.F. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J. Neurochem. 1997, 69, 2064–2074. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, W.A.; Fu, W.; Keller, J.N.; Markesbery, W.R.; Appel, S.; Smith, R.G.; Kasarskis, E.; Mattson, M.P. Protein modification by the lipid peroxidation product 4-hydroxynonenal in the spinal cords of amyotrophic lateral sclerosis patients. Ann. Neurol. 1998, 44, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Simpson, E.P.; Henry, Y.K.; Henkel, J.S.; Smith, R.G.; Appel, S.H. Increased lipid peroxidation in sera of als patients: A potential biomarker of disease burden. Neurology 2004, 62, 1758–1765. [Google Scholar] [CrossRef]
- Barker, A.T.; Jalinous, R.; Freeston, I.L. Non-invasive magnetic stimulation of human motor cortex. Lancet 1985, 1, 1106–1107. [Google Scholar] [CrossRef]
- Di Lazzaro, V.; Bella, R.; Benussi, A.; Bologna, M.; Borroni, B.; Capone, F.; Chen, K.S.; Chen, R.; Chistyakov, A.V.; Classen, J.; et al. Diagnostic contribution and therapeutic perspectives of transcranial magnetic stimulation in dementia. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2021, 132, 2568–2607. [Google Scholar] [CrossRef]
- Di Lazzaro, V.; Profice, P.; Ranieri, F.; Capone, F.; Dileone, M.; Oliviero, A.; Pilato, F. I-wave origin and modulation. Brain Stimul. 2012, 5, 512–525. [Google Scholar] [CrossRef]
- Di Lazzaro, V.; Ranieri, F.; Profice, P.; Pilato, F.; Mazzone, P.; Capone, F.; Insola, A.; Oliviero, A. Transcranial direct current stimulation effects on the excitability of corticospinal axons of the human cerebral cortex. Brain Stimul. 2013, 6, 641–643. [Google Scholar] [CrossRef]
- Geevasinga, N.; Van den Bos, M.; Menon, P.; Vucic, S. Utility of transcranial magnetic simulation in studying upper motor neuron dysfunction in amyotrophic lateral sclerosis. Brain Sci. 2021, 11, 906. [Google Scholar] [CrossRef]
- Van den Bos, M.A.J.; Geevasinga, N.; Higashihara, M.; Menon, P.; Vucic, S. Pathophysiology and diagnosis of als: Insights from advances in neurophysiological techniques. Int. J. Mol. Sci. 2019, 20, 2818. [Google Scholar] [CrossRef] [Green Version]
- Vucic, S.; Rutkove, S.B. Neurophysiological biomarkers in amyotrophic lateral sclerosis. Curr. Opin. Neurol. 2018, 31, 640–647. [Google Scholar] [CrossRef]
- Wassermann, E.M. Variation in the response to transcranial magnetic brain stimulation in the general population. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2002, 113, 1165–1171. [Google Scholar] [CrossRef]
- Siniatchkin, M.; Groppa, S.; Siebner, H.; Stephani, U. A single dose of sulthiame induces a selective increase in resting motor threshold in human motor cortex: A transcranial magnetic stimulation study. Epilepsy Res. 2006, 72, 18–24. [Google Scholar] [CrossRef]
- Di Lazzaro, V.; Oliviero, A.; Profice, P.; Pennisi, M.A.; Pilato, F.; Zito, G.; Dileone, M.; Nicoletti, R.; Pasqualetti, P.; Tonali, P.A. Ketamine increases human motor cortex excitability to transcranial magnetic stimulation. J. Physiol. 2003, 547, 485–496. [Google Scholar] [CrossRef]
- Chen, R.; Corwell, B.; Yaseen, Z.; Hallett, M.; Cohen, L.G. Mechanisms of cortical reorganization in lower-limb amputees. J. Neurosci. Off. J. Soc. Neurosci. 1998, 18, 3443–3450. [Google Scholar] [CrossRef] [Green Version]
- Eisen, A.; Weber, M. The motor cortex and amyotrophic lateral sclerosis. Muscle Nerve 2001, 24, 564–573. [Google Scholar] [CrossRef]
- Brown, W.F.; Ebers, G.C.; Hudson, A.J.; Pringle, C.E.; Veitch, J. Motor-evoked responses in primary lateral sclerosis. Muscle Nerve 1992, 15, 626–629. [Google Scholar] [CrossRef]
- Liu, J.; Li, Y.; Niu, J.; Zhang, L.; Fan, J.; Guan, Y.; Cui, L.; Liu, M. Fasciculation differences between als and non-als patients: An ultrasound study. BMC Neurol. 2021, 21, 441. [Google Scholar] [CrossRef]
- Ma, J.; Wen, Q.; Pang, X.; Huang, S.; Zhang, J.; Wang, J.; Chang, X.; Guo, J.; Zhang, W. Fasciculation score: A sensitive biomarker in amyotrophic lateral sclerosis. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2021, 42, 4657–4666. [Google Scholar] [CrossRef]
- Rajula, R.R.; Saini, J.; Unnikrishnan, G.; Vengalil, S.; Nashi, S.; Bardhan, M.; Huddar, A.; Chawla, T.; Sindhu, D.M.; Ganaraja, V.H.; et al. Muscle ultrasonography in detecting fasciculations: A noninvasive diagnostic tool for amyotrophic lateral sclerosis. J. Clin. Ultrasound 2021, 50, 286–291. [Google Scholar] [CrossRef]
- Wannop, K.; Bashford, J.; Wickham, A.; Iniesta, R.; Drakakis, E.; Boutelle, M.; Mills, K.; Shaw, C. Fasciculation analysis reveals a novel parameter that correlates with predicted survival in amyotrophic lateral sclerosis. Muscle Nerve 2021, 63, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Cros, D.; Curra, A.; Di Lazzaro, V.; Lefaucheur, J.P.; Magistris, M.R.; Mills, K.; Rösler, K.M.; Triggs, W.J.; Ugawa, Y.; et al. The clinical diagnostic utility of transcranial magnetic stimulation: Report of an ifcn committee. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2008, 119, 504–532. [Google Scholar] [CrossRef] [PubMed]
- Vucic, S.; Nicholson, G.A.; Kiernan, M.C. Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain 2008, 131, 1540–1550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertel, O.; Malessa, S.; Sluga, E.; Hornykiewicz, O. Amyotrophic lateral sclerosis: Changes of noradrenergic and serotonergic transmitter systems in the spinal cord. Brain Res. 1991, 566, 54–60. [Google Scholar] [CrossRef]
- Chew, S.; Atassi, N. Positron emission tomography molecular imaging biomarkers for amyotrophic lateral sclerosis. Front. Neurol. 2019, 10, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spreux-Varoquaux, O.; Bensimon, G.; Lacomblez, L.; Salachas, F.; Pradat, P.F.; Le Forestier, N.; Marouan, A.; Dib, M.; Meininger, V. Glutamate levels in cerebrospinal fluid in amyotrophic lateral sclerosis: A reappraisal using a new hplc method with coulometric detection in a large cohort of patients. J. Neurol. Sci. 2002, 193, 73–78. [Google Scholar] [CrossRef]
- Inghilleri, M.; Berardelli, A.; Cruccu, G.; Manfredi, M. Silent period evoked by transcranial stimulation of the human cortex and cervicomedullary junction. J. Physiol. 1993, 466, 521–534. [Google Scholar] [PubMed]
- Cantello, R.; Gianelli, M.; Civardi, C.; Mutani, R. Magnetic brain stimulation: The silent period after the motor evoked potential. Neurology 1992, 42, 1951–1959. [Google Scholar]
- Attarian, S.; Azulay, J.P.; Lardillier, D.; Verschueren, A.; Pouget, J. Transcranial magnetic stimulation in lower motor neuron diseases. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2005, 116, 35–42. [Google Scholar] [CrossRef]
- Garg, N.; Park, S.B.; Vucic, S.; Yiannikas, C.; Spies, J.; Howells, J.; Huynh, W.; Matamala, J.M.; Krishnan, A.V.; Pollard, J.D.; et al. Differentiating lower motor neuron syndromes. J. Neurol. Neurosurg. Psychiatry 2017, 88, 474–483. [Google Scholar] [CrossRef] [Green Version]
- Mills, K.R. The natural history of central motor abnormalities in amyotrophic lateral sclerosis. Brain 2003, 126, 2558–2566. [Google Scholar] [CrossRef] [Green Version]
- Tankisi, H.; Howells, J.; Cengiz, B.; Samusyte, G.; Koltzenburg, M.; Bostock, H. Conventional and threshold-tracking transcranial magnetic stimulation tests for single-handed operation. J. Vis. Exp. 2021, 174, e62787. [Google Scholar] [CrossRef]
- Vucic, S.; Nicholson, G.A.; Kiernan, M.C. Cortical excitability in hereditary motor neuronopathy with pyramidal signs: Comparison with als. J. Neurol. Neurosurg. Psychiatry 2010, 81, 97–100. [Google Scholar] [CrossRef]
- Mrachacz-Kersting, N.; Stevenson, A.J.T.; Ziemann, U. Short-interval intracortical inhibition and facilitation targeting upper and lower limb muscles. Sci. Rep. 2021, 11, 21993. [Google Scholar] [CrossRef]
- Nielsen, C.S.; Samusyte, G.; Pugdahl, K.; Blicher, J.U.; Fuglsang-Frederiksen, A.; Cengiz, B.; Tankisi, H. Test-retest reliability of short-interval intracortical inhibition assessed by threshold-tracking and automated conventional techniques. eNeuro 2021, 8, 0103-21. [Google Scholar] [CrossRef]
- Agarwal, S.; Highton-Williamson, E.; Caga, J.; Howells, J.; Dharmadasa, T.; Matamala, J.M.; Ma, Y.; Shibuya, K.; Hodges, J.R.; Ahmed, R.M.; et al. Motor cortical excitability predicts cognitive phenotypes in amyotrophic lateral sclerosis. Sci. Rep. 2021, 11, 2172. [Google Scholar] [CrossRef]
- Dharmadasa, T.; Matamala, J.M.; Howells, J.; Vucic, S.; Kiernan, M.C. Early focality and spread of cortical dysfunction in amyotrophic lateral sclerosis: A regional study across the motor cortices. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2020, 131, 958–966. [Google Scholar] [CrossRef]
- Tankisi, H.; Nielsen, C.S.; Howells, J.; Cengiz, B.; Samusyte, G.; Koltzenburg, M.; Blicher, J.U.; Møller, A.T.; Pugdahl, K.; Fuglsang-Frederiksen, A.; et al. Early diagnosis of amyotrophic lateral sclerosis by threshold tracking and conventional transcranial magnetic stimulation. Eur. J. Neurol. 2021, 28, 3030–3039. [Google Scholar] [CrossRef]
- Shibuya, K.; Simon, N.G.; Geevasinga, N.; Menon, P.; Howells, J.; Park, S.B.; Huynh, W.; Noto, Y.I.; Vucic, S.; Kiernan, M.C. The evolution of motor cortical dysfunction in amyotrophic lateral sclerosis. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2017, 128, 1075–1082. [Google Scholar] [CrossRef]
- Menon, P.; Geevasinga, N.; Yiannikas, C.; Howells, J.; Kiernan, M.C.; Vucic, S. Sensitivity and specificity of threshold tracking transcranial magnetic stimulation for diagnosis of amyotrophic lateral sclerosis: A prospective study. Lancet Neurol. 2015, 14, 478–484. [Google Scholar] [CrossRef]
- Geevasinga, N.; Menon, P.; Yiannikas, C.; Kiernan, M.C.; Vucic, S. Diagnostic utility of cortical excitability studies in amyotrophic lateral sclerosis. Eur. J. Neurol. 2014, 21, 1451–1457. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Bokuda, K.; Kimura, H.; Kamiyama, T.; Nakayama, Y.; Kawata, A.; Isozaki, E.; Ugawa, Y. Sensory cortex hyperexcitability predicts short survival in amyotrophic lateral sclerosis. Neurology 2018, 90, e1578–e1587. [Google Scholar] [CrossRef] [PubMed]
- Nardone, R.; Golaszewski, S.; Thomschewski, A.; Sebastianelli, L.; Versace, V.; Brigo, F.; Orioli, A.; Saltuari, L.; Höller, Y.; Trinka, E. Disinhibition of sensory cortex in patients with amyotrophic lateral sclerosis. Neurosci. Lett. 2020, 722, 134860. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, S.; Rothman, D.L. 1H magnetic resonance spectroscopy to understand the biological basis of als, diagnose patients earlier, and monitor disease progression. Front. Neurol. 2021, 12, 701170. [Google Scholar] [CrossRef] [PubMed]
- Weerasekera, A.; Peeters, R.; Sima, D.; Dresselaers, T.; Sunaert, S.; De Vocht, J.; Claeys, K.; Van Huffel, S.; Van Damme, P.; Himmelreich, U. Motor cortex metabolite alterations in amyotrophic lateral sclerosis assessed in vivo using edited and non-edited magnetic resonance spectroscopy. Brain Res. 2019, 1718, 22–31. [Google Scholar] [CrossRef]
- Cheong, I.; Deelchand, D.K.; Eberly, L.E.; Marjańska, M.; Manousakis, G.; Guliani, G.; Walk, D.; Öz, G. Neurochemical correlates of functional decline in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2019, 90, 294–301. [Google Scholar] [CrossRef]
- Foerster, B.R.; Pomper, M.G.; Callaghan, B.C.; Petrou, M.; Edden, R.A.; Mohamed, M.A.; Welsh, R.C.; Carlos, R.C.; Barker, P.B.; Feldman, E.L. An imbalance between excitatory and inhibitory neurotransmitters in amyotrophic lateral sclerosis revealed by use of 3-t proton magnetic resonance spectroscopy. JAMA Neurol. 2013, 70, 1009–1016. [Google Scholar] [CrossRef]
- Foerster, B.R.; Callaghan, B.C.; Petrou, M.; Edden, R.A.; Chenevert, T.L.; Feldman, E.L. Decreased motor cortex γ-aminobutyric acid in amyotrophic lateral sclerosis. Neurology 2012, 78, 1596–1600. [Google Scholar] [CrossRef] [Green Version]
- Kassubek, J.; Pagani, M. Imaging in amyotrophic lateral sclerosis: Mri and pet. Curr. Opin. Neurol. 2019, 32, 740–746. [Google Scholar] [CrossRef]
- Turner, M.R.; Hammers, A.; Al-Chalabi, A.; Shaw, C.E.; Andersen, P.M.; Brooks, D.J.; Leigh, P.N. Cortical involvement in four cases of primary lateral sclerosis using [11C]-flumazenil pet. J. Neurol. 2007, 254, 1033–1036. [Google Scholar] [CrossRef]
- Turner, M.R.; Hammers, A.; Al-Chalabi, A.; Shaw, C.E.; Andersen, P.M.; Brooks, D.J.; Leigh, P.N. Distinct cerebral lesions in sporadic and ‘D90A’ SOD1 ALS: Studies with [11C]flumazenil PET. Brain A J. Neurol. 2005, 128, 1323–1329. [Google Scholar] [CrossRef] [Green Version]
- Ikawa, M.; Okazawa, H.; Tsujikawa, T.; Matsunaga, A.; Yamamura, O.; Mori, T.; Hamano, T.; Kiyono, Y.; Nakamoto, Y.; Yoneda, M. Increased oxidative stress is related to disease severity in the als motor cortex: A pet study. Neurology 2015, 84, 2033–2039. [Google Scholar] [CrossRef]
- Corona, J.C.; Tapia, R. Ampa receptor activation, but not the accumulation of endogenous extracellular glutamate, induces paralysis and motor neuron death in rat spinal cord in vivo. J. Neurochem. 2004, 89, 988–997. [Google Scholar] [CrossRef]
- Konen, L.M.; Wright, A.L.; Royle, G.A.; Morris, G.P.; Lau, B.K.; Seow, P.W.; Zinn, R.; Milham, L.T.; Vaughan, C.W.; Vissel, B. A new mouse line with reduced glua2 q/r site rna editing exhibits loss of dendritic spines, hippocampal ca1-neuron loss, learning and memory impairments and nmda receptor-independent seizure vulnerability. Mol. Brain 2020, 13, 27. [Google Scholar] [CrossRef] [Green Version]
- Takuma, H.; Kwak, S.; Yoshizawa, T.; Kanazawa, I. Reduction of glur2 rna editing, a molecular change that increases calcium influx through ampa receptors, selective in the spinal ventral gray of patients with amyotrophic lateral sclerosis. Ann. Neurol. 1999, 46, 806–815. [Google Scholar] [CrossRef]
- Allaman, I.; Bélanger, M.; Magistretti, P.J. Astrocyte-neuron metabolic relationships: For better and for worse. Trends Neurosci. 2011, 34, 76–87. [Google Scholar] [CrossRef]
- Tanaka, K.; Watase, K.; Manabe, T.; Yamada, K.; Watanabe, M.; Takahashi, K.; Iwama, H.; Nishikawa, T.; Ichihara, N.; Kikuchi, T.; et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter glt-1. Science 1997, 276, 1699–1702. [Google Scholar] [CrossRef]
- Guo, H.; Lai, L.; Butchbach, M.E.; Stockinger, M.P.; Shan, X.; Bishop, G.A.; Lin, C.L. Increased expression of the glial glutamate transporter eaat2 modulates excitotoxicity and delays the onset but not the outcome of als in mice. Hum. Mol. Genet. 2003, 12, 2519–2532. [Google Scholar] [CrossRef]
- Sasaki, S.; Komori, T.; Iwata, M. Excitatory amino acid transporter 1 and 2 immunoreactivity in the spinal cord in amyotrophic lateral sclerosis. Acta Neuropathol. 2000, 100, 138–144. [Google Scholar] [CrossRef]
- Rao, S.D.; Yin, H.Z.; Weiss, J.H. Disruption of glial glutamate transport by reactive oxygen species produced in motor neurons. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 2627–2633. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.L.; Bristol, L.A.; Jin, L.; Dykes-Hoberg, M.; Crawford, T.; Clawson, L.; Rothstein, J.D. Aberrant rna processing in a neurodegenerative disease: The cause for absent eaat2, a glutamate transporter, in amyotrophic lateral sclerosis. Neuron 1998, 20, 589–602. [Google Scholar] [CrossRef] [Green Version]
- Walker, A.K.; Atkin, J.D. Stress signaling from the endoplasmic reticulum: A central player in the pathogenesis of amyotrophic lateral sclerosis. IUBMB Life 2011, 63, 754–763. [Google Scholar] [CrossRef]
- Zhang, I.X.; Raghavan, M.; Satin, L.S. The endoplasmic reticulum and calcium homeostasis in pancreatic beta cells. Endocrinology 2020, 161, bqz028. [Google Scholar] [CrossRef]
- Bahar, E.; Kim, H.; Yoon, H. Er stress-mediated signaling: Action potential and ca(2+) as key players. Int. J. Mol. Sci. 2016, 17, 1558. [Google Scholar] [CrossRef]
- Milanese, M.; Zappettini, S.; Onofri, F.; Musazzi, L.; Tardito, D.; Bonifacino, T.; Messa, M.; Racagni, G.; Usai, C.; Benfenati, F.; et al. Abnormal exocytotic release of glutamate in a mouse model of amyotrophic lateral sclerosis. J. Neurochem. 2011, 116, 1028–1042. [Google Scholar] [CrossRef]
- Jara, J.H.; Sheets, P.L.; Nigro, M.J.; Perić, M.; Brooks, C.; Heller, D.B.; Martina, M.; Andjus, P.R.; Ozdinler, P.H. The electrophysiological determinants of corticospinal motor neuron vulnerability in als. Front. Mol. Neurosci. 2020, 13, 73. [Google Scholar] [CrossRef]
- Andreadou, E.; Kapaki, E.; Kokotis, P.; Paraskevas, G.P.; Katsaros, N.; Libitaki, G.; Petropoulou, O.; Zis, V.; Sfagos, C.; Vassilopoulos, D. Plasma glutamate and glycine levels in patients with amyotrophic lateral sclerosis. In Vivo 2008, 22, 137–141. [Google Scholar]
- Cieslarova, Z.; Lopes, F.S.; do Lago, C.L.; França, M.C., Jr.; Colnaghi Simionato, A.V. Capillary electrophoresis tandem mass spectrometry determination of glutamic acid and homocysteine’s metabolites: Potential biomarkers of amyotrophic lateral sclerosis. Talanta 2017, 170, 63–68. [Google Scholar] [CrossRef]
- Shibuya, K.; Misawa, S.; Sekiguchi, Y.; Beppu, M.; Amino, H.; Suichi, T.; Suzuki, Y.I.; Tsuneyama, A.; Kuwabara, S. Prodromal muscle cramps predict rapid motor functional decline in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2018, 90, 242–243. [Google Scholar] [CrossRef]
- De Carvalho, M.; Kiernan, M.C.; Swash, M. Fasciculation in amyotrophic lateral sclerosis: Origin and pathophysiological relevance. J. Neurol. Neurosurg. Psychiatry 2017, 88, 773–779. [Google Scholar] [CrossRef]
- Duarte, M.L.; Iared, W.; Oliveira, A.S.B.; Dos Santos, L.R.; Peccin, M.S. Ultrasound versus electromyography for the detection of fasciculation in amyotrophic lateral sclerosis: Systematic review and meta-analysis. Radiol. Bras. 2020, 53, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Hobson-Webb, L.D.; Simmons, Z. Ultrasound in the diagnosis and monitoring of amyotrophic lateral sclerosis: A review. Muscle Nerve 2019, 60, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Tamborska, A.; Bashford, J.; Wickham, A.; Iniesta, R.; Masood, U.; Cabassi, C.; Planinc, D.; Hodson-Tole, E.; Drakakis, E.; Boutelle, M.; et al. Non-invasive measurement of fasciculation frequency demonstrates diagnostic accuracy in amyotrophic lateral sclerosis. Brain Commun. 2020, 2, fcaa141. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, Y.; Noto, Y.I.; Kitaoji, T.; Kojima, Y.; Mizuno, T. Difference in distribution of fasciculations between multifocal motor neuropathy and amyotrophic lateral sclerosis. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2020, 131, 2804–2808. [Google Scholar] [CrossRef]
- Tsuji, Y.; Noto, Y.I.; Shiga, K.; Teramukai, S.; Nakagawa, M.; Mizuno, T. A muscle ultrasound score in the diagnosis of amyotrophic lateral sclerosis. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2017, 128, 1069–1074. [Google Scholar] [CrossRef]
- De Carvalho, M.; Dengler, R.; Eisen, A.; England, J.D.; Kaji, R.; Kimura, J.; Mills, K.; Mitsumoto, H.; Nodera, H.; Shefner, J.; et al. Electrodiagnostic criteria for diagnosis of als. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2008, 119, 497–503. [Google Scholar] [CrossRef]
- Shefner, J.M.; Al-Chalabi, A.; Baker, M.R.; Cui, L.Y.; de Carvalho, M.; Eisen, A.; Grosskreutz, J.; Hardiman, O.; Henderson, R.; Matamala, J.M.; et al. A proposal for new diagnostic criteria for als. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2020, 131, 1975–1978. [Google Scholar] [CrossRef]
- De Carvalho, M. Electrodiagnosis of amyotrophic lateral sclerosis: A review of existing guidelines. J. Clin. Neurophysiol. Off. Publ. Am. Electroencephalogr. Soc. 2020, 37, 294–298. [Google Scholar] [CrossRef]
- Swash, M.; Czesnik, D.; de Carvalho, M. Muscular cramp: Causes and management. Eur. J. Neurol. 2019, 26, 214–221. [Google Scholar] [CrossRef]
- Mitsumoto, H.; Chiuzan, C.; Gilmore, M.; Zhang, Y.; Ibagon, C.; McHale, B.; Hupf, J.; Oskarsson, B. A novel muscle cramp scale (mcs) in amyotrophic lateral sclerosis (als). Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 328–335. [Google Scholar] [CrossRef]
- Caress, J.B.; Ciarlone, S.L.; Sullivan, E.A.; Griffin, L.P.; Cartwright, M.S. Natural history of muscle cramps in amyotrophic lateral sclerosis. Muscle Nerve 2016, 53, 513–517. [Google Scholar] [CrossRef] [Green Version]
- Tomlinson, S.E.; Howells, J.; Burke, D. In vivo assessment of neurological channelopathies: Application of peripheral nerve excitability studies. Neuropharmacology 2018, 132, 98–107. [Google Scholar] [CrossRef]
- Vucic, S.; Kiernan, M.C. Axonal excitability properties in amyotrophic lateral sclerosis. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2006, 117, 1458–1466. [Google Scholar] [CrossRef]
- Iwai, Y.; Shibuya, K.; Misawa, S.; Sekiguchi, Y.; Watanabe, K.; Amino, H.; Kuwabara, S. Axonal dysfunction precedes motor neuronal death in amyotrophic lateral sclerosis. PLoS ONE 2016, 11, e0158596. [Google Scholar] [CrossRef]
- Park, S.B.; Kiernan, M.C.; Vucic, S. Axonal excitability in amyotrophic lateral sclerosis: Axonal excitability in als. Neurother. J. Am. Soc. Exp. NeuroTherapeutics 2017, 14, 78–90. [Google Scholar] [CrossRef] [Green Version]
- Shibuya, K.; Misawa, S.; Uzawa, A.; Sawai, S.; Tsuneyama, A.; Suzuki, Y.I.; Suichi, T.; Kojima, Y.; Nakamura, K.; Kano, H.; et al. Split hand and motor axonal hyperexcitability in spinal and bulbar muscular atrophy. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1189–1194. [Google Scholar] [CrossRef]
- Hu, N.; Wang, J.; Liu, M. Split hand in amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas 2021, 90, 293–301. [Google Scholar] [CrossRef]
- Hannaford, A.; Higashihara, M.; Pavey, N.; van den Bos, M.; Geevasinga, N.; Vucic, S.; Menon, P. Split-hand index: A diagnostic and prognostic marker in amyotrophic lateral sclerosis across varying regions of onset. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2021, 132, 2130–2135. [Google Scholar] [CrossRef]
- Corcia, P.; Bede, P.; Pradat, P.F.; Couratier, P.; Vucic, S.; de Carvalho, M. Split-hand and split-limb phenomena in amyotrophic lateral sclerosis: Pathophysiology, electrophysiology and clinical manifestations. J. Neurol. Neurosurg. Psychiatry 2021, 92, 1126–1130. [Google Scholar] [CrossRef]
- Shibuya, K.; Misawa, S.; Nasu, S.; Sekiguchi, Y.; Mitsuma, S.; Beppu, M.; Ohmori, S.; Iwai, Y.; Ito, S.; Kanai, K.; et al. Split hand syndrome in amyotrophic lateral sclerosis: Different excitability changes in the thenar and hypothenar motor axons. J. Neurol. Neurosurg. Psychiatry 2013, 84, 969–972. [Google Scholar] [CrossRef]
- Matamala, J.M.; Howells, J.; Dharmadasa, T.; Huynh, W.; Park, S.B.; Burke, D.; Kiernan, M.C. Excitability of sensory axons in amyotrophic lateral sclerosis. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2018, 129, 1472–1478. [Google Scholar] [CrossRef]
- Jiang, Y.M.; Yamamoto, M.; Kobayashi, Y.; Yoshihara, T.; Liang, Y.; Terao, S.; Takeuchi, H.; Ishigaki, S.; Katsuno, M.; Adachi, H.; et al. Gene expression profile of spinal motor neurons in sporadic amyotrophic lateral sclerosis. Ann. Neurol. 2005, 57, 236–251. [Google Scholar] [CrossRef]
- Shibuya, K.; Misawa, S.; Arai, K.; Nakata, M.; Kanai, K.; Yoshiyama, Y.; Ito, K.; Isose, S.; Noto, Y.; Nasu, S.; et al. Markedly reduced axonal potassium channel expression in human sporadic amyotrophic lateral sclerosis: An immunohistochemical study. Exp. Neurol. 2011, 232, 149–153. [Google Scholar] [CrossRef]
- Shibuya, K.; Park, S.B.; Geevasinga, N.; Menon, P.; Howells, J.; Simon, N.G.; Huynh, W.; Noto, Y.; Götz, J.; Kril, J.J.; et al. Motor cortical function determines prognosis in sporadic als. Neurology 2016, 87, 513–520. [Google Scholar] [CrossRef]
- Kanai, K.; Shibuya, K.; Sato, Y.; Misawa, S.; Nasu, S.; Sekiguchi, Y.; Mitsuma, S.; Isose, S.; Fujimaki, Y.; Ohmori, S.; et al. Motor axonal excitability properties are strong predictors for survival in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2012, 83, 734–738. [Google Scholar] [CrossRef] [Green Version]
- Shibuya, K.; Misawa, S.; Kimura, H.; Noto, Y.-I.; Sekiguchi, Y.; Iwai, Y.; Shimizu, T.; Mizuno, T.; Nakagawa, M.; Kuwabara, S. Increased motor axonal persistent sodium currents predict rapid functional declines in amyotrophic lateral sclerosis. Neurol. Clin. Neurosci. 2016, 4, 108–111. [Google Scholar] [CrossRef] [Green Version]
- Geevasinga, N.; Menon, P.; Ng, K.; Van Den Bos, M.; Byth, K.; Kiernan, M.C.; Vucic, S. Riluzole exerts transient modulating effects on cortical and axonal hyperexcitability in als. Amyotroph. Lateral Scler. Front. Degener. 2016, 17, 580–588. [Google Scholar] [CrossRef]
- Nakagawa, H.; Munakata, T.; Sunami, A. Mexiletine block of voltage-gated sodium channels: Isoform- and state-dependent drug-pore interactions. Mol. Pharmacol. 2019, 95, 236–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibuya, K.; Misawa, S.; Kimura, H.; Noto, Y.; Sato, Y.; Sekiguchi, Y.; Iwai, Y.; Mitsuma, S.; Beppu, M.; Watanabe, K.; et al. A single blind randomized controlled clinical trial of mexiletine in amyotrophic lateral sclerosis: Efficacy and safety of sodium channel blocker phase ii trial. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 353–358. [Google Scholar] [CrossRef]
- Weiss, M.D.; Macklin, E.A.; Simmons, Z.; Knox, A.S.; Greenblatt, D.J.; Atassi, N.; Graves, M.; Parziale, N.; Salameh, J.S.; Quinn, C.; et al. A randomized trial of mexiletine in als: Safety and effects on muscle cramps and progression. Neurology 2016, 86, 1474–1481. [Google Scholar] [CrossRef] [Green Version]
- Weiss, M.D.; Macklin, E.A.; McIlduff, C.E.; Vucic, S.; Wainger, B.J.; Kiernan, M.C.; Goutman, S.A.; Goyal, N.A.; Rutkove, S.B.; Ladha, S.S.; et al. Effects of mexiletine on hyperexcitability in sporadic amyotrophic lateral sclerosis: Preliminary findings from a small phase II randomized controlled trial. Muscle Nerve 2021, 63, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Park, S.B.; Vucic, S.; Cheah, B.C.; Lin, C.S.; Kirby, A.; Mann, K.P.; Zoing, M.C.; Winhammar, J.; Kiernan, M.C. Flecainide in amyotrophic lateral sclerosis as a neuroprotective strategy (fans): A randomized placebo-controlled trial. EBioMedicine 2015, 2, 1916–1922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, M.K. Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med. Res. Rev. 2019, 39, 733–748. [Google Scholar] [CrossRef]
- Wainger, B.J.; Macklin, E.A.; Vucic, S.; McIlduff, C.E.; Paganoni, S.; Maragakis, N.J.; Bedlack, R.; Goyal, N.A.; Rutkove, S.B.; Lange, D.J.; et al. Effect of ezogabine on cortical and spinal motor neuron excitability in amyotrophic lateral sclerosis: A randomized clinical trial. JAMA Neurol. 2021, 78, 186–196. [Google Scholar] [CrossRef]
- Oskarsson, B.; Mauricio, E.A.; Shah, J.S.; Li, Z.; Rogawski, M.A. Cortical excitability threshold can be increased by the ampa blocker perampanel in amyotrophic lateral sclerosis. Muscle Nerve 2021, 64, 215–219. [Google Scholar] [CrossRef]
- Hotait, M.; Ismail, H.H.; Saab, G.E.; Salameh, J.S. An open label pilot study of the safety and tolerability of perampanel in amyotrophic lateral sclerosis. Muscle Nerve 2021, 64, 504–508. [Google Scholar] [CrossRef]
- Aizawa, H.; Kato, H.; Oba, K.; Kawahara, T.; Okubo, Y.; Saito, T.; Naito, M.; Urushitani, M.; Tamaoka, A.; Nakamagoe, K.; et al. Randomized phase 2 study of perampanel for sporadic amyotrophic lateral sclerosis. J. Neurol. 2021, 269, 885–896. [Google Scholar] [CrossRef]
- Cappella, M.; Pradat, P.F.; Querin, G.; Biferi, M.G. Beyond the traditional clinical trials for amyotrophic lateral sclerosis and the future impact of gene therapy. J. Neuromuscul. Dis. 2021, 8, 25–38. [Google Scholar] [CrossRef]
- Cook, S.F.; Rhodes, T.; Schlusser, C.; Han, S.; Chen, C.; Zach, N.; Murthy, V.; Davé, S. A descriptive review of global real world evidence efforts to advance drug discovery and clinical development in amyotrophic lateral sclerosis. Front. Neurol. 2021, 12, 770001. [Google Scholar] [CrossRef]
- Ranieri, F.; Mariotto, S.; Dubbioso, R.; Di Lazzaro, V. Brain stimulation as a therapeutic tool in amyotrophic lateral sclerosis: Current status and interaction with mechanisms of altered cortical excitability. Front. Neurol. 2020, 11, 605335. [Google Scholar] [CrossRef]
- Edmond, E.C.; Stagg, C.J.; Turner, M.R. Therapeutic non-invasive brain stimulation in amyotrophic lateral sclerosis: Rationale, methods and experience. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Oriuwa, C.; Mollica, A.; Feinstein, A.; Giacobbe, P.; Lipsman, N.; Perez, D.L.; Burke, M.J. Neuromodulation for the treatment of functional neurological disorder and somatic symptom disorder: A systematic review. J. Neurol. Neurosurg. Psychiatry 2022, 93, 280–290. [Google Scholar] [CrossRef]
- Di Lazzaro, V.; Ranieri, F.; Capone, F.; Musumeci, G.; Dileone, M. Direct current motor cortex stimulation for amyotrophic lateral sclerosis: A proof of principle study. Brain Stimul. 2013, 6, 969–970. [Google Scholar] [CrossRef]
- Madhavan, S.; Sivaramakrishnan, A.; Bond, S.; Jiang, Q.M. Safety and feasibility of transcranial direct current stimulation in amyotrophic lateral sclerosis—A pilot study with a single subject experimental design. Physiother. Theory Pract. 2019, 35, 458–463. [Google Scholar] [CrossRef]
- Sivaramakrishnan, A.; Datta, A.; Bikson, M.; Madhavan, S. Remotely supervised transcranial direct current stimulation: A feasibility study for amyotrophic lateral sclerosis. NeuroRehabilitation 2019, 45, 369–378. [Google Scholar] [CrossRef]
- Munneke, M.A.; Stegeman, D.F.; Hengeveld, Y.A.; Rongen, J.J.; Schelhaas, H.J.; Zwarts, M.J. Transcranial direct current stimulation does not modulate motor cortex excitability in patients with amyotrophic lateral sclerosis. Muscle Nerve 2011, 44, 109–114. [Google Scholar] [CrossRef] [Green Version]
- Benussi, A.; Alberici, A.; Cotelli, M.S.; Dell’Era, V.; Cantoni, V.; Bonetta, E.; Manenti, R.; Filosto, M.; Morini, R.; Datta, A.; et al. Cortico-spinal tdcs in als: A randomized, double-blind, sham-controlled trial. Brain Stimul. 2019, 12, 1332–1334. [Google Scholar] [CrossRef]
- George, M.S.; Caulfield, K.A.; Wiley, M. Shaping plasticity with non-invasive brain stimulation in the treatment of psychiatric disorders: Present and future. Handb. Clin. Neurol. 2022, 184, 497–507. [Google Scholar]
- Arrington, C.N.; Ossowski, A.E.; Baig, H.; Persichetti, E.; Morris, R. The impact of transcranial magnetic stimulation on reading processes: A systematic review. Neuropsychol. Rev. 2022. [Google Scholar] [CrossRef]
- Tang, V.M.; Le Foll, B.; Blumberger, D.M.; Voineskos, D. Repetitive transcranial magnetic stimulation for comorbid major depressive disorder and alcohol use disorder. Brain Sci. 2021, 12, 48. [Google Scholar] [CrossRef]
- Di Lazzaro, V.; Oliviero, A.; Saturno, E.; Pilato, F.; Dileone, M.; Sabatelli, M.; Tonali, P.A. Motor cortex stimulation for amyotrophic lateral sclerosis. Time for a therapeutic trial? Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2004, 115, 1479–1485. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, F.; Oliviero, A.; Pilato, F.; Saturno, E.; Dileone, M.; Versace, V.; Musumeci, G.; Batocchi, A.P.; Tonali, P.A.; Di Lazzaro, V. Transcranial magnetic stimulation and bdnf plasma levels in amyotrophic lateral sclerosis. Neuroreport 2004, 15, 717–720. [Google Scholar] [CrossRef] [PubMed]
- Zanette, G.; Forgione, A.; Manganotti, P.; Fiaschi, A.; Tamburin, S. The effect of repetitive transcranial magnetic stimulation on motor performance, fatigue and quality of life in amyotrophic lateral sclerosis. J. Neurol. Sci. 2008, 270, 18–22. [Google Scholar] [CrossRef]
- Di Lazzaro, V.; Dileone, M.; Pilato, F.; Profice, P.; Ranieri, F.; Musumeci, G.; Angelucci, F.; Sabatelli, M.; Tonali, P.A. Repetitive transcranial magnetic stimulation for als. A preliminary controlled study. Neurosci. Lett. 2006, 408, 135–140. [Google Scholar] [CrossRef]
- Di Lazzaro, V.; Pilato, F.; Profice, P.; Ranieri, F.; Musumeci, G.; Florio, L.; Beghi, E.; Frisullo, G.; Capone, F.; Sabatelli, M.; et al. Motor cortex stimulation for als: A double blind placebo-controlled study. Neurosci. Lett. 2009, 464, 18–21. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shibuya, K.; Otani, R.; Suzuki, Y.-i.; Kuwabara, S.; Kiernan, M.C. Neuronal Hyperexcitability and Free Radical Toxicity in Amyotrophic Lateral Sclerosis: Established and Future Targets. Pharmaceuticals 2022, 15, 433. https://doi.org/10.3390/ph15040433
Shibuya K, Otani R, Suzuki Y-i, Kuwabara S, Kiernan MC. Neuronal Hyperexcitability and Free Radical Toxicity in Amyotrophic Lateral Sclerosis: Established and Future Targets. Pharmaceuticals. 2022; 15(4):433. https://doi.org/10.3390/ph15040433
Chicago/Turabian StyleShibuya, Kazumoto, Ryo Otani, Yo-ichi Suzuki, Satoshi Kuwabara, and Matthew C. Kiernan. 2022. "Neuronal Hyperexcitability and Free Radical Toxicity in Amyotrophic Lateral Sclerosis: Established and Future Targets" Pharmaceuticals 15, no. 4: 433. https://doi.org/10.3390/ph15040433
APA StyleShibuya, K., Otani, R., Suzuki, Y.-i., Kuwabara, S., & Kiernan, M. C. (2022). Neuronal Hyperexcitability and Free Radical Toxicity in Amyotrophic Lateral Sclerosis: Established and Future Targets. Pharmaceuticals, 15(4), 433. https://doi.org/10.3390/ph15040433