
Hybridization Approach to Identify Salicylanilides as Inhibitors of Tubulin Polymerization and Signal Transducers and Activators of Transcription 3 (STAT3)

 , , and
, , and 
Abstract
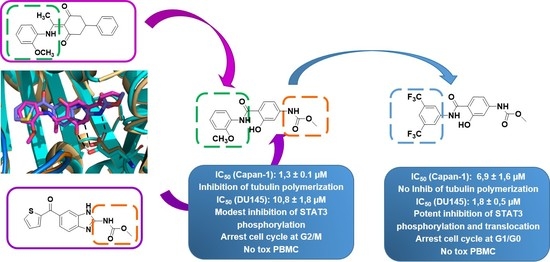
:
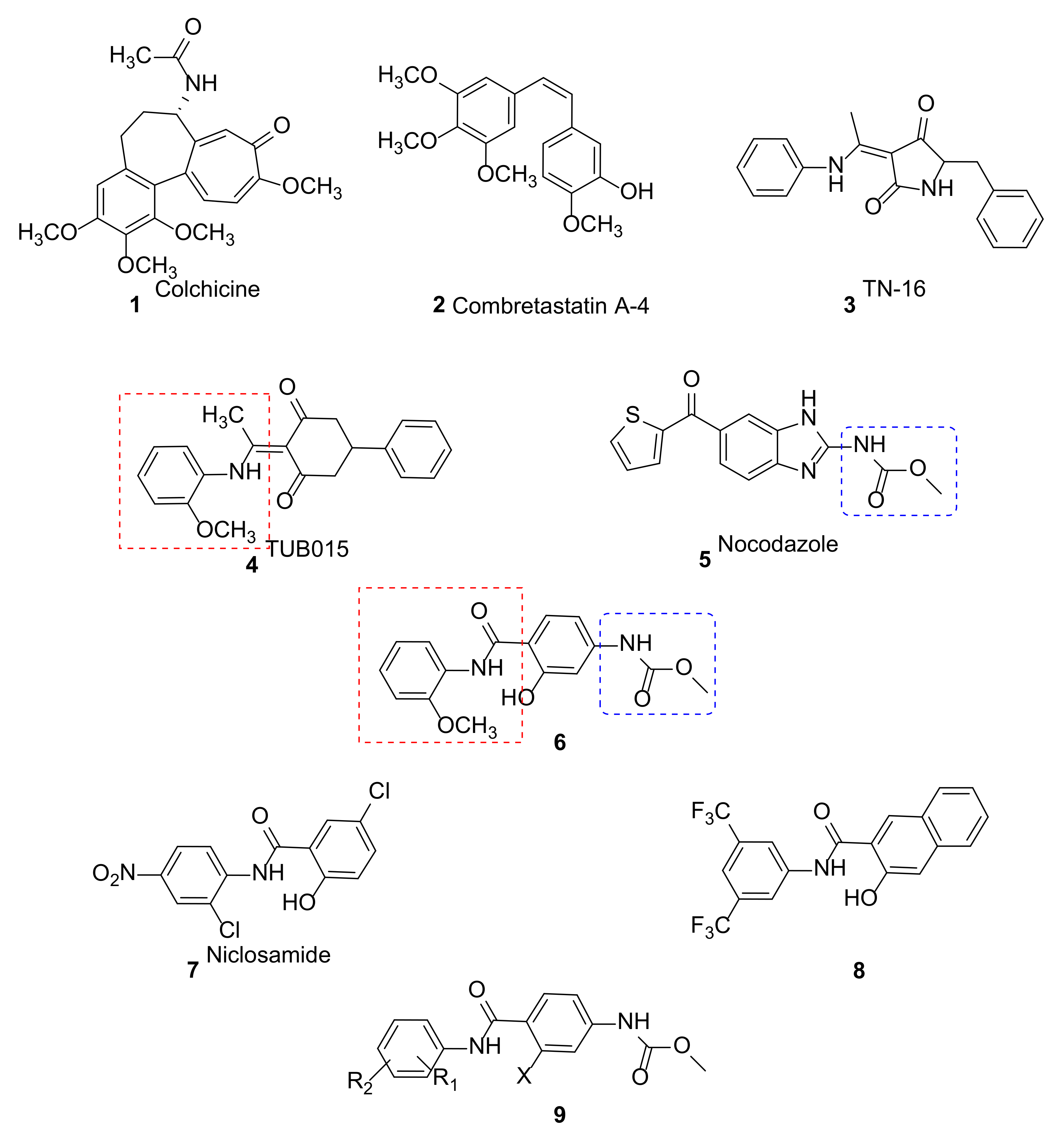
1. Introduction
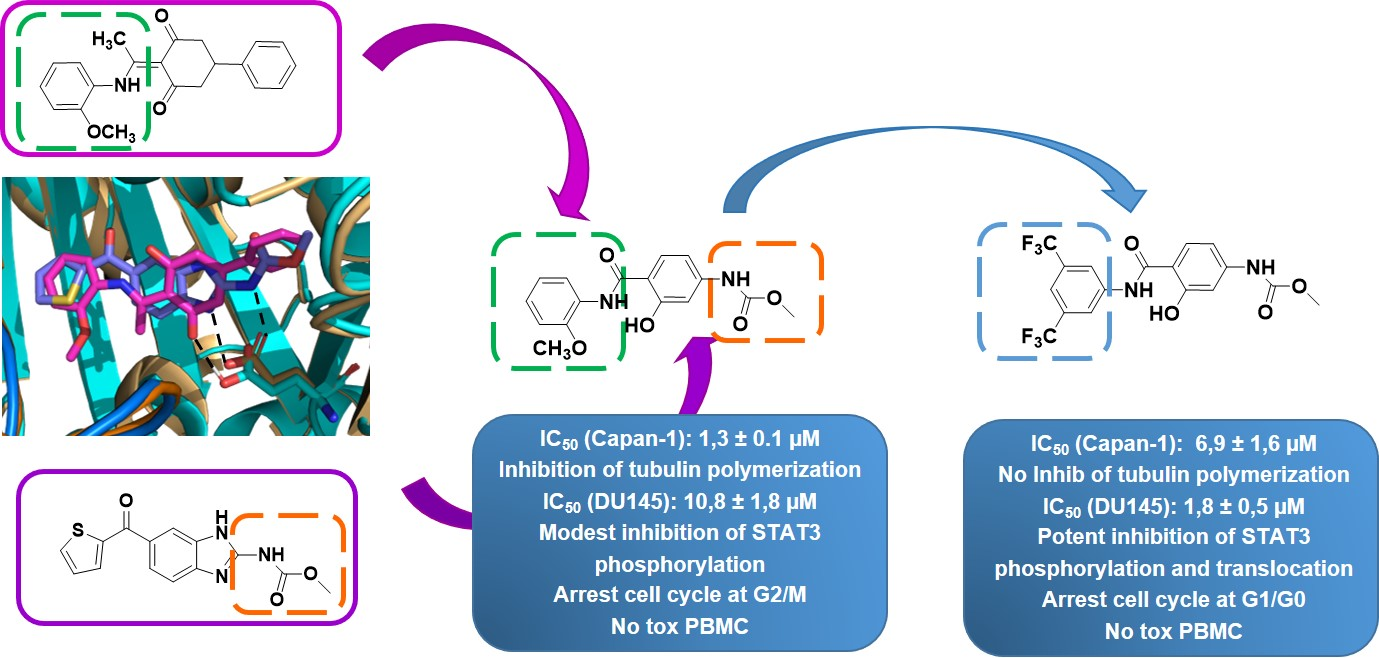
2. Results and Discussion
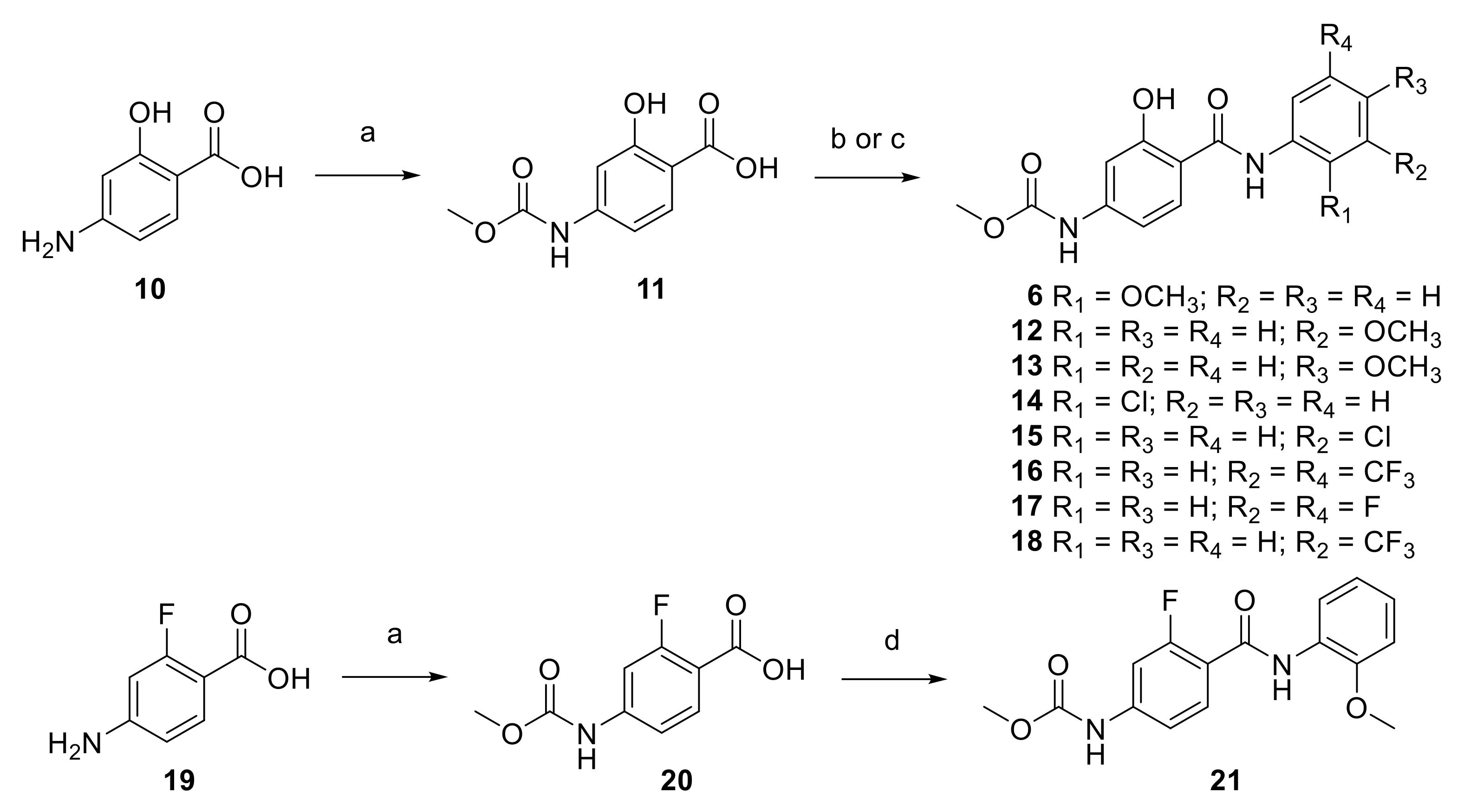
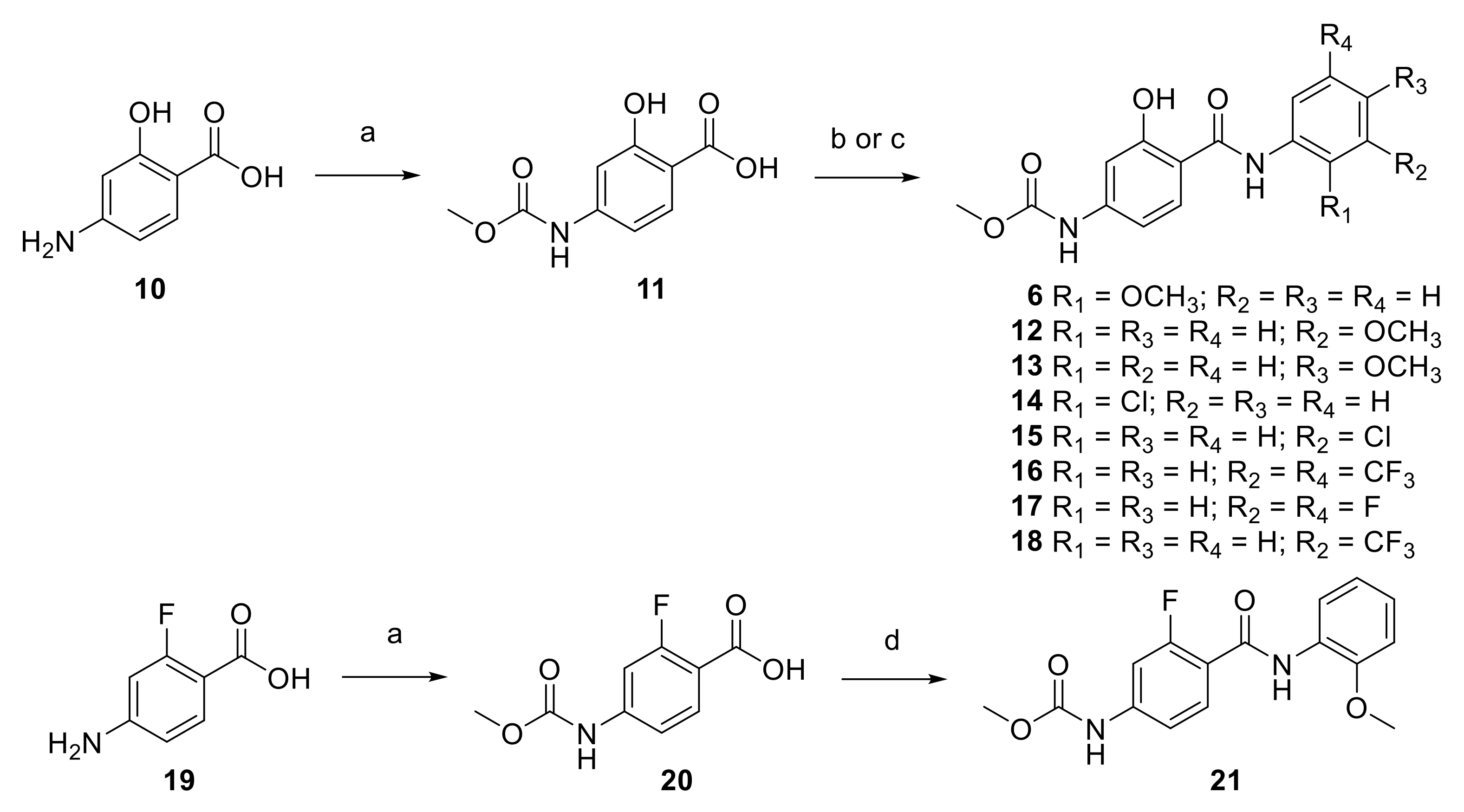
2.1. Synthesis
2.2. Biological Evaluation
2.2.1. Antiproliferative Activity
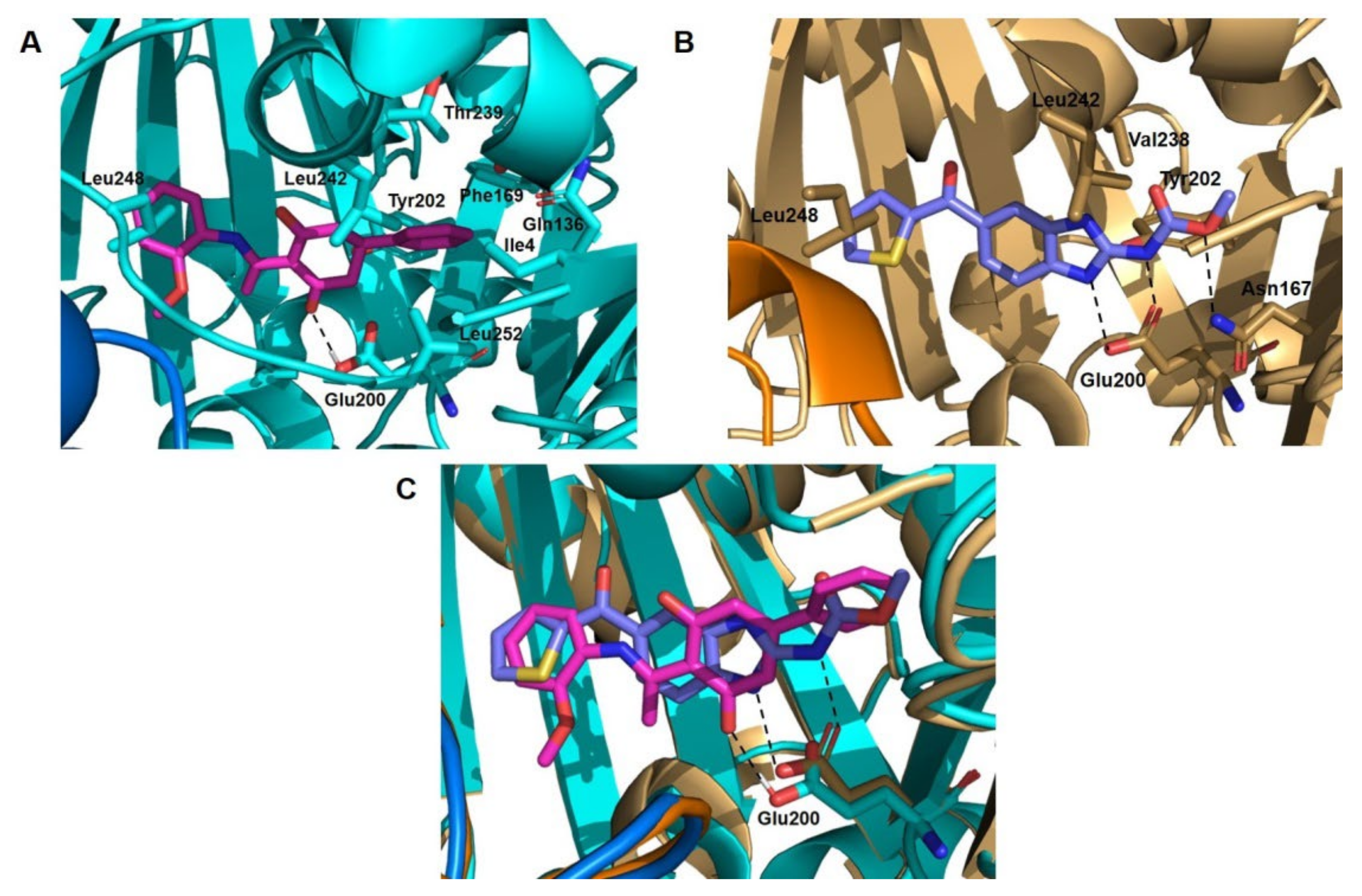
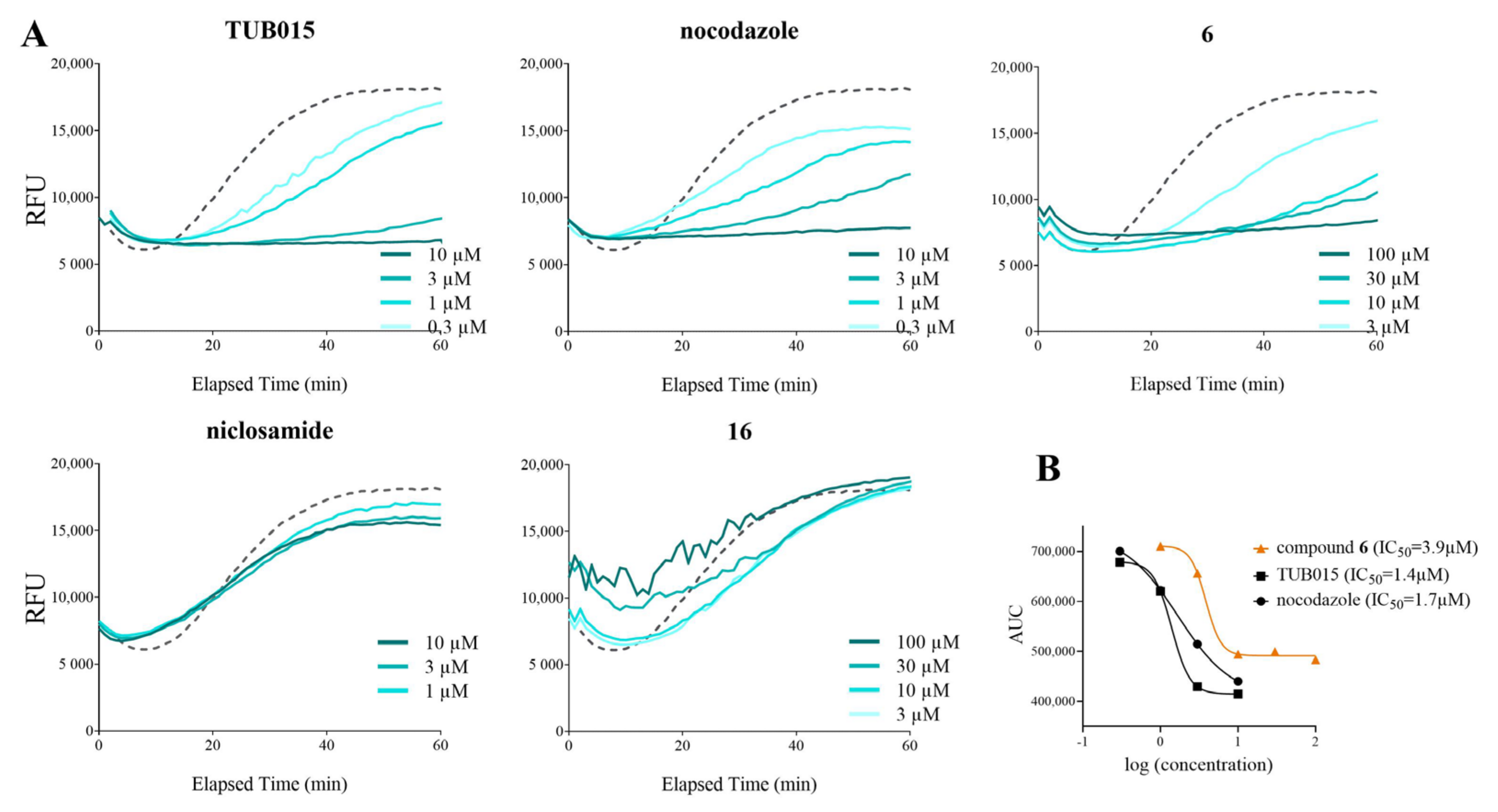
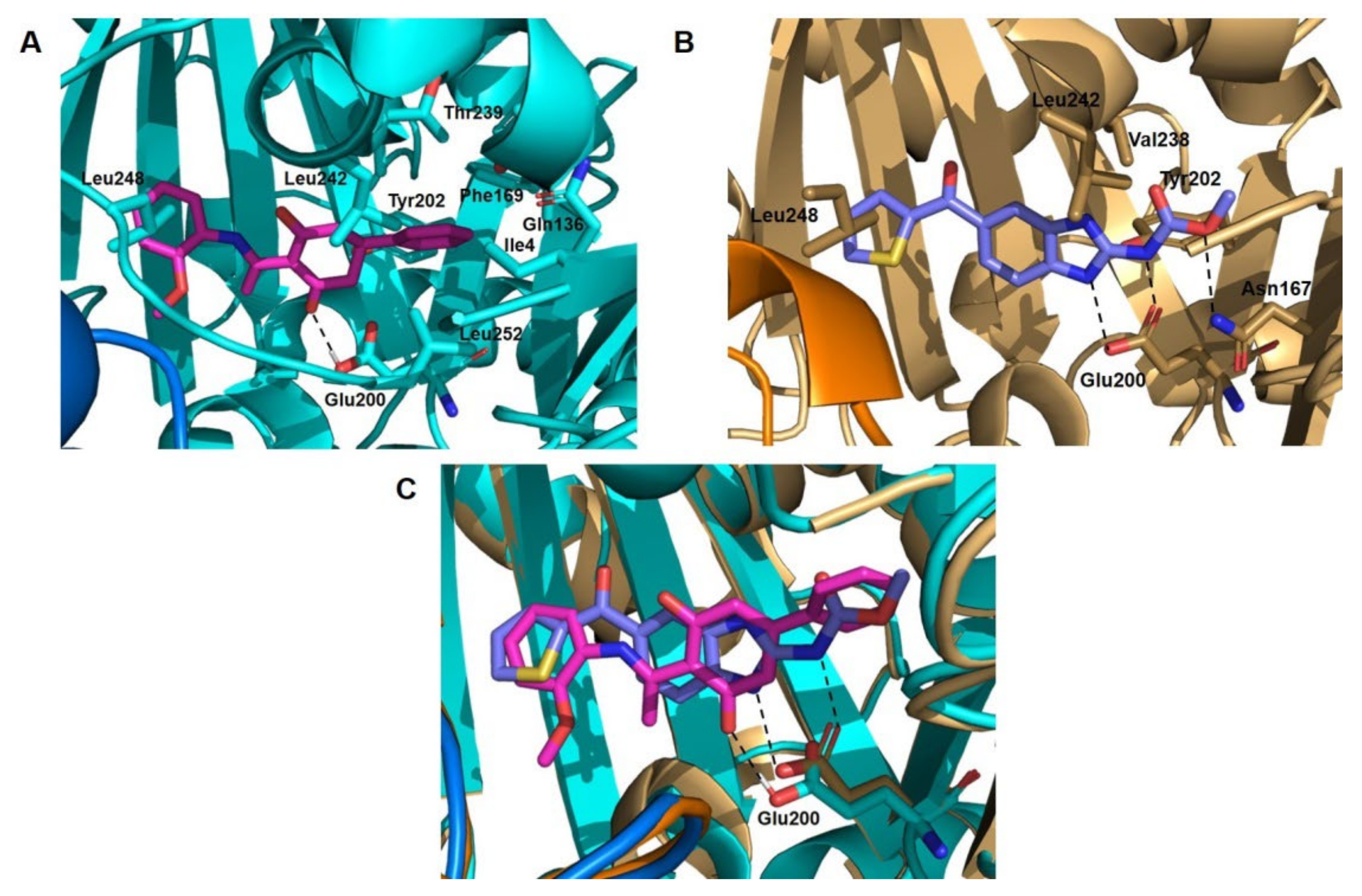
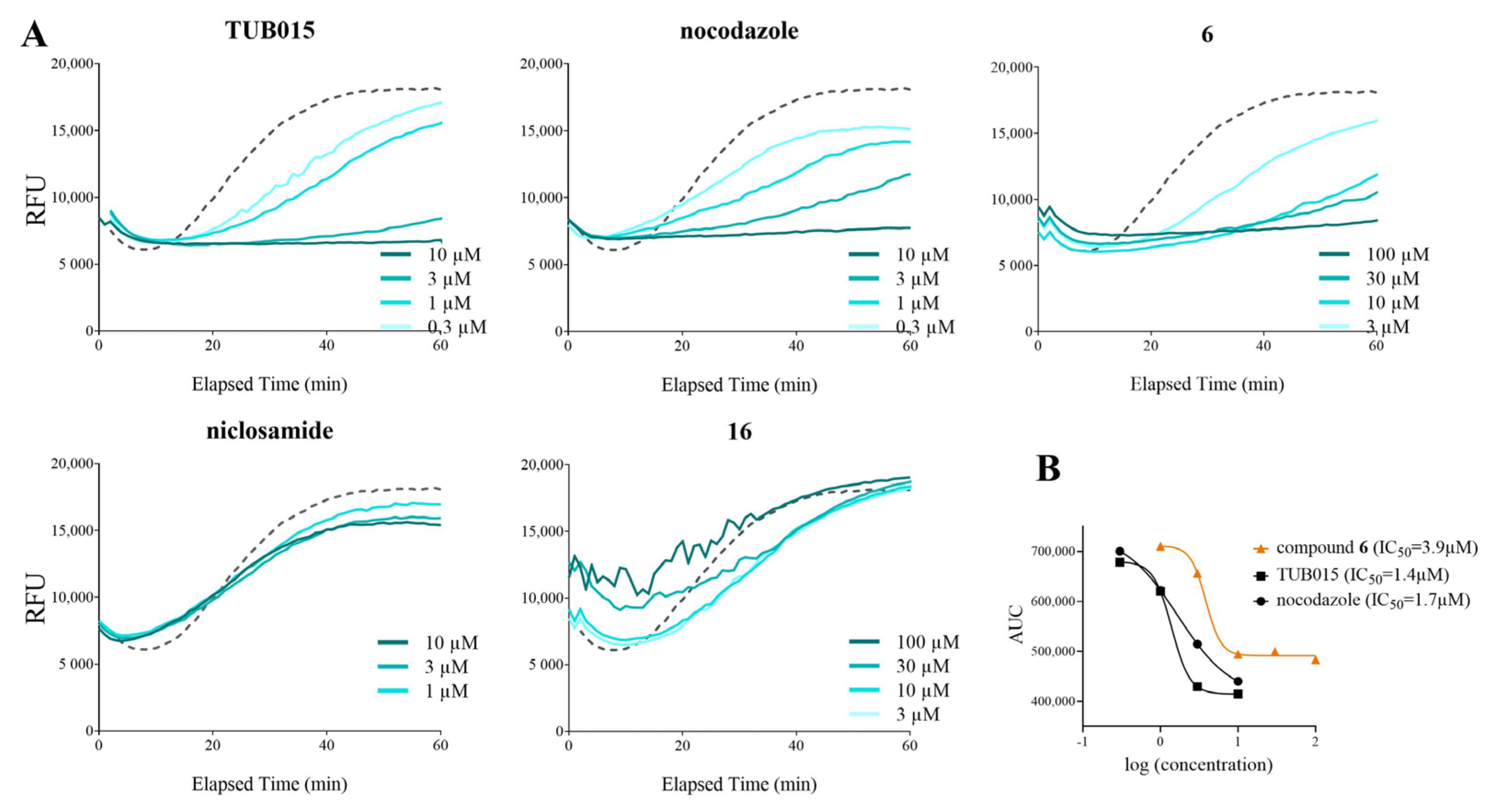
2.2.2. Inhibition of Tubulin Polymerization
2.2.3. Antiproliferative Activity in Human Prostate Cancer Cells DU145
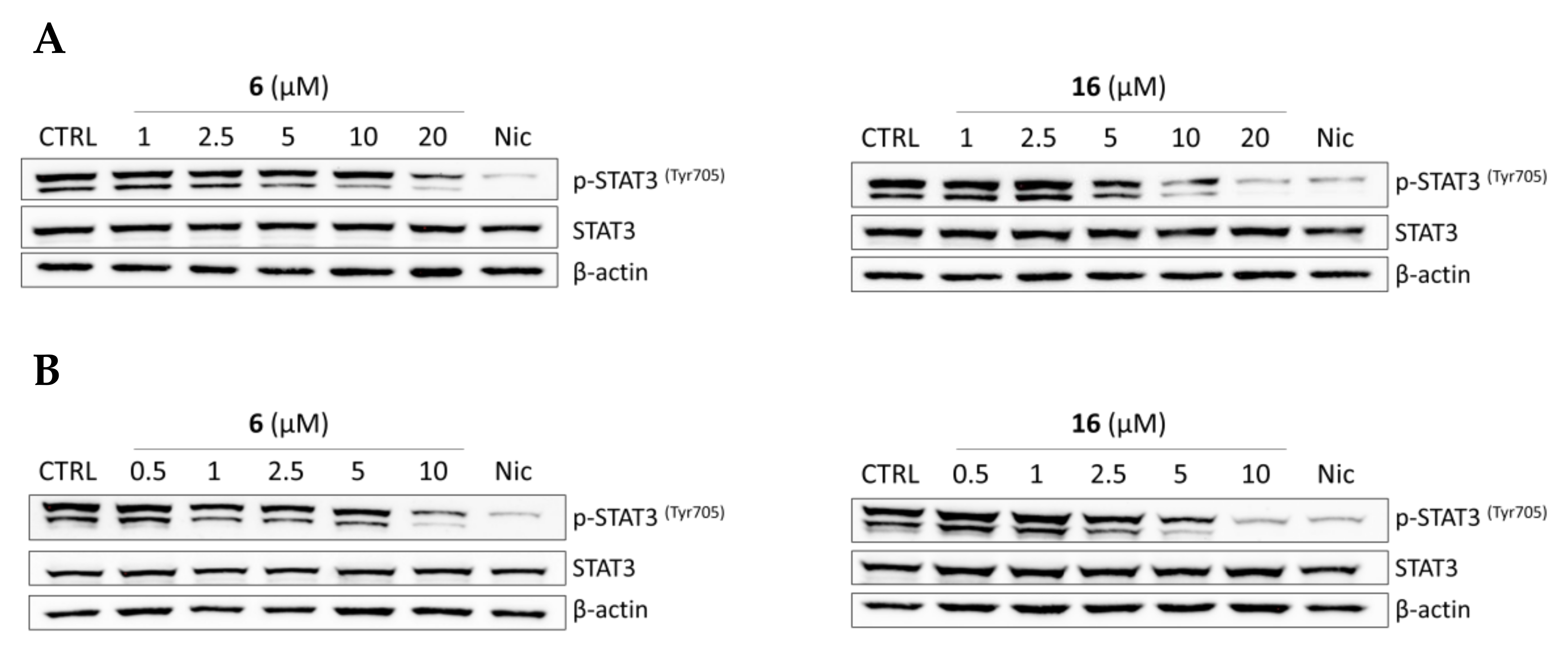
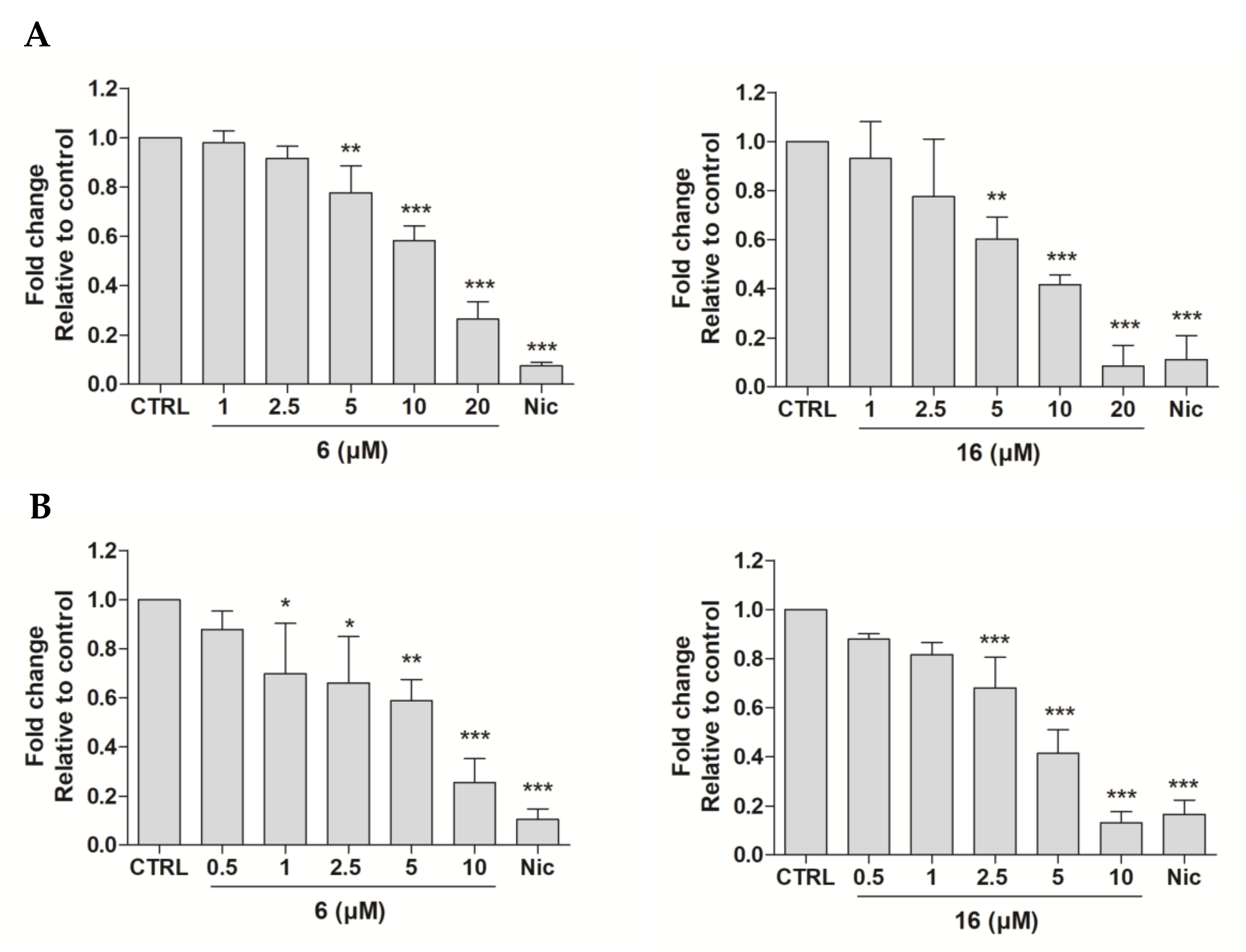
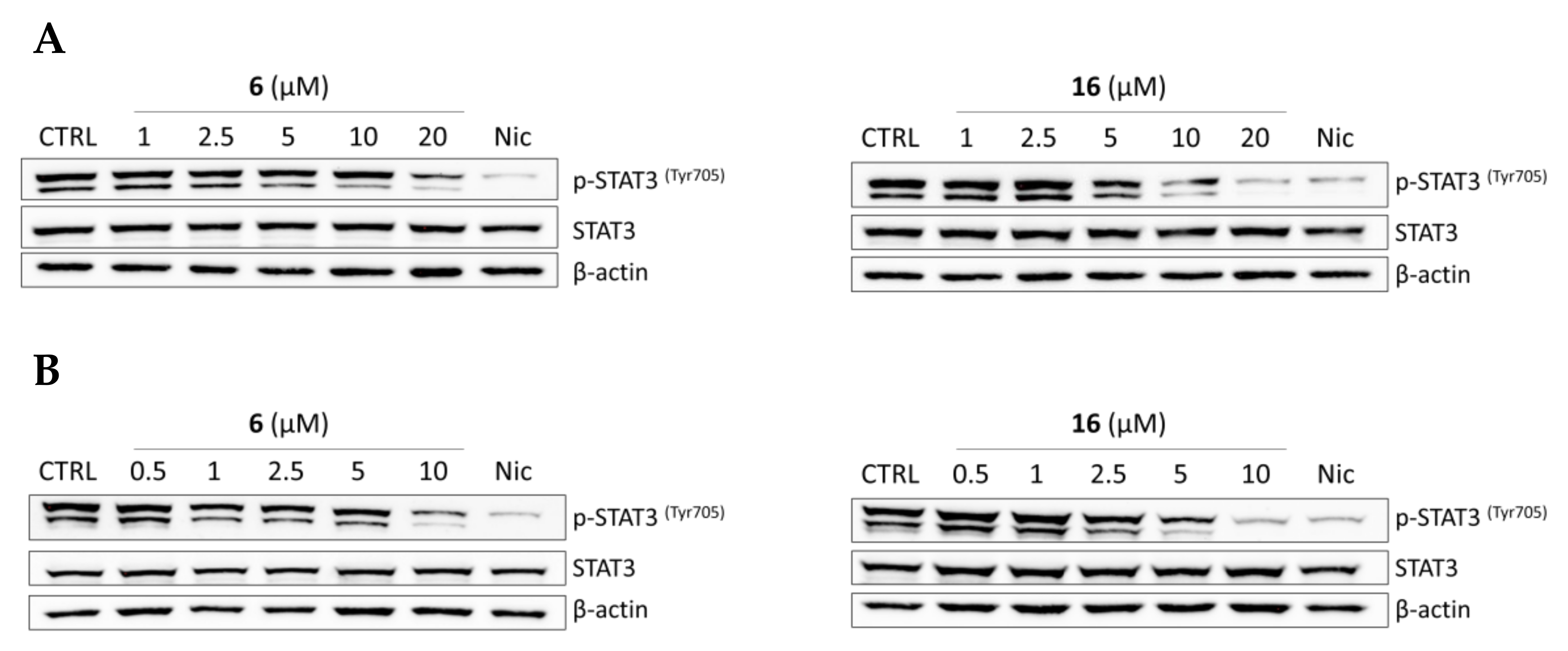
2.2.4. Effects of Compounds 6 and 16 in Regulating STAT3 Signaling Pathway In Vitro
2.2.5. STAT3 Translocation Experiments
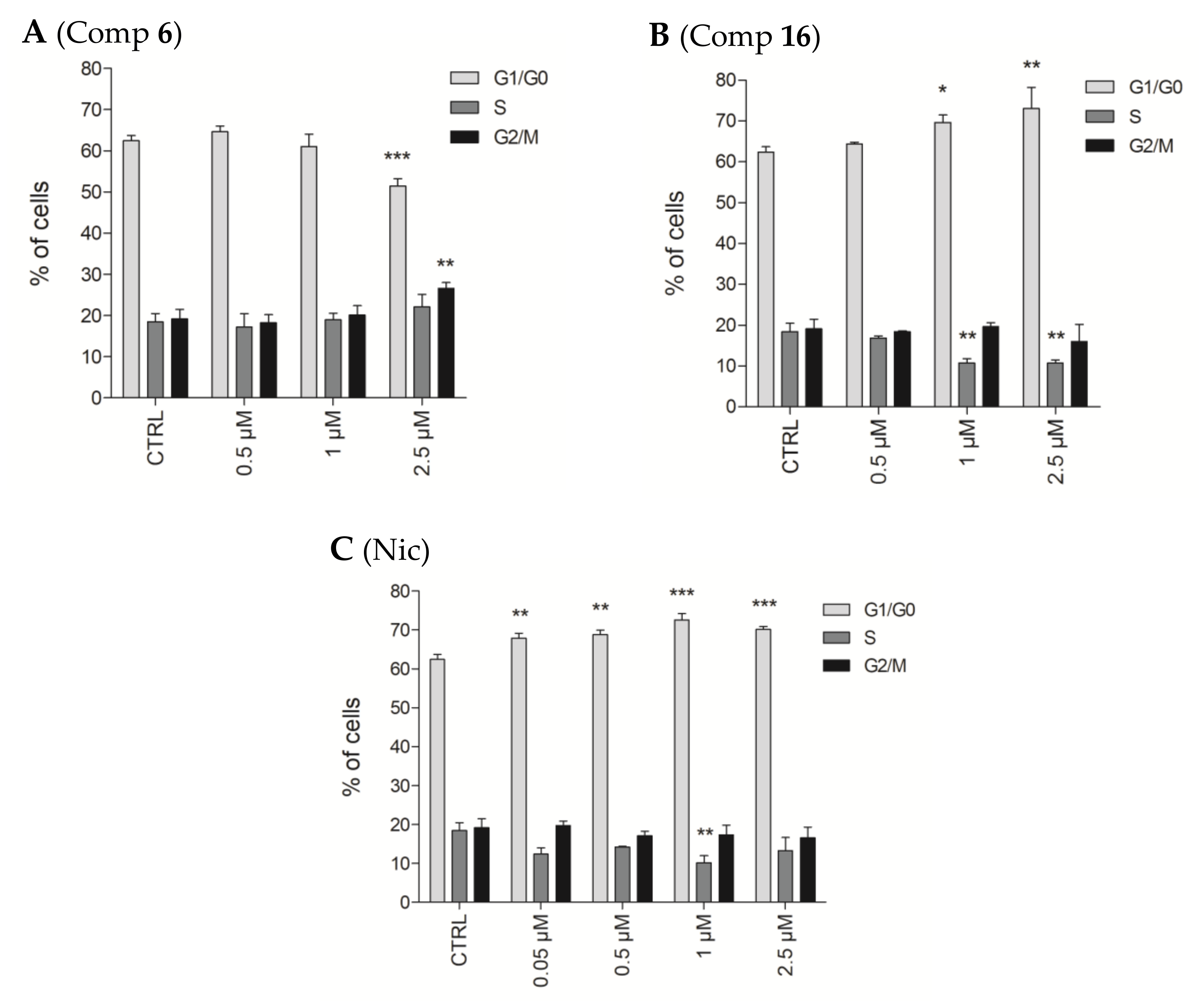
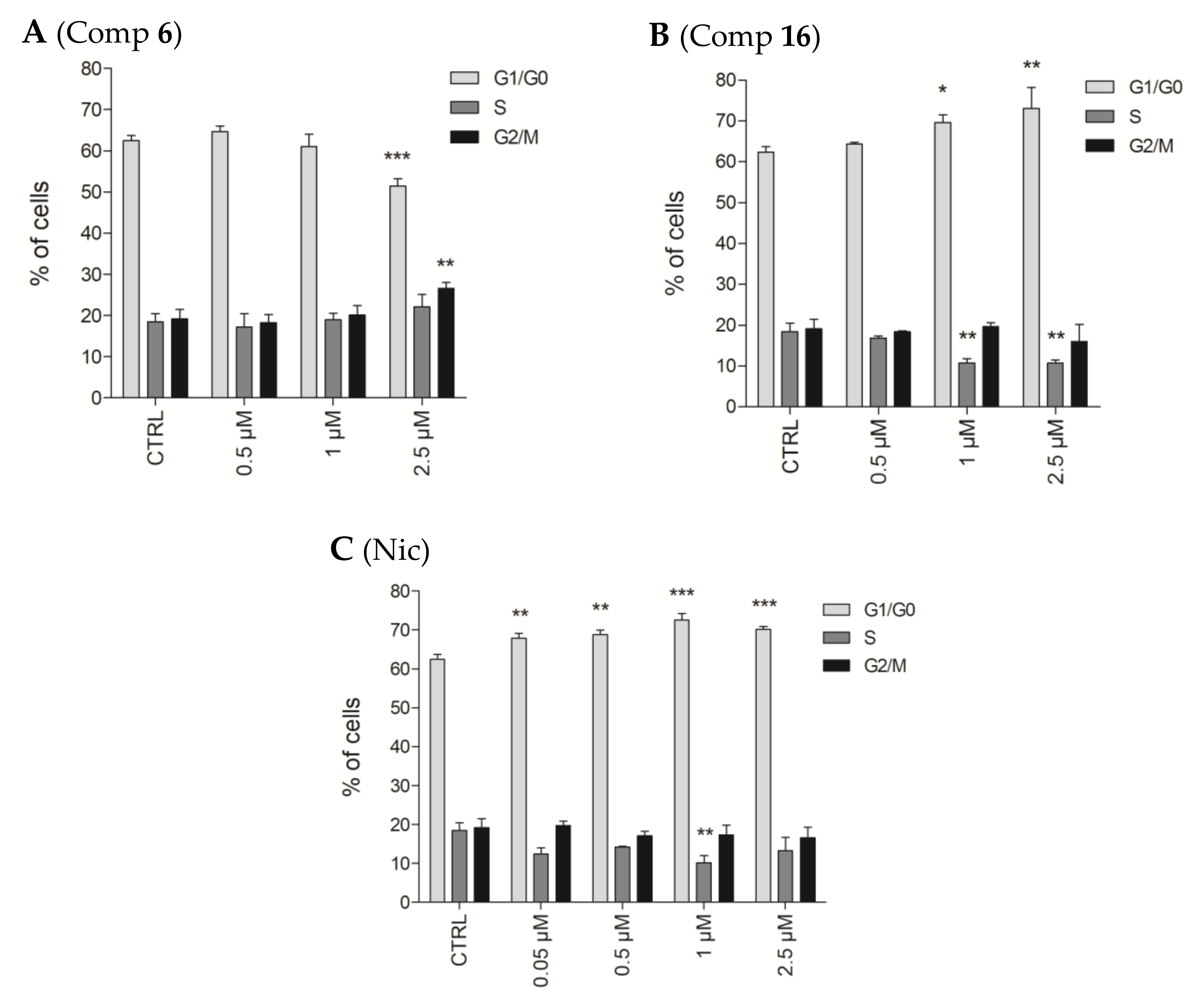
2.2.6. Effects of Compounds 6 and 16 in Cell Cycle Progression
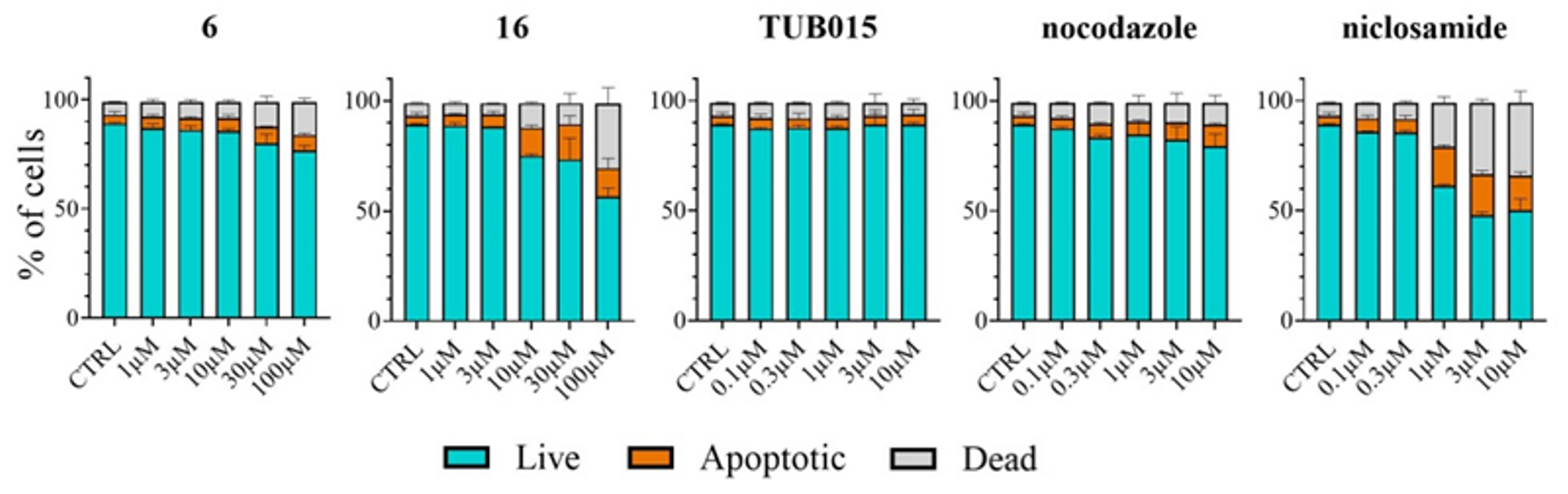
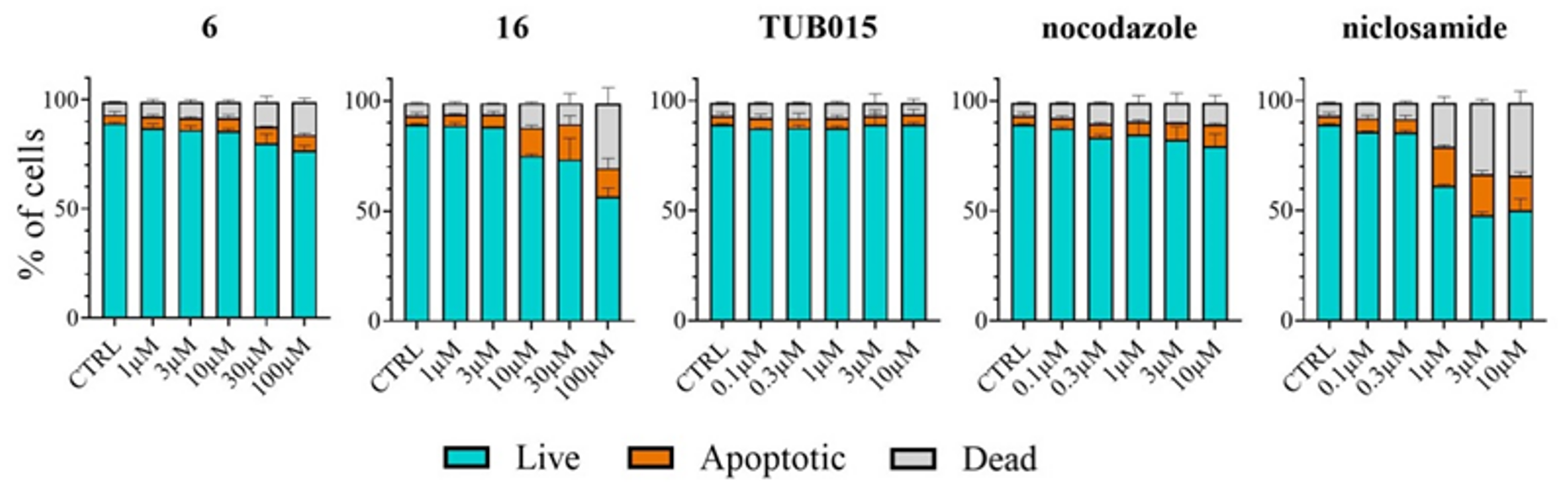
2.2.7. Analysis of Apoptosis Induction in PBMC
3. Materials and Methods
3.1. Chemistry Procedures
3.2. Biological Assays
3.2.1. Cell Culture and Reference Compounds
3.2.2. Cell Proliferation Assays
3.2.3. WST-1 Analysis of Cell Proliferation
3.2.4. In Vitro Tubulin Polymerization Assay
3.2.5. Tubulin Immunofluorescence Staining
3.2.6. Western Blotting
3.2.7. Analysis of STAT3 Nuclear Translocation
3.2.8. Cell Cycle Analysis
3.2.9. Statistical Analysis
3.2.10. Apoptosis Induction Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bates, D.; Eastman, A. Microtubule Destabilising Agents: Far More than Just Antimitotic Anticancer Drugs. Br. J. Clin. Pharmacol. 2017, 83, 255–268. [Google Scholar] [CrossRef] [Green Version]
- Leung, Y.Y.; Yao Hui, L.L.; Kraus, V.B. Colchicine—Update on Mechanisms of Action and Therapeutic Uses. Semin. Arthritis Rheum. 2015, 45, 341–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes, A.Z.; Hu, K.A.; Teperman, J.; Wampler Muskardin, T.L.; Tardif, J.-C.; Shah, B.; Pillinger, M.H. Anti-Inflammatory Therapy for COVID-19 Infection: The Case for Colchicine. Ann. Rheum. Dis. 2021, 80, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Colchicine. COVID-19 Treatment Guidelines. Available online: https://www.covid19treatmentguidelines.nih.gov/therapies/immunomodulators/colchicine/ (accessed on 22 February 2022).
- Greene, L.M.; Meegan, M.J.; Zisterer, D.M. Combretastatins: More than Just Vascular Targeting Agents? J. Pharmacol. Exp. Ther. 2015, 355, 212–227. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Pérez, M.-J.; Priego, E.-M.; Bueno, O.; Martins, M.S.; Canela, M.-D.; Liekens, S. Blocking Blood Flow to Solid Tumors by Destabilizing Tubulin: An Approach to Targeting Tumor Growth. J. Med. Chem. 2016, 59, 8685–8711. [Google Scholar] [CrossRef] [Green Version]
- McLoughlin, E.C.; O’Boyle, N.M. Colchicine-Binding Site Inhibitors from Chemistry to Clinic: A Review. Pharmaceuticals 2020, 13, 8. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Chen, J.; Xiao, M.; Li, W.; Miller, D.D. An Overview of Tubulin Inhibitors That Interact with the Colchicine Binding Site. Pharm. Res. 2012, 29, 2943–2971. [Google Scholar] [CrossRef] [Green Version]
- Canela, M.-D.; Pérez-Pérez, M.-J.; Noppen, S.; Sáez-Calvo, G.; Díaz, J.F.F.; Camarasa, M.-J.; Liekens, S.; Priego, E.-M. Novel Colchicine-Site Binders with a Cyclohexanedione Scaffold Identified through a Ligand-Based Virtual Screening Approach. J. Med. Chem. 2014, 57, 3924–3938. [Google Scholar] [CrossRef] [Green Version]
- Bueno, O.; Estévez Gallego, J.; Martins, S.; Prota, A.E.; Gago, F.; Gómez-Sanjuan, A.; Camarasa, M.J.; Barasoain, I.; Steinmetz, M.O.; Díaz, J.F.; et al. High-Affinity Ligands of the Colchicine Domain in Tubulin Based on a Structure-Guided Design. Sci. Rep. 2018, 8, 4242. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, H.; Gigant, B.; Yu, Y.; Wu, Y.; Chen, X.; Lai, Q.; Yang, Z.; Chen, Q.; Yang, J. Structures of a Diverse Set of Colchicine Binding Site Inhibitors in Complex with Tubulin Provide a Rationale for Drug Discovery. FEBS J. 2016, 283, 102–111. [Google Scholar] [CrossRef]
- Satoh, K.; Zhang, L.; Zhang, Y.; Chelluri, R.; Boufraqech, M.; Nilubol, N.; Patel, D.; Shen, M.; Kebebew, E. Identification of Niclosamide as a Novel Anticancer Agent for Adrenocortical Carcinoma. Clin. Cancer Res. 2016, 22, 3458–3466. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Mook, R.A., Jr.; Premont, R.T.; Wang, J. Niclosamide: Beyond an Antihelminthic Drug. Cell Signal 2018, 41, 89–96. [Google Scholar] [CrossRef]
- Ren, X.; Duan, L.; He, Q.; Zhang, Z.; Zhou, Y.; Wu, D.; Pan, J.; Pei, D.; Ding, K. Identification of Niclosamide as a New Small-Molecule Inhibitor of the STAT3 Signaling Pathway. ACS Med. Chem. Lett. 2010, 1, 454–459. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; You, S.; Hu, Z.; Chen, Z.G.; Sica, G.L.; Khuri, F.R.; Curran, W.J.; Shin, D.M.; Deng, X. Inhibition of STAT3 by Niclosamide Synergizes with Erlotinib against Head and Neck Cancer. PLoS ONE 2013, 8, e74670. [Google Scholar] [CrossRef]
- Kusaba, T.; Nakayama, T.; Yamazumi, K.; Yakata, Y.; Yoshizaki, A.; Inoue, K.; Nagayasu, T.; Sekine, I. Activation of STAT3 Is a Marker of Poor Prognosis in Human Colorectal Cancer. Oncol. Rep. 2006, 15, 1445–1451. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Zhong, Y.; Dong, H.; Zheng, Q.; Shi, S.; Zhu, K.; Qu, X.; Hu, W.; Zhang, X.; Wang, Y. Revisiting Signal Transducer and Activator of Transcription 3 (STAT3) as an Anticancer Target and Its Inhibitor Discovery: Where Are We and Where Should We Go? Eur. J. Med. Chem. 2020, 187, 111922. [Google Scholar] [CrossRef]
- Luo, F.; Luo, M.; Rong, Q.-X.; Zhang, H.; Chen, Z.; Wang, F.; Zhao, H.-Y.; Fu, L.-W. Niclosamide, an Antihelmintic Drug, Enhances Efficacy of PD-1/PD-L1 Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. J. Immunother. Cancer 2019, 7, 245. [Google Scholar] [CrossRef]
- Zou, S.; Tong, Q.; Liu, B.; Huang, W.; Tian, Y.; Fu, X. Targeting STAT3 in Cancer Immunotherapy. Mol. Cancer 2020, 19, 145. [Google Scholar] [CrossRef]
- Kauerová, T.; Goněc, T.; Jampílek, J.; Hafner, S.; Gaiser, A.-K.; Syrovets, T.; Fedr, R.; Souček, K.; Kollar, P. Ring-Substituted 1-Hydroxynaphthalene-2-Carboxanilides Inhibit Proliferation and Trigger Mitochondria-Mediated Apoptosis. Int. J. Mol. Sci. 2020, 21, 3416. [Google Scholar] [CrossRef]
- Maly, D.J.; Leonetti, F.; Backes, B.J.; Dauber, D.S.; Harris, J.L.; Craik, C.S.; Ellman, J.A. Expedient Solid-Phase Synthesis of Fluorogenic Protease Substrates Using the 7-Amino-4-Carbamoylmethylcoumarin (ACC) Fluorophore. J. Org. Chem. 2002, 67, 910–915. [Google Scholar] [CrossRef]
- Dahlgren, M.K.; Kauppi, A.M.; Olsson, I.-M.; Linusson, A.; Elofsson, M. Design, Synthesis, and Multivariate Quantitative Structure−Activity Relationship of Salicylanilides Potent Inhibitors of Type III Secretion in Yersinia. J. Med. Chem. 2007, 50, 6177–6188. [Google Scholar] [CrossRef]
- Singh, M.; Singh, S.K.; Gangwar, M.; Nath, G.; Singh, S.K. Design, Synthesis and Mode of Action of Some Benzothiazole Derivatives Bearing an Amide Moiety as Antibacterial Agents. RSC Adv. 2014, 4, 19013–19023. [Google Scholar] [CrossRef]
- Kang, S.; Min, H.-J.; Kang, M.-S.; Jung, M.-G.; Kim, S. Discovery of Novel 2-Hydroxydiarylamide Derivatives as TMPRSS4 Inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 1748–1751. [Google Scholar] [CrossRef]
- Li, C.; Ren, S.-F.; Hou, J.-L.; Yi, H.-P.; Zhu, S.-Z.; Jiang, X.-K.; Li, Z.-T. F⋯H–N Hydrogen Bonding Driven Foldamers: Efficient Receptors for Dialkylammonium Ions. Angew. Chem. Int. Ed. 2005, 44, 5725–5729. [Google Scholar] [CrossRef]
- Racine, E.; Monnier, F.; Vors, J.-P.; Taillefer, M. A Simple Copper-Catalyzed Synthesis of Tertiary Acyclic Amides. Org. Lett. 2011, 13, 2818–2821. [Google Scholar] [CrossRef]
- Shin, D.-S.; Kim, H.-N.; Shin, K.D.; Yoon, Y.J.; Kim, S.-J.; Han, D.C.; Kwon, B.-M. Cryptotanshinone Inhibits Constitutive Signal Transducer and Activator of Transcription 3 Function through Blocking the Dimerization in DU145 Prostate Cancer Cells. Cancer Res. 2009, 69, 193–202. [Google Scholar] [CrossRef] [Green Version]
- Furtek, S.L.; Backos, D.S.; Matheson, C.J.; Reigan, P. Strategies and Approaches of Targeting STAT3 for Cancer Treatment. ACS Chem. Biol. 2016, 11, 308–318. [Google Scholar] [CrossRef]
- Kauerova, T.; Kos, J.; Gonec, T.; Jampilek, J.; Kollar, P. Antiproliferative and Pro-Apoptotic Effect of Novel Nitro-Substituted Hydroxynaphthanilides on Human Cancer Cell Lines. Int. J. Mol. Sci. 2016, 17, 1219. [Google Scholar] [CrossRef]
- Kollár, P.; Bárta, T.; Závalová, V.; Smejkal, K.; Hampl, A. Geranylated Flavanone Tomentodiplacone B Inhibits Proliferation of Human Monocytic Leukaemia (THP-1) Cells. Br. J. Pharmacol. 2011, 162, 1534–1541. [Google Scholar] [CrossRef] [Green Version]
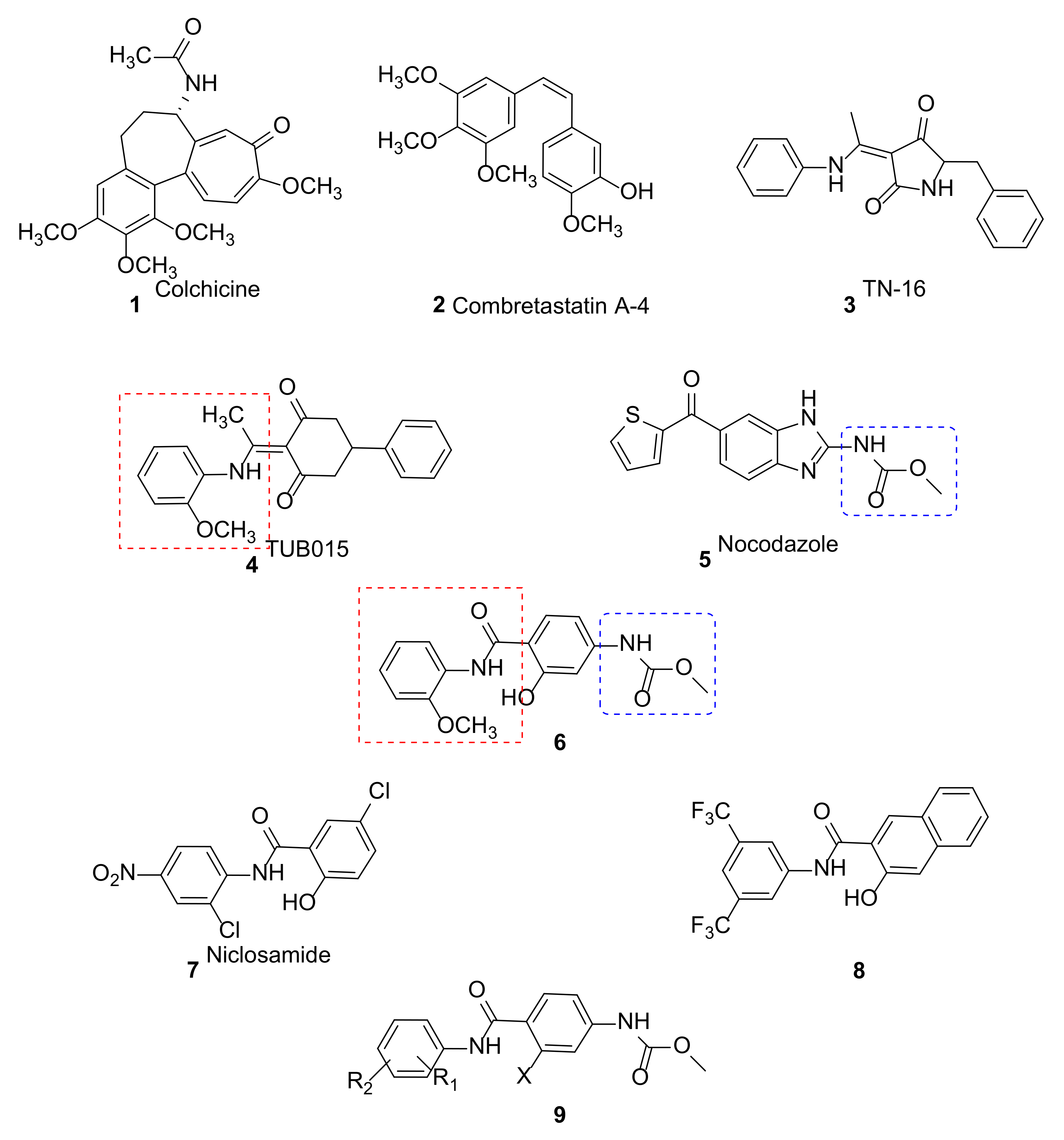


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (µM) a | |||||||
|---|---|---|---|---|---|---|---|
| Comp | Capan-1 b | HCT-116 b | NCI-H460 b | DND-41 b | HL-60 b | K-562 b | Z-138 b |
| 6 | 1.3 ± 0.1 | 1.4 ± 3.5 | 2.8 ± 1.6 | 1.3 ± 0.5 | 1.5 ± 0.1 | 1.3 ± 0.1 | 1.3 ± 0.7 |
| 12 | >100 | >100 | >100 | >100 | >100 | ≥84.5 | >100 |
| 13 | 35.3 ± 20.1 | 39.8 ± 25.0 | 33.3 ± 0.2 | ≥39.4 | 37.3 ± 20.4 | ≥67.8 | >100 |
| 14 | 2.5 ± 0.6 | 10.4 ± 1.6 | 15.2 ± 6.3 | 3.8 ± 0.2 | 9.1 ± 1.9 | 2.9 ± 1.1 | 13.5 ± 3.3 |
| 15 | >100 | >100 | >100 | ≥74.1 | ≥89.6 | ≥75.8 | ≥88.4 |
| 16 | 6.9 ± 1.6 | 4.6 ± 0.07 | 9.2 ± 3.9 | 8.7 ± 1.2 | 4.8 ± 4.5 | 6.7 ± 2.4 | 12.2 ± 4.0 |
| 17 | 49.9 ± 28.5 | ≥47.1 | >100 | >100 | 3.3 ± 1.1 | 35.7 ± 21.0 | >100 |
| 18 | 28.0 ± 0.7 | 71.4 ± 40.4 | 62.3 ± 12.0 | 29.9 ± 3.0 | 68.0 ± 1.4 | 26.3 ± 13.3 | 60.2 ± 35.9 |
| 21 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| TUB015 | 0.06 ± 0.003 | 1.1 ± 0.3 | 0.30 ± 0.01 | 0.40 ± 0.007 | 0.6 ± 0.1 | 0.5 ± 0.1 | 0.1 ± 0.06 |
| Nic b | 1.35 ± 0.6 | 0.4 ± 0.3 | 0.9 ± 0.3 | 0.45 ± 0.4 | 2.1 ± 0.6 | 0.7 ± 0.7 | 0.3 ± 0.4 |
| Noc b | 0.018 ± 0.01 | 0.070 ± 0.04 | 0.20 ± 0.10 | 0.21 ± 0.16 | 0.13 ± 0.09 | 0.095 ± 0.04 | 0.088 ± 0.04 |
| Compound | Incubation Time (h) | IC50 (µM) a |
|---|---|---|
| 6 | 24 | >30 |
| 48 | 11.87 ± 0.69 | |
| 72 | 10.81 ± 1.83 | |
| 16 | 24 | 13.67 ± 1.82 |
| 48 | 1.95 ± 0.63 | |
| 72 | 1.83 ± 0.49 | |
| Niclosamide | 24 | 0.94 ± 0.13 |
| 48 | 0.36 ± 0.12 | |
| 72 | 0.25 ± 0.04 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gargantilla, M.; Persoons, L.; Kauerová, T.; del Río, N.; Daelemans, D.; Priego, E.-M.; Kollar, P.; Pérez-Pérez, M.-J. Hybridization Approach to Identify Salicylanilides as Inhibitors of Tubulin Polymerization and Signal Transducers and Activators of Transcription 3 (STAT3). Pharmaceuticals 2022, 15, 835. https://doi.org/10.3390/ph15070835
Gargantilla M, Persoons L, Kauerová T, del Río N, Daelemans D, Priego E-M, Kollar P, Pérez-Pérez M-J. Hybridization Approach to Identify Salicylanilides as Inhibitors of Tubulin Polymerization and Signal Transducers and Activators of Transcription 3 (STAT3). Pharmaceuticals. 2022; 15(7):835. https://doi.org/10.3390/ph15070835
Chicago/Turabian StyleGargantilla, Marta, Leentje Persoons, Tereza Kauerová, Natalia del Río, Dirk Daelemans, Eva-María Priego, Peter Kollar, and María-Jesús Pérez-Pérez. 2022. "Hybridization Approach to Identify Salicylanilides as Inhibitors of Tubulin Polymerization and Signal Transducers and Activators of Transcription 3 (STAT3)" Pharmaceuticals 15, no. 7: 835. https://doi.org/10.3390/ph15070835
APA StyleGargantilla, M., Persoons, L., Kauerová, T., del Río, N., Daelemans, D., Priego, E.-M., Kollar, P., & Pérez-Pérez, M.-J. (2022). Hybridization Approach to Identify Salicylanilides as Inhibitors of Tubulin Polymerization and Signal Transducers and Activators of Transcription 3 (STAT3). Pharmaceuticals, 15(7), 835. https://doi.org/10.3390/ph15070835