
New Genetic Bomb Trigger: Design, Synthesis, Molecular Dynamics Simulation, and Biological Evaluation of Novel BIBR1532-Related Analogs Targeting Telomerase against Non-Small Cell Lung Cancer

 ,
,  ,
,  , and
, and
Abstract
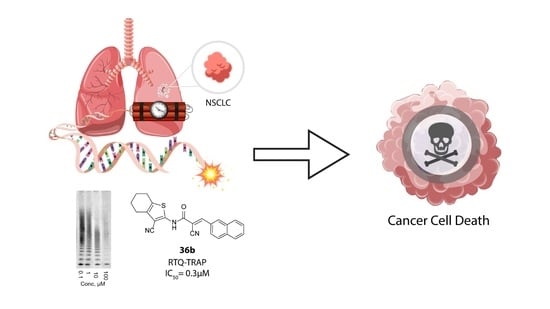
:
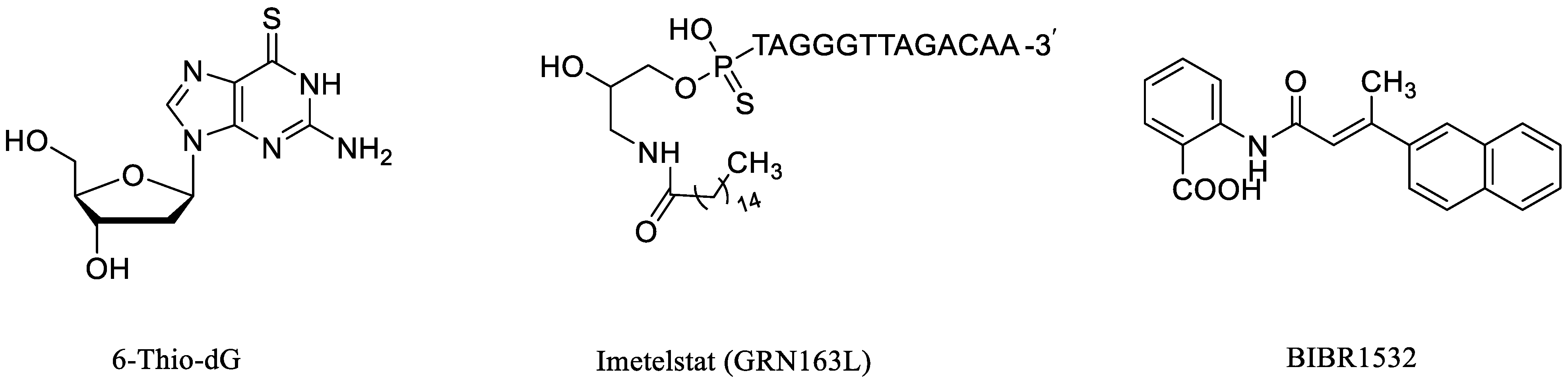
1. Introduction
2. Results and Discussion
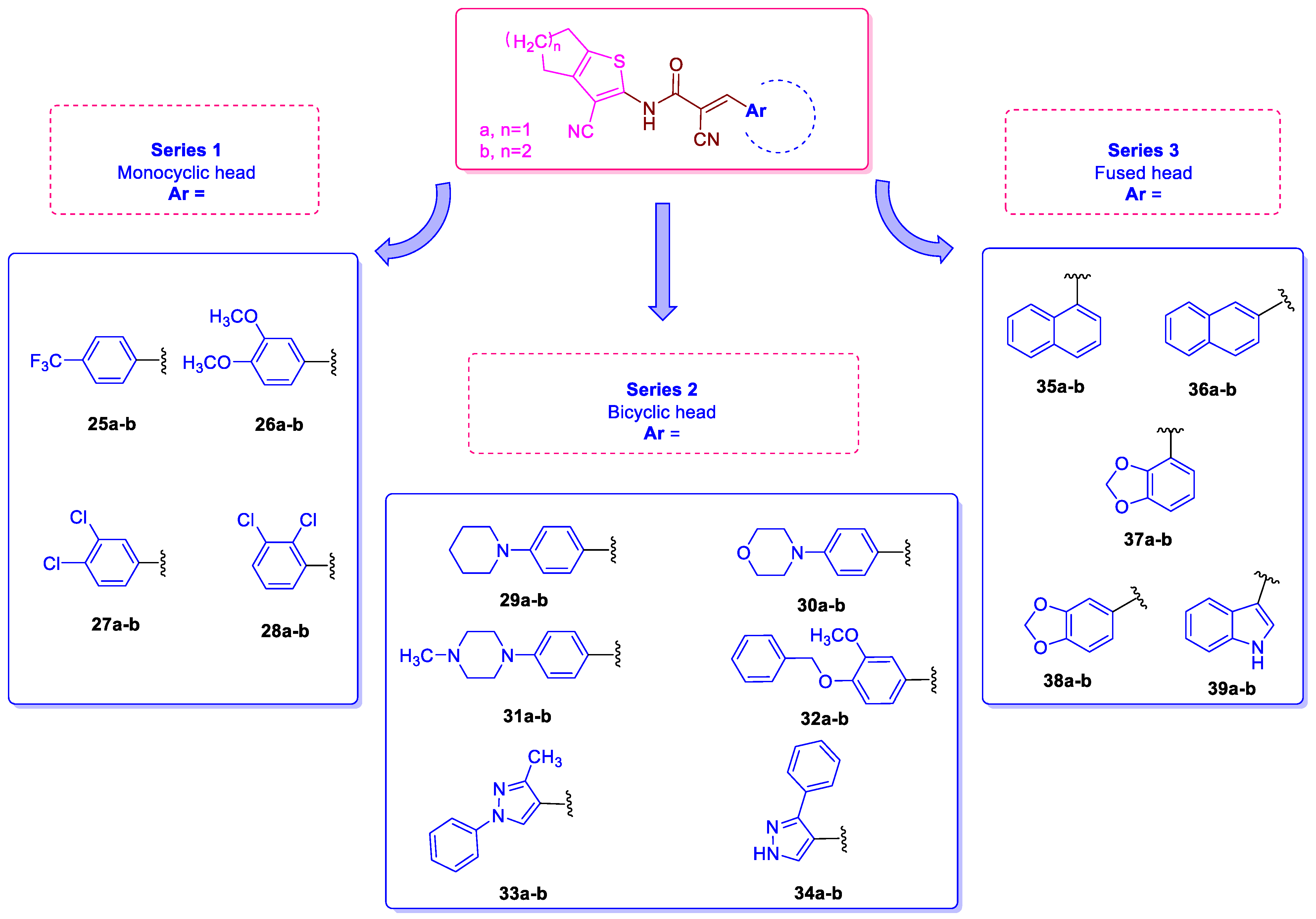
2.1. Rational Design
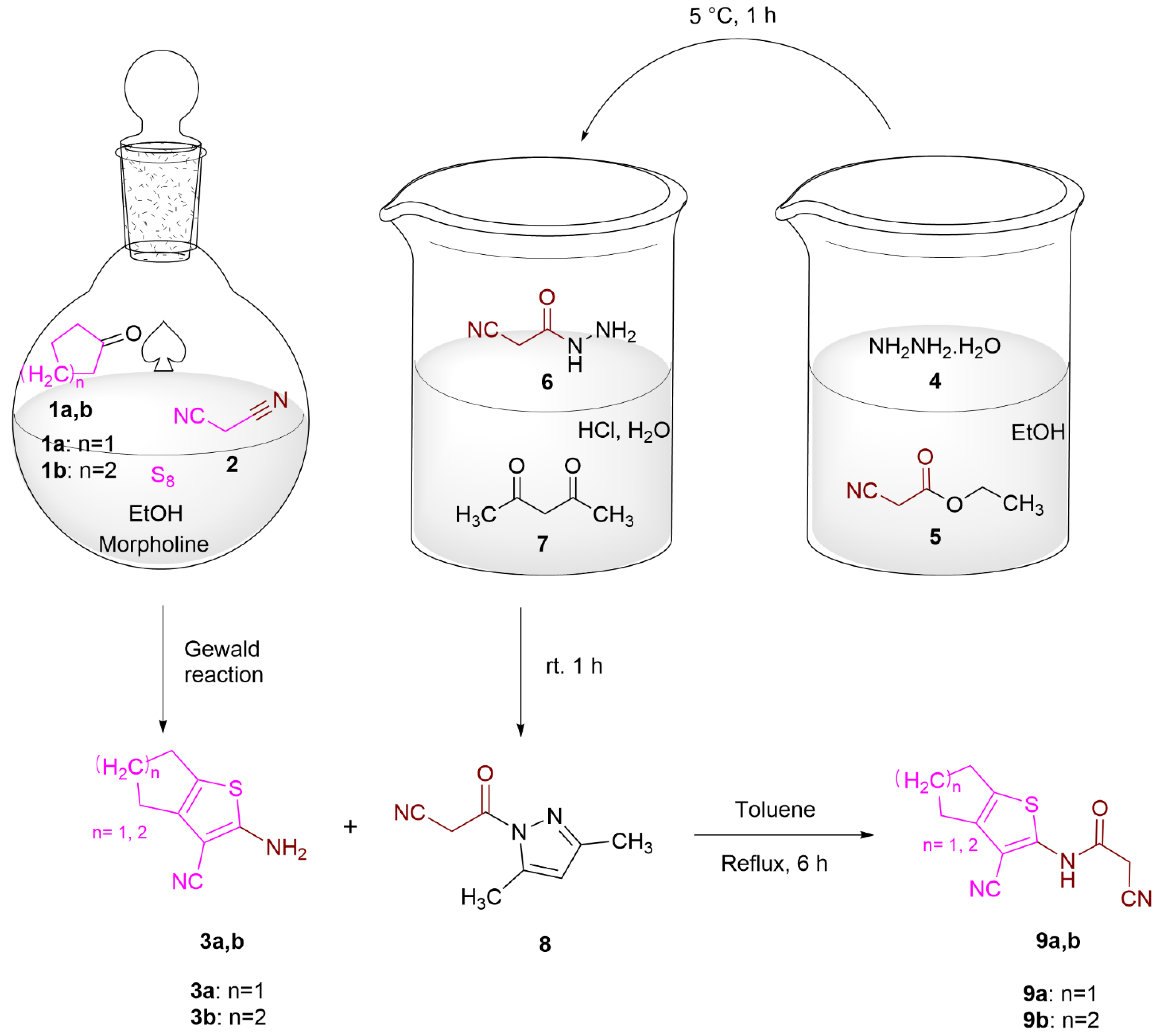
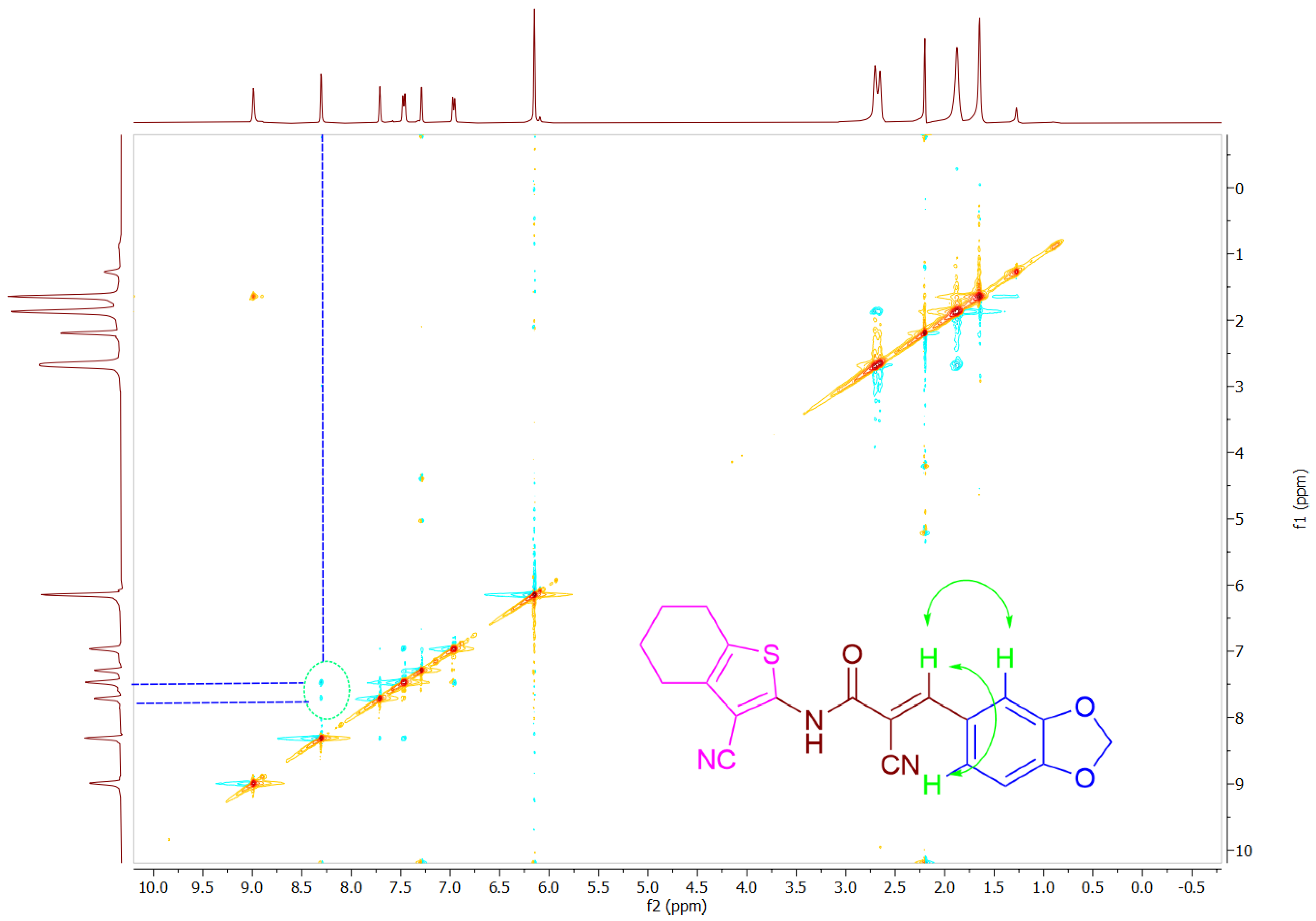
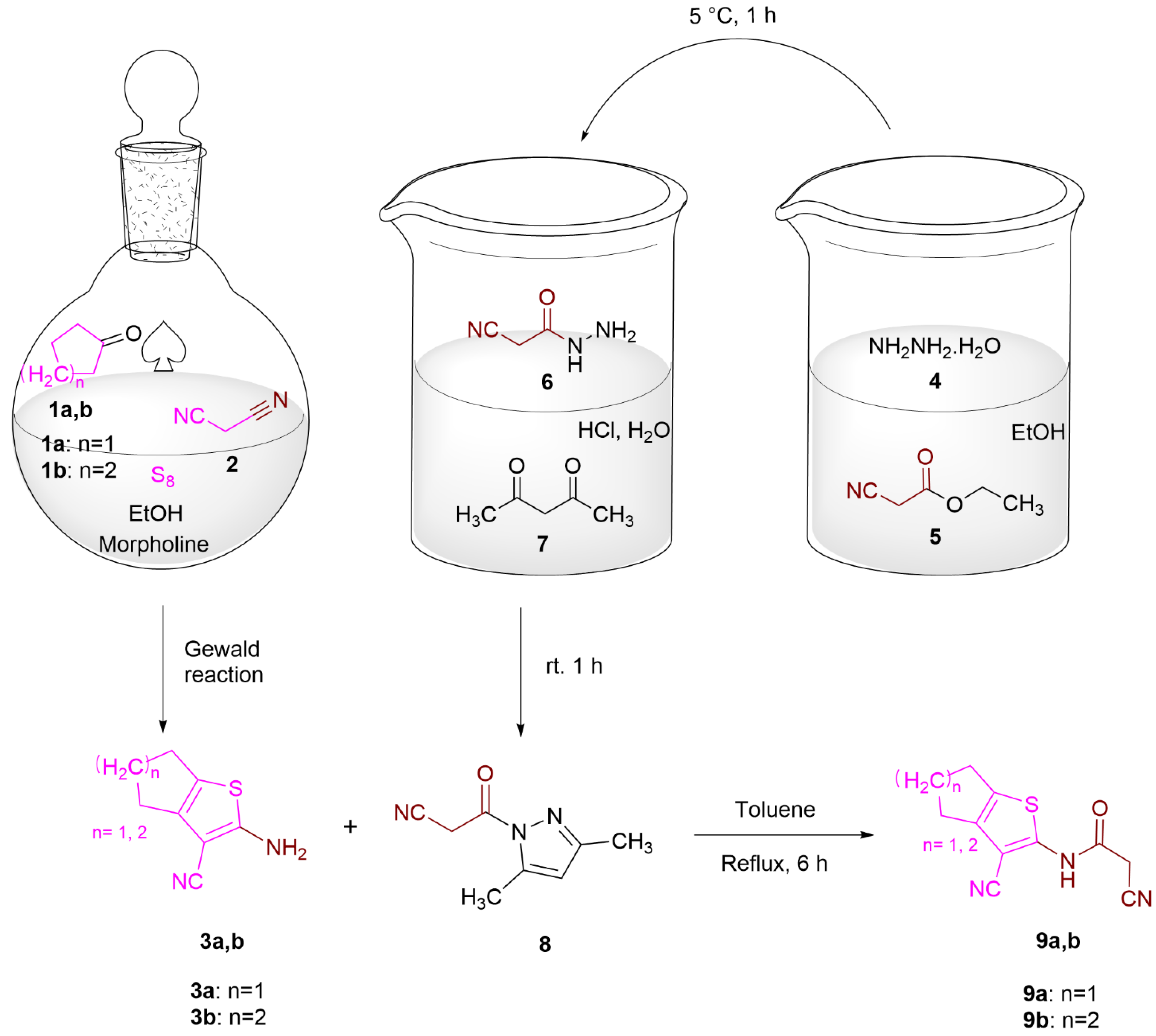
2.2. Chemistry
2.3. Biological Studies
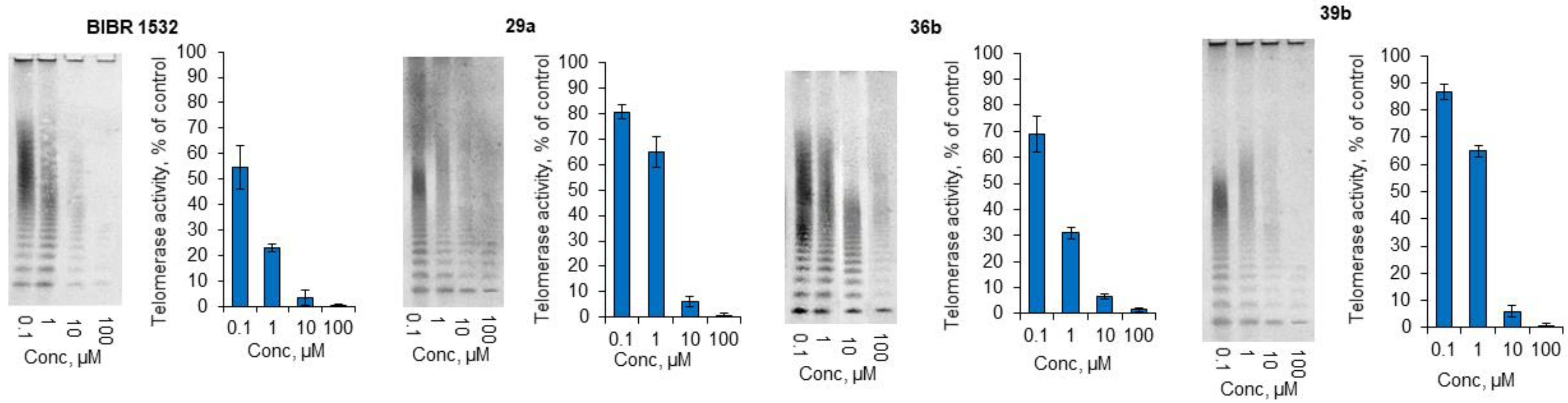
2.3.1. In Vitro Inhibition of Telomerase Enzyme
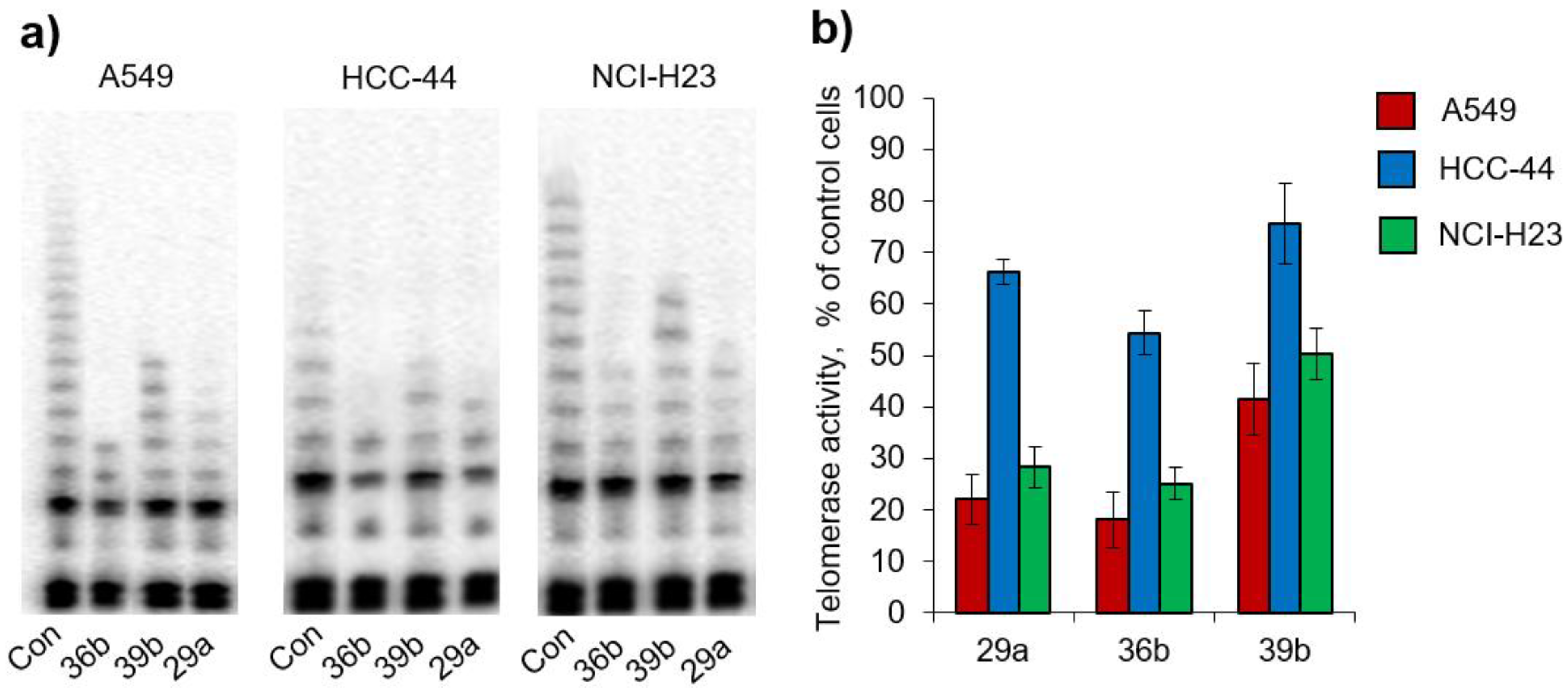
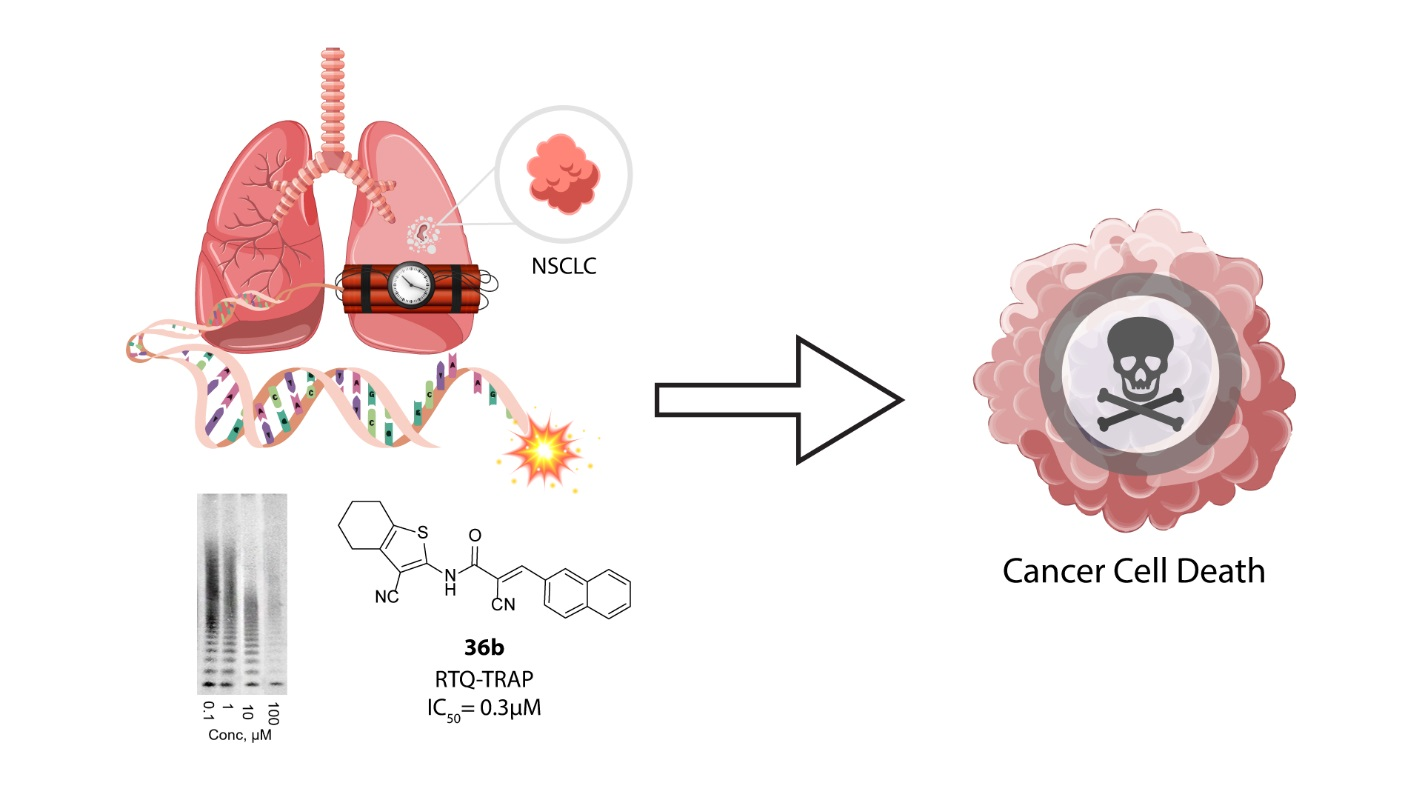
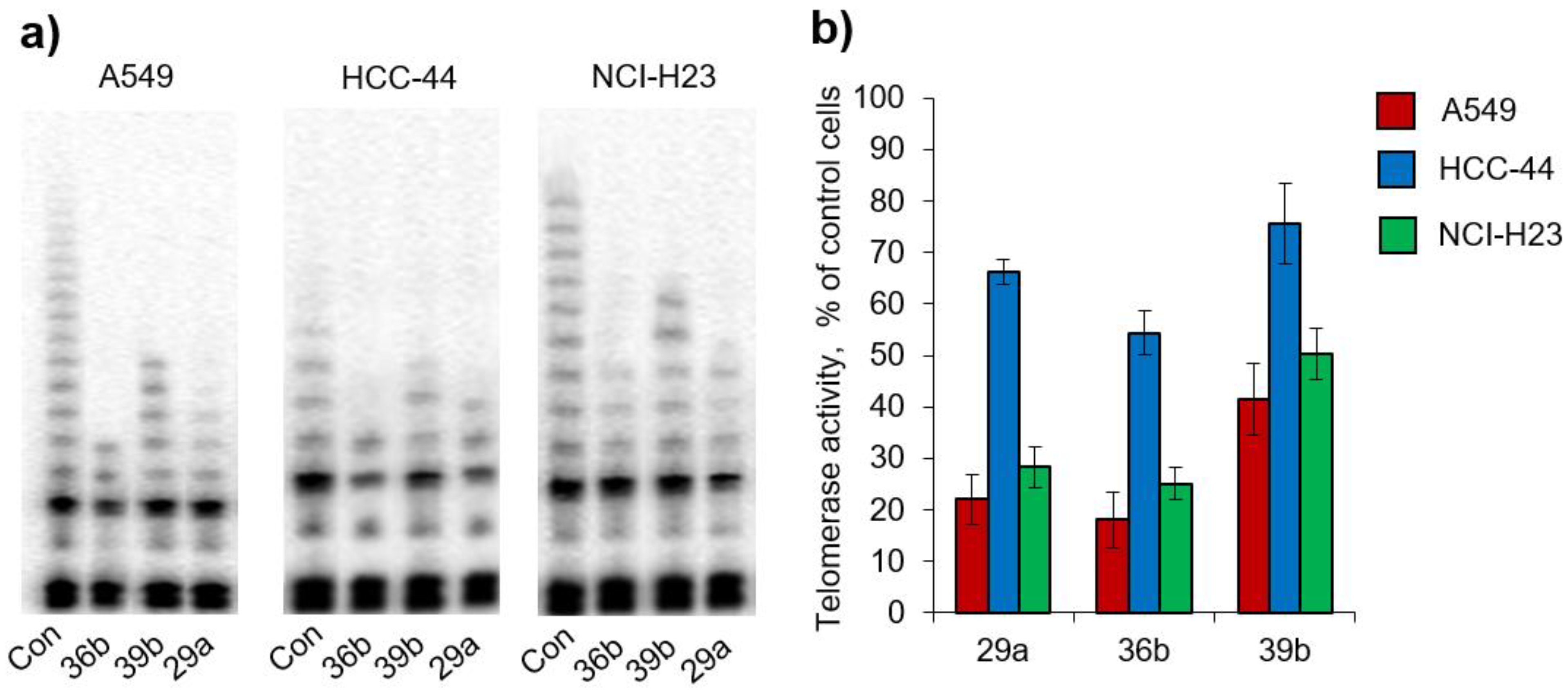
2.3.2. Living-Cell TRAP Assay
2.3.3. Telomerase Selectivity and Safety
2.4. In Silico Study
2.4.1. Molecular Docking
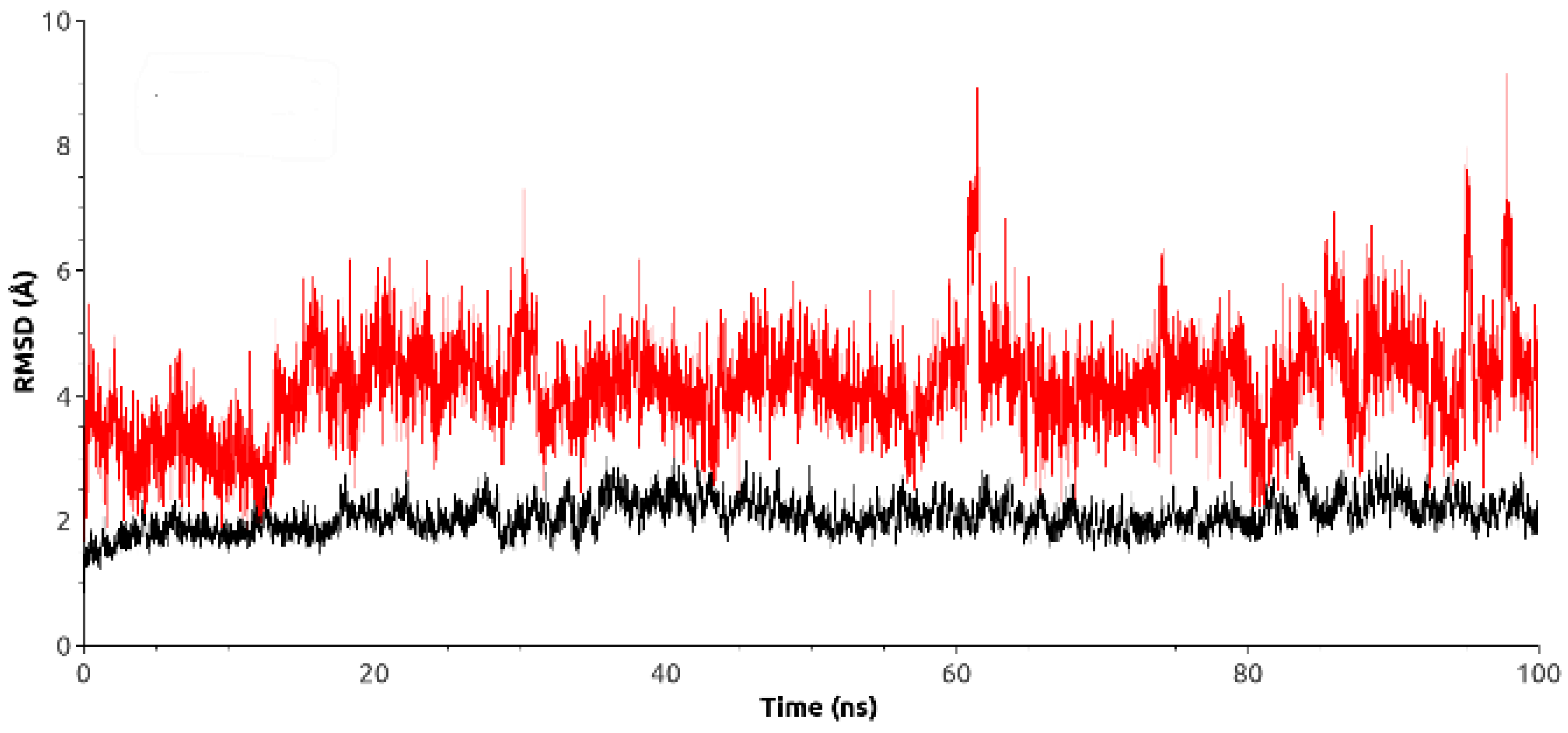
2.4.2. Molecular Dynamics Simulation
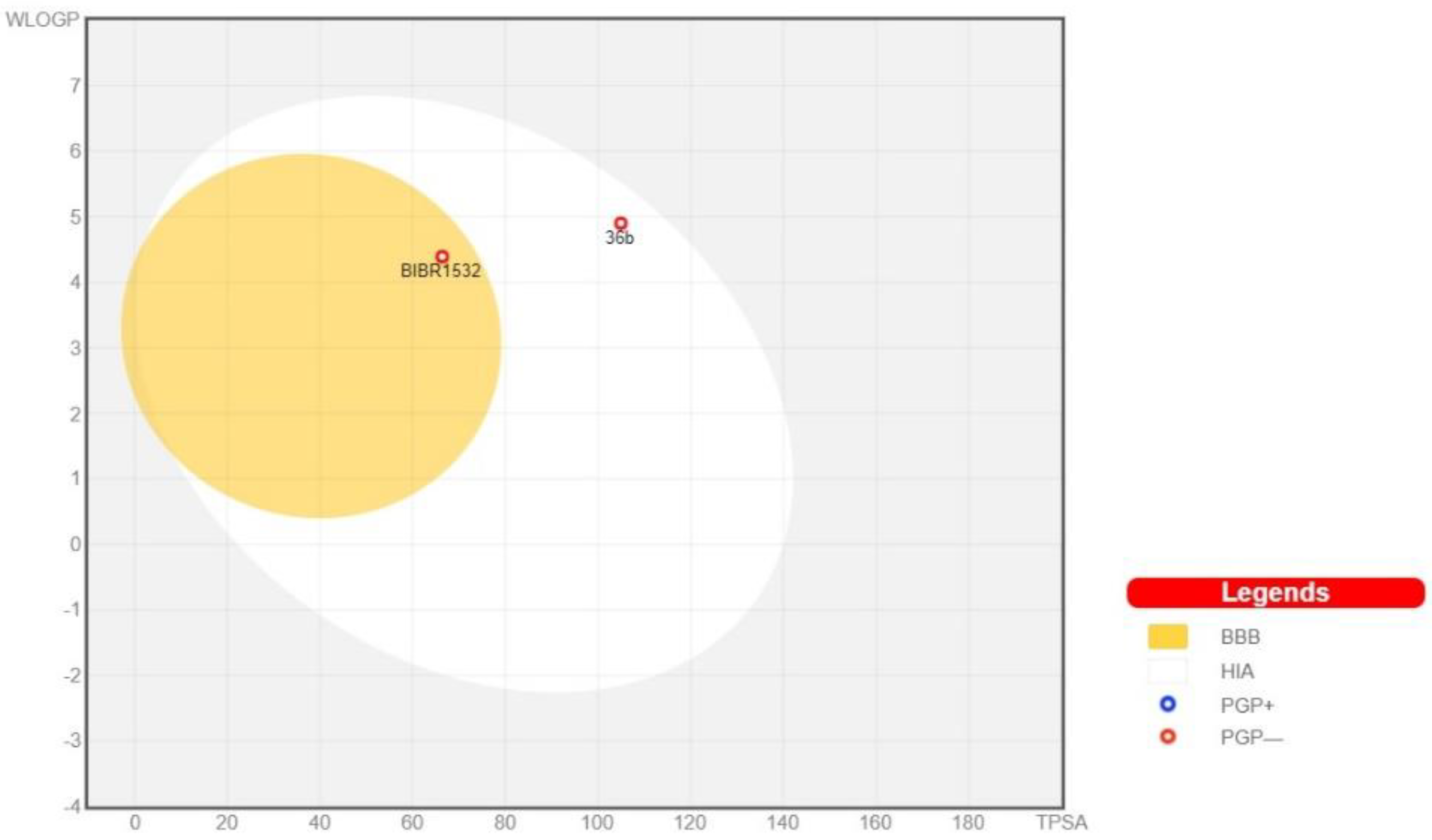
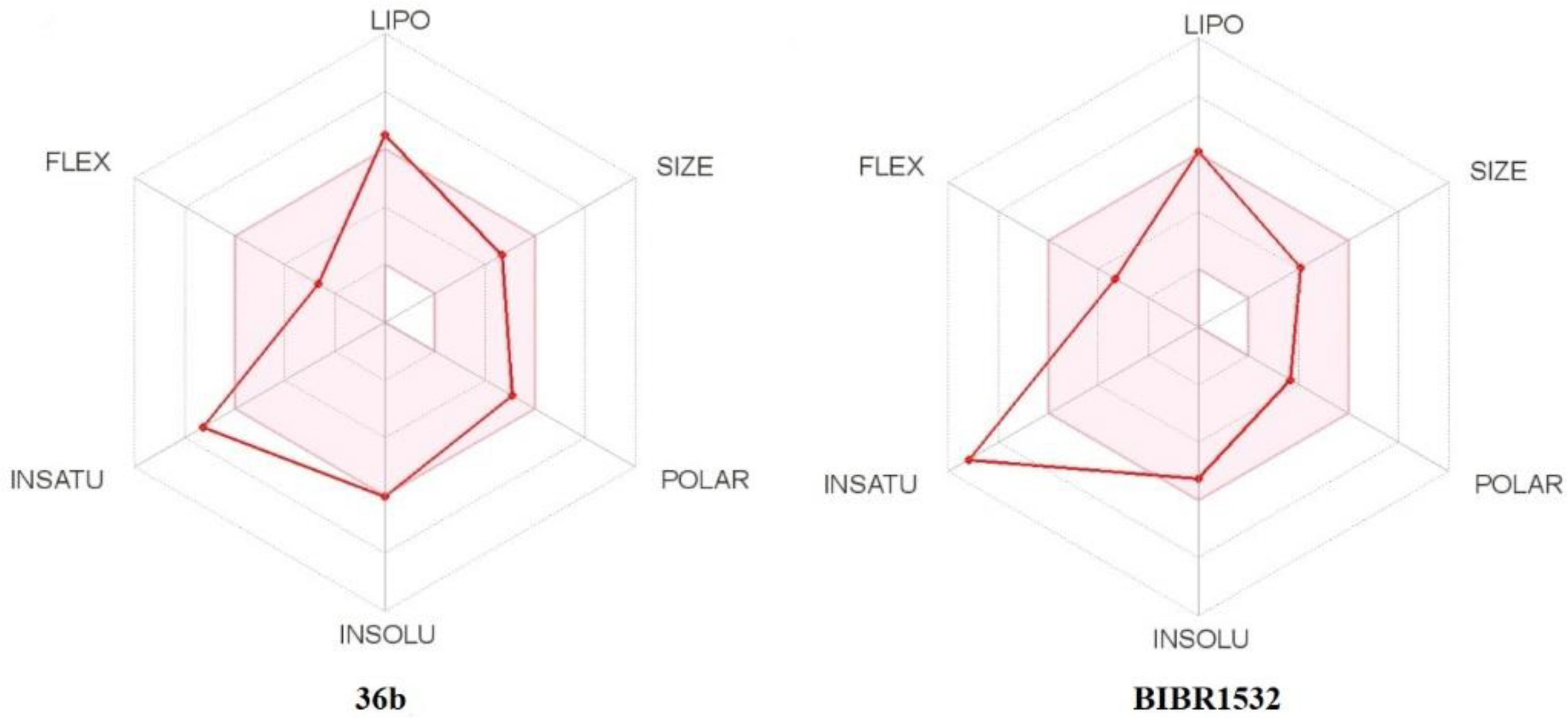
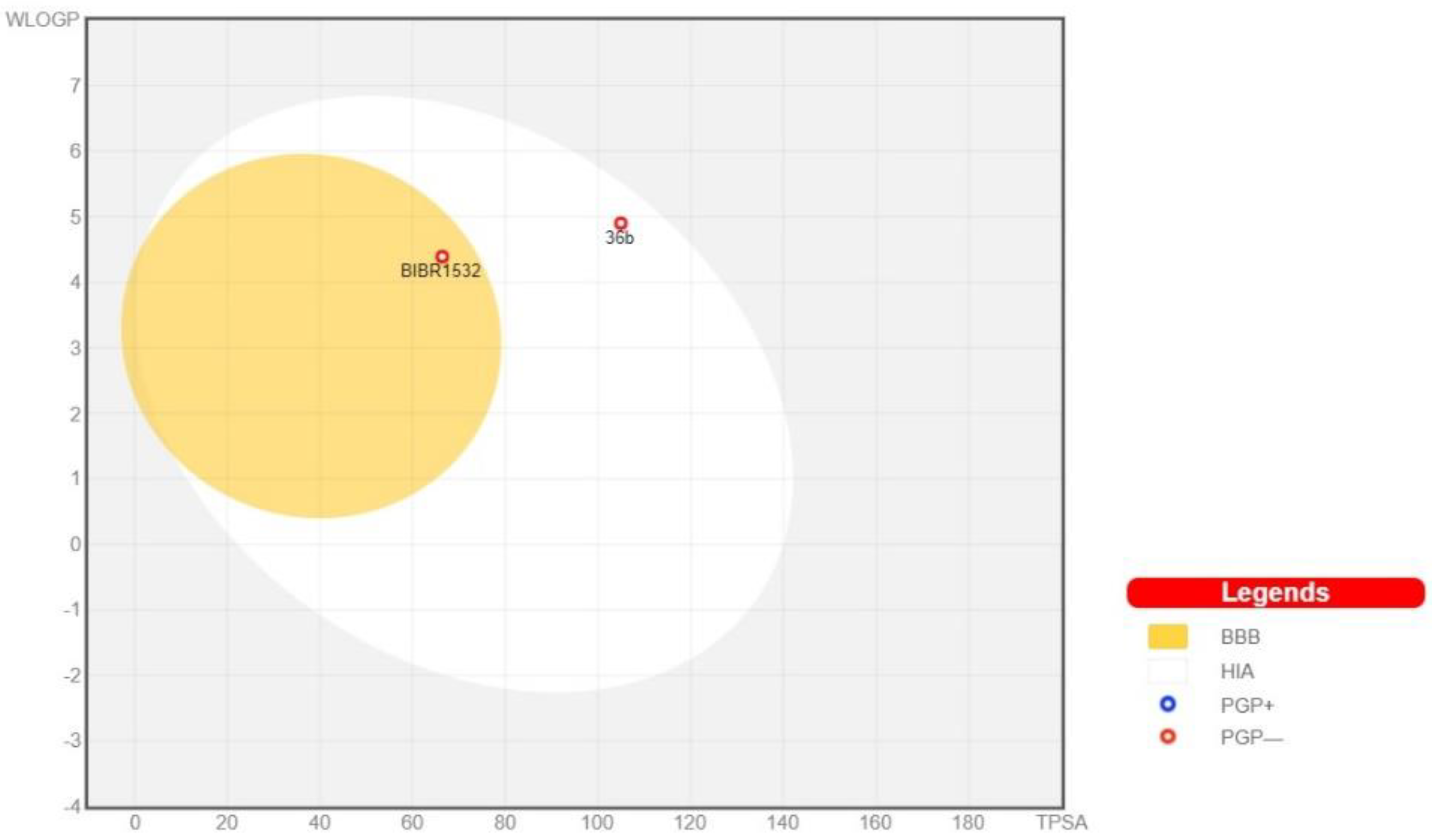
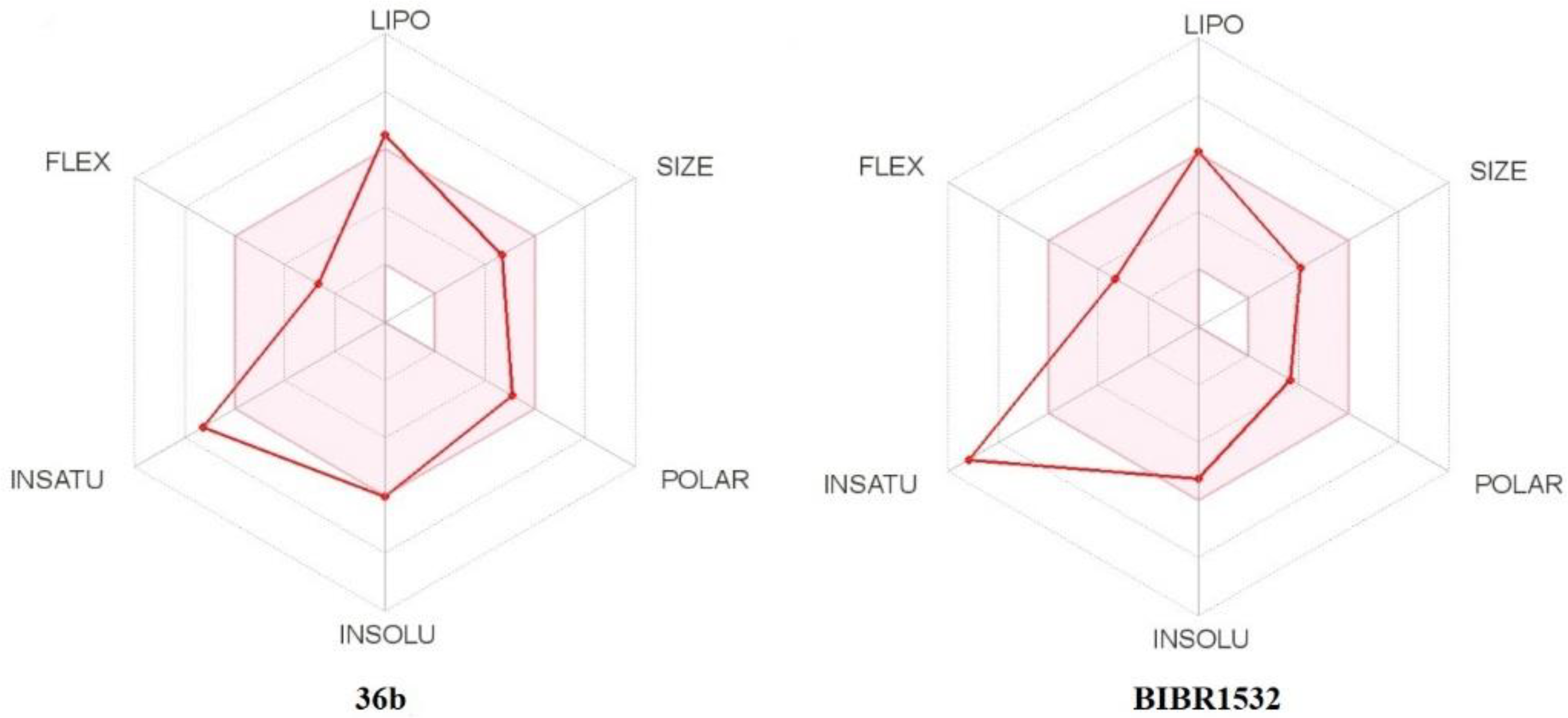
2.4.3. In Silico Pharmacokinetic, Physicochemical Prediction, and PAINS Filters
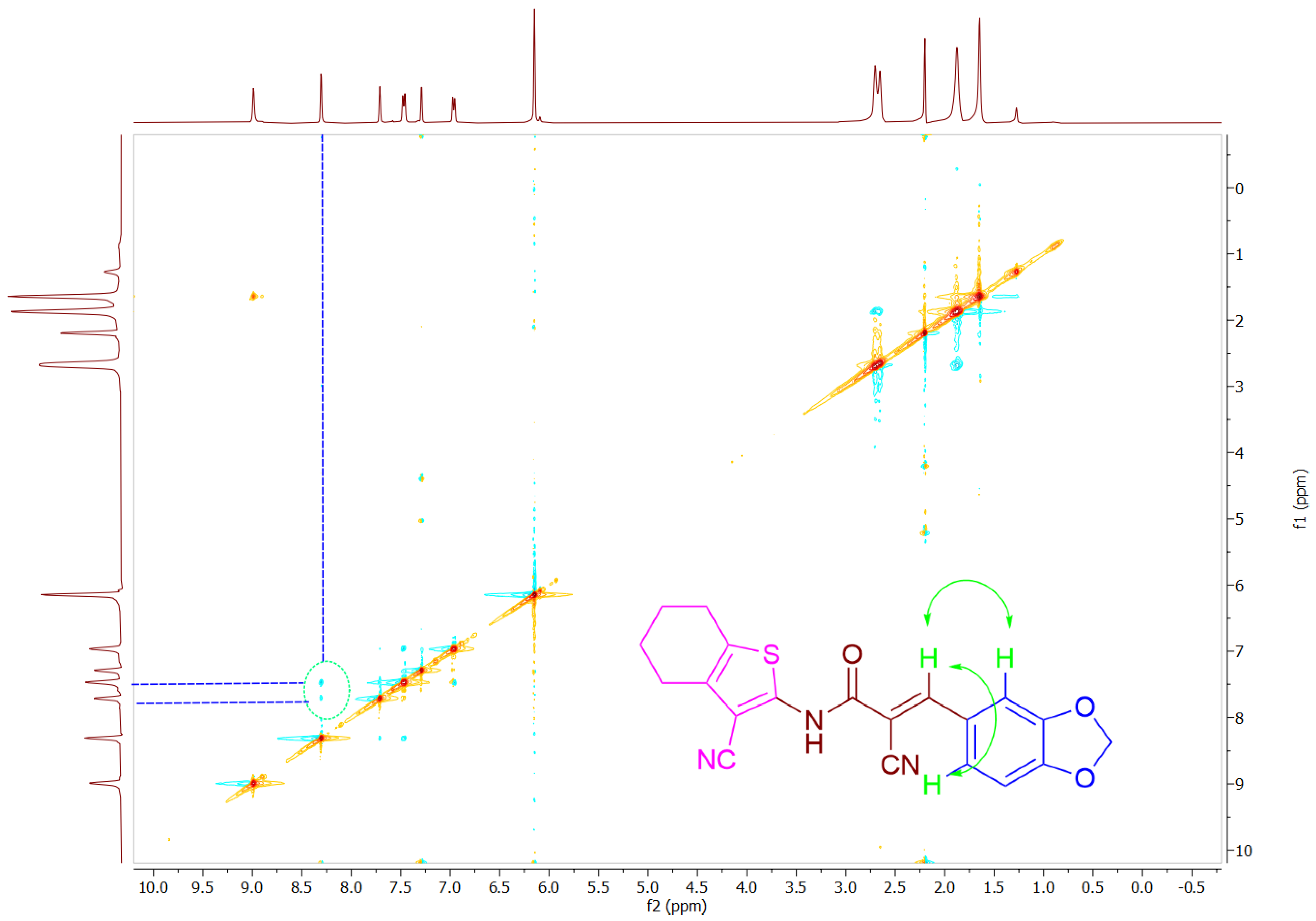
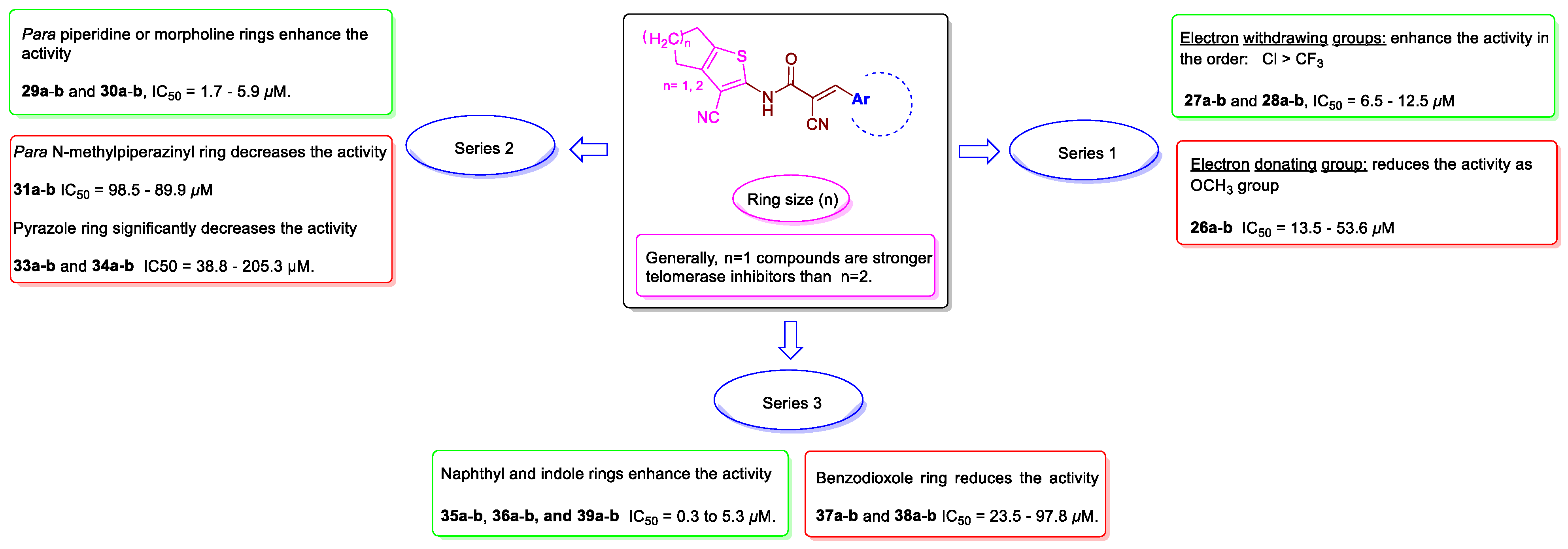
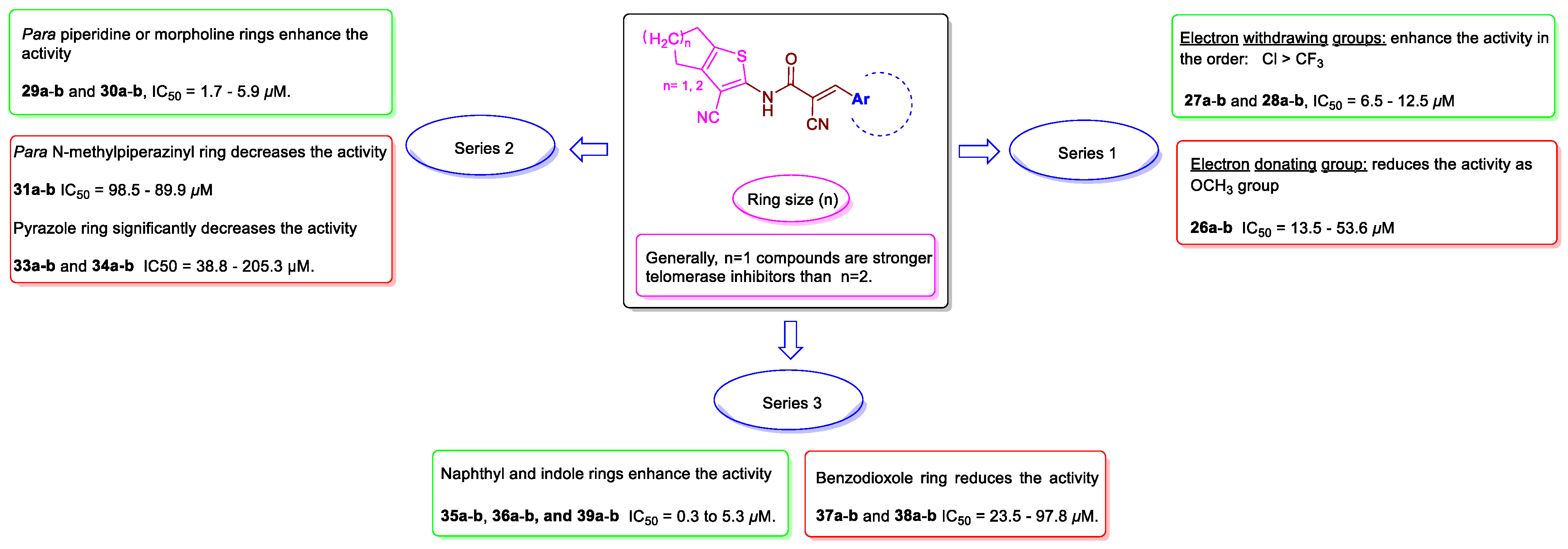
2.5. Structure-Activity Relationship (SAR)
3. Materials and Methods
3.1. Chemistry
3.1.1. General Procedure for the Preparation of 3a,b
3.1.2. Cyanoacetic Acid Hydrazide (6)
3.1.3. 1-Cyanoacetyl-3,5-dimethylpyrazole (8)
3.1.4. General Procedure for the Preparation of 9a,b
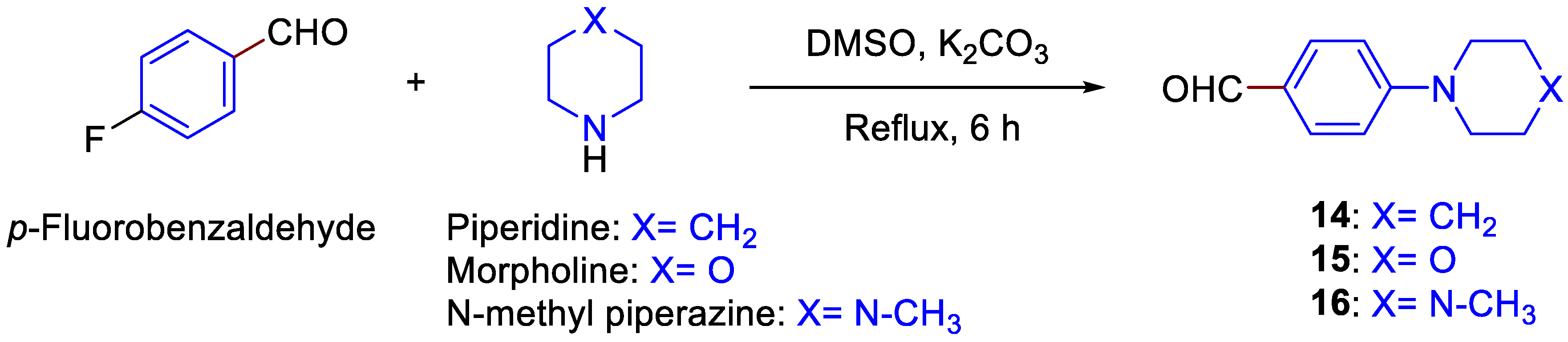
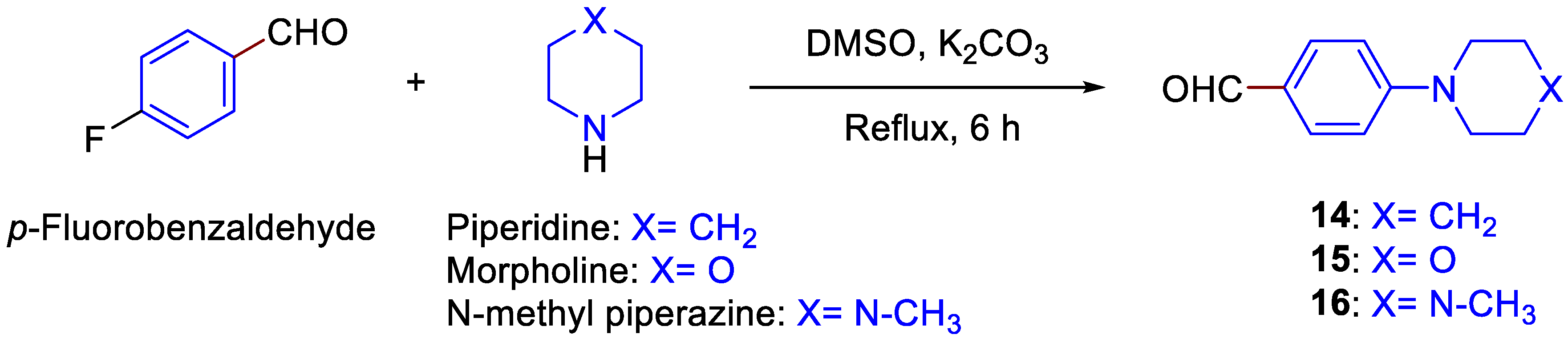
3.1.5. General Procedure for the Preparation of 14–16
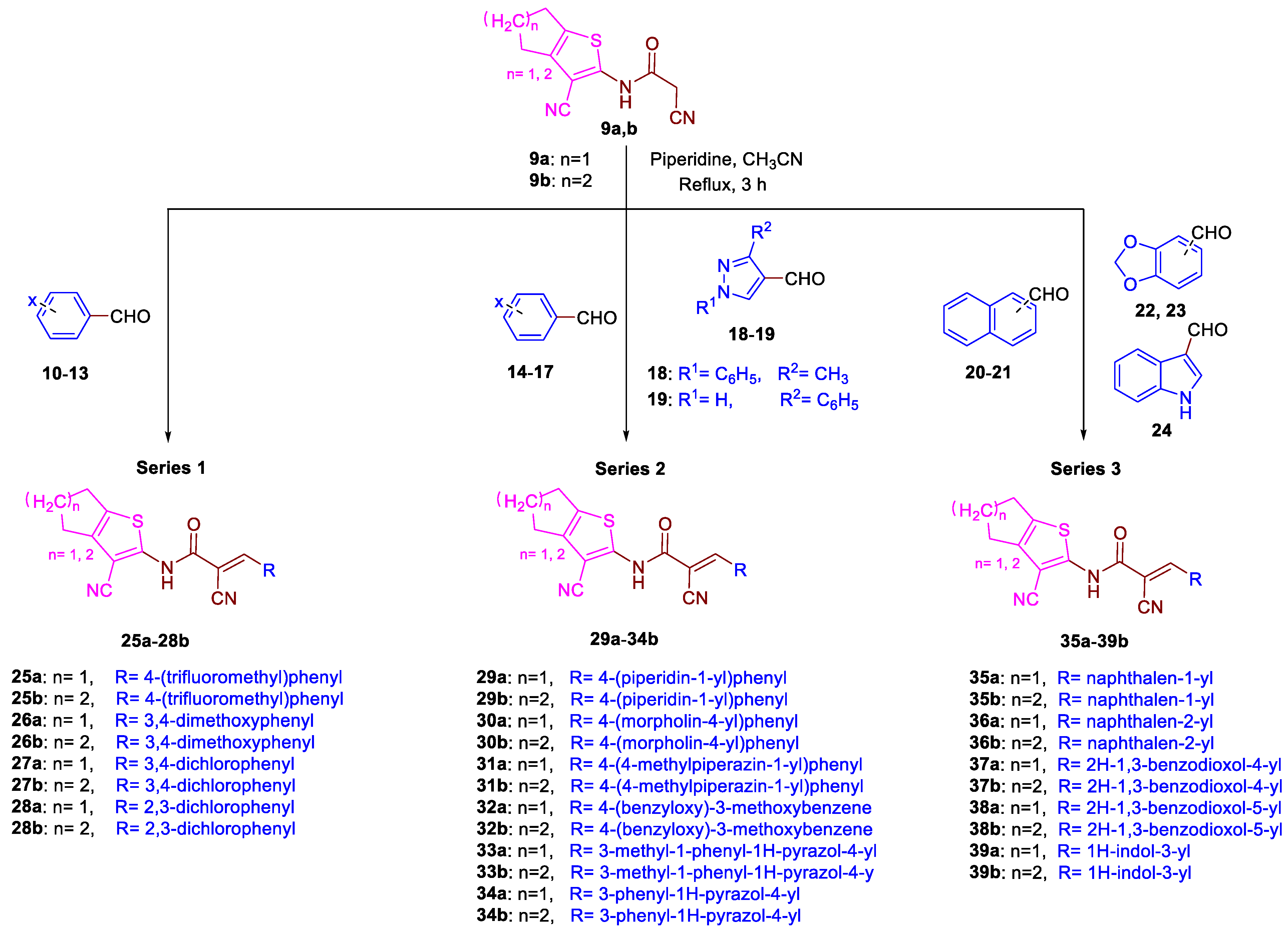
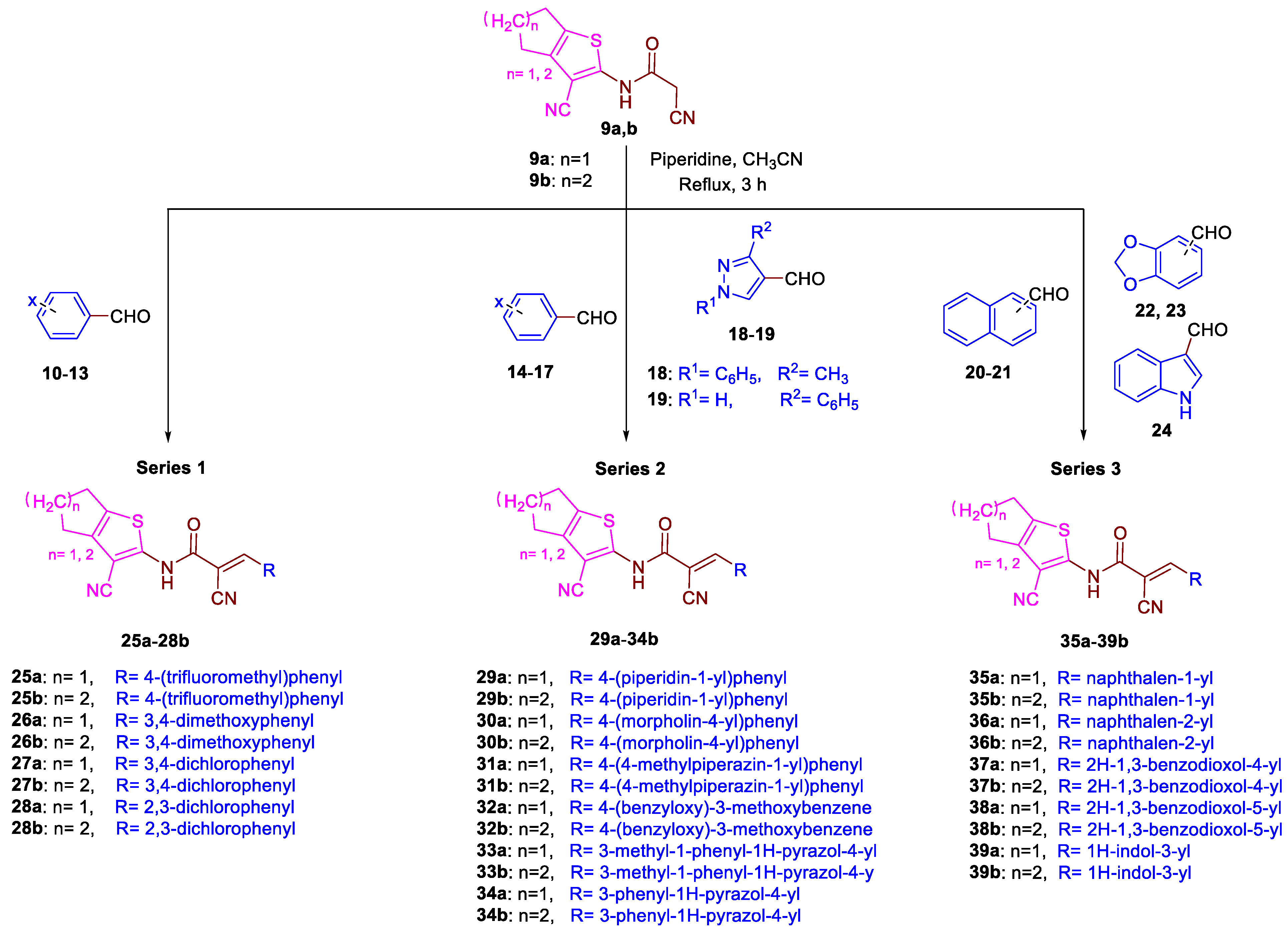
3.1.6. General Procedure for the Preparation of 25a–39b
3.2. In Vitro Telomerase Activity Assay
3.3. Cell Lines and Incubation with the Compounds
3.4. In Vitro Anticancer Screening
3.5. Statistical Analysis
3.6. Molecular Docking
3.7. Molecular Dynamics Simulation
3.8. In Silico Pharmacokinetic; Physicochemical Prediction and PAINS Filters
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nassour, J.; Schmidt, T.T.; Karlseder, J. Telomeres and Cancer: Resolving the Paradox. Annu. Rev. Cancer Biol. 2021, 5, 59–77. [Google Scholar] [CrossRef] [PubMed]
- Seimiya, H.; Nagasawa, K.; Shin-Ya, K. Chemical targeting of G-quadruplexes in telomeres and beyond for molecular cancer therapeutics. J. Antibiot. 2021, 74, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Gao, T.; Shi, L.; Chen, T.; Xiang, Y.; Li, Y.; Li, G. Molecular imaging of telomerase and the enzyme activity-triggered drug release by using a conformation-switchable nanoprobe in cancerous cells. Sci. Rep. 2018, 8, 16341. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, S.; Kumar, M.S.; Peters, G.J.; Mayur, Y.C. Targeting telomerase for its advent in cancer therapeutics. Med. Res. Rev. 2020, 40, 1871–1919. [Google Scholar] [CrossRef]
- Wu, L.; Fidan, K.; Um, J.Y.; Ahn, K.S. Telomerase: Key regulator of inflammation and cancer. Pharmacol. Res. 2020, 155, 1–12. [Google Scholar] [CrossRef]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef]
- Nassrally, M.S.; Lau, A.; Wise, K.; John, N.; Kotecha, S.; Lee, K.L.; Brooks, R.F. Cell cycle arrest in replicative senescence is not an immediate consequence of telomere dysfunction. Mech. Ageing Dev. 2019, 179, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Harley, C.B. Telomere loss: Mitotic clock or genetic time bomb? Mutat. Res./DNAging 1991, 256, 271–282. [Google Scholar] [CrossRef]
- Harley, C.B. Telomerase and cancer therapeutics. Nat. Rev. Cancer 2008, 8, 167–179. [Google Scholar] [CrossRef]
- Azarm, K.; Bhardwaj, A.; Kim, E.; Smith, S. Persistent telomere cohesion protects aged cells from premature senescence. Nat. Commun. 2020, 11, 3321. [Google Scholar] [CrossRef]
- Graham, M.K.; Meeker, A. Telomeres and telomerase in prostate cancer development and therapy. Nat. Rev. Urol. 2017, 14, 607–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaul, Z.; Cheung, C.T.Y.; Bhargava, P.; Sari, A.N.; Yu, Y.; Huifu, H.; Bid, H.; Henson, J.D.; Groden, J.; Reddel, R.R.; et al. Functional characterization of miR-708 microRNA in telomerase positive and negative human cancer cells. Sci. Rep. 2021, 11, 17052. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Yu, Y.L.; Ma, D.; Zhang, Z.Y.; Liu, X.H. Synthesis, telomerase inhibitory and anticancer activity of new 2-phenyl-4H-chromone derivatives containing 1,3,4-oxadiazole moiety. J. Enzym. Inhib. Med. Chem. 2021, 36, 344–360. [Google Scholar] [CrossRef] [PubMed]
- Asamitsu, S.; Shioda, N.; Sugiyama, H. Switching Off Cancer-Causing Telomerase Using Small Molecules. Cell Chem. Biol. 2019, 26, 1045–1047. [Google Scholar] [CrossRef]
- Maciejowski, J.; de Lange, T. Telomeres in cancer: Tumour suppression and genome instability. Nat. Rev. Mol. Cell Biol. 2017, 18, 175–186. [Google Scholar] [CrossRef] [Green Version]
- Vasina, D.A.; Zhdanov, D.D.; Orlova, E.V.; Orlova, V.S.; Pokrovskaya, M.V.; Aleksandrova, S.S.; Sokolov, N.N. Apoptotic endonuclease EndoG inhibits telomerase activity and induces malignant transformation of human CD4+ T cells. Biochemistry 2017, 82, 24–37. [Google Scholar] [CrossRef]
- Camp, E.R.; Summy, J.; Bauer, T.W.; Liu, W.; Gallick, G.E.; Ellis, L.M. Molecular Mechanisms of Resistance to Therapies Targeting the Epidermal Growth Factor Receptor. Clin. Cancer Res. 2005, 11, 397–405. [Google Scholar] [CrossRef]
- Damm, K.; Hemmann, U.; Garin-Chesa, P.; Hauel, N.; Kauffmann, I.; Priepke, H.; Niestroj, C.; Daiber, C.; Enenkel, B.; Guilliard, B.; et al. A highly selective telomerase inhibitor limiting human cancer cell proliferation. EMBO J. 2001, 20, 6958–6968. [Google Scholar] [CrossRef]
- Burchett, K.M.; Etekpo, A.; Batra, S.K.; Yan, Y.; Ouellette, M.M. Inhibitors of telomerase and poly(ADP-ribose) polymerases synergize to limit the lifespan of pancreatic cancer cells. Oncotarget 2017, 8, 83754–83767. [Google Scholar] [CrossRef] [Green Version]
- Ma, F.; Wang, T.T.; Jiang, L.; Zhang, C.Y. Ultrasensitive detection of telomerase activity in lung cancer cells with quencher-free molecular beacon-assisted quadratic signal amplification. Anal. Chim. Acta 2019, 1053, 122–130. [Google Scholar] [CrossRef]
- American Cancer Society. What Is Lung Cancer? 1 October 2019. Available online: https://www.cancer.org/cancer/lung-cancer/about/what-is.html (accessed on 17 March 2022).
- World Health Organization. Cancer. 3 February 2022. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 17 March 2022).
- Chen, K.Y.; Lee, L.N.; Yu, C.J.; Lee, Y.C.; Kuo, S.H.; Yang, P.C. Elevation of telomerase activity positively correlates to poor prognosis of patients with non-small cell lung cancer. Cancer Lett. 2006, 240, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, N.; Wang, M.; Zhang, Y.H.; Yan, L.D.; Zhou, W.; Yu, Z.Q.; Peng, X.C.; Cai, J. Tumorigenic effect of TERT and its potential therapeutic target in NSCLC (Review). Oncol. Rep. 2021, 46, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.Q.; Cutz, J.C.; Liu, N.; Lau, D.; Shepherd, F.A.; Squire, J.A.; Tsao, M.S. Amplification of telomerase (hTERT) gene is a poor prognostic marker in non-small-cell lung cancer. Br. J. Cancer 2006, 94, 1452–1459. [Google Scholar] [CrossRef]
- Mender, I.; LaRanger, R.; Luitel, K.; Peyton, M.; Girard, L.; Lai, T.P.; Batten, K.; Cornelius, C.; Dalvi, M.P.; Ramirez, M.; et al. Telomerase-Mediated Strategy for Overcoming Non-Small Cell Lung Cancer Targeted Therapy and Chemotherapy Resistance. Neoplasia 2018, 20, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Cheng, J.; Pang, Q.; Wei, X.; Zhang, X.; Wang, P.; Yuan, Z.; Qian, D. BIBR1532, a Selective Telomerase Inhibitor, Enhances Radiosensitivity of Non-Small Cell Lung Cancer Through Increasing Telomere Dysfunction and ATM/CHK1 Inhibition. Int. J. Radiat. Oncol. Biol. Phys. 2019, 105, 861–874. [Google Scholar] [CrossRef] [Green Version]
- Plyasova, A.A.; Berrino, E.; Khan, I.I.; Veselovsky, A.V.; Pokrovsky, V.S.; Angeli, A.; Ferraroni, M.; Supuran, C.T.; Pokrovskaya, M.V.; Alexandrova, S.S.; et al. Mechanisms of the Antiproliferative and Antitumor Activity of Novel Telomerase–Carbonic Anhydrase Dual-Hybrid Inhibitors. J. Med. Chem. 2021, 64, 11432–11444. [Google Scholar] [CrossRef]
- Berrino, E.; Angeli, A.; Zhdanov, D.D.; Kiryukhina, A.P.; Milaneschi, A.; De Luca, A.; Bozdag, M.; Carradori, S.; Selleri, S.; Bartolucci, G.; et al. Azidothymidine “Clicked” into 1,2,3-Triazoles: First Report on Carbonic Anhydrase-Telomerase Dual-Hybrid Inhibitors. J. Med. Chem. 2020, 63, 7392–7409. [Google Scholar] [CrossRef]
- Guterres, A.N.; Villanueva, J. Targeting telomerase for cancer therapy. Oncogene 2020, 39, 5811–5824. [Google Scholar] [CrossRef]
- Huppert, J.L.; Balasubramanian, S. Prevalence of quadruplexes in the human genome. Nucleic Acids Res. 2005, 33, 2908–2916. [Google Scholar] [CrossRef] [Green Version]
- Sanford, S.L.; Welfer, G.A.; Freudenthal, B.D.; Opresko, P.L. Mechanisms of telomerase inhibition by oxidized and therapeutic dNTPs. Nat. Commun. 2020, 11, 5288. [Google Scholar] [CrossRef]
- Chiappori, A.A.; Kolevska, T.; Spigel, D.R.; Hager, S.; Rarick, M.; Gadgeel, S.; Blais, N.; Von Pawel, J.; Hart, L.; Reck, M.; et al. A randomized phase II study of the telomerase inhibitor imetelstat as maintenance therapy for advanced non-small-cell lung cancer. Ann. Oncol. 2015, 26, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Pascolo, E.; Wenz, C.; Lingner, J.; Hauel, N.; Priepke, H.; Kauffmann, I.; Garin-Chesa, P.; Rettig, W.J.; Damm, K.; Schnapp, A. Mechanism of human telomerase inhibition by BIBR1532, a synthetic, non-nucleosidic drug candidate. J. Biol. Chem. 2002, 277, 15566–15572. [Google Scholar] [CrossRef] [Green Version]
- Parsch, D.; Brassat, U.; Brümmendorf, T.H.; Fellenberg, J. Consequences of Telomerase Inhibition by BIBR1532 on Proliferation and Chemosensitivity of Chondrosarcoma Cell Lines. Cancer Investig. 2008, 26, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhong, D.; Li, Y.; Wu, H.; Xu, X.; Yang, J.; Gu, Z. Tumor-Oriented Telomerase-Terminated Nanoplatform as Versatile Strategy for Multidrug Resistance Reversal in Cancer Treatment. Adv. Healthc. Mater. 2020, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Barma, D.K.; Elayadi, A.; Falck, J.R.; Corey, D.R. Inhibition of telomerase by BIBR 1532 and related analogues. Bioorg. Med. Chem. Lett. 2003, 13, 1333–1336. [Google Scholar] [CrossRef]
- Bryan, C.; Rice, C.; Hoffman, H.; Harkisheimer, M.; Sweeney, M.; Skordalakes, E. Structural Basis of Telomerase Inhibition by the Highly Specific BIBR1532. Structure 2015, 23, 1934–1942. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Zhou, H.; Sheng, X.B.; Liu, X.H.; Chen, F.H. Design, synthesis and SARs of novel telomerase inhibitors based on BIBR1532. Bioorg. Chem. 2020, 102, 1–16. [Google Scholar] [CrossRef]
- Mohamed, M.; Attia, Y.; Shouman, S.; Abdelhamid, I. Anticancer Activities of New N-hetaryl-2-cyanoacetamide Derivatives Incorporating 4,5,6,7-Tetrahydrobenzo[b]thiophene Moiety. Anticancer Agents Med. Chem. 2017, 17, 1084–1092. [Google Scholar] [CrossRef]
- Shams, H.Z.; Mohareb, R.M.; Helal, M.H.; Mahmoud, A.E. Novel Synthesis and Antitumor Evaluation of Polyfunctionally Substituted Heterocyclic Compounds Derived from 2-Cyano-N-(3-cyano-4,5,6,7-tetrahydrobenzo[b]thiophen-2-yl)-acetamide. Molecules 2011, 16, 52. [Google Scholar] [CrossRef]
- Mohareb, R.; Ibrahim, R.; Wardakhan, W. Synthesis of pyridine, pyran and thiazole containing thiophene derivatives and their anti-tumor evaluations. Med. Chem. Res. 2016, 25, 2187–2204. [Google Scholar] [CrossRef]
- Punetha, A.; Ngo, H.X.; Holbrook, S.Y.L.; Green, K.D.; Willby, M.J.; Bonnett, S.A.; Krieger, K.; Dennis, E.K.; Posey, J.E.; Parish, T.; et al. Structure-Guided Optimization of Inhibitors of Acetyltransferase Eis from Mycobacterium tuberculosis. ACS Chem. Biol. 2020, 15, 1581–1594. [Google Scholar] [CrossRef]
- Rodrigues, K.A.; Dias, C.N.; Neris, P.L.; Rocha Jda, C.; Scotti, M.T.; Scotti, L.; Mascarenhas, S.R.; Veras, R.C.; de Medeiros, I.A.; Keesen Tde, S.; et al. 2-Amino-thiophene derivatives present antileishmanial activity mediated by apoptosis and immunomodulation in vitro. Eur. J. Med. Chem. 2015, 106, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Elrayess, R.; Abdel Aziz, Y.M.; Elgawish, M.S.; Elewa, M.; Elshihawy, H.A.; Said, M.M. Pharmacophore modeling, 3D-QSAR, synthesis, and anti-lung cancer evaluation of novel thieno[2,3-d][1,2,3]triazines targeting EGFR. Arch. Pharm. 2020, 353, 1–15. [Google Scholar] [CrossRef]
- Assiri, M.A.; Al-Sehemi, A.G.; Pannipara, M. AIE based “on-off” fluorescence probe for the detection of Cu2+ ions in aqueous media. Inorg. Chem. Commun. 2019, 99, 11–15. [Google Scholar] [CrossRef]
- Hosseini, H.; Bayat, M. Cyanoacetohydrazides in Synthesis of Heterocyclic Compounds. Top. Curr. Chem. 2018, 376, 1–67. [Google Scholar] [CrossRef] [PubMed]
- Iraji, A.; Firuzi, O.; Khoshneviszadeh, M.; Tavakkoli, M.; Mahdavi, M.; Nadri, H.; Edraki, N.; Miri, R. Multifunctional iminochromene-2H-carboxamide derivatives containing different aminomethylene triazole with BACE1 inhibitory, neuroprotective and metal chelating properties targeting Alzheimer’s disease. Eur. J. Med. Chem. 2017, 141, 690–702. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, E.Z.; Mahmoud, W.R.; George, R.F.; Hassan, G.S.; Omar, F.A.; Georgey, H.H. Synthesis, in vitro anticancer activity and in silico studies of certain pyrazole-based derivatives as potential inhibitors of cyclin dependent kinases (CDKs). Bioorg. Chem. 2021, 116, 105347. [Google Scholar] [CrossRef]
- Almarhoon, Z.; Al Rasheed, H.H.; El-Faham, A. Ultrasonically Assisted N-Cyanoacylation and Synthesis of Alkyl(4-(3-cyano-4,6-dimethyl-2-oxopyridin-1(2H)-yl)benzoyl)amino Acid Ester Derivatives. ACS Omega 2020, 5, 30671–30678. [Google Scholar] [CrossRef]
- Gewald, K.; Schäfer, H.; Sattler, K. Synthesen von 4-Amino-thieno[2,3—b]pyridinen. Monatsh. Chem. 1979, 110, 1189–1196. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE); 2020.09; Chemical Computing Group ULC: Montreal, QC, Canada, 2022.
- El Bialy, S.A.A.; Gouda, M.A. Cyanoacetamide in heterocyclic chemistry: Synthesis, antitumor and antioxidant activities of some new benzothiophenes. J. Heterocycl. Chem. 2011, 48, 1280–1286. [Google Scholar] [CrossRef]
- Schoen, B.W.; Lira, C.T.; Lee, A. Separation and Solubility of Cis and Trans Isomers in Nanostructured Double-Decker Silsequioxanes. J. Chem. Eng. Data 2014, 59, 1483–1493. [Google Scholar] [CrossRef]
- Hassan, A.S.; Askar, A.A.; Naglah, A.M.; Almehizia, A.A.; Ragab, A. Discovery of New Schiff Bases Tethered Pyrazole Moiety: Design, Synthesis, Biological Evaluation, and Molecular Docking Study as Dual Targeting DHFR/DNA Gyrase Inhibitors with Immunomodulatory Activity. Molecules 2020, 25, 2593. [Google Scholar] [CrossRef] [PubMed]
- Allam, H.A.; Fahim, S.H.; Abo-Ashour, M.F.; Nocentini, A.; Elbakry, M.E.; Abdelrahman, M.A.; Eldehna, W.M.; Ibrahim, H.S.; Supuran, C.T. Application of hydrazino and hydrazido linkers to connect benzenesulfonamides with hydrophilic/phobic tails for targeting the middle region of human carbonic anhydrases active site: Selective inhibitors of hCA IX. Eur. J. Med. Chem. 2019, 179, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New Colorimetric Cytotoxicity Assay for Anticancer-Drug Screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Zoete, V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [Green Version]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Peter, L.C.H.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific Association of Human Telomerase Activity with Immortal Cells and Cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef]
- Kovalenko, N.A.; Zhdanov, D.D.; Bibikova, M.V.; Gotovtseva, V.Y. The influence of compound aITEL1296 on telomerase activity and growth of cancer cells. Biochem. Suppl. B. Biomed. Chem. 2012, 6, 48–54. [Google Scholar] [CrossRef]
- Zhdanov, D.D.; Vasina, D.A.; Grachev, V.A.; Orlova, E.V.; Orlova, V.S.; Pokrovskaya, M.V.; Alexandrova, S.S.; Sokolov, N.N. Alternative splicing of telomerase catalytic subunit hTERT generated by apoptotic endonuclease EndoG induces human CD4+ T cell death. Eur. J. Cell Biol. 2017, 96, 653–664. [Google Scholar] [CrossRef]
- Hou, M.; Dw, X.; Björkholm, M.; Gruber, A. Real-Time Quantitative Telomeric Repeat Amplification Protocol Assay for the Detection of Telomerase Activity. Clin. Chem. 2001, 47, 519–524. [Google Scholar] [CrossRef] [Green Version]
- Sebaugh, J.L. Guidelines for accurate EC50/IC50 estimation. Pharm. Stat. 2011, 10, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015, 43, 443–447. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. iLOGP: A Simple, Robust, and Efficient Description of n-Octanol/Water Partition Coefficient for Drug Design Using the GB/SA Approach. J. Chem. Inf. Model. 2014, 54, 3284–3301. [Google Scholar] [CrossRef] [PubMed]
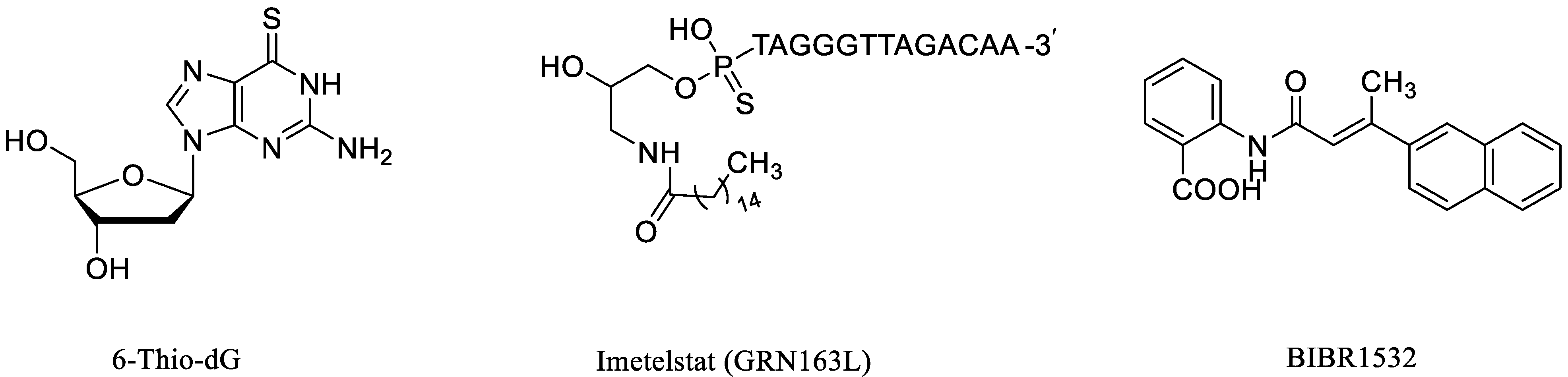
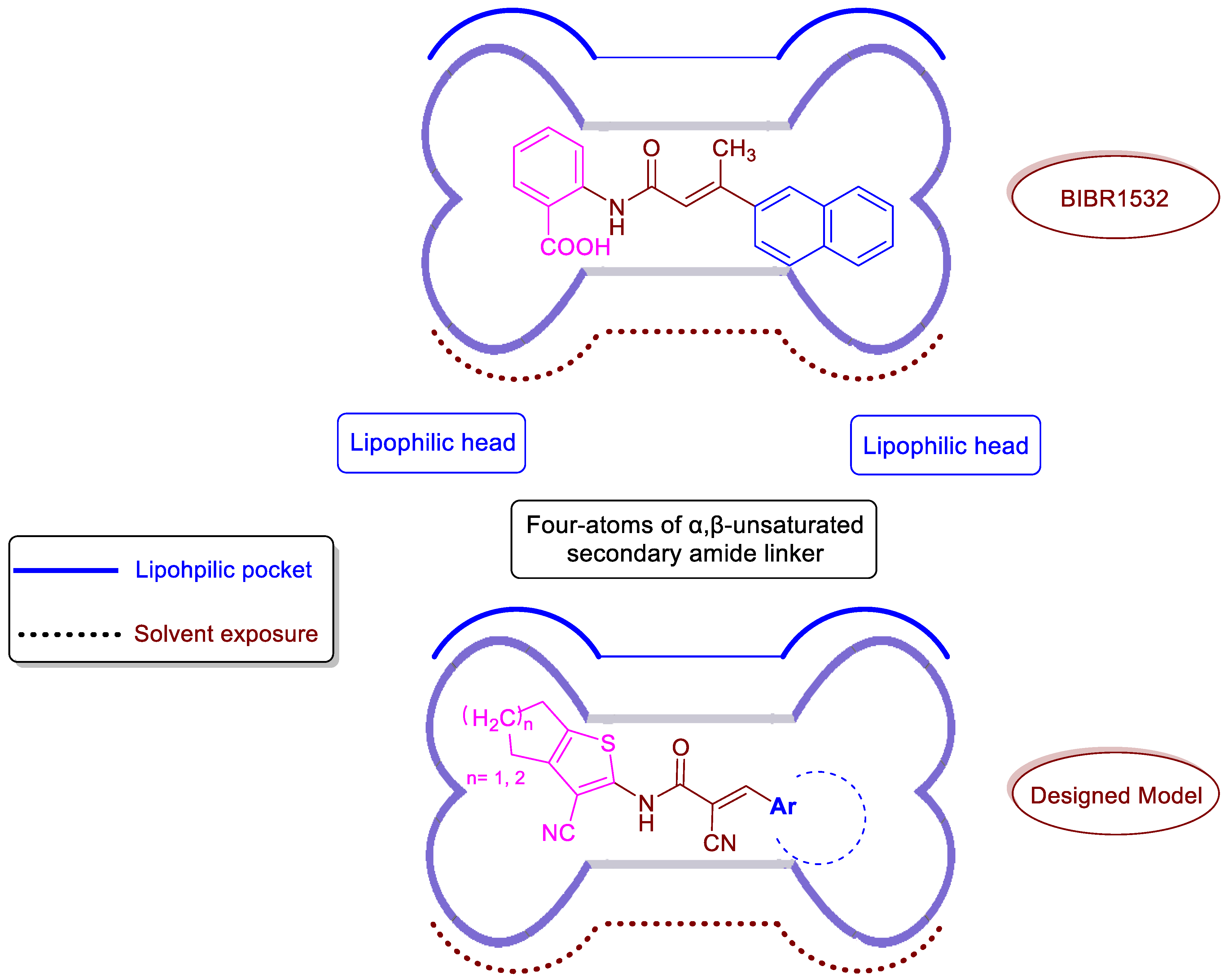





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (μM) | IC90 (μM) | Compound | IC50 (μM) | IC90 (μM) |
|---|---|---|---|---|---|
| 25a | 20.9 ± 0.7 | 108.7 ± 3.2 | 33a | 205.3 ± 4.0 | 643.0 |
| 25b | 25.8 ± 1.0 | 140.2 ± 4.7 | 33b | 177.5 ± 3.3 | 573.8 |
| 26a | 13.5 ± 0.6 | 153.5 | 34a | 188.8 ± 6.4 | 1078.1 |
| 26b | 53.6 ± 1.8 | 750.8 | 34b | 38.8 ± 1.0 | 284.2 ± 6.6 |
| 27a | 6.5 ± 0.3 | 163.6 | 35a | 5.1 ± 0.2 | 31.1 ± 1.1 |
| 27b | 12.5 ± 0.6 | 138.0 | 35b | 5.3 ± 0.2 | 18.6 ± 0.9 |
| 28a | 6.7 ± 0.1 | 67.8 | 36a | 5.2 ± 0.2 | 34.9 ± 1.6 |
| 28b | 8.3 ± 0.3 | 57.4 | 36b | 0.3 ± 0.01 | 6.2 ± 0.3 |
| 29a | 1.7 ± 0.04 | 6.3 ± 0.3 | 37a | 23.5 ± 0.9 | 136.6 ± 2.6 |
| 29b | 4.8 ± 0.2 | 32.8 ± 1.5 | 37b | 97.8 ± 2.6 | 173.9 |
| 30a | 5.2 ± 0.2 | 112.5 ± 3.6 | 38a | 25.4 ± 1.0 | 282.4 |
| 30b | 5.9 ± 0.2 | 119.4 ± 4.6 | 38b | 40.3 ± 1.7 | 339.4 |
| 31a | 98.5 ± 3.8 | 200.5 | 39a | 2.5 ± 0.1 | 9.5 ± 0.3 |
| 31b | 89.9 ± 3.6 | 165.2 | 39b | 2.0 ± 0.03 | 7.1 ± 0.3 |
| 32a | 6.4 ± 0.2 | 177.4 | BIBR1532 | 0.2 ± 0.01 | 5.6 ± 0.02 |
| 32b | 109.7 ± 3.7 | 180.8 |
| % Telomerase Activity in Cancer Cells | |||
|---|---|---|---|
| Compound | A549 | HCC44 | NCI-H23 |
| 29a | 22.1 ± 4.8% | 66.1 ± 2.4% | 28.2 ± 3.9% |
| 36b | 18.1 ± 5.4% | 54.4 ± 4.4% | 25.1 ± 3.2% |
| 39b | 41.5 ± 6.9% | 75.6 ± 7.9% | 50.3 ± 5.1% |
| Cell Line | Growth Percent | Cell Line | Growth Percent | Cell Line | Growth Percent |
|---|---|---|---|---|---|
| Leukemia | KM12 | 101.43 | OVCAR-5 | 104.98 | |
| CCRF-CEM | 104.69 | SW-620 | 103.69 | OVCAR-8 | 102.91 |
| HL-60(TB) | 99.47 | CNS cancer | NCI/ADR-RES | 99.44 | |
| K-562 | 99.98 | SF-268 | 104.45 | SK-OV-3 | 89.18 |
| MOLT-4 | 100.37 | SF-295 | 95.53 | Renal cancer | |
| RPMI-8226 | 99.85 | SF-539 | 98.71 | 786-0 | 101.2 |
| SR | 95.72 | SNB-19 | 100.30 | A498 | 99.38 |
| Lung cancer (NSCLC) | SNB-75 | 101.09 | ACHN | 103.29 | |
| A549/ATCC | 100.18 | U251 | 98.73 | CAKI-1 | 94.28 |
| EKVX | 92.47 | Melanoma | RXF 393 | 111.16 | |
| HOP-62 | 98.36 | LOX IMVI | 102.88 | SN12C | 100.72 |
| HOP-92 | 90.75 | MALME-3M | 109.97 | TK-10 | 113.35 |
| NCI-H226 | 89.78 | M14 | 94.57 | UO-31 | 81.95 |
| NCI-H23 | 94.56 | MDA-MB-435 | 101.92 | Prostate cancer | |
| NCI-H322M | 98.58 | SK-MEL-2 | 100.31 | PC-3 | 104.77 |
| NCI-H460 | 102.88 | SK-MEL-28 | 102.31 | DU-145 | 99.89 |
| NCI-H522 | 94.31 | SK-MEL-5 | 97.65 | Breast cancer | |
| Colon cancer | UACC-257 | 102.32 | MCF7 | 92.41 | |
| COLO 205 | 103.10 | UACC-62 | 91.73 | MDA-MB-231/ATCC | 92.15 |
| HCC-2998 | 114.98 | Ovarian cancer | HS 578T | 103.54 | |
| HCT-116 | 100.18 | IGROV1 | 103.06 | BT-549 | 107.71 |
| HCT-15 | 99.95 | OVCAR-3 | 98.78 | T-47D | 97.26 |
| HT29 | 112.60 | OVCAR-4 | 100.16 | MDA-MB-468 | 90.36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tawfik, H.O.; El-Hamaky, A.A.; El-Bastawissy, E.A.; Shcherbakov, K.A.; Veselovsky, A.V.; Gladilina, Y.A.; Zhdanov, D.D.; El-Hamamsy, M.H. New Genetic Bomb Trigger: Design, Synthesis, Molecular Dynamics Simulation, and Biological Evaluation of Novel BIBR1532-Related Analogs Targeting Telomerase against Non-Small Cell Lung Cancer. Pharmaceuticals 2022, 15, 481. https://doi.org/10.3390/ph15040481
Tawfik HO, El-Hamaky AA, El-Bastawissy EA, Shcherbakov KA, Veselovsky AV, Gladilina YA, Zhdanov DD, El-Hamamsy MH. New Genetic Bomb Trigger: Design, Synthesis, Molecular Dynamics Simulation, and Biological Evaluation of Novel BIBR1532-Related Analogs Targeting Telomerase against Non-Small Cell Lung Cancer. Pharmaceuticals. 2022; 15(4):481. https://doi.org/10.3390/ph15040481
Chicago/Turabian StyleTawfik, Haytham O., Anwar A. El-Hamaky, Eman A. El-Bastawissy, Kirill A. Shcherbakov, Alexander V. Veselovsky, Yulia A. Gladilina, Dmitry D. Zhdanov, and Mervat H. El-Hamamsy. 2022. "New Genetic Bomb Trigger: Design, Synthesis, Molecular Dynamics Simulation, and Biological Evaluation of Novel BIBR1532-Related Analogs Targeting Telomerase against Non-Small Cell Lung Cancer" Pharmaceuticals 15, no. 4: 481. https://doi.org/10.3390/ph15040481
APA StyleTawfik, H. O., El-Hamaky, A. A., El-Bastawissy, E. A., Shcherbakov, K. A., Veselovsky, A. V., Gladilina, Y. A., Zhdanov, D. D., & El-Hamamsy, M. H. (2022). New Genetic Bomb Trigger: Design, Synthesis, Molecular Dynamics Simulation, and Biological Evaluation of Novel BIBR1532-Related Analogs Targeting Telomerase against Non-Small Cell Lung Cancer. Pharmaceuticals, 15(4), 481. https://doi.org/10.3390/ph15040481