
Differential Effects of a Novel Opioid Ligand UTA1003 on Antinociceptive Tolerance and Motor Behaviour

 , ,
, ,  , and
, and 
Abstract
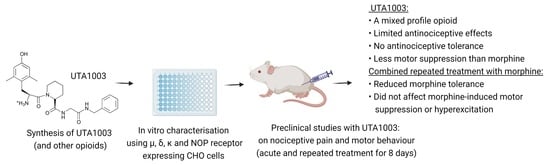
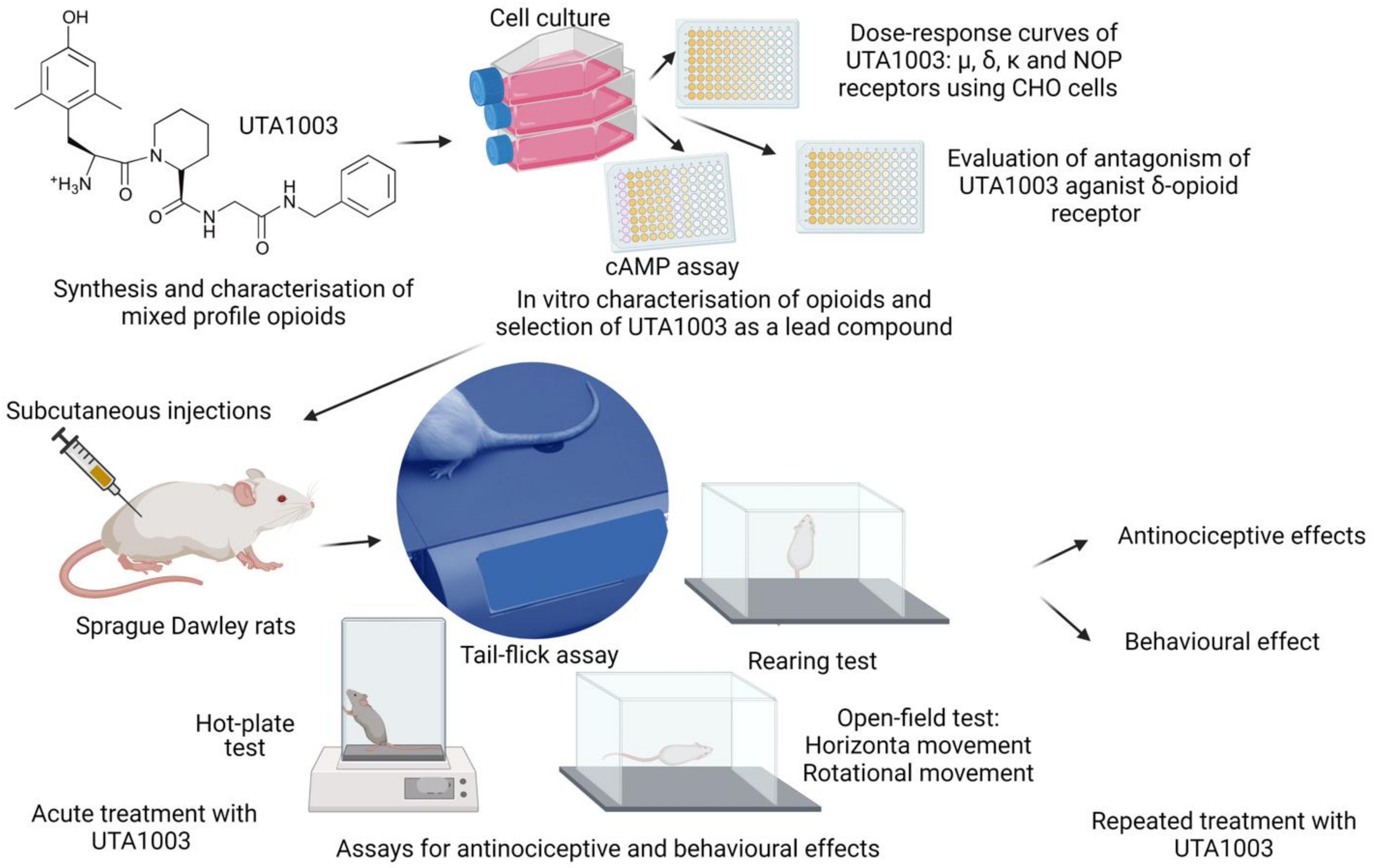
1. Introduction
2. Results
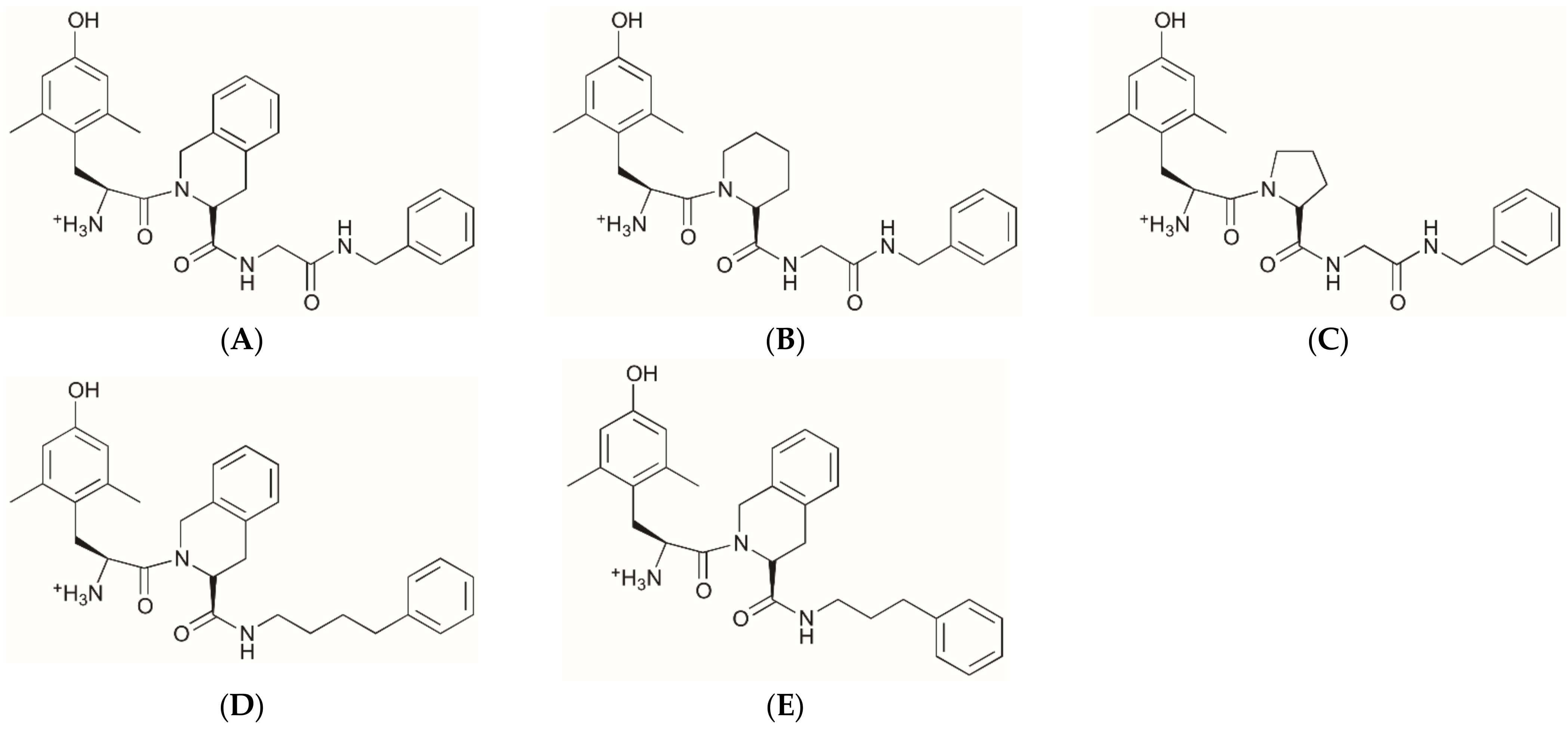
2.1. Structures of Novel UTA-Opioids
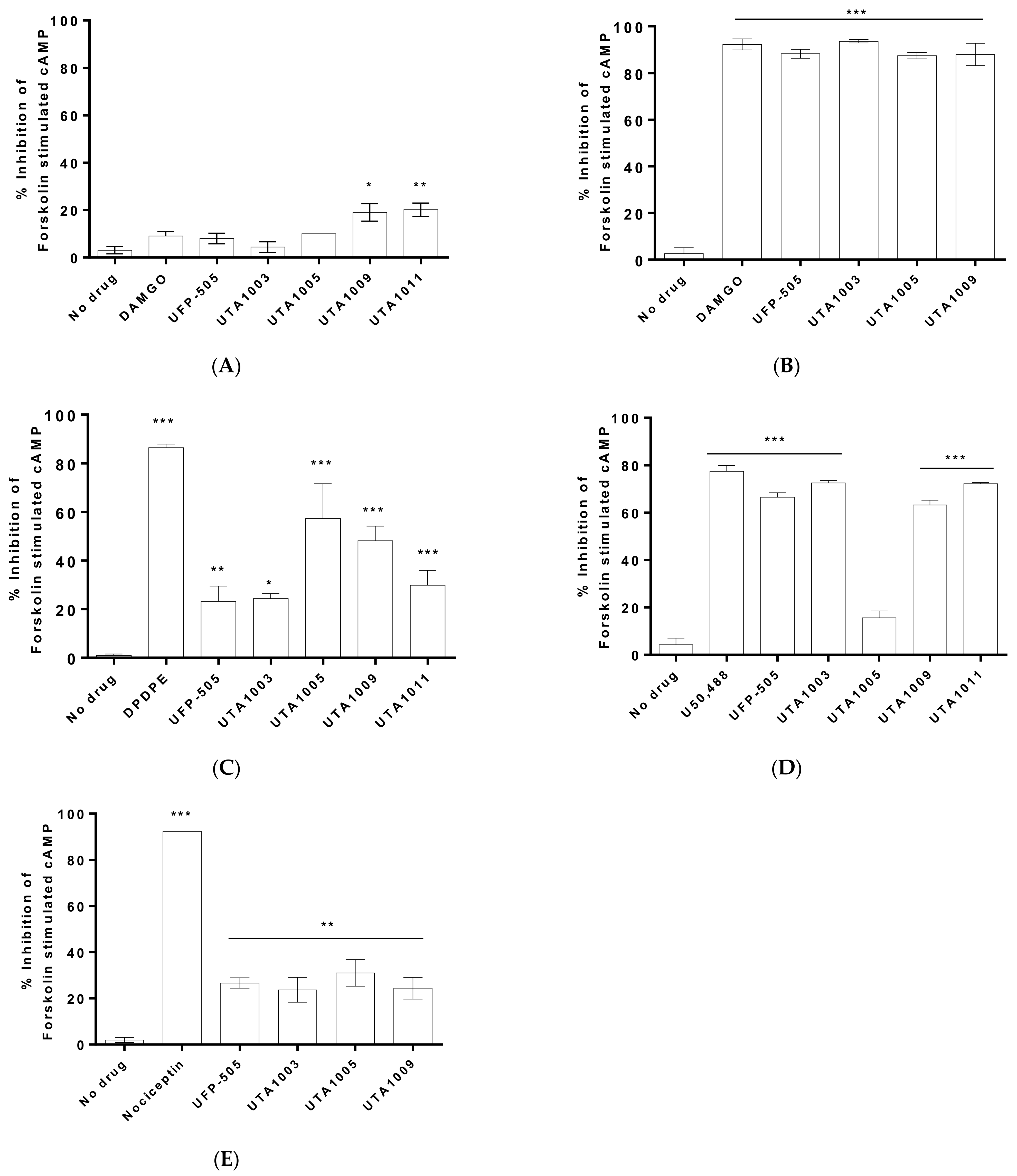
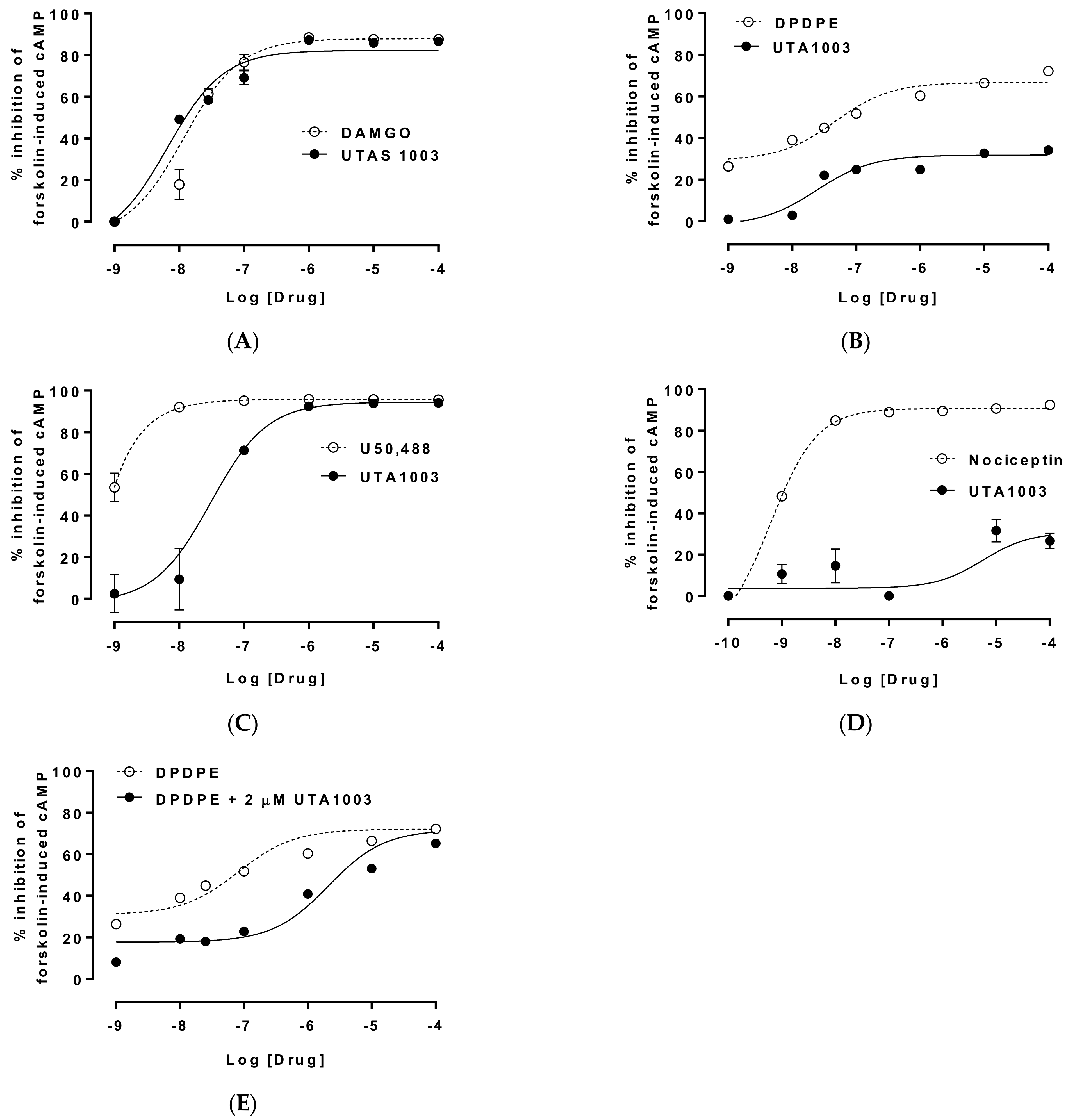
2.2. Specificity of Novel UTA-Opioids on Different Opioid Receptors
2.3. Detailed Agonistic Effects of UTA1003 towards Different Opioid Receptors
2.4. Antagonistic Effect of UTA1003 on DOP Receptors
2.5. Toxicity of UTA1003
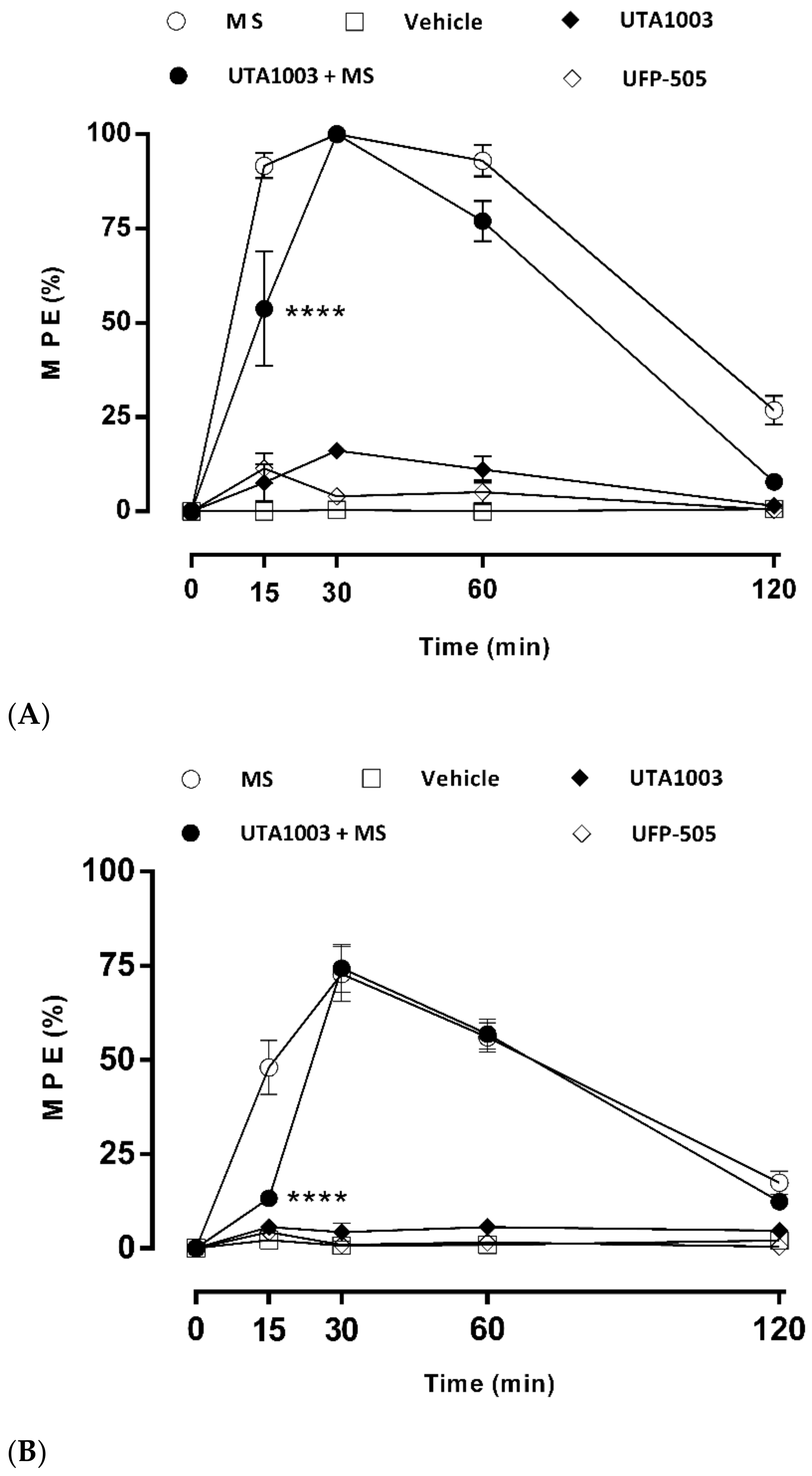
2.6. Antinociceptive Effect of UTA1003 after Acute Administration
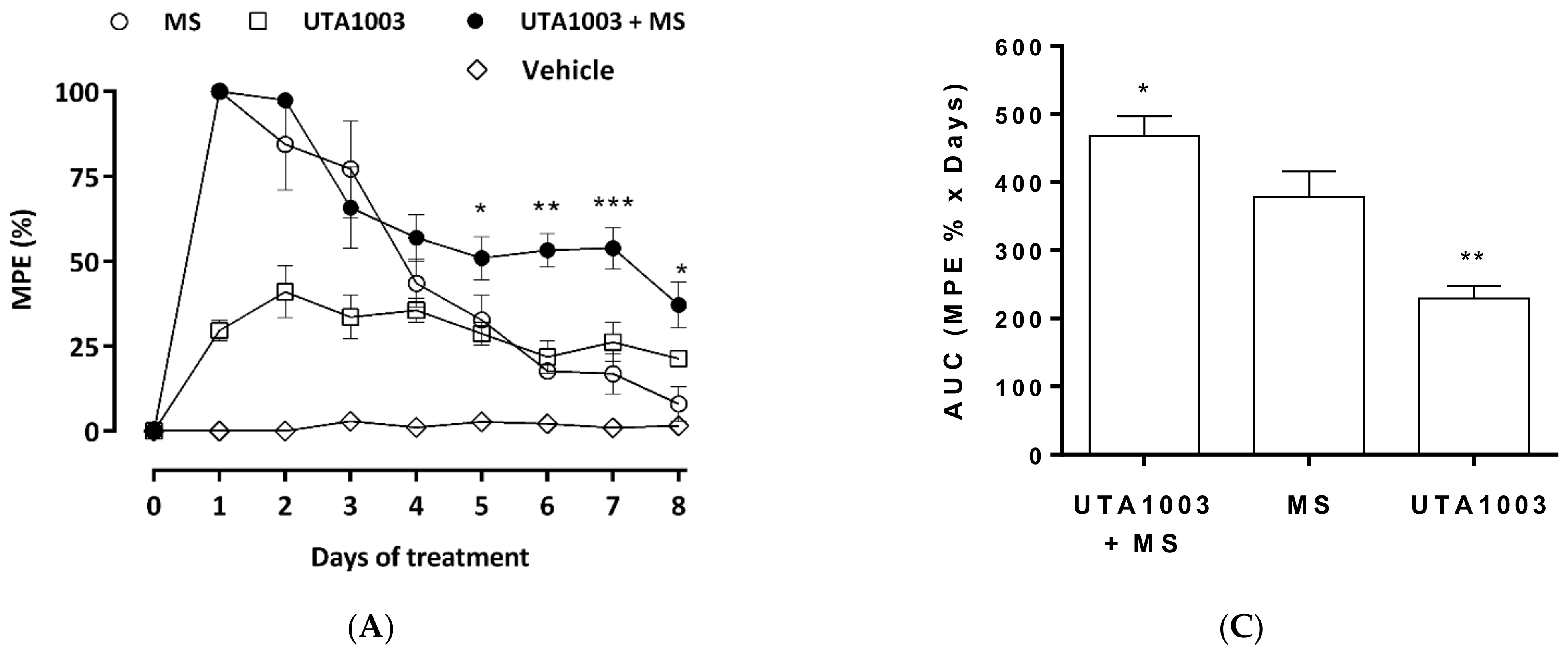
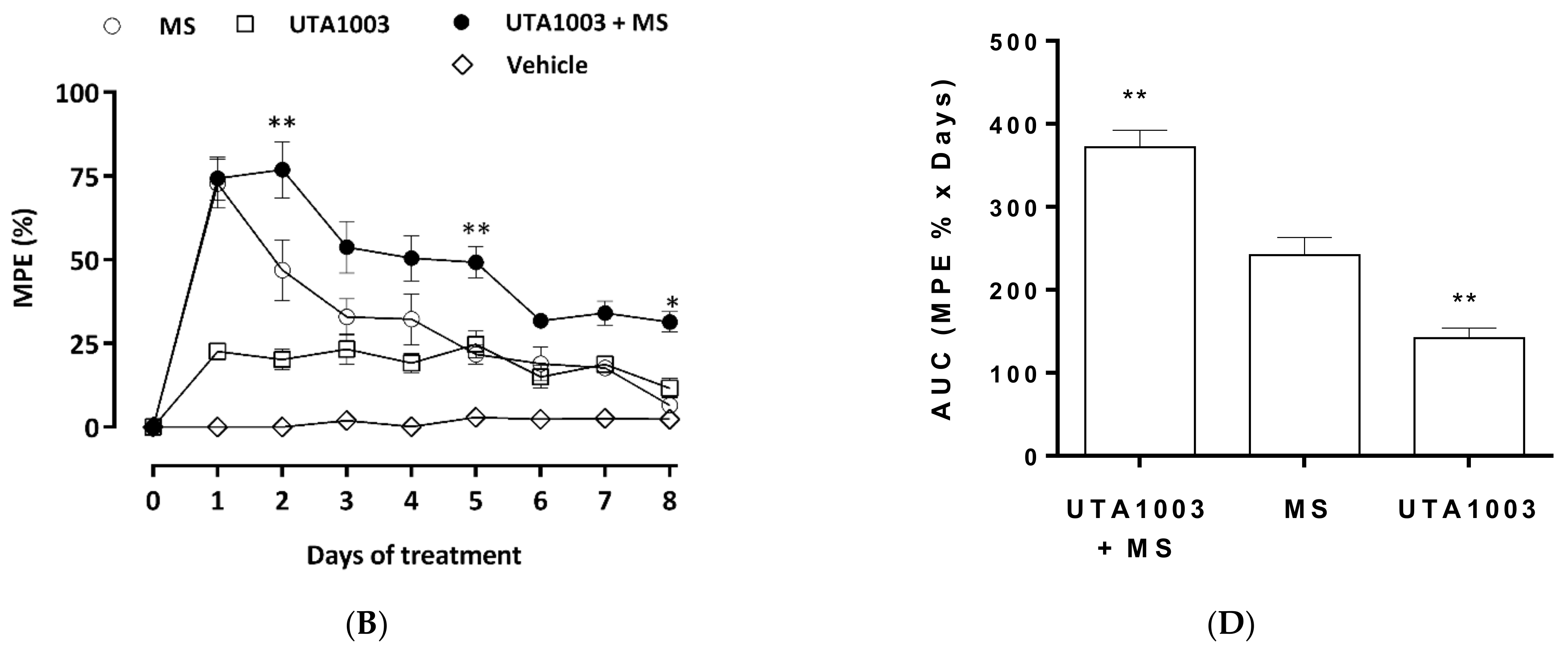
2.7. Effect of UTA1003 on Morphine-Induced Antinociceptive Tolerance
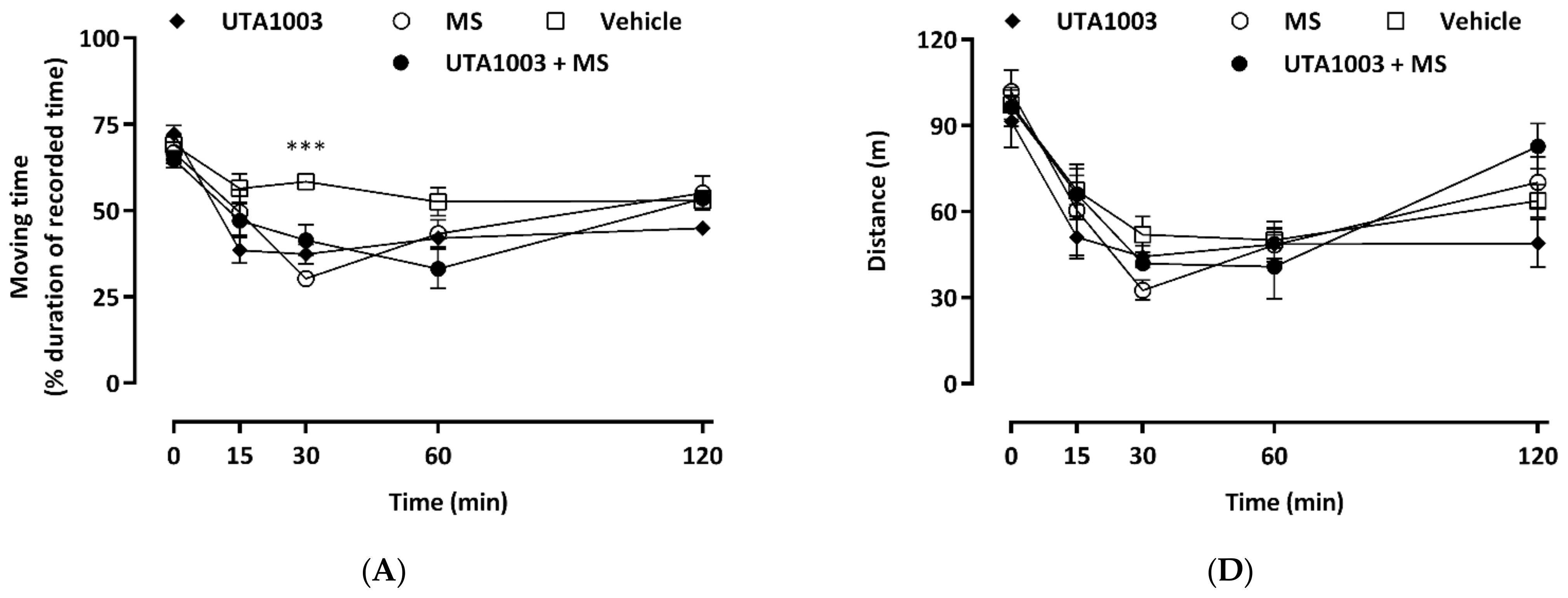
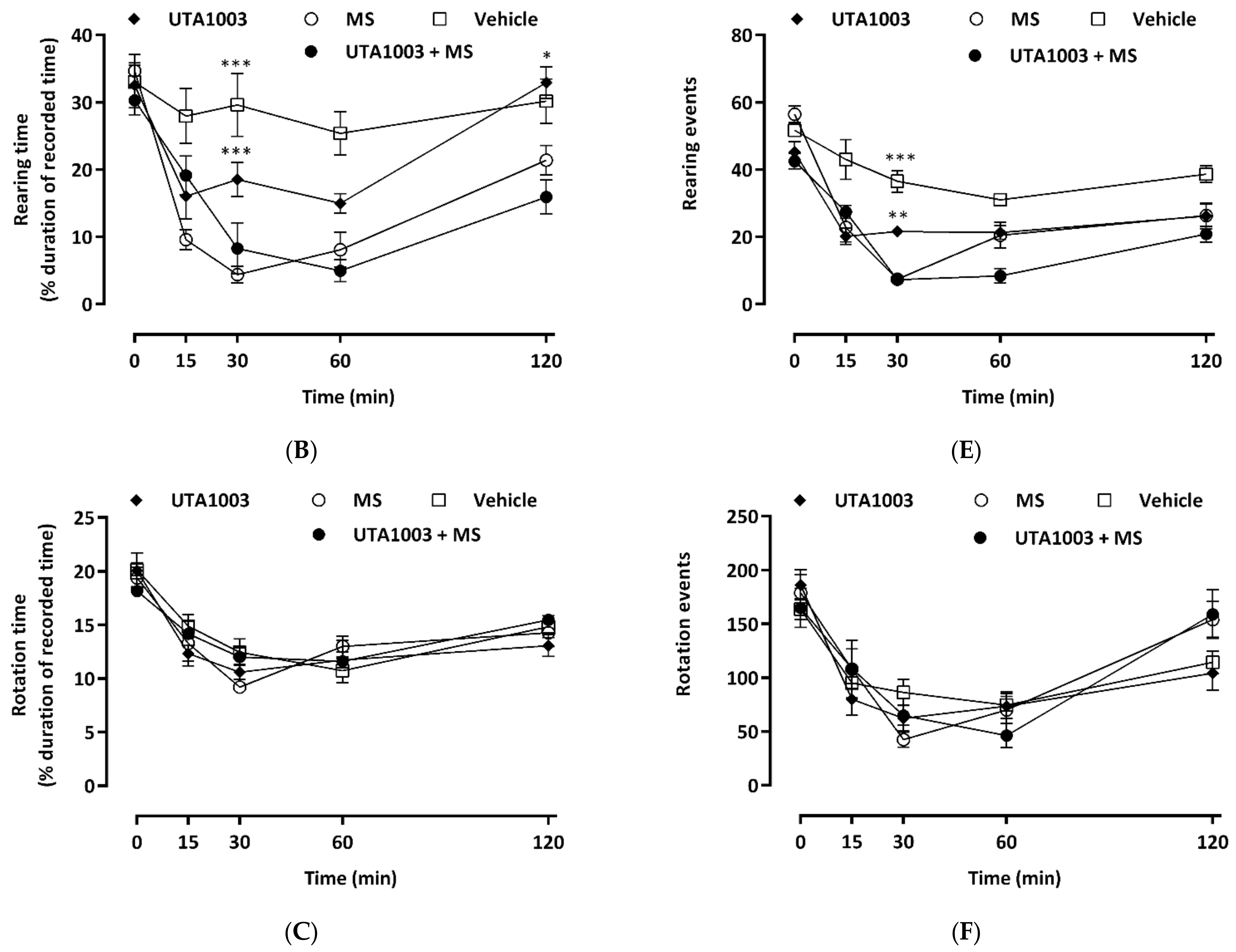
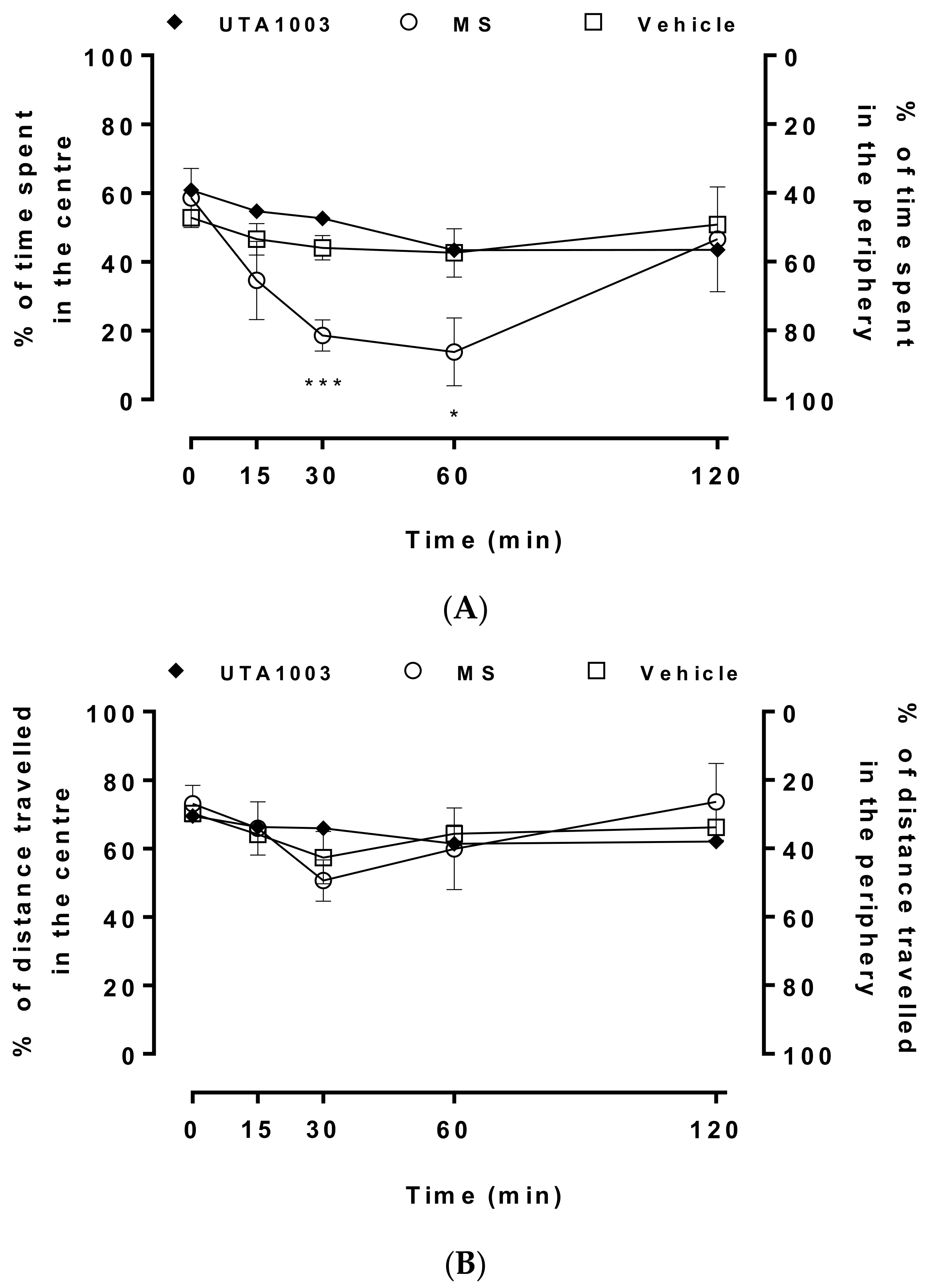
2.8. Effect of Acute Administration of UTA1003 on Motor Behaviour
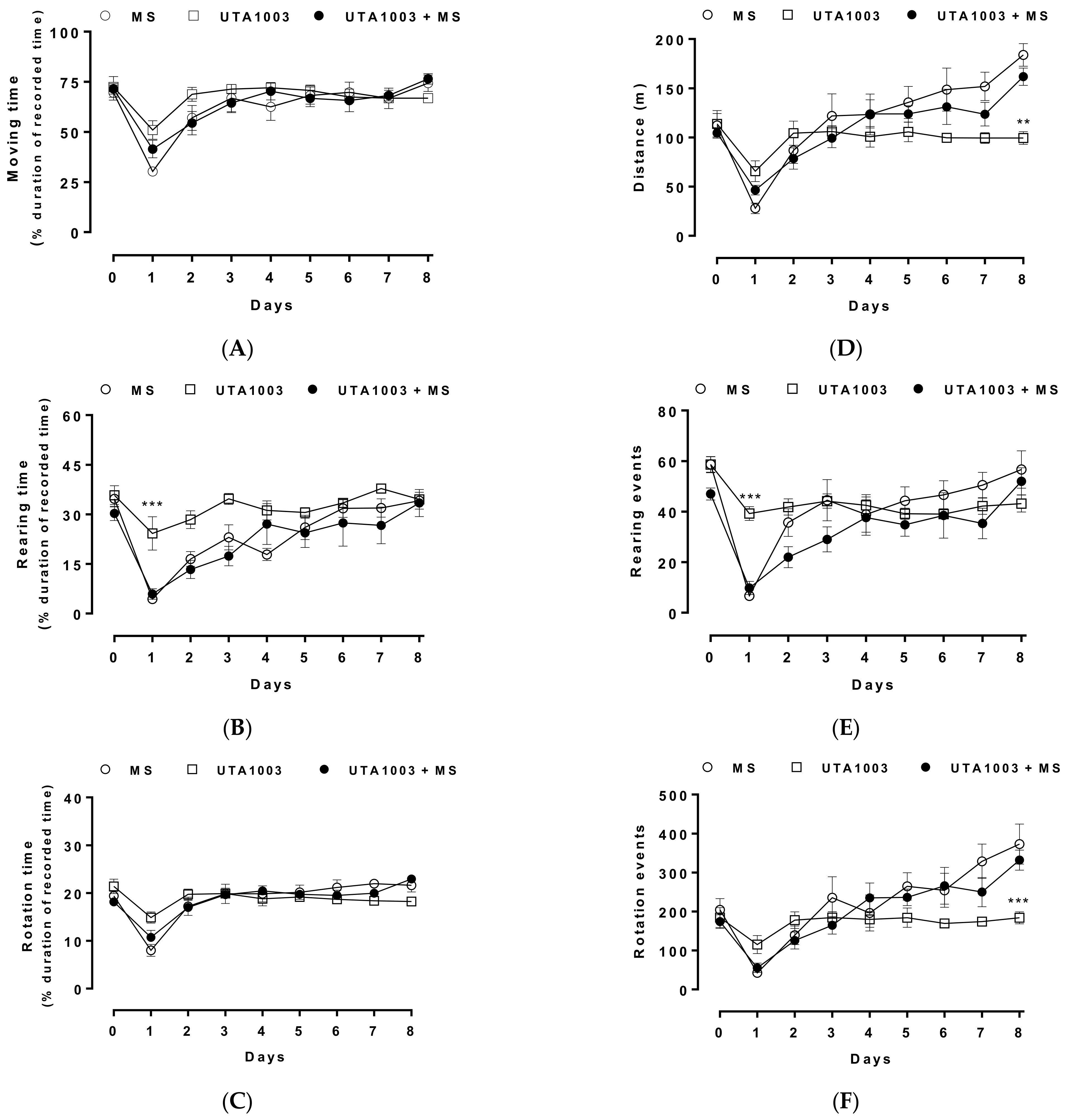
2.9. Effect of Chronic UTA1003 Treatment on Motor Behaviour
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Chemical Synthesis
4.3. cAMP Assay
4.4. Animal Maintenance and Care
4.5. Preclinical Treatment Protocol
4.6. Assessment of Antinociception
4.7. Behavioural Measurements
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Corbett, A.D.; Henderson, G.; McKnight, A.T.; Paterson, S.J. 75 years of opioid research: The exciting but vain quest for the holy grail. Br. J. Pharmacol. 2006, 147, S153–S162. [Google Scholar] [CrossRef] [PubMed]
- Stein, C. Opioids, sensory systems and chronic pain. Eur. J. Pharmacol. 2013, 716, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Portenoy, R.K.; Ahmed, E. Principles of opioid use in cancer pain. J. Clin. Oncol. 2014, 32, 1662–1670. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC). Prescription Opioids: Side Effects. Available online: https://www.cdc.gov/drugoverdose/opioids/prescribed.html (accessed on 29 December 2020).
- Therapeutic Guidelines Ltd. Principles of Nonsteroidal Anti-Inflammatory Drug Use for Musculoskeletal Conditions in Adults. In eTG Complete; Therapeutic Guidelines Ltd.: West Melbourne, VIC, Australia, 2017; Available online: https://tgldcdp.tg.org.au/index (accessed on 29 December 2020).
- World Health Organization (WHO). Guidelines for the Psychosocially Assisted Pharmacological Treatment of Opioid Dependence; World Health Organization: Geneva, Switzerland, 2009; Available online: http://apps.who.int/iris/bitstream/10665/43948/1/9789241547543_eng.Pdf (accessed on 14 March 2017).
- Mohammed, W.; Alhaddad, H.; Marie, N.; Tardy, F.; Lamballais, F.; Risede, P.; Noble, F.; Baud, F.J.; Megarbane, B. Comparison of tolerance to morphine-induced respiratory and analgesic effects in mice. Toxicol. Lett. 2013, 217, 251–259. [Google Scholar] [CrossRef]
- Paronis, C.A.; Woods, J.H. Ventilation in morphine-maintained rhesus monkeys. Ii: Tolerance to the antinociceptive but not the ventilatory effects of morphine. J. Pharmacol. Exp. Ther. 1997, 282, 355–362. [Google Scholar]
- Emery, M.J.; Groves, C.C.; Kruse, T.N.; Shi, C.; Terman, G.W. Ventilation and the response to hypercapnia after morphine in opioid-naive and opioid-tolerant rats. Anesthesiology 2016, 124, 945–957. [Google Scholar] [CrossRef]
- Paul, A.K.; Gueven, N.; Dietis, N. Morphine dosing strategy plays a key role in the generation and duration of the produced antinociceptive tolerance. Neuropharmacology 2017, 121, 158–166. [Google Scholar] [CrossRef]
- Dominguez, J.E.; Habib, A.S. Prophylaxis and treatment of the side-effects of neuraxial morphine analgesia following cesarean delivery. Curr. Opin. Anaesthesiol. 2013, 26, 288–295. [Google Scholar] [CrossRef]
- Girgin, N.K.; Gurbet, A.; Turker, G.; Aksu, H.; Gulhan, N. Intrathecal morphine in anesthesia for cesarean delivery: Dose-response relationship for combinations of low-dose intrathecal morphine and spinal bupivacaine. J. Clin. Anesth. 2008, 20, 180–185. [Google Scholar] [CrossRef]
- Raffaeli, W.; Marconi, G.; Fanelli, G.; Taddei, S.; Borghi, G.B.; Casati, A. Opioid-related side-effects after intrathecal morphine: A prospective, randomized, double-blind dose-response study. Eur. J. Anaesthesiol. 2006, 23, 605–610. [Google Scholar] [CrossRef]
- Paul, A.K.; Lewis, R.J. Pain management in older adults: Facts to consider. Pain 2022, 163, e497–e498. [Google Scholar] [CrossRef]
- Abdelhamid, E.E.; Sultana, M.; Portoghese, P.; Takemori, A. Selective blockage of delta opioid receptors prevents the development of morphine tolerance and dependence in mice. J. Pharmacol. Exp. Ther. 1991, 258, 299–303. [Google Scholar]
- Fundytus, M.E.; Schiller, P.W.; Shapiro, M.; Weltrowska, G.; Coderre, T.J. Attenuation of morphine tolerance and dependence with the highly selective delta-opioid receptor antagonist tipp[psi]. Eur. J. Pharmacol. 1995, 286, 105–108. [Google Scholar] [CrossRef]
- Hepburn, M.J.; Little, P.J.; Gingras, J.; Kuhn, C.M. Differential effects of naltrindole on morphine-induced tolerance and physical dependence in rats. J. Pharmacol. Exp. Ther. 1997, 281, 1350–1356. [Google Scholar]
- Roy, S.; Guo, X.; Kelschenbach, J.; Liu, Y.; Loh, H.H. In vivo activation of a mutant mu-opioid receptor by naltrexone produces a potent analgesic effect but no tolerance: Role of mu-receptor activation and delta-receptor blockade in morphine tolerance. J. Neurosci. 2005, 25, 3229–3233. [Google Scholar] [CrossRef]
- Abul-Husn, N.S.; Sutak, M.; Milne, B.; Jhamandas, K. Augmentation of spinal morphine analgesia and inhibition of tolerance by low doses of mu- and delta-opioid receptor antagonists. Br. J. Pharmacol. 2007, 151, 877–887. [Google Scholar] [CrossRef]
- McNaull, B.; Trang, T.; Sutak, M.; Jhamandas, K. Inhibition of tolerance to spinal morphine antinociception by low doses of opioid receptor antagonists. Eur. J. Pharmacol. 2007, 560, 132–141. [Google Scholar] [CrossRef]
- Dietis, N.; Guerrini, R.; Calo, G.; Salvadori, S.; Rowbotham, D.J.; Lambert, D.G. Simultaneous targeting of multiple opioid receptors: A strategy to improve side-effect profile. Br. J. Anaesth. 2009, 103, 38–49. [Google Scholar] [CrossRef]
- Schiller, P.W.; Fundytus, M.E.; Merovitz, L.; Weltrowska, G.; Nguyen, T.M.; Lemieux, C.; Chung, N.N.; Coderre, T.J. The opioid mu agonist/delta antagonist dipp-nh(2)[psi] produces a potent analgesic effect, no physical dependence, and less tolerance than morphine in rats. J. Med. Chem. 1999, 42, 3520–3526. [Google Scholar] [CrossRef]
- Schiller, P.W. Bi- or multifunctional opioid peptide drugs. Life Sci. 2010, 86, 598–603. [Google Scholar] [CrossRef]
- Healy, J.R.; Bezawada, P.; Shim, J.; Jones, J.W.; Kane, M.A.; MacKerell, A.D., Jr.; Coop, A.; Matsumoto, R.R. Synthesis, modeling, and pharmacological evaluation of umb 425, a mixed mu agonist/delta antagonist opioid analgesic with reduced tolerance liabilities. ACS Chem. Neurosci. 2013, 4, 1256–1266. [Google Scholar] [CrossRef]
- Ananthan, S.; Saini, S.K.; Dersch, C.M.; Xu, H.; McGlinchey, N.; Giuvelis, D.; Bilsky, E.J.; Rothman, R.B. 14-alkoxy- and 14-acyloxypyridomorphinans: Mu agonist/delta antagonist opioid analgesics with diminished tolerance and dependence side effects. J. Med. Chem. 2012, 55, 8350–8363. [Google Scholar] [CrossRef]
- Wells, J.L.; Bartlett, J.L.; Ananthan, S.; Bilsky, E.J. In vivo pharmacological characterization of sori 9409, a nonpeptidic opioid mu-agonist/delta-antagonist that produces limited antinociceptive tolerance and attenuates morphine physical dependence. J. Pharmacol. Exp. Ther. 2001, 297, 597–605. [Google Scholar]
- Anand, J.P.; Boyer, B.T.; Mosberg, H.I.; Jutkiewicz, E.M. The behavioral effects of a mixed efficacy antinociceptive peptide, VRP26, following chronic administration in mice. Psychopharmacology 2016, 233, 2479–2487. [Google Scholar] [CrossRef]
- Lowery, J.J.; Raymond, T.J.; Giuvelis, D.; Bidlack, J.M.; Polt, R.; Bilsky, E.J. In vivo characterization of mmp-2200, a mixed delta/mu opioid agonist, in mice. J. Pharmacol. Exp. Ther. 2011, 336, 767–778. [Google Scholar] [CrossRef]
- Guillemyn, K.; Kleczkowska, P.; Novoa, A.; Vandormael, B.; Van den Eynde, I.; Kosson, P.; Asim, M.F.; Schiller, P.W.; Spetea, M.; Lipkowski, A.W.; et al. In vivo antinociception of potent mu opioid agonist tetrapeptide analogues and comparison with a compact opioid agonist-neurokinin 1 receptor antagonist chimera. Mol. Brain 2012, 5, 4. [Google Scholar] [CrossRef]
- Li, N.; Han, Z.L.; Wang, Z.L.; Xing, Y.H.; Sun, Y.L.; Li, X.H.; Song, J.J.; Zhang, T.; Zhang, R.; Zhang, M.N.; et al. BN-9, a chimeric peptide with mixed opioid and neuropeptide ff receptor agonistic properties, produces nontolerance-forming antinociception in mice. Br. J. Pharmacol. 2016, 173, 1864–1880. [Google Scholar] [CrossRef]
- Matsumoto, K.; Takayama, H.; Narita, M.; Nakamura, A.; Suzuki, M.; Suzuki, T.; Murayama, T.; Wongseripipatana, S.; Misawa, K.; Kitajima, M.; et al. MGM-9 [(e)-methyl 2-(3-ethyl-7a,12a-(epoxyethanoxy)-9-fluoro-1,2,3,4,6,7,12,12b-octahydro-8-methoxy indolo[2,3-a]quinolizin-2-yl)-3-methoxyacrylate], a derivative of the indole alkaloid mitragynine: A novel dual-acting mu- and kappa-opioid agonist with potent antinociceptive and weak rewarding effects in mice. Neuropharmacology 2008, 55, 154–165. [Google Scholar] [CrossRef]
- Mello, N.K.; Mendelson, J.H.; Sholar, M.B.; Jaszyna-Gasior, M.; Goletiani, N.; Siegel, A.J. Effects of the mixed mu/kappa opioid nalbuphine on cocaine-induced changes in subjective and cardiovascular responses in men. Neuropsychopharmacology 2005, 30, 618–632. [Google Scholar] [CrossRef]
- Toll, L.; Khroyan, T.V.; Polgar, W.E.; Jiang, F.; Olsen, C.; Zaveri, N.T. Comparison of the antinociceptive and antirewarding profiles of novel bifunctional nociceptin receptor/mu-opioid receptor ligands: Implications for therapeutic applications. J. Pharmacol. Exp. Ther. 2009, 331, 954–964. [Google Scholar] [CrossRef]
- Khroyan, T.V.; Polgar, W.E.; Orduna, J.; Jiang, F.; Olsen, C.; Toll, L.; Zaveri, N.T. Activity of new NOP receptor ligands in a rat peripheral mononeuropathy model: Potentiation of morphine anti-allodynic activity by NOP receptor antagonists. Eur. J. Pharmacol. 2009, 610, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Dietis, N.; Niwa, H.; Tose, R.; McDonald, J.; Ruggieri, V.; Filaferro, M.; Vitale, G.; Micheli, L.; Ghelardini, C.; Salvadori, S.; et al. In vitro and in vivo characterisation of the bifunctional mop/dop ligand UFP-505. Br. J. Pharmacol. 2018, 175, 2881–2896. [Google Scholar] [CrossRef] [PubMed]
- Mosberg, H.I.; Yeomans, L.; Anand, J.P.; Porter, V.; Sobczyk-Kojiro, K.; Traynor, J.R.; Jutkiewicz, E.M. Development of a bioavailable mu opioid receptor (MOPr) agonist, delta opioid receptor (DOPr) antagonist peptide that evokes antinociception without development of acute tolerance. J. Med. Chem. 2014, 57, 3148–3153. [Google Scholar] [CrossRef] [PubMed]
- Dietis, N.; McDonald, J.; Molinari, S.; Calo, G.; Guerrini, R.; Rowbotham, D.J.; Lambert, D.G. Pharmacological characterization of the bifunctional opioid ligand h-dmt-tic-gly-nh-bzl (UFP-505). Br. J. Anaesth. 2012, 108, 262–270. [Google Scholar] [CrossRef]
- Purington, L.C.; Sobczyk-Kojiro, K.; Pogozheva, I.D.; Traynor, J.R.; Mosberg, H.I. Development and in vitro characterization of a novel bifunctional mu-agonist/delta-antagonist opioid tetrapeptide. ACS Chem. Biol 2011, 6, 1375–1381. [Google Scholar] [CrossRef]
- Gharagozlou, P.; Demirci, H.; Clark, J.D.; Lameh, J. Activation profiles of opioid ligands in HEK cells expressing delta opioid receptors. BMC Neurosci. 2002, 3, 19. [Google Scholar] [CrossRef]
- Liu, X.Y.; Liu, Z.C.; Sun, Y.G.; Ross, M.; Kim, S.; Tsai, F.F.; Li, Q.F.; Jeffry, J.; Kim, J.Y.; Loh, H.H.; et al. Unidirectional cross-activation of GRPR by MOR1D uncouples itch and analgesia induced by opioids. Cell 2011, 147, 447–458. [Google Scholar] [CrossRef]
- Chinachoti, T.; Nilrat, P.; Samarnpiboonphol, P. Nausea, vomiting and pruritus induced by intrathecal morphine. J. Med. Assoc. Thail. 2013, 96, 589–594. [Google Scholar]
- Miyamoto, T.; Patapoutian, A. Why does morphine make you itch? Cell 2011, 147, 261–262. [Google Scholar] [CrossRef]
- Hina, N.; Fletcher, D.; Poindessous-Jazat, F.; Martinez, V. Hyperalgesia induced by low-dose opioid treatment before orthopaedic surgery: An observational case-control study. Eur. J. Anaesthesiol. 2015, 32, 255–261. [Google Scholar] [CrossRef]
- Chu, L.F.; D’Arcy, N.; Brady, C.; Zamora, A.K.; Young, C.A.; Kim, J.E.; Clemenson, A.M.; Angst, M.S.; Clark, J.D. Analgesic tolerance without demonstrable opioid-induced hyperalgesia: A double-blinded, randomized, placebo-controlled trial of sustained-release morphine for treatment of chronic nonradicular low-back pain. Pain 2012, 153, 1583–1592. [Google Scholar] [CrossRef]
- Elhabazi, K.; Ayachi, S.; Ilien, B.; Simonin, F. Assessment of morphine-induced hyperalgesia and analgesic tolerance in mice using thermal and mechanical nociceptive modalities. J. Vis. Exp. 2014, 89, e51264. [Google Scholar] [CrossRef]
- Wala, E.P.; Sloan, P.A.; Holtman, J.R., Jr. Effect of prior treatment with ultra-low-dose morphine on opioid- and nerve injury-induced hyperalgesia in rats. J. Opioid Manag. 2011, 7, 377–389. [Google Scholar] [CrossRef]
- Swartjes, M.; Mooren, R.A.; Waxman, A.R.; Arout, C.; van de Wetering, K.; den Hartigh, J.; Beijnen, J.H.; Kest, B.; Dahan, A. Morphine induces hyperalgesia without involvement of mu-opioid receptor or morphine-3-glucuronide. Mol. Med. 2012, 18, 1320–1326. [Google Scholar] [CrossRef]
- Song, L.; Wu, C.; Zuo, Y. Melatonin prevents morphine-induced hyperalgesia and tolerance in rats: Role of protein kinase c and n-methyl-d-aspartate receptors. BMC Anesth. 2015, 15, 12. [Google Scholar] [CrossRef]
- Alves, R.; Carvalho, J.G.B.d.; Venditti, M.A.C. High-and low-rearing rats differ in the brain excitability controlled by the allosteric benzodiazepine site in the GABAa receptor. J. Behav. Brain Sci. 2012, 2, 315–325. [Google Scholar] [CrossRef]
- Lever, C.; Burton, S.; O’Keefe, J. Rearing on hind legs, environmental novelty, and the hippocampal formation. Rev. Neurosci. 2006, 17, 111–133. [Google Scholar] [CrossRef]
- Rodriguiz, R.M.; Wetsel, W.C. Frontiers in Neuroscience Assessments of Cognitive Deficits in Mutant Mice. In Animal Models of Cognitive Impairment; Levin, E.D., Buccafusco, J.J., Eds.; CRC Press/Taylor & Francis Group, LLC: Boca Raton, FL, USA, 2006. [Google Scholar]
- Noda, Y.; Mamiya, T.; Nabeshima, T. The mechanisms of morphine dependence and it’s withdrawal syndrome: Study in mutant mice. Nihon Yakurigaku Zasshi 2001, 117, 21–26. [Google Scholar] [CrossRef][Green Version]
- Loscher, W. Abnormal circling behavior in rat mutants and its relevance to model specific brain dysfunctions. Neurosci. Biobehav. Rev. 2010, 34, 31–49. [Google Scholar] [CrossRef]
- Prut, L.; Belzung, C. The open field as a paradigm to measure the effects of drugs on anxiety-like behaviors: A review. Eur. J. Pharmacol. 2003, 463, 3–33. [Google Scholar] [CrossRef]
- Sestakova, N.; Puzserova, A.; Kluknavsky, M.; Bernatova, I. Determination of motor activity and anxiety-related behaviour in rodents: Methodological aspects and role of nitric oxide. Interdiscip. Toxicol. 2013, 6, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.T.; Ingram, S.L.; Henderson, G.; Chavkin, C.; von Zastrow, M.; Schulz, S.; Koch, T.; Evans, C.J.; Christie, M.J. Regulation of mu-opioid receptors: Desensitization, phosphorylation, internalization, and tolerance. Pharmacol. Rev. 2013, 65, 223–254. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Shibasaki, Y.; Matsumoto, K.; Shibasaki, M.; Hasegawa, M.; Wang, E.; Masukawa, D.; Yoshizawa, K.; Horie, S.; Suzuki, T. Mechanisms that underlie mu-opioid receptor agonist-induced constipation: Differential involvement of mu-opioid receptor sites and responsible regions. J. Pharmacol. Exp. Ther. 2013, 347, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Romberg, R.; Sarton, E.; Teppema, L.; Matthes, H.W.; Kieffer, B.L.; Dahan, A. Comparison of morphine-6-glucuronide and morphine on respiratory depressant and antinociceptive responses in wild type and mu-opioid receptor deficient mice. Br. J. Anaesth. 2003, 91, 862–870. [Google Scholar] [CrossRef]
- Dietis, N. Strategies to Reduce Morphine Tolerance in Cancer: Evaluation of the Bifunctional Opioid UFP-505. Ph.D. Thesis, University of Leicester, Leicester, UK, 2012. [Google Scholar]
- Balboni, G.; Guerrini, R.; Salvadori, S.; Bianchi, C.; Rizzi, D.; Bryant, S.D.; Lazarus, L.H. Evaluation of the dmt-tic pharmacophore: Conversion of a potent delta-opioid receptor antagonist into a potent delta agonist and ligands with mixed properties. J. Med. Chem. 2002, 45, 713–720. [Google Scholar] [CrossRef]
- Balboni, G.; Fiorini, S.; Baldisserotto, A.; Trapella, C.; Sasaki, Y.; Ambo, A.; Marczak, E.D.; Lazarus, L.H.; Salvadori, S. Further studies on lead compounds containing the opioid pharmacophore dmt-tic. J. Med. Chem. 2008, 51, 5109–5117. [Google Scholar] [CrossRef]
- Balboni, G.; Salvadori, S.; Marczak, E.D.; Knapp, B.I.; Bidlack, J.M.; Lazarus, L.H.; Peng, X.; Si, Y.G.; Neumeyer, J.L. Opioid bifunctional ligands from morphine and the opioid pharmacophore dmt-tic. Eur. J. Med. Chem. 2011, 46, 799–803. [Google Scholar] [CrossRef][Green Version]
- Balboni, G.; Salvadori, S.; Trapella, C.; Knapp, B.I.; Bidlack, J.M.; Lazarus, L.H.; Peng, X.; Neumeyer, J.L. Evolution of the bifunctional lead mu agonist/delta antagonist containing the dmt-tic opioid pharmacophore. ACS Chem. Neurosci. 2010, 1, 155–164. [Google Scholar] [CrossRef]
- Salvadori, S.; Balboni, G.; Guerrini, R.; Tomatis, R.; Bianchi, C.; Bryant, S.D.; Cooper, P.S.; Lazarus, L.H. Evolution of the dmt-tic pharmacophore: N-terminal methylated derivatives with extraordinary delta opioid antagonist activity. J. Med. Chem. 1997, 40, 3100–3108. [Google Scholar] [CrossRef]
- Follenfant, R.L.; Hardy, G.W.; Lowe, L.A.; Schneider, C.; Smith, T.W. Antinociceptive effects of the novel opioid peptide bw443c compared with classical opiates; peripheral versus central actions. Br. J. Pharmacol. 1988, 93, 85–92. [Google Scholar] [CrossRef]
- Yin, X. Pharmacological Modulation of Mood, Behaviour and Cognition. Ph.D. Thesis, University of Tasmania, Hobart, TAS, Australia, 2018. [Google Scholar]
- Aldrich, J.V.; McLaughlin, J.P. Opioid peptides: Potential for drug development. Drug Discov. Today Technol. 2012, 9, e23–e31. [Google Scholar] [CrossRef]
- Le Bars, D.; Gozariu, M.; Cadden, S.W. Animal models of nociception. Pharmacol. Rev. 2001, 53, 597–652. [Google Scholar]
- Gardmark, M.; Hoglund, A.U.; Hammarlund-Udenaes, M. Aspects on tail-flick, hot-plate and electrical stimulation tests for morphine antinociception. Pharmacol. Toxicol. 1998, 83, 252–258. [Google Scholar] [CrossRef]
- Porreca, F.; Mosberg, H.I.; Hurst, R.; Hruby, V.J.; Burks, T.F. Roles of mu, delta and kappa opioid receptors in spinal and supraspinal mediation of gastrointestinal transit effects and hot-plate analgesia in the mouse. J. Pharmacol. Exp. Ther. 1984, 230, 341–348. [Google Scholar]
- Dong, Y.P.; Sun, L.; Liu, X.Y.; Liu, R.S. Switching from morphine to fentanyl attenuates the decline of micro-opioid receptor expression in periaqueductal gray of rats with morphine tolerance. Chin. Med. J. 2013, 126, 3712–3716. [Google Scholar]
- Healy, J.R.; Bezawada, P.; Griggs, N.W.; Devereaux, A.L.; Matsumoto, R.R.; Traynor, J.R.; Coop, A.; Cunningham, C.W. Benzylideneoxymorphone: A new lead for development of bifunctional mu/delta opioid receptor ligands. Bioorg. Med. Chem. Lett. 2017, 27, 666–669. [Google Scholar] [CrossRef]
- Mansouri, M.T.; Khodayar, M.J.; Tabatabaee, A.; Ghorbanzadeh, B.; Naghizadeh, B. Modulation of morphine antinociceptive tolerance and physical dependence by co-administration of simvastatin. Pharmacol. Biochem. Behav. 2015, 137, 38–43. [Google Scholar] [CrossRef]
- Enquist, J.; Kim, J.A.; Bartlett, S.; Ferwerda, M.; Whistler, J.L. A novel knock-in mouse reveals mechanistically distinct forms of morphine tolerance. J. Pharmacol. Exp. Ther. 2011, 338, 633–640. [Google Scholar] [CrossRef]
- Rawls, S.M.; Zielinski, M.; Patel, H.; Sacavage, S.; Baron, D.A.; Patel, D. Beta-lactam antibiotic reduces morphine analgesic tolerance in rats through GLT-1 transporter activation. Drug Alcohol Depend. 2010, 107, 261–263. [Google Scholar] [CrossRef]
- Dogrul, A.; Seyrek, M. Systemic morphine produce antinociception mediated by spinal 5-HT7, but not 5-HT1A and 5-HT2 receptors in the spinal cord. Br. J. Pharmacol. 2006, 149, 498–505. [Google Scholar] [CrossRef]
- Advokat, C.; Mosser, H.; Hutchinson, K. Morphine and dextrorphan lose antinociceptive activity but exhibit an antispastic action in chronic spinal rats. Physiol. Behav. 1997, 62, 799–804. [Google Scholar] [CrossRef]
- Heinricher, M.M.; Schouten, J.C.; Jobst, E.E. Activation of brainstem n-methyl-d-aspartate receptors is required for the analgesic actions of morphine given systemically. Pain 2001, 92, 129–138. [Google Scholar] [CrossRef]
- Matos, F.F.; Rollema, H.; Taiwo, Y.O.; Levine, J.D.; Basbaum, A.I. Relationship between analgesia and extracellular morphine in brain and spinal cord in awake rats. Brain Res. 1995, 693, 187–195. [Google Scholar] [CrossRef]
- Ramabadran, K. Effects of n-methylnaloxone and n-methylnaltrexone on nociception and precipitated abstinence in mice. Life Sci. 1982, 31, 1253–1256. [Google Scholar] [CrossRef]
- Patti, C.L.; Frussa-Filho, R.; Silva, R.H.; Carvalho, R.C.; Kameda, S.R.; Takatsu-Coleman, A.L.; Cunha, J.L.; Abilio, V.C. Behavioral characterization of morphine effects on motor activity in mice. Pharmacol. Biochem. Behav. 2005, 81, 923–927. [Google Scholar] [CrossRef]
- Kahveci, N.; Gulec, G.; Ozluk, K. Effects of intracerebroventricularly-injected morphine on anxiety, memory retrieval and locomotor activity in rats: Involvement of vasopressinergic system and nitric oxide pathway. Pharmacol. Biochem. Behav. 2006, 85, 859–867. [Google Scholar] [CrossRef]
- Szekely, J.I.; Miglecz, E.; Ronai, A.Z. Biphasic effects of a potent enkephalin analogue (d-Met2,Pro5)-enkephalinamide and morphine on locomotor activity in mice. Psychopharmacology 1980, 71, 299–301. [Google Scholar]
- Paul, A.K.; Smith, C.M.; Rahmatullah, M.; Nissapatorn, V.; Wilairatana, P.; Spetea, M.; Gueven, N.; Dietis, N. Opioid analgesia and opioid-induced adverse effects: A review. Pharmaceuticals 2021, 14, 1091. [Google Scholar] [CrossRef]
- Hollais, A.W.; Patti, C.L.; Zanin, K.A.; Fukushiro, D.F.; Berro, L.F.; Carvalho, R.C.; Kameda, S.R.; Frussa-Filho, R. Effects of acute and long-term typical or atypical neuroleptics on morphine-induced behavioural effects in mice. Clin. Exp. Pharmacol. Physiol. 2014, 41, 255–263. [Google Scholar] [CrossRef]
- Babbini, M.; Davis, W.M. Time-dose relationships for locomotor activity effects of morphine after acute or repeated treatment. Br. J. Pharmacol. 1972, 46, 213–224. [Google Scholar] [CrossRef]
- Rodriguez-Arias, M.; Broseta, I.; Aguilar, M.A.; Minarro, J. Lack of specific effects of selective D(1) and D(2) dopamine antagonists vs. Risperidone on morphine-induced hyperactivity. Pharmacol. Biochem. Behav. 2000, 66, 189–197. [Google Scholar] [CrossRef]
- Paul, A.K.; Gueven, N.; Dietis, N. Profiling the effects of repetitive morphine administration on motor behavior in rats. Molecules 2021, 26, 4355. [Google Scholar] [CrossRef] [PubMed]
- Paul, A. Strategies to Reduce the Impact of Opioid-Induced Adverse Effects. Ph.D. Thesis, University of Tasmania, Hobart, TAS, Australia, 2018; pp. 111–142. [Google Scholar]
- Paul, A.K.; Gueven, N.; Dietis, N. Age-dependent antinociception and behavioral inhibition by morphine. Pharmacol. Biochem. Behav. 2018, 168, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Walsh, R.N.; Cummins, R.A. The open-field test: A critical review. Psychol. Bull. 1976, 83, 482–504. [Google Scholar] [CrossRef]
- Kuzmin, A.; Sandin, J.; Terenius, L.; Ogren, S.O. Dose- and time-dependent bimodal effects of kappa-opioid agonists on locomotor activity in mice. J. Pharmacol. Exp. Ther. 2000, 295, 1031–1042. [Google Scholar]
- Wilkinson, V.E.; Jackson, M.L.; Westlake, J.; Stevens, B.; Barnes, M.; Swann, P.; Rajaratnam, S.M.; Howard, M.E. The accuracy of eyelid movement parameters for drowsiness detection. J. Clin. Sleep Med. 2013, 9, 1315–1324. [Google Scholar] [CrossRef]
- Bounes, V.; Charriton-Dadone, B.; Levraut, J.; Delangue, C.; Carpentier, F.; Mary-Chalon, S.; Houze-Cerfon, V.; Sommet, A.; Houze-Cerfon, C.H.; Ganetsky, M. Predicting morphine related side effects in the ED: An international cohort study. Am. J. Emerg. Med. 2017, 35, 531–535. [Google Scholar] [CrossRef]
- Rezayof, A.; Assadpour, S.; Alijanpour, S. Morphine-induced anxiolytic-like effect in morphine-sensitized mice: Involvement of ventral hippocampal nicotinic acetylcholine receptors. Pharmacol. Biochem. Behav. 2013, 103, 460–466. [Google Scholar] [CrossRef]
- Gholami, M.; Saboory, E.; Khalkhali, H.R. Chronic morphine and tramadol re-exposure induced an anti-anxiety effect in prepubertal rats exposed neonatally to the same drugs. Clin. Exp. Pharmacol. Physiol. 2014, 41, 838–843. [Google Scholar] [CrossRef]
- Besson, A.; Privat, A.M.; Eschalier, A.; Fialip, J. Reversal of learned helplessness by morphine in rats: Involvement of a dopamine mediation. Pharmacol. Biochem. Behav. 1998, 60, 519–525. [Google Scholar] [CrossRef]
- Zomkowski, A.D.; Santos, A.R.; Rodrigues, A.L. Evidence for the involvement of the opioid system in the agmatine antidepressant-like effect in the forced swimming test. Neurosci. Lett. 2005, 381, 279–283. [Google Scholar] [CrossRef]
- Papp, M.; Moryl, E.; Willner, P. Pharmacological validation of the chronic mild stress model of depression. Eur. J. Pharmacol. 1996, 296, 129–136. [Google Scholar] [CrossRef]
- Blundell, C.; Crossman, A.R.; Slater, P. The effect of morphine on turning behaviour in rats and mice with unilateral 6-hydroxydopamine lesions [proceedings]. Br. J. Pharmacol. 1976, 57, 456P. [Google Scholar]
- Jacquet, Y.F.; Carol, M.; Russell, I.S. Morphine-induced rotation in naive, nonlesioned rats. Science 1976, 192, 261–263. [Google Scholar] [CrossRef]
- Barber, D.L.; Blackburn, T.P.; Greenwood, D.T. An automatic apparatus for recording rotational behaviour in rats with brain lesions. Physiol. Behav. 1973, 11, 117–120. [Google Scholar] [CrossRef]
- Christie, J.E.; Crow, T.J. Turning behaviour as an index of the action of amphetamines and ephedrines on central dopamine-containing neurones. Br. J. Pharmacol. 1971, 43, 658–667. [Google Scholar] [CrossRef]
- Szewczak, M.R.; Spoerlein, M.T. Opiate-induced turning in rats after injection into the ventral tegmental area. Pharmacol. Biochem. Behav. 1986, 25, 959–965. [Google Scholar] [CrossRef]
- Colucci, M.; Maione, F.; Bonito, M.C.; Piscopo, A.; Di Giannuario, A.; Pieretti, S. New insights of dimethyl sulphoxide effects (dmso) on experimental in vivo models of nociception and inflammation. Pharmacol. Res. 2008, 57, 419–425. [Google Scholar] [CrossRef]
- Choleris, E.; Thomas, A.W.; Kavaliers, M.; Prato, F.S. A detailed ethological analysis of the mouse open field test: Effects of diazepam, chlordiazepoxide and an extremely low frequency pulsed magnetic field. Neurosci. Biobehav. Rev. 2001, 25, 235–260. [Google Scholar] [CrossRef]
- Ramos, A.; Berton, O.; Mormede, P.; Chaouloff, F. A multiple-test study of anxiety-related behaviours in six inbred rat strains. Behav. Brain Res. 1997, 85, 57–69. [Google Scholar] [CrossRef]
- Jahkel, M.; Rilke, O.; Koch, R.; Oehler, J. Open field locomotion and neurotransmission in mice evaluated by principal component factor analysis-effects of housing condition, individual activity disposition and psychotropic drugs. Prog. Neuropsychopharmacol. Biol. Psychiatry 2000, 24, 61–84. [Google Scholar] [CrossRef]
- Bailey, K.R.; Crawley, J.N. Frontiers in neuroscience. Anxiety-related Behaviors in mice. In Methods of Behavior Analysis in Neuroscience; Buccafusco, J.J., Ed.; CRC Press/Taylor & Francis Group, LLC: Boca Raton, FL, USA, 2009. [Google Scholar]
- Holter, S.M.; Einicke, J.; Sperling, B.; Zimprich, A.; Garrett, L.; Fuchs, H.; Gailus-Durner, V.; Hrabe de Angelis, M.; Wurst, W. Tests for anxiety-related behavior in mice. Curr. Protoc. Mouse Biol. 2015, 5, 291–309. [Google Scholar] [CrossRef] [PubMed]
- Fossum, E.N.; Lisowski, M.J.; Macey, T.A.; Ingram, S.L.; Morgan, M.M. Microinjection of the vehicle dimethyl sulfoxide (DMSO) into the periaqueductal gray modulates morphine antinociception. Brain Res. 2008, 1204, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Haigler, H.J.; Spring, D.D. Comparison of the analgesic effects of dimethyl sulfoxide and morphine. Ann. N. Y. Acad. Sci. 1983, 411, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.H.; Majluf-Cruz, A.; Omburo, G.A. Cyclic AMP-specific phosphodiesterase inhibitor rolipram and RO-20-1724 promoted apoptosis in HL60 promyelocytic leukemic cells via cyclic AMP-independent mechanism. Life Sci. 1998, 63, 265–274. [Google Scholar] [CrossRef]
- Alasbahi, R.H.; Melzig, M.F. Forskolin and derivatives as tools for studying the role of camp. Pharmazie 2012, 67, 5–13. [Google Scholar]
- Woolley, K. Synthesis of Naphthoquinones and Peptides. Honours Thesis, University of Tasmania, Hobart, TAS, Australia, 2013. [Google Scholar]
- Craft, R.M.; Stratmann, J.A.; Bartok, R.E.; Walpole, T.I.; King, S.J. Sex differences in development of morphine tolerance and dependence in the rat. Psychopharmacology 1999, 143, 1–7. [Google Scholar] [CrossRef]
- National Health and Medical Research Council (NHMRC). Australian Code for the Care and Use of Animals for Scientific Purposes, 8th ed.; NHMRC: Canberra, ACT, Australia, 2013.
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef]
- Festing, M.F.; Altman, D.G. Guidelines for the design and statistical analysis of experiments using laboratory animals. ILAR J. 2002, 43, 244–258. [Google Scholar] [CrossRef]
- Harris, L.S.; Pierson, A.K. Some narcotic antagonists in the benzomorphan series. J. Pharmacol. Exp. Ther. 1964, 143, 141–148. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cells | EC50 of UTA1003 | pEC50 of UTA1003 | Emax (%) | Ref. Compound | EC50 of Ref. Compound | pEC50 of Reference | Emax (%) | Antagonist Effect (Kd) |
|---|---|---|---|---|---|---|---|---|
| CHO-MOP | 6.89 nM | 8.16 | 82.44 | DAMGO | 12.5 nM | 7.90 | 88.76 | |
| CHO-DOP | 26.6 nM | 7.58 | 28.20 | DPDPE | 20.7 nM | 7.68 | 68.18 | 47.8 nM |
| CHO-KOP | 30.9 nM | 7.51 | 94.47 | U50,488 | 0.20 pM | 12.70 | 95.94 | |
| CHO-NOP | 1.79 µM | 5.75 | 28.34 | Nociceptin | 0.69 nM | 9.16 | 90.73 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paul, A.K.; Woolley, K.L.; Rahmatullah, M.; Wilairatana, P.; Smith, J.A.; Gueven, N.; Dietis, N. Differential Effects of a Novel Opioid Ligand UTA1003 on Antinociceptive Tolerance and Motor Behaviour. Pharmaceuticals 2022, 15, 789. https://doi.org/10.3390/ph15070789
Paul AK, Woolley KL, Rahmatullah M, Wilairatana P, Smith JA, Gueven N, Dietis N. Differential Effects of a Novel Opioid Ligand UTA1003 on Antinociceptive Tolerance and Motor Behaviour. Pharmaceuticals. 2022; 15(7):789. https://doi.org/10.3390/ph15070789
Chicago/Turabian StylePaul, Alok K., Krystel L. Woolley, Mohammed Rahmatullah, Polrat Wilairatana, Jason A. Smith, Nuri Gueven, and Nikolas Dietis. 2022. "Differential Effects of a Novel Opioid Ligand UTA1003 on Antinociceptive Tolerance and Motor Behaviour" Pharmaceuticals 15, no. 7: 789. https://doi.org/10.3390/ph15070789
APA StylePaul, A. K., Woolley, K. L., Rahmatullah, M., Wilairatana, P., Smith, J. A., Gueven, N., & Dietis, N. (2022). Differential Effects of a Novel Opioid Ligand UTA1003 on Antinociceptive Tolerance and Motor Behaviour. Pharmaceuticals, 15(7), 789. https://doi.org/10.3390/ph15070789