
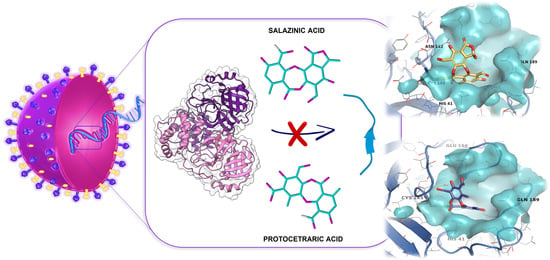
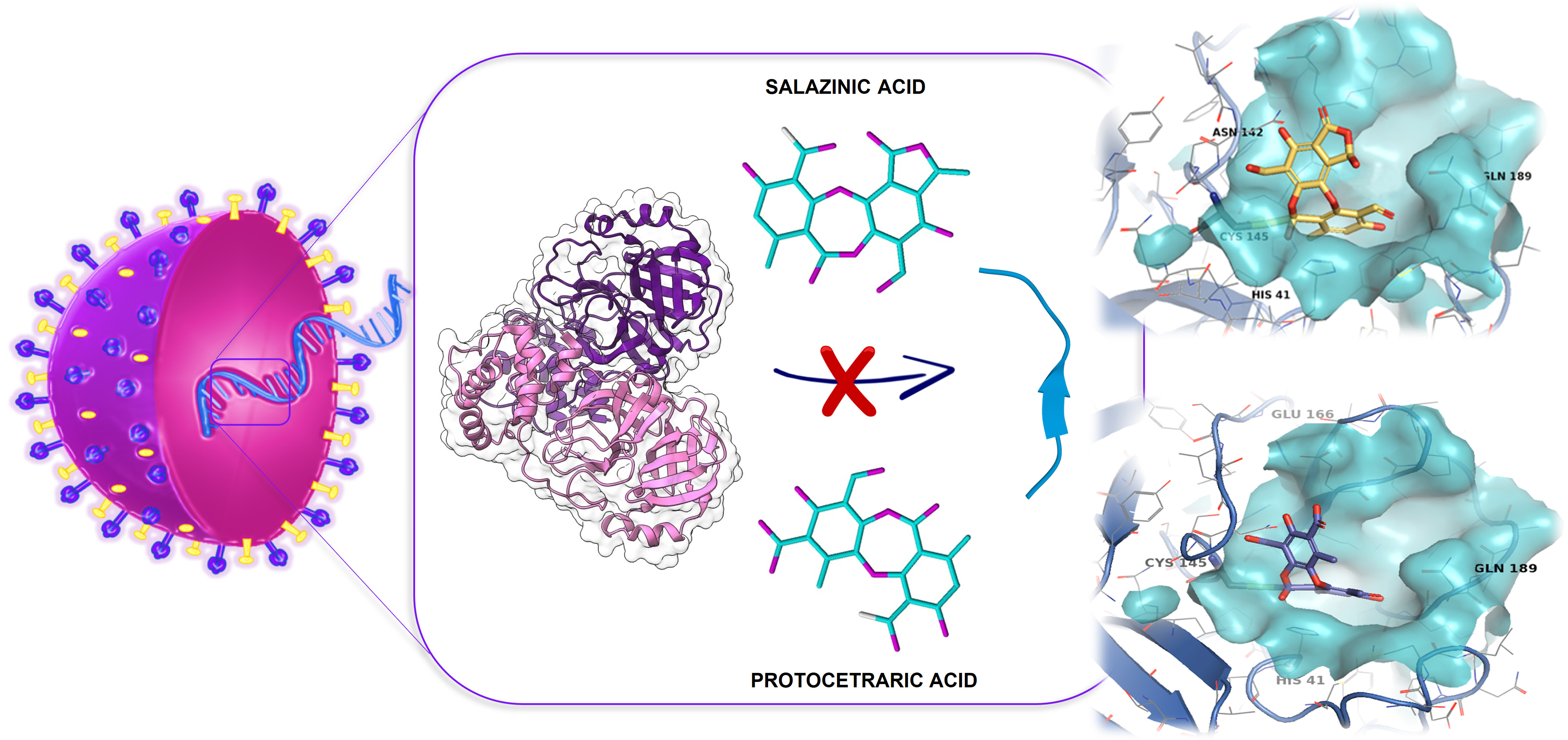
Protocetraric and Salazinic Acids as Potential Inhibitors of SARS-CoV-2 3CL Protease: Biochemical, Cytotoxic, and Computational Characterization of Depsidones as Slow-Binding Inactivators

 , ,
, ,  , , ,
, , ,  and
and
Abstract
:
1. Introduction
2. Results
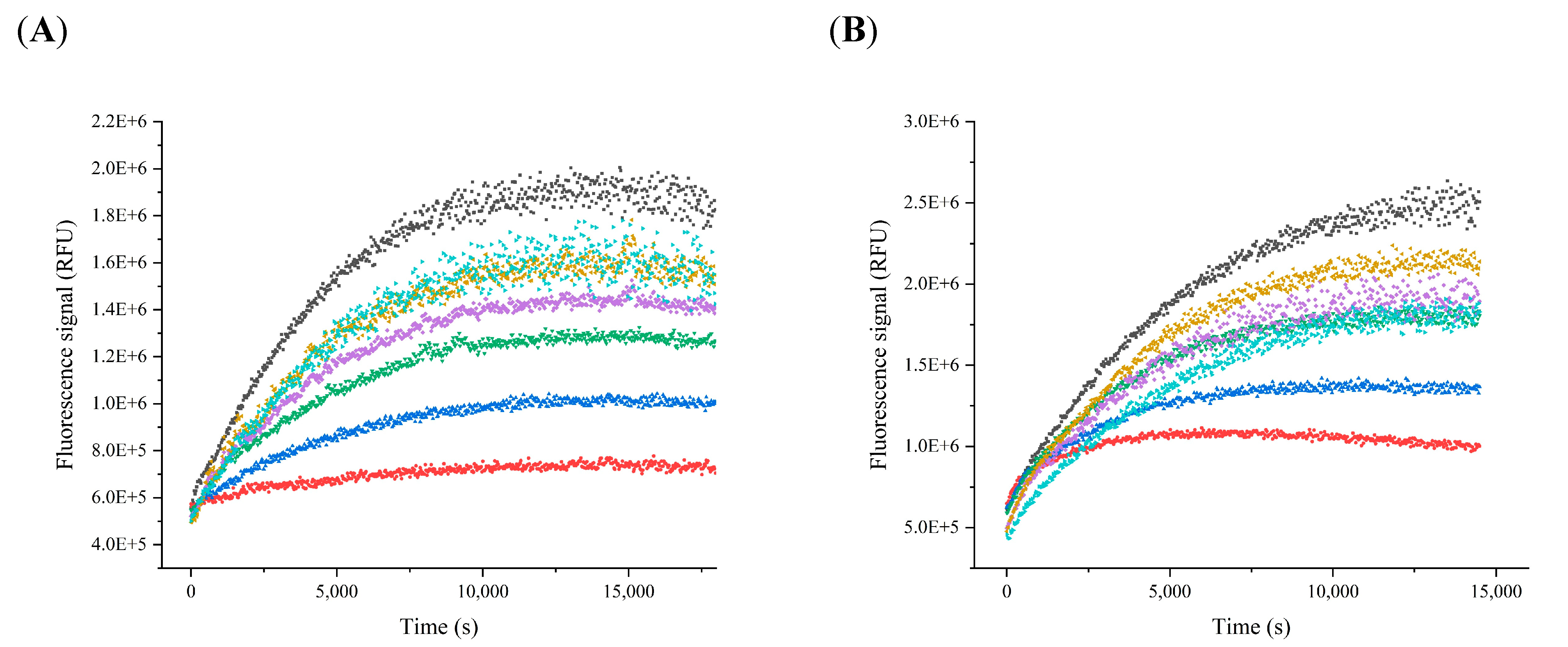
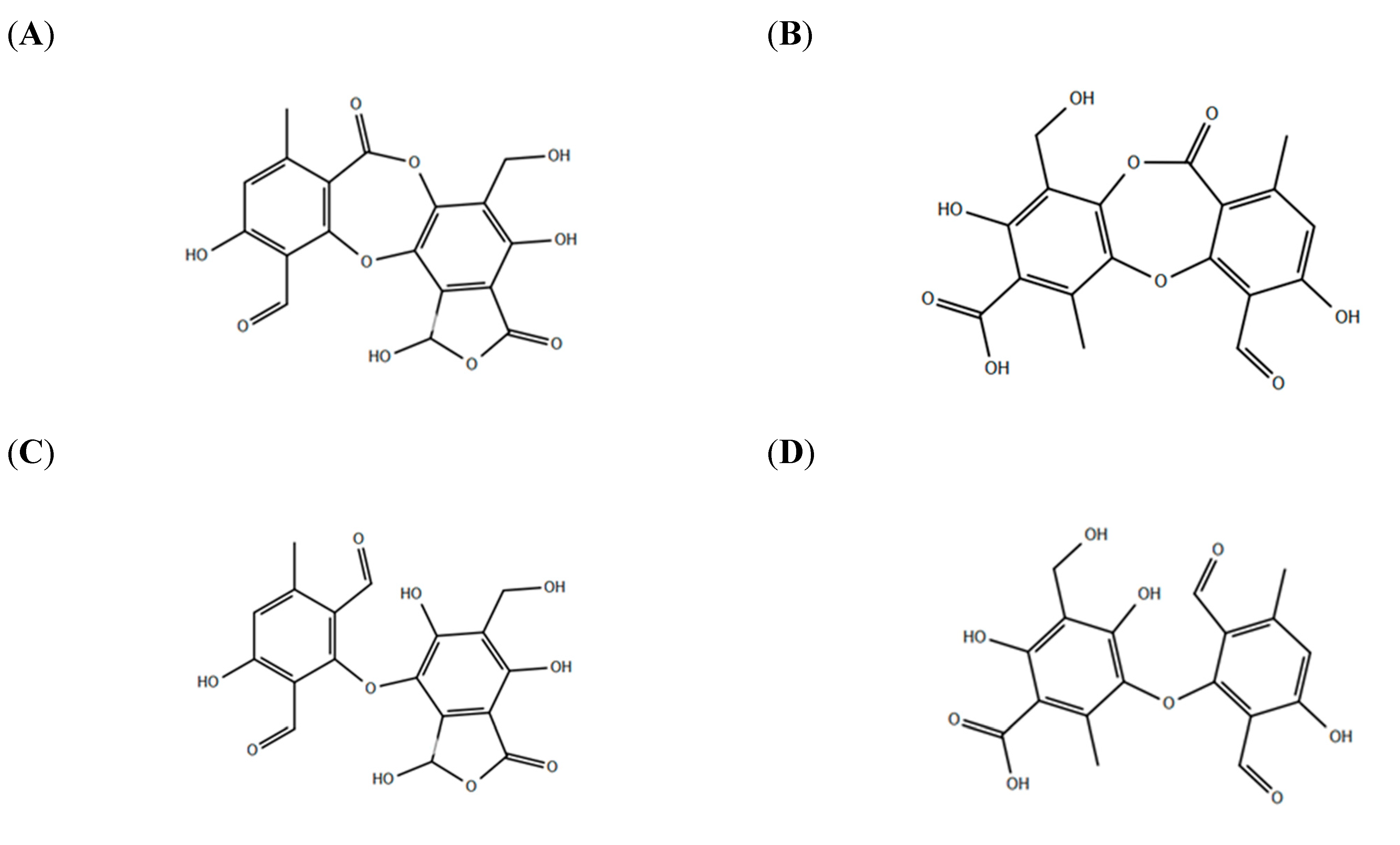
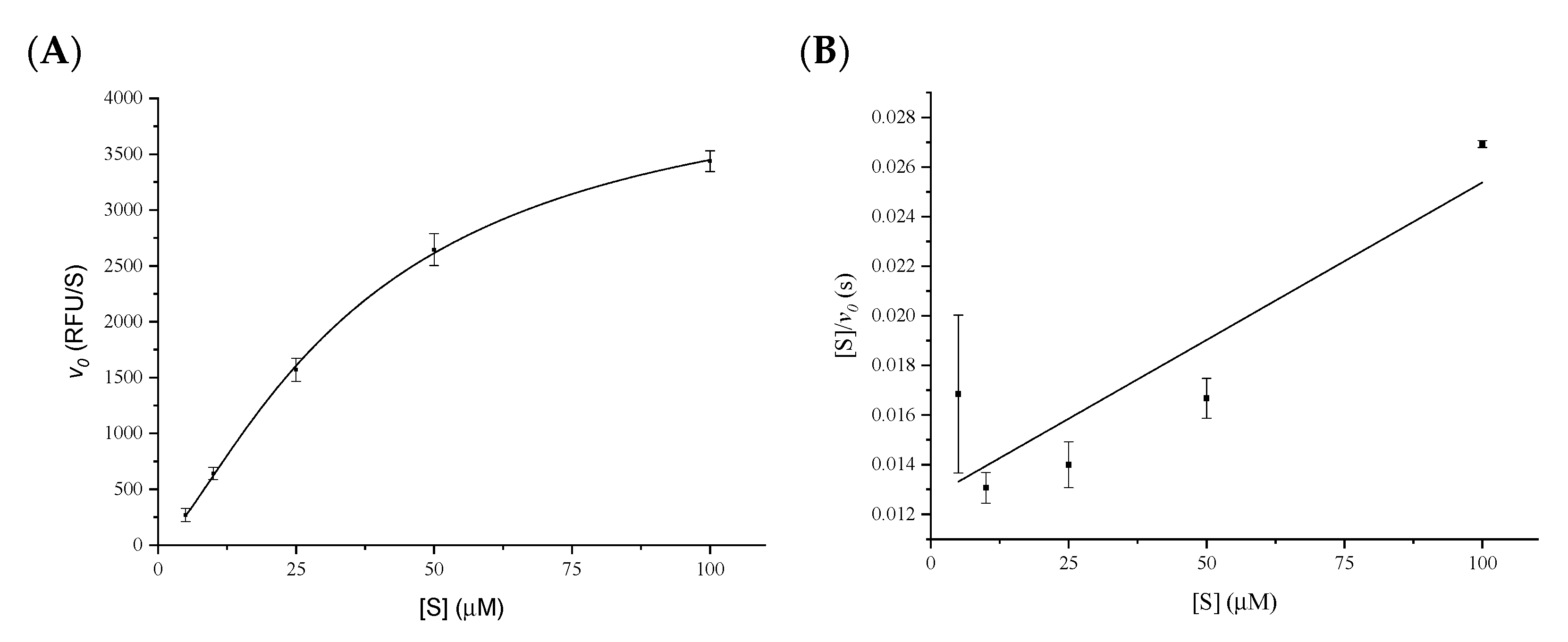
2.1. Determination of the Kinetic Parameters
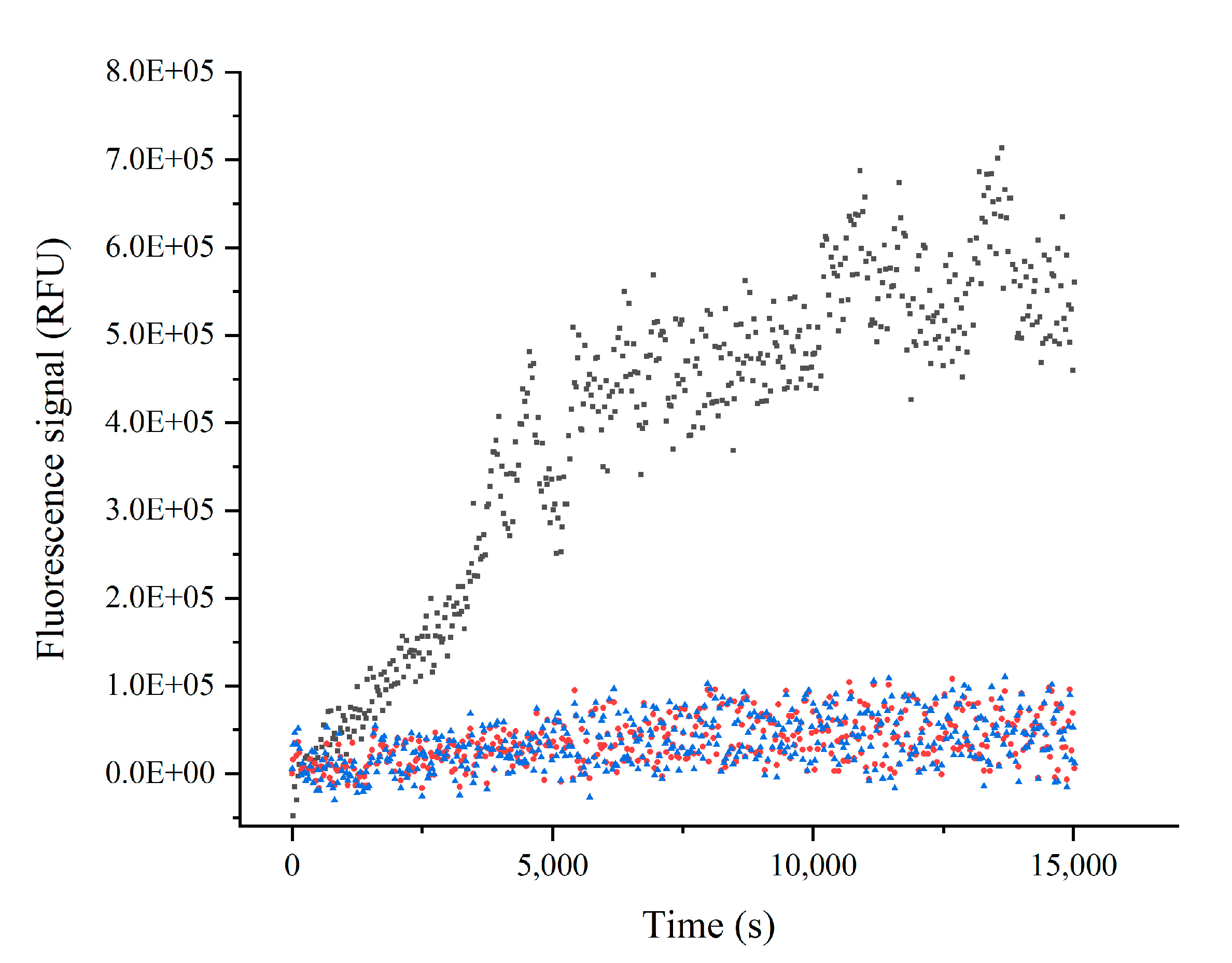
2.2. Inhibition Assay
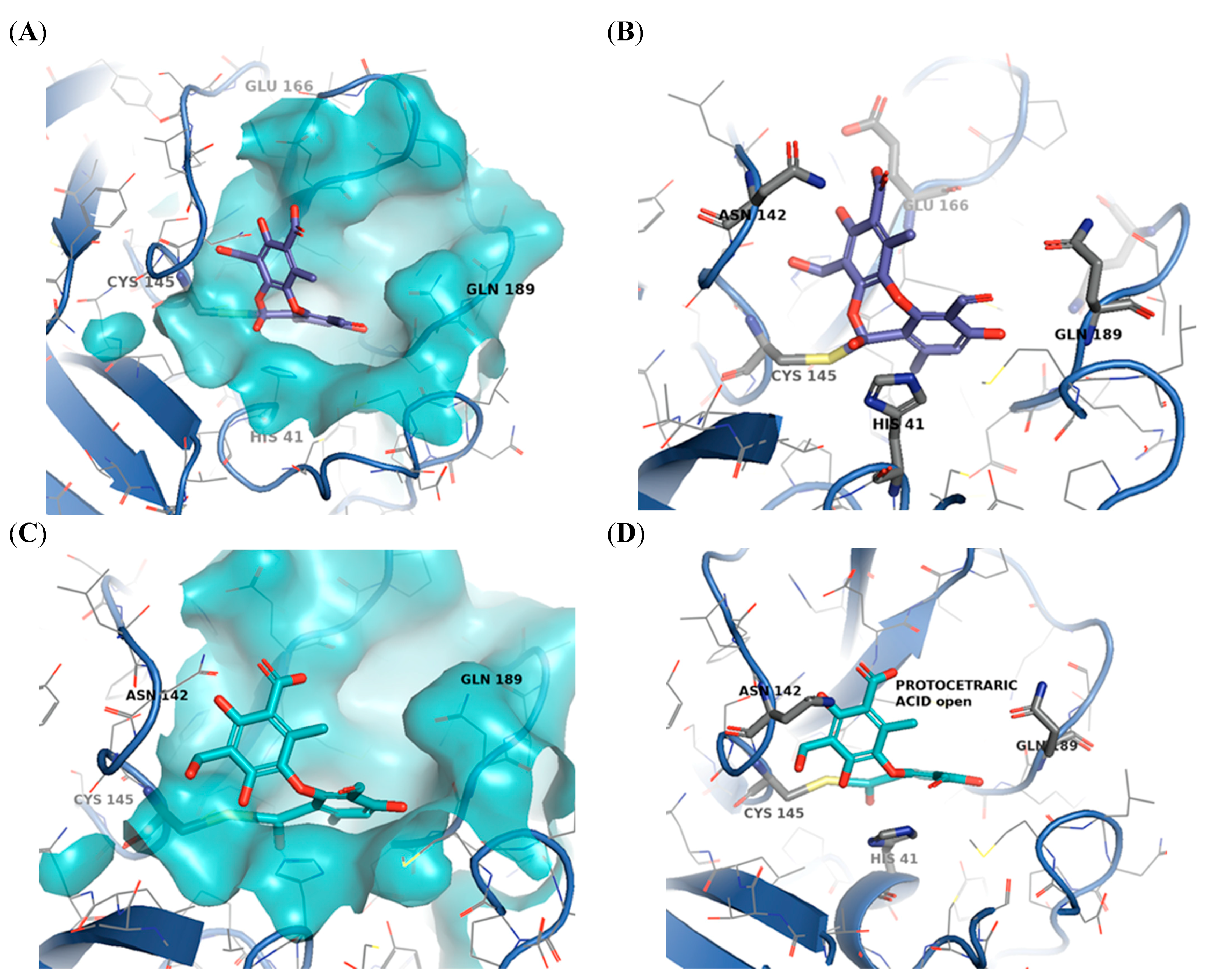
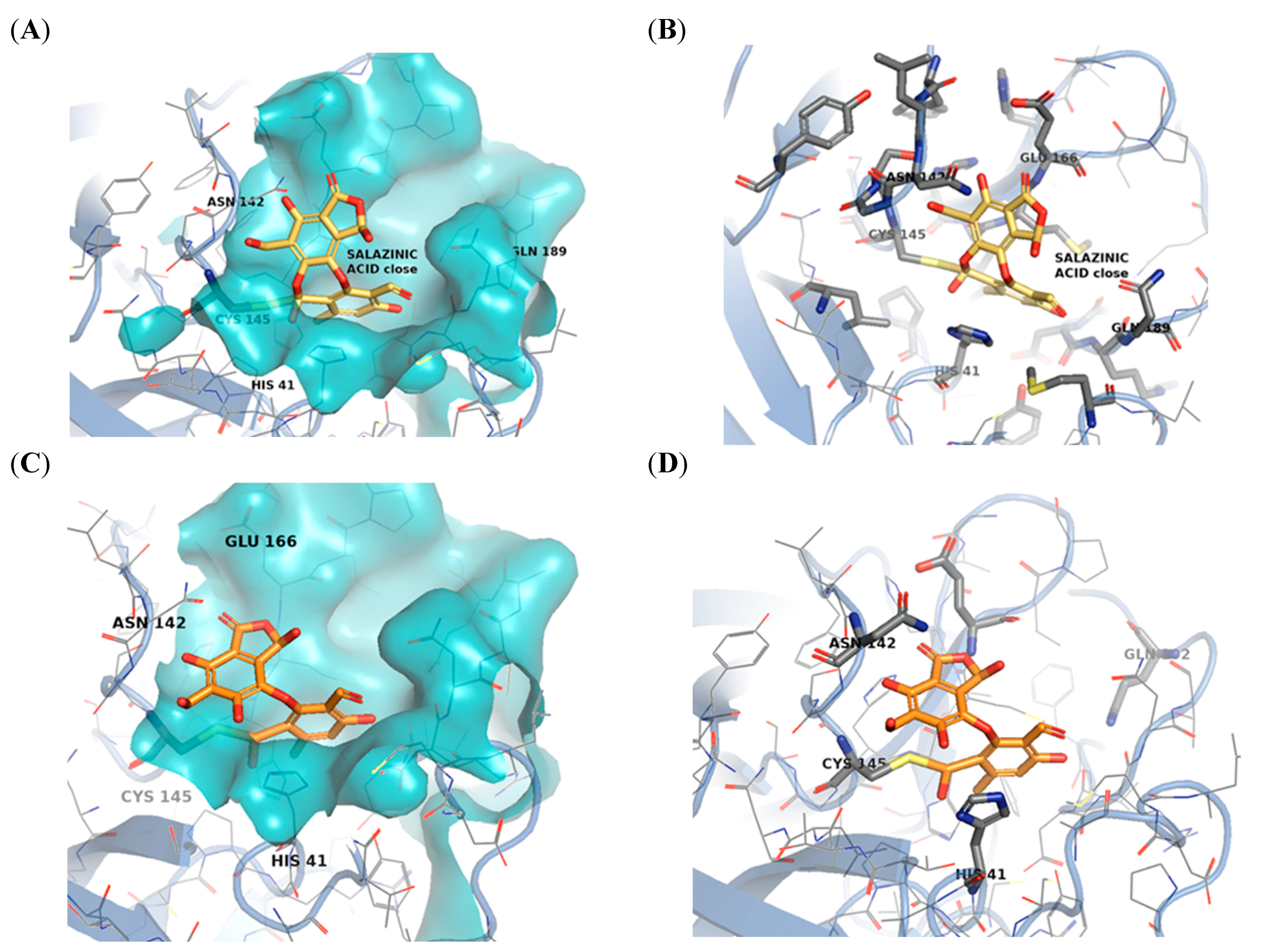
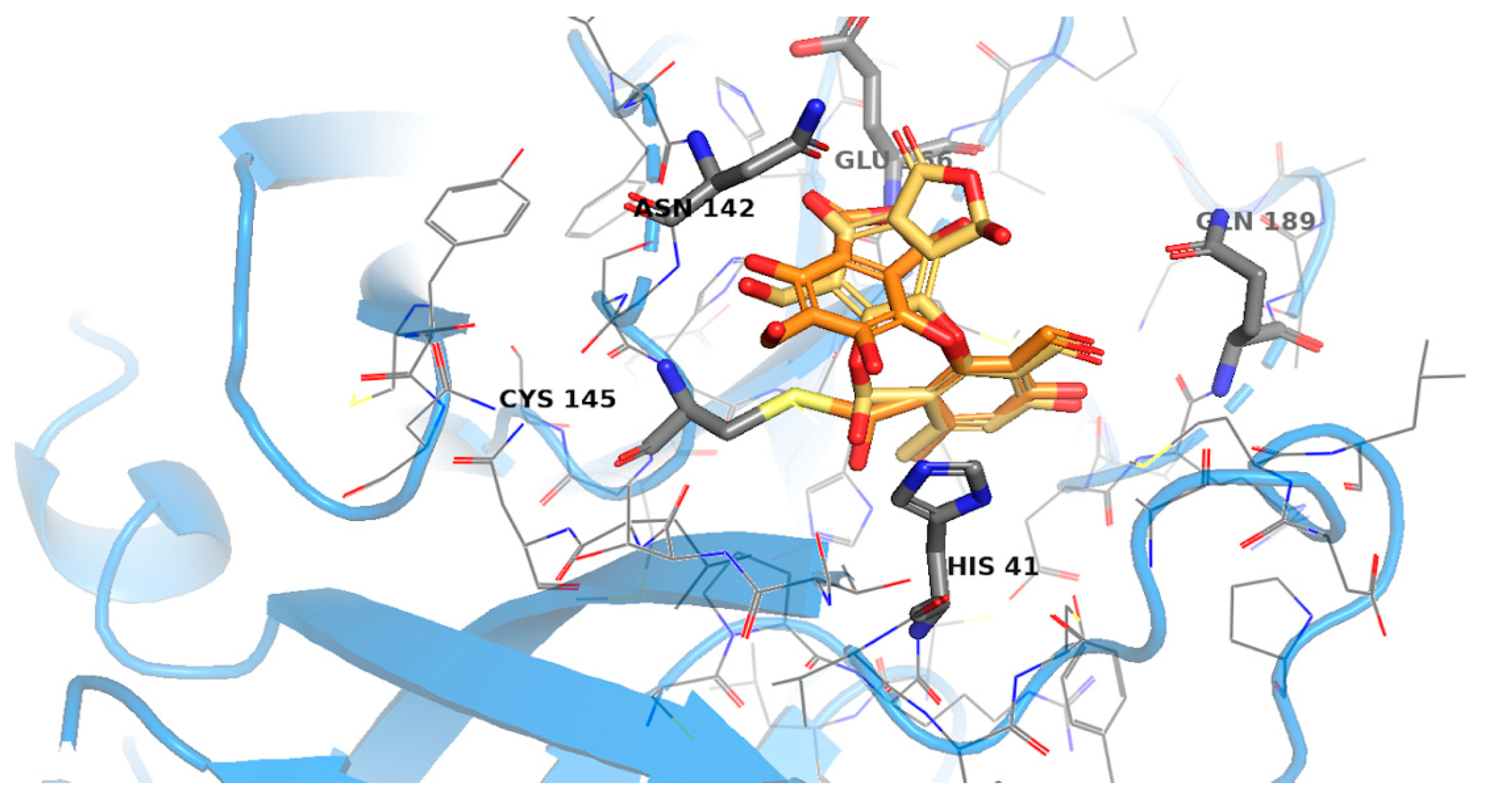
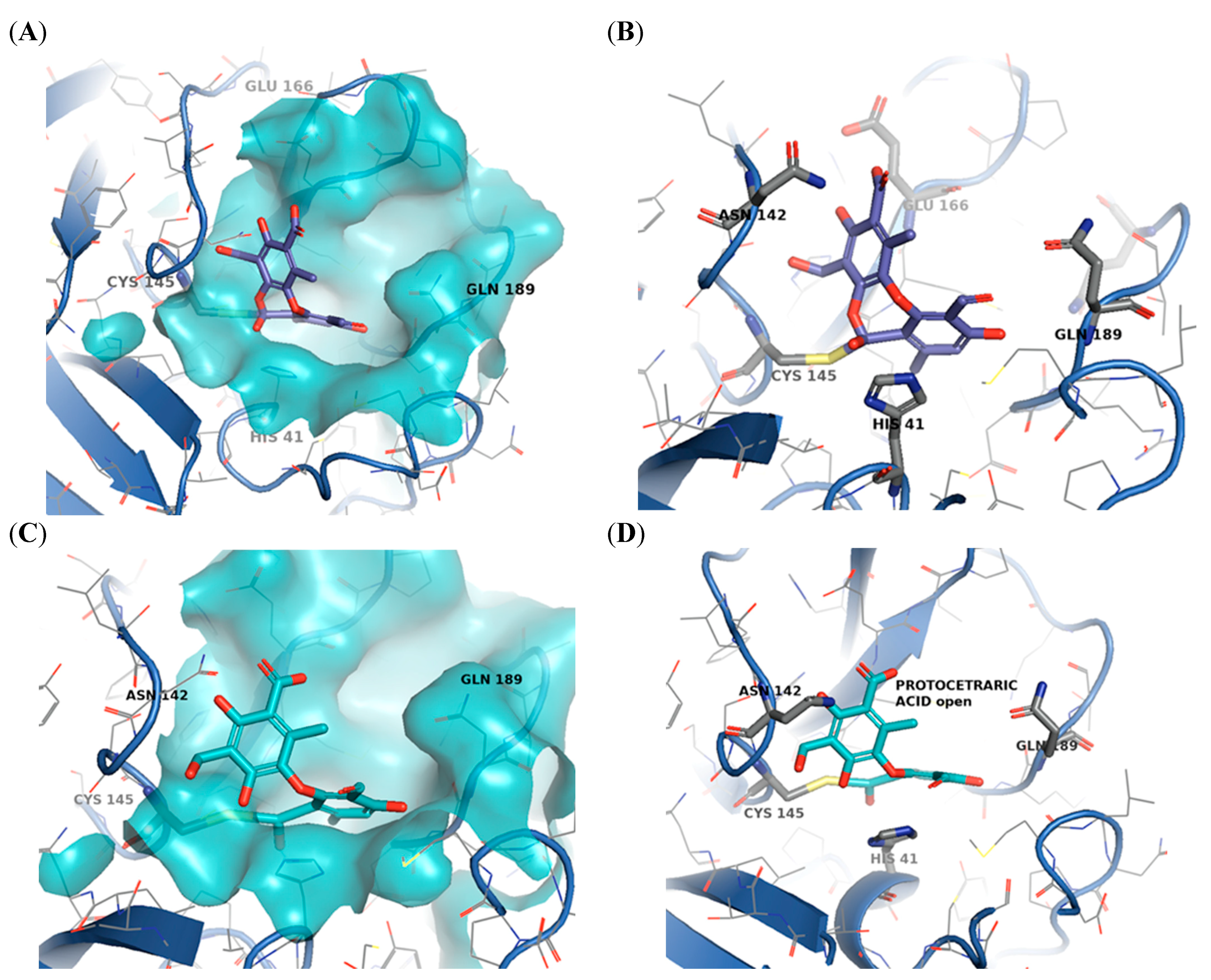
2.3. Molecular Docking
2.4. Cell Proliferation and Viability in TM4 Epithelial Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Spectrofluorimetric Assay
4.3. Determination of Kinetic Parameters
4.4. Inhibition Assays
4.5. Determination of the Mechanism of Inhibition
4.6. Molecular Docking
4.6.1. Docking Protocol Validation
4.6.2. Extra Precision (XP) and Covalent Docking (CovDOCK)
4.7. Cell Culture and Viability Assay
4.8. Statistics and Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Topley, W.W.C.; William, W.C. Topley & Wilson’s Microbiology & Microbial Infections; Hodder Arnold: London, UK, 2005; ISBN 0 340 88563 7. [Google Scholar]
- Jo, S.; Kim, S.; Shin, D.H.; Kim, M.S. Inhibition of SARS-CoV 3CL protease by flavonoids. J. Enzym. Inhib. Med. Chem. 2020, 35, 145–151. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Xu, Y.; Bao, L.; Zhang, L.; Yu, P.; Qu, Y.; Zhu, H.; Zhao, W.; Han, Y.; Qin, C. From SARS to MERS, Thrusting Coronaviruses into the Spotlight. Viruses 2019, 11, 59. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J.; et al. Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef] [Green Version]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) Structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, R.; Chaudhary, J.K.; Jain, N.; Chaudhary, P.K.; Khanra, S.; Dhamija, P.; Sharma, A.; Kumar, A.; Handu, S. Role of Structural and Non-Structural Proteins and Therapeutic Targets of SARS-CoV-2 for COVID-19. Cells 2021, 10, 821. [Google Scholar] [CrossRef] [PubMed]
- Graziano, V.; McGrath, W.J.; Yang, L.; Mangel, W.F. SARS CoV Main Proteinase: The Monomer−Dimer Equilibrium Dissociation Constant. Biochemistry 2006, 45, 14632–14641. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wei, P.; Huang, C.; Tan, L.; Liu, Y.; Lai, L. Only one protomer is active in the dimer of SARS 3C-like proteinase. J. Biol. Chem. 2006, 281, 13894–13898. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Song, J. The catalysis of the SARS 3C-like protease is under extensive regulation by its extra domain. FEBS J. 2006, 273, 1035–1045. [Google Scholar] [CrossRef]
- Needle, D.; Lountos, G.T.; Waugh, D.S. Structures of the Middle East respiratory syndrome coronavirus 3C-like protease reveal insights into substrate specificity. Acta Cryst. 2015, D71, 1102–1111. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Sivaraman, J.; Song, J. Mechanism for Controlling the Dimer-Monomer Switch and Coupling Dimerization to Catalysis of the Severe Acute Respiratory Syndrome Coronavirus 3C-Like Protease. J. Virol. 2008, 82, 4620–4629. [Google Scholar] [CrossRef] [Green Version]
- Pillaiyar, T.; Manickam, M.; Namasivayam, V.; Hayashi, Y.; Jung, S.H. An Overview of Severe Acute Respiratory Syndrome-Coronavirus (SARS-CoV) 3CL Protease Inhibitors: Peptidomimetics and Small Molecule Chemotherapy. J. Med. Chem. 2016, 59, 6595–6628. [Google Scholar] [CrossRef] [PubMed]
- Dubanevics, I.; McLeish, T.C.B. Computational analysis of dynamic allostery and control in the SARS-CoV-2 main protease. J. R. Soc. Interface 2021, 18, 20200591. [Google Scholar] [CrossRef]
- Fishbane, S.; Hirsch, J.S.; Nair, V. Special Considerations for Paxlovid Treatment Among Transplant Recipients With SARS-CoV-2 Infection. Am. J. Kidney Dis. 2022, 79, 480–482. [Google Scholar] [CrossRef] [PubMed]
- Mahase, E. Covid-19: Pfizer’s paxlovid is 89% effective in patients at risk of serious illness, company reports. BMJ 2021, 375, n2713. [Google Scholar] [CrossRef] [PubMed]
- Eberle, R.J.; Olivier, D.S.; Amaral, M.S.; Gering, I.; Willbold, D.; Arni, R.K.; Coronado, M.A. The Repurposed Drugs Suramin and Quinacrine Cooperatively Inhibit SARS-CoV-2 3CL pro In Vitro. Viruses 2021, 13, 873. [Google Scholar] [CrossRef]
- Liu, J.; Cao, R.; Xu, M.; Wang, X.; Zhang, H.; Hu, H.; Li, Y.; Hu, Z.; Zhong, W.; Wang, M. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Malcolm, B.A.; Liu, R.; Lahser, F.; Agrawal, S.; Belanger, B.; Butkiewicz, N.; Chase, R.; Gheyas, F.; Hart, A.; Hesk, D.; et al. SCH 503034, a mechanism-based inhibitor of hepatitis C virus NS3 protease, suppresses polyprotein maturation and enhances the antiviral activity of alpha interferon in replicon cells. Antimicrob. Agents Chemother. 2006, 50, 1013–1020. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.T.H.; Woo, H.J.; Kang, H.K.; Nguyen, V.D.; Kim, Y.M.; Kim, D.W.; Ahn, S.A.; Xia, Y.; Kim, D. Flavonoid-mediated inhibition of SARS coronavirus 3C-like protease expressed in Pichia pastoris. Biotechnol. Lett. 2012, 34, 831–838. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, S.; Sauter, D.; Wang, K.; Zhang, R.; Sun, B.; Karioti, A.; Bilia, A.R.; Efferth, T.; Schwarz, W. Kaempferol derivatives as antiviral drugs against the 3a channel protein of coronavirus. Planta Med. 2014, 80, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Müller, K. Pharmaceutically relevant metabolites from lichens. Appl. Microbiol. Biotechnol. 2001, 56, 9–16. [Google Scholar] [CrossRef]
- Boustie, J.; Tomasi, S.; Grube, M. Bioactive lichen metabolites: Alpine habitats as an untapped source. Phytochem. Rev. 2010, 10, 287–307. [Google Scholar] [CrossRef]
- Celenza, G.; Segatore, B.; Setacci, D.; Bellio, P.; Brisdelli, F.; Piovano, M.; Garbarino, J.A.; Nicoletti, M.; Perilli, M.; Amicosante, G. In vitro antimicrobial activity of pannarin alone and in combination with antibiotics against methicillin-resistant Staphylococcus aureus clinical isolates. Phytomedicine 2012, 19, 596–602. [Google Scholar] [CrossRef]
- Segatore, B.; Bellio, P.; Setacci, D.; Brisdelli, F.; Piovano, M.; Garbarino, J.A.; Nicoletti, M.; Amicosante, G.; Perilli, M.; Celenza, G. In vitro interaction of usnic acid in combination with antimicrobial agents against methicillin-resistant Staphylococcus aureus clinical isolates determined by FICI and ΔE model methods. Phytomedicine 2012, 19, 341–347. [Google Scholar] [CrossRef]
- Bellio, P.; Segatore, B.; Mancini, A.; Di Pietro, L.; Bottoni, C.; Sabatini, A.; Brisdelli, F.; Piovano, M.; Nicoletti, M.; Amicosante, G.; et al. Interaction between lichen secondary metabolites and antibiotics against clinical isolates methicillin-resistant Staphylococcus aureus strains. Phytomedicine 2015, 22, 223–230. [Google Scholar] [CrossRef]
- Brisdelli, F.; Perilli, M.; Sellitri, D.; Bellio, P.; Bozzi, A.; Amicosante, G.; Nicoletti, M.; Piovano, M.; Celenza, G. Protolichesterinic acid enhances doxorubicin-induced apoptosis in HeLa cells in vitro. Life Sci. 2016, 158, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Molnár, K.; Farkas, E. Current results on biological activities of lichen secondary metabolites: A review. Z. Naturforsch. C. 2010, 65, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Hemaiswarya, S.; Kruthiventi, A.K.; Doble, M. Synergism between natural products and antibiotics against infectious diseases. Phytomedicine 2008, 15, 639–652. [Google Scholar] [CrossRef] [PubMed]
- Stocker-Wörgötter, E.; Cordeiro, L.M.C.; Iacomini, M. Accumulation of Potential Pharmaceutically Relevant Lichen Metabolites in Lichens and Cultured Lichen Symbionts. Stud. Nat. Prod. Chem. 2013, 39, 337–380. [Google Scholar] [CrossRef]
- Ureña-Vacas, I.; González-Burgos, E.; Divakar, K.; Pilar Gómez-Serranillos, M.; González Burgos, E. Lichen Depsidones with Biological Interest Reviews. Planta Med. 2021. [Google Scholar] [CrossRef]
- Singh, H.; Husain, T.; Agnihotri, P.; Pande, P.C.; Khatoon, S. An ethnobotanical study of medicinal plants used in sacred groves of Kumaon Himalaya, Uttarakhand, India. J. Ethnopharmacol. 2014, 154, 98–108. [Google Scholar] [CrossRef]
- Devkota, S.; Chaudhary, R.P.; Werth, S.; Scheidegger, C. Indigenous knowledge and use of lichens by the lichenophilic communities of the Nepal Himalaya. J. Ethnobiol. Ethnomed. 2017, 13, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellio, P.; Di Pietro, L.; Mancini, A.; Piovano, M.; Nicoletti, M.; Brisdelli, F.; Tondi, D.; Cendron, L.; Franceschini, N.; Amicosante, G.; et al. SOS response in bacteria: Inhibitory activity of lichen secondary metabolites against Escherichia coli RecA protein. Phytomedicine 2017, 29, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.T.; Sbrana, E.; Iwata-Yoshikawa, N.; Newman, P.C.; Garron, T.; Atmar, R.L.; Peters, C.J.; Couch, R.B. Immunization with SARS coronavirus vaccines leads to pulmonary immunopathology on challenge with the SARS virus. PLoS ONE 2012, 7, e35421. [Google Scholar] [CrossRef]
- WHO|World Health Organization. Available online: https://www.who.int/ (accessed on 30 January 2022).
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Holshue, M.L.; DeBolt, C.; Lindquist, S.; Lofy, K.H.; Wiesman, J.; Bruce, H.; Spitters, C.; Ericson, K.; Wilkerson, S.; Tural, A.; et al. First Case of 2019 Novel Coronavirus in the United States. N. Engl. J. Med. 2020, 382, 929–936. [Google Scholar] [CrossRef]
- Phan, L.T.; Nguyen, T.V.; Luong, Q.C.; Nguyen, T.V.; Nguyen, H.T.; Le, H.Q.; Nguyen, T.T.; Cao, T.M.; Pham, Q.D. Importation and Human-to-Human Transmission of a Novel Coronavirus in Vietnam. N. Engl. J. Med. 2020, 382, 872–874. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Ye, F.; Feng, Y.; Yu, F.; Wang, Q.; Wu, Y.; Zhao, C.; Sun, H.; Huang, B.; Niu, P.; et al. Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun. 2020, 11, 4417. [Google Scholar] [CrossRef]
- Kim, Y.; Lovell, S.; Tiew, K.-C.; Mandadapu, S.R.; Alliston, K.R.; Battaile, K.P.; Groutas, W.C.; Chang, K.-O. Broad-spectrum antivirals against 3C or 3C-like proteases of picornaviruses, noroviruses, and coronaviruses. J. Virol. 2012, 86, 11754–11762. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, N.C.; Kim, Y.; Liu, H.; Galasiti Kankanamalage, A.C.; Eckstrand, C.; Groutas, W.C.; Bannasch, M.; Meadows, J.M.; Chang, K.O. Efficacy of a 3C-like protease inhibitor in treating various forms of acquired feline infectious peritonitis. J. Feline Med. Surg. 2018, 20, 378–392. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, L. In the age of Omicron variant: Paxlovid raises new hopes of COVID-19 recovery. J. Med. Virol. 2021, 94, 1766–1767. [Google Scholar] [CrossRef]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 M pro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Pfizer’s Novel COVID-19 Oral Antiviral Treatment Candidate Reduced Risk of Hospitalization or Death by 89% in Interim Analysis of Phase 2/3 EPIC-HR Study|Pfizer. Available online: https://www.pfizer.com/news/press-release/press-release-detail/pfizers-novel-covid-19-oral-antiviral-treatment-candidate (accessed on 31 January 2022).
- Zhao, Y.; Fang, C.; Zhang, Q.; Zhang, R.; Zhao, X.; Duan, Y.; Wang, H.; Zhu, Y.; Feng, L.; Zhao, J.; et al. Crystal structure of SARS-CoV-2 main protease in complex with protease inhibitor PF-07321332. Protein Cell 2021, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lockbaum, G.J.; Reyes, A.C.; Lee, J.M.; Tilvawala, R.; Nalivaika, E.A.; Ali, A.; Yilmaz, N.K.; Thompson, P.R.; Schiffer, C.A. Crystal Structure of SARS-CoV-2 Main Protease in Complex with the Non-Covalent Inhibitor ML188. Viruses 2021, 13, 174. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Sacco, M.D.; Hurst, B.; Townsend, J.A.; Hu, Y.; Szeto, T.; Zhang, X.; Tarbet, B.; Marty, M.T.; Chen, Y.; et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020, 30, 678–692. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, B.; Jiang, X.M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [Green Version]
- Reis, M.M.S.; Moreira, A.C.; Sousa, M.; Mathur, P.P.; Oliveira, P.F.; Alves, M.G. Sertoli cell as a model in male reproductive toxicology: Advantages and disadvantages. J. Appl. Toxicol. 2015, 35, 870–883. [Google Scholar] [CrossRef]
- Petricca, S.; Flati, V.; Celenza, G.; Di Gregorio, J.; Lizzi, A.R.; Luzi, C.; Cristiano, L.; Cinque, B.; Rossi, G.; Festuccia, C.; et al. Tebuconazole and econazole act synergistically in mediating mitochondrial stress, energy imbalance, and sequential activation of autophagy and apoptosis in mouse Sertoli TM4 cells: Possible role of AMPK/ULK1 axis. Toxicol. Sci. 2019, 169, 209–223. [Google Scholar] [CrossRef]
- Ingelfinger, R.; Henke, M.; Roser, L.; Ulshöfer, T.; Calchera, A.; Singh, G.; Parnham, M.J.; Geisslinger, G.; Fürst, R.; Schmitt, I.; et al. Unraveling the Pharmacological Potential of Lichen Extracts in the Context of Cancer and Inflammation With a Broad Screening Approach. Front. Pharmacol. 2020, 11, 1322. [Google Scholar] [CrossRef]
- Manojlovic, N.T.; Vasiljevic, P.J.; Maskovic, P.Z.; Juskovic, M.; Bogdanovic-Dusanovic, G. Chemical composition, antioxidant, and antimicrobial activities of lichen Umbilicaria cylindrica (L.) delise (Umbilicariaceae). Evid.-Based Complement. Altern. Med. 2012, 2012, 452431. [Google Scholar] [CrossRef] [Green Version]
- Brandão, L.F.G.; Alcantara, G.B.; De Fátima Cepa Matos, M.; Bogo, D.; Dos Santos Freitas, D.; Oyama, N.M.; Honda, N.K. Cytotoxic evaluation of phenolic compounds from lichens against melanoma cells. Chem. Pharm. Bull. 2013, 61, 176–183. [Google Scholar] [CrossRef] [Green Version]
- Alexandrino, C.A.F.; Honda, N.K.; Matos, M.D.F.C.; Portugal, L.C.; de Souza, P.R.B.; Perdomo, R.T.; Guimarães, R.D.C.A.; Kadri, M.C.T.; Silva, M.C.B.L.; Bogo, D. Antitumor effect of depsidones from lichens on tumor cell lines and experimental murine melanoma. Rev. Bras. Farmacogn. 2019, 29, 449–456. [Google Scholar] [CrossRef]
- Amo de Paz, G.; Raggio, J.; Gómez-Serranillos, M.P.; Palomino, O.M.; González-Burgos, E.; Carretero, M.E.; Crespo, A. HPLC isolation of antioxidant constituents from Xanthoparmelia spp. J. Pharm. Biomed. Anal. 2010, 53, 165–171. [Google Scholar] [CrossRef]
- Bacha, U.; Barrila, J.; Velazquez-Campoy, A.; Leavitt, S.A.; Freire, E. Identification of novel inhibitors of the SARS coronavirus main protease 3CLpro. Biochemistry 2004, 43, 4906–4912. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Kang, X. Activation and maturation of SARS-CoV main protease. Protein Cell 2011, 2, 282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muramatsu, T.; Takemoto, C.; Kim, Y.-T.; Wang, H.; Nishii, W.; Terada, T.; Shirouzu, M.; Yokoyama, S. SARS-CoV 3CL protease cleaves its C-terminal autoprocessing site by novel subsite cooperativity. Proc. Natl. Acad. Sci. USA 2016, 113, 12997–13002. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.C.; Chang, G.G.; Chou, C.Y. Mutation of Glu-166 blocks the substrate-induced dimerization of SARS coronavirus main protease. Biophys. J. 2010, 98, 1327–1336. [Google Scholar] [CrossRef] [Green Version]
- Morrison, J.F.; Walsh, C.T. The Behavior and Significance of Slow-Binding Enzyme Inhibitors. Adv. Enzymol. Relat. Areas Mol. Biol. 1988, 61, 201–301. [Google Scholar] [CrossRef]
- Waley, S.G. The kinetics of slow-binding and slow, tight-binding inhibition: The effects of substrate depletion. Biochem. J. 1993, 294, 195–200. [Google Scholar] [CrossRef]
- Brooks, W.H.; Daniel, K.G.; Sung, S.S.; Guida, W.C. Computational validation of the importance of absolute stereochemistry in virtual screening. J. Chem. Inf. Model. 2008, 48, 639–645. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Raghavaiah, J.; Shahabi, D.; Yadav, M.; Anson, B.J.; Lendy, E.K.; Hattori, S.I.; Higashi-Kuwata, N.; Mitsuya, H.; Mesecar, A.D. Indole Chloropyridinyl Ester-Derived SARS-CoV-2 3CLpro Inhibitors: Enzyme Inhibition, Antiviral Efficacy, Structure-Activity Relationship, and X-ray Structural Studies. J. Med. Chem. 2021, 64, 14702–14714. [Google Scholar] [CrossRef] [PubMed]
- wwPDB: 7RC1. Available online: https://www.wwpdb.org/pdb?id=pdb_00007rc1 (accessed on 21 March 2022).
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Toledo Warshaviak, D.; Golan, G.; Borrelli, K.W.; Zhu, K.; Kalid, O. Structure-based virtual screening approach for discovery of covalently bound ligands. J. Chem. Inf. Model. 2014, 54, 1941–1950. [Google Scholar] [CrossRef]
- PyMOL|pymol.org. Available online: https://pymol.org/2/ (accessed on 21 March 2022).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | kinact (s−1) | Kiapp (µM) | Ki (µM) | kinact/Ki (s−1 µM−1) |
|---|---|---|---|---|
| Protocetraric acid | 1.19 × 10−4 ± 2.1 × 10−6 | 5.22 ± 0.07 | 3.95 | 3.01 × 10−5 |
| Salazinic acid | 1.33 × 10−4 ± 3.0 × 10−6 | 4.98 ± 0.09 | 3.77 | 3.53 × 10−5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fagnani, L.; Nazzicone, L.; Bellio, P.; Franceschini, N.; Tondi, D.; Verri, A.; Petricca, S.; Iorio, R.; Amicosante, G.; Perilli, M.; et al. Protocetraric and Salazinic Acids as Potential Inhibitors of SARS-CoV-2 3CL Protease: Biochemical, Cytotoxic, and Computational Characterization of Depsidones as Slow-Binding Inactivators. Pharmaceuticals 2022, 15, 714. https://doi.org/10.3390/ph15060714
Fagnani L, Nazzicone L, Bellio P, Franceschini N, Tondi D, Verri A, Petricca S, Iorio R, Amicosante G, Perilli M, et al. Protocetraric and Salazinic Acids as Potential Inhibitors of SARS-CoV-2 3CL Protease: Biochemical, Cytotoxic, and Computational Characterization of Depsidones as Slow-Binding Inactivators. Pharmaceuticals. 2022; 15(6):714. https://doi.org/10.3390/ph15060714
Chicago/Turabian StyleFagnani, Lorenza, Lisaurora Nazzicone, Pierangelo Bellio, Nicola Franceschini, Donatella Tondi, Andrea Verri, Sabrina Petricca, Roberto Iorio, Gianfranco Amicosante, Mariagrazia Perilli, and et al. 2022. "Protocetraric and Salazinic Acids as Potential Inhibitors of SARS-CoV-2 3CL Protease: Biochemical, Cytotoxic, and Computational Characterization of Depsidones as Slow-Binding Inactivators" Pharmaceuticals 15, no. 6: 714. https://doi.org/10.3390/ph15060714
APA StyleFagnani, L., Nazzicone, L., Bellio, P., Franceschini, N., Tondi, D., Verri, A., Petricca, S., Iorio, R., Amicosante, G., Perilli, M., & Celenza, G. (2022). Protocetraric and Salazinic Acids as Potential Inhibitors of SARS-CoV-2 3CL Protease: Biochemical, Cytotoxic, and Computational Characterization of Depsidones as Slow-Binding Inactivators. Pharmaceuticals, 15(6), 714. https://doi.org/10.3390/ph15060714