
Recent Progress in the Synthesis of Drugs and Bioactive Molecules Incorporating Nitro(het)arene Core



Abstract
1. Introduction
2. Nitrobenzene Derivatives
3. Nitro Heterocycles
3.1. Nitrofurans
3.2. 2-Nitroimidazoles
3.3. 4(5)-Nitroimidazoles
3.4. Miscelanous Nitroheterocycles
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nepali, K.; Lee, H.-Y.; Liou, J.-P. Nitro-Group-Containing Drugs. J. Med. Chem. 2019, 62, 2851–2893. [Google Scholar] [CrossRef] [PubMed]
- Parry, R.; Nishino, S.; Spain, J. Naturally-occurring nitro compounds. Nat. Prod. Rep. 2011, 28, 152–167. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, P.; Chaudhary, R.; Rani, N.; Sakshi; Venugopalan, P. 3-(Ethoxycarbonyl)-1-(5-methyl-5-(nitrosooxy)hexyl)pyridin-1-ium cation: A green alternative to tert-butyl nitrite for synthesis of nitrogroup-containing arenes and drugs at room temperature. Tetrahedron Lett. 2020, 61, 151529. [Google Scholar] [CrossRef]
- Patterson, S.; Wyllie, S. Nitro drugs for the treatment of trypanosomatid diseases: Past, present, and future prospects. Trends Parasitol. 2014, 30, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Patterson, S.; Fairlam, A.H. Current and Future Prospects of Nitro-compounds as Drugs for Trypanosomiasis and Leishmaniasis. Curr. Med. Chem. 2019, 26, 4454–4475. [Google Scholar] [CrossRef]
- Chung, M.C.; Bosquesi, P.L.; dos Santos, J.L. Prodrug Approach to Improve the Physico-Chemical Properties and Decrease the Genotoxicity of Nitro Compounds. Curr. Pharm. Des. 2011, 17, 3515–3526. [Google Scholar] [CrossRef]
- Dong, J.; Du, D.-M. Highly enantioselective synthesis of Warfarin and its analogs catalysed by primary amine–phosphinamide bifunctional catalysts. Org. Biomol. Chem. 2012, 10, 8125–8131. [Google Scholar] [CrossRef]
- Isık, M.; Akkoca, H.U.; Akhmedov, I.M.; Tanyeli, C. A bis-Lewis basic 2-aminoDMAP/prolinamide organocatalyst for application to the enantioselective synthesis of Warfarin and derivatives. Tetrahedron Asymm. 2016, 27, 384–388. [Google Scholar] [CrossRef]
- Ueda, A.; Umeno, T.; Doi, M.; Akagawa, K.; Kudo, K.; Tanaka, M. Helical-Peptide-Catalyzed Enantioselective Michael Addition Reactions and Their Mechanistic Insights. J. Org. Chem. 2016, 81, 6343–6356. [Google Scholar] [CrossRef]
- Rogozin’ska-Szymczak, M.; Mlynarski, J. Asymmetric synthesis of warfarin and its analogues on water. Tetrahedron Asymm. 2014, 25, 813–820. [Google Scholar] [CrossRef]
- Lim, J.Y.; Kim, D.Y. Enantioselective Conjugate Addition of 4-Hydroxycoumarin to Enones Catalyzed by Binaphthyl-Modified Primary Amine Organocatalyst. Bull. Korean Chem. Soc. 2012, 33, 1825–1826. [Google Scholar] [CrossRef][Green Version]
- Alonzi, M.; Bracciale, M.P.; Broggi, A.; Lanari, D.; Marrocchi, A.; Santarelli, M.L.; Vaccaro, L. Synthesis and characterization of novel polystyrene-supported TBD catalysts and their use in the Michael addition for the synthesis of Warfarin and its analogues. J. Catal. 2014, 309, 260–267. [Google Scholar] [CrossRef]
- Liu, J.; Li, Y.; Ke, M.; Liu, M.; Zhan, P.; Xiao, Y.-C.; Chen, F. Unified Strategy to Amphenicol Antibiotics: Asymmetric Synthesis of (−)-Chloramphenicol, (−)-Azidamphenicol, and (+)-Thiamphenicol and Its (+)-3-Floride. J. Org. Chem. 2020, 85, 15360–15367. [Google Scholar] [CrossRef]
- Xia, Y.; Jiang, M.; Liu, M.; Zhang, Y.; Qu, H.; Xiong, T.; Huang, H.; Cheng, D.; Chen, F. Catalytic Syn-Selective Nitroaldol Approach to Amphenicol Antibiotics: Evolution of a Unified Asymmetric Synthesis of (−)-Chloramphenicol, (−)-Azidamphenicol, (+)-Thiamphenicol, and (+)-Florfenicol. J. Org. Chem. 2021, 86, 11557–11570. [Google Scholar] [CrossRef]
- Lu, H.; Chanco, E.; Zhao, H. CmlI is an N-oxygenase in the biosynthesis of chloramphenicol. Tetrahedron 2012, 68, 7651–7654. [Google Scholar] [CrossRef]
- Makris, T.M.; Vu, V.V.; Meier, K.K.; Komor, A.J.; Rivard, B.S.; Münck, E.; Que, L.; Lipscomb, J.D. An Unusual Peroxo Intermediate of the Arylamine Oxygenase of the Chloramphenicol Biosynthetic Pathway. J. Am. Chem. Soc. 2015, 137, 1608–1617. [Google Scholar] [CrossRef]
- Franchino, A.; Jakubec, P.; Dixon, D.J. Enantioselective synthesis of (−)-chloramphenicol via silver-catalysed asymmetric isocyanoacetate aldol reaction. Org. Biomol. Chem. 2016, 14, 93–96. [Google Scholar] [CrossRef]
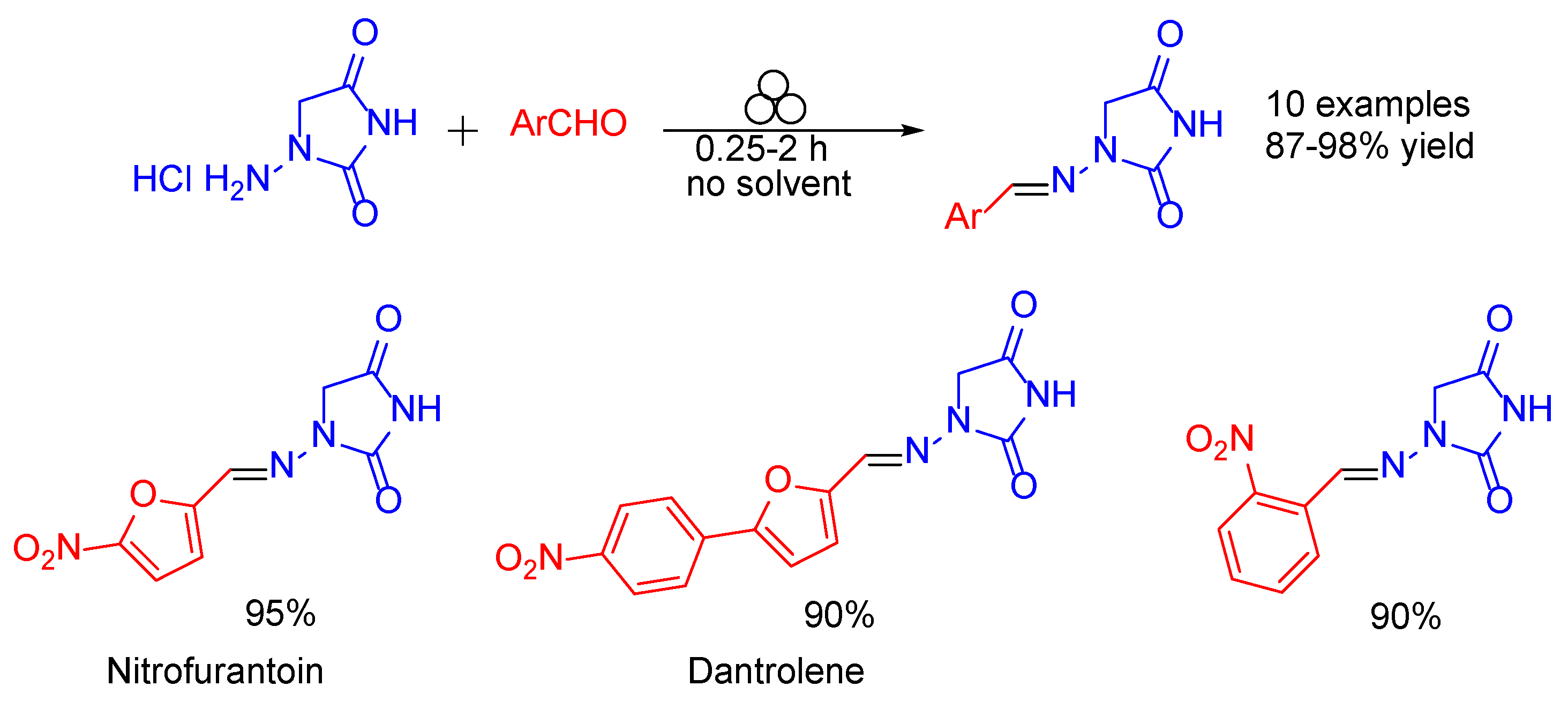
- Ellis, K.O.; Castellion, A.W.; Honkomp, L.J.; Wessels, F.L.; Carpenter, J.E.; Halliday, R.P. Dantrolene, a direct acting skeletal muscle relaxant. J. Pharm. Sci. 1973, 62, 948–951. [Google Scholar] [CrossRef]
- Snyder, H.R.; Davis, C.S.; Bickerton, R.K.; Halliday, R.P. 1-[5-Arylfurfurylidene)amino]hydantoins. A New Class of Muscle Relaxants. J. Med. Chem. 1967, 10, 807–810. [Google Scholar] [CrossRef]
- Ahmed, J.; Sau, S.C.; Sreejyothi, P.; Hota, P.K.; Vardhanapu, P.K.; Vijaykumar, G.; Mandal, S.K. Direct C-H Arylation of Heteroarenes with Aryl Chlorides using Abnormal NHC Coordinated Palladium Catalyst. Eur. J. Org. Chem. 2017, 5, 1004–1011. [Google Scholar] [CrossRef]
- Song, A.-X.; Zeng, X.-X.; Ma, B.-B.; Xu, C.; Liu, F.-S. Direct (Hetero)arylation of Heteroarenes Catalyzed by Unsymmetrical Pd-PEPPSI-NHC Complexes under Mild Conditions. Organometallics 2021, 39, 3524–3534. [Google Scholar] [CrossRef]
- Crisostomo, F.P.; Martin, T.; Carrillo, R. Ascorbic Acid as an Initiator for the Direct C-H Arylation of (Hetero)arenes with Anilines Nitrosated in situ. Angew. Chem. Int. Ed. 2014, 53, 2181–2185. [Google Scholar] [CrossRef] [PubMed]
- Harisha, A.S.; Nayak, S.P.; Pavan, M.S.; Shridhara, K.; Rao, S.K.; Rajendra, K.; Pari, K.D.; Sivaramkrishnan, H.; Row, T.N.G.; Nagarajan, K. A new synthesis of Entacapone and report on related studies. J. Chem. Sci. 2015, 127, 1977–1991. [Google Scholar] [CrossRef]
- Harisha, A.S.; Nayak, S.P.; Nagarajan, K.; Row, T.N.G.; Hosamani, A.A. Novel triethylamine mediated thermal reactions of 3-aryl-2-cyanoprop-2-enoic acid derivatives—Demethylation, reduction and vinylogation. Tetrahedron Lett. 2015, 56, 1427–1431. [Google Scholar] [CrossRef]
- Guo, M.; Mao, S. Entacapone Preparation Method. CN Patent 108440340 A, 24 August 2017. [Google Scholar]
- Fu, Y.; Zhong, H.; Lin, F.; Xiao, H.; Zheng, Y.; Lin, X.; Tang, X. Method for Producing Entacapone. CN Patent 112624941 A, 9 April 2021. [Google Scholar]
- Srikanth, G.; Ray, U.K.; Rao, D.V.N.S.; Gupta, P.B.; Lavanya, P.; Islam, A. Efficient approach to pure entacapone and related compounds. Synth. Commun. 2012, 42, 1359–1366. [Google Scholar] [CrossRef]
- Veerareddy, A.; Reddy, G.S. Synthesis of entacapone by Pd-catalyzed Heck coupling reaction. Synth. Commun. 2014, 44, 1274–1278. [Google Scholar] [CrossRef]
- Baker, J.W.; Bachman, G.L.; Schumacher, I.; Roman, D.P.; Tharp, A.L. Synthesis and bacteriostatic activity of some nitrofluoro methylanilides. J. Med. Chem. 1967, 10, 93–95. [Google Scholar] [CrossRef]
- Neri, R.O.; Topliss, J.G. Substituted Anilides as Anti-Androgens. US Patent 4144270 A, 13 March 1979. [Google Scholar]
- Peer, L.; Maye, J. Process for Nitrating Anilides. US Patent 4302599 A, 24 November 1981. [Google Scholar]
- Bandgar, B.P.; Sawant, S.S. Novel and Gram-Scale Green Synthesis of Flutamide. Synth. Commun. 2006, 36, 859–864. [Google Scholar] [CrossRef]
- Ghaffarzadeh, M.; Rahbar, S. A novel method for synthesis of flutamide on the bench-scale. J. Chem. Res. 2014, 38, 200–201. [Google Scholar] [CrossRef]
- McPherson, C.G.; Livingstone, K.; Jamieson, C.; Simpson, I. Palladium-Catalyzed Synthesis of Aryl Amides through Silanoate-Mediated Hydrolysis of Nitriles. Synlett 2016, 27, 88–92. [Google Scholar] [CrossRef]
- Tan, B.Y.-H.; Teo, Y.-C. Mild and Efficient Cobalt-Catalyzed Cross-Coupling of Aliphatic Amides and Aryl Iodides in Water. Synlett 2015, 26, 1697–1701. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, X.; Wan, Z.; Ren, F. TCDA: Practical synthesis and application in trifluoromethylation of arenes and heteroarenes. Org. Proc. Res. Dev. 2016, 20, 836–839. [Google Scholar] [CrossRef]
- Divi, M.; Krishna, P.; Rao, M.A.N.; Rajuri, V. Process for the Preparation of 4-iodo-3-nitrobenzamide. US Patent 2013172618 A1, 4 July 2013. [Google Scholar]
- Zhang, L.; Zhao, X.; Cui, Z.; Ma, T. Method of Preparing 4-iodo-3-nitrobenzamide. CN Patent 106366012 A, 1 February 2016. [Google Scholar]
- Cant, A.A.; Bhalla, R.; Pimlott, S.L.; Sutherland, A. Nickel-catalysed aromatic Finkelstein reaction of aryl and heteroarylbromides. Chem. Commun. 2012, 48, 3993–3995. [Google Scholar] [CrossRef]
- Prasanthi, G.; Prasad, K.V.S.R.G.; Bharathi, K. Synthesis, anticonvulsant activity and molecular properties prediction of dialkyl 1-(di(ethoxycarbonyl)methyl)-2,6-dimethyl-4-substituted-1,4-dihydropyridine-3,5-dicarboxylates. Eur. J. Med. Chem. 2014, 73, 97–104. [Google Scholar] [CrossRef]
- Subudhi, B.B.; Sahoo, S.P. Synthesis and Evaluation of Antioxidant, Anti-inflammatory and Antiulcer Activity of Conjugates of Amino Acids with Nifedipine. Chem. Pharm. Bull. 2011, 59, 1153–1156. [Google Scholar] [CrossRef][Green Version]
- Sliskovic, D.R. Cardiovascular Drugs. In Drug Discovery: Practices, Processes, and Perspectives; Li, J.J., Corey, E.J., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2013; pp. 141–204. [Google Scholar]
- Siddaiah, V.; Basha, G.M.; Rao, G.P.; Prasad, U.V.; Rao, R.S. PEG-mediated catalyst-free synthesis of Hantzsch 1,4-dihydropyridines and polyhydroquinoline derivatives. Synth. Commun. 2012, 42, 627–634. [Google Scholar] [CrossRef]
- Affeldt, R.F.; Benvenutti, E.V.; Russowsky, D. A new In–SiO2 composite catalyst in the solvent-free multicomponent synthesis of Ca2+ channel blockers nifedipine and nemadipine B. New J. Chem. 2012, 36, 1502–1511. [Google Scholar] [CrossRef]
- Wu, X.Y. Facile and green synthesis of 1,4-dihydropyridine derivatives in n-butyl pyridinium tetraflouroborate. Synth. Commun. 2012, 42, 454–459. [Google Scholar] [CrossRef]
- Katakami, T.; Yokoyama, T.; Miyamoto, M.; Mori, H.; Kawauchi, N.; Nobori, T.; Sannohe, K.; Kamiya, J.; Ishii, M.; Yoshihara, K. Pyrimidinedione Compounds, Method of Producing the Same and Antiarrythmic Agents Containing the Same. US Patent 5008267 A, 16 April 1991. [Google Scholar]
- Zhu, Y.; Wang, Y.; Xiao, J.; Li, H. Preparation Method of High-Purity Nifekalant Hydrochloride. CN Patent 103288751 A, 11 September 2013. [Google Scholar]
- Elks, J.; Ganellin, C.R. The Dictionary of Drugs: Chemical Data, Structures and Bibliographies; Springer: Boston, MA, USA, 2014; pp. 845–891. [Google Scholar]
- Kreipl, A.; Boege, N.; Thiel, W.R.; Zylum, B.P. Method for Coupling Halgen-Substituted Aromatic Compounds with Organic Compounds Comprising Trialkylsilyl-Sustituted Heteroatoms. US Patent 2012142937 A1, 7 June 2012. [Google Scholar]
- Yamamoto, Y.; Hisa, T.; Arai, J.; Saito, Y.; Yamamoto, F.; Mukai, T.; Ohshima, T.; Maeda, M.; Ohkubo, Y. Isomeric methoxy analog of nimesulide for development of brain cyclooxygenase-2 (COX-2)-targeted imaging agents: Synthesis, in vitro COX-2 inhibitory potency, and cellular transport properties. Bioorg. Med. Chem. 2015, 23, 6807–6814. [Google Scholar] [CrossRef]
- Attolono, E. Crystalline Inhibitor of 4-hydroxyphenylpyruvate dioxygenase, and a Process of Synthesis and Crystallization Thereof. US Patent 9783485 B1, 10 October 2017. [Google Scholar]
- El-Moselhy, T.F.; Sidhom, P.A.; Esmat, E.A.; El-Mahdy, N.A. Synthesis, Docking Simulation, Biological Evaluations and 3D-QSAR Study of 1,4-Dihydropyridines as Calcium Channel Blockers. Chem. Pharm. Bull. 2017, 65, 893–903. [Google Scholar] [CrossRef]
- Zhou, K.; Wang, X.; Zhao, Y.; Cao, Y.; Fu, Q.; Zhang, S. Synthesis and antihypertensive activity evaluation in spontaneously hypertensive rats of nitrendipine analogues. Med. Chem. Res. 2011, 20, 1325–1330. [Google Scholar] [CrossRef]
- Palermo, V.; Sathicq, A.G.; Constantieux, T.; Rodrıguez, J.; Vazquez, P.G.; Romanelli, G.P. First Report About the Use of Micellar Keggin Heteropolyacids as Catalysts in the Green Multicomponent Synthesis of Nifedipine Derivatives. Catal. Lett. 2016, 146, 1634–1647. [Google Scholar] [CrossRef]
- Fedorova, O.V.; Koryakova, O.V.; Valova, M.S.; Ovchinnikova, I.G.; Titova, Y.A.; Rusinov, G.L.; Charushin, V.N. Catalytic Effect of Nanosized Metal Oxides on the Hantzsch Reaction. Kinet. Catal. 2010, 51, 566–572. [Google Scholar] [CrossRef]
- Wang, B.; Zhu, G.; Wei, Q.; Qu, J.; Dai, Q. Nitrocefin Synthesis Method. CN Patent 106083894 A, 9 November 2016. [Google Scholar]
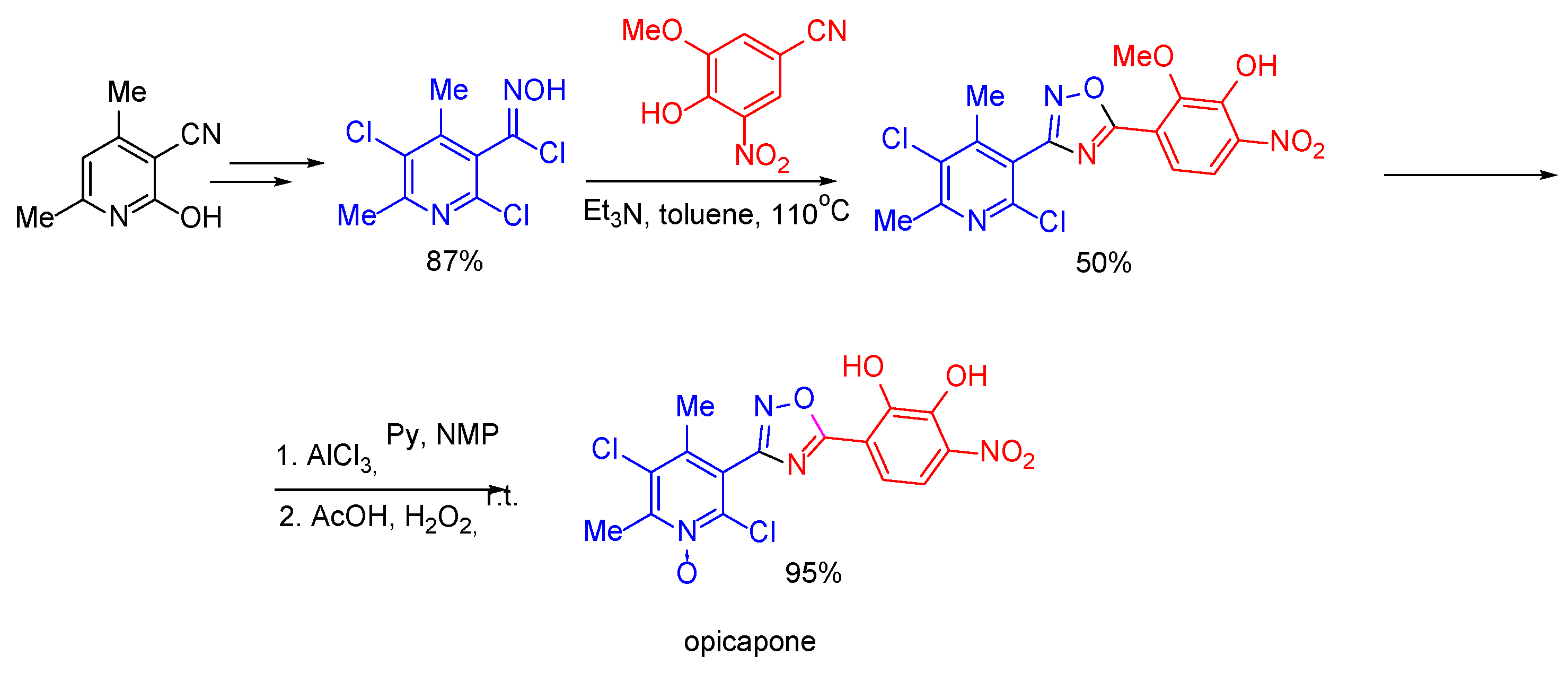
- Nabold, C.F.; Aebersold, C.; Grieko, P.G.; Aeschbacher, R.G. Route of Synthesis for Opicapone. US Patent 2018370958 A1, 27 December 2018. [Google Scholar]
- Sathe, D.G.; Das, A.; Gawas, D.V.; Chowkekar, S.B.; Jagtap, R.S. Process for the Preparation of Opicapone and Intermediates Thereof. WO Patent 2019123066 A1, 27 June 2019. [Google Scholar]
- Kiss, L.E.; Ferreira, H.S.; Torrao, L.; Bonifacio, M.J.; Palma, P.N.; Soares-da-Silva, P.; Learmonth, D.A. Discovery of a Long-Acting, Peripherally Selective Inhibitor of Catechol-O-methyltransferase. J. Med. Chem. 2010, 53, 3396–3411. [Google Scholar] [CrossRef]
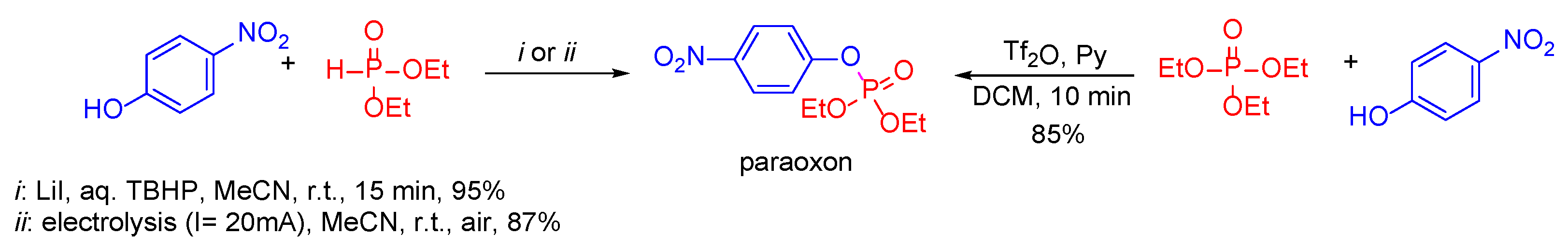
- Zhong, Z.; Xu, P.; Zhou, A. Electrochemical phosphorylation of arenols and anilines leading to organophosphates and phosphoramidates. Org. Biomol. Chem. 2021, 19, 5342–5347. [Google Scholar] [CrossRef]
- Anitha, T.; Ashalu, K.C.; Sandeep, M.; Mohd, A.; Wencel-Delord, J.; Colobert, F.; Reddy, K.R. LiI/TBHP Mediated Oxidative Cross-Coupling of P(O)–H Compounds with Phenols and Various Nucleophiles: Direct Access to the Synthesis of Organophosphates. Eur. J. Org. Chem. 2019, 45, 7463–7474. [Google Scholar] [CrossRef]
- Huang, H.; Ash, J.; Kang, J.Y. Tf2O-Promoted Activating Strategy of Phosphate Analogues: Synthesis of Mixed Phosphates and Phosphinate. Org. Lett. 2018, 20, 4938–4941. [Google Scholar] [CrossRef]
- Li, Y.; Wang, M.; Jiang, X. Straightforward Sulfonamidation via Metabisulfite-Mediated Cross Coupling of Nitroarenes and Boronic Acids under Transition-Metal-Free Conditions. Chin. J. Chem. 2020, 38, 1521–1525. [Google Scholar] [CrossRef]
- Gallardo-Garrido, C.; Cho, Y.; Cortes-Rios, J.; Vasquez, D.; Pessoa-Mahana, C.D.; Araya-Maturana, R.; Pessoa-Mahana, H.; Faundez, M. Nitrofuran Drugs Beyond Redox Cycling: Evidence of Nitroreduction-independent Cytotoxicity Mechanism. Toxicol. Appl. Pharmacol. 2020, 401, 115104. [Google Scholar] [CrossRef]
- Alhalaweh, A.; George, S.; Basavoju, S.; Childs, S.L.; Rizvi, S.A.A.; Velaga, S.P. Pharmaceutical Cocrystals of Nitrofurantoin: Screening, Characterization and Crystal Structure Analysis. CrystEngComm 2012, 14, 5078–5088. [Google Scholar] [CrossRef]
- Colacino, E.; Porcheddu, A.; Halasz, I.; Charnay, C.; Delogu, F.; Guerra, R.; Fullenwarth, J. Mechanochemistry for “no solvent, no base” preparation of hydantoin-based active pharmaceutical ingredients: Nitrofurantoin and dantrolene. Green Chem. 2018, 20, 2973–2977. [Google Scholar] [CrossRef]
- Alsaeedi, H.S.; Aljaber, N.A.; Ara, I. Synthesis and Investigation of Antimicrobal Activity of Some Nifuroxazide Analogs. Asian J. Chem. 2015, 27, 3639–3646. [Google Scholar] [CrossRef]
- Shuai, F.; Wang, X.; Zhang, J. Method for synthesizing furacilin under catalysis of supported catalyst. CN Patent 108101874 A, 1 June 2018. [Google Scholar]
- Fesenko, A.A.; Yankov, A.N.; Shutalev, A.D. A General and Convenient Synthesis of 4-(Tosylmethyl)semicarbazones and Their Use in Amidoalkylation of Hydrogen, Heteroatom and Carbon Nucleophiles. Tetrahedron 2019, 75, 130527. [Google Scholar] [CrossRef]
- da Cruz, A.C.N.; Brondani, D.J.; de Santana, T.I.; da Silva, L.O.; Borba, E.F.D.; de Faria, A.R.; de Albuquerque, J.F.C.; Piessard, S.; Ximenes, R.M.; Baratte, B.; et al. Biological Evaluation of Arylsemicarbazone Derivatives as Potential Anticancer Agents. Pharmaceuticals 2019, 12, 169. [Google Scholar] [CrossRef]
- Grueneberg, R.N.; Leakey, A. Treatment of Candidal Urinary Tract Infection with Nifuratel. Br. Med. J. 1976, 2, 908–910. [Google Scholar] [CrossRef]
- Cai, Z.; An, F.; He, Z. Preparation Method of Compound Nifuratel as Shown in Formula E. CN Patent 104402874 A, 11 March 2015. [Google Scholar]
- Hu, Z.; Li, F.; Jing, R. Industrial Preparation Method of Nifuratel. CN Patent 112745307 A, 4 May 2021. [Google Scholar]
- Yi, M.; Sun, B.; Xu, L.; Ma, Q.; Zhang, X.; Wang, X.; Zhang, N. Preparation Process of Anti-Infective Drug Nifuratel. CN Patent 107987069 A, 4 May 2018. [Google Scholar]
- Gagliardi, S.; Consonni, A.; Mailland, F.; Bulgheroni, A. (R)-Nifuratel and synthesis of (R)- and (S)-Nifuratel. EP Patent 2662371 A1, 13 November 2013. [Google Scholar]
- Coleman, C.N. Modulating the Radiation Response. Oncologist 1996, 1, 227–231. [Google Scholar] [CrossRef]
- Sree Kumar, K.; Weiss, J.F. Inhibition of Glutathione Peroxidase and Glutathione Transferase in Mouse Liver by Misonidazole. Biochem. Pharmacol. 1986, 35, 3143–3146. [Google Scholar] [CrossRef]
- Wilde, F.; Chamseddin, C.; Lemmerhirt, H.; Bednarski, P.J.; Jira, T.; Link, A. Evaluation of (S)- and (R)-Misonidazole as GPX Inhibitors: Synthesis, Characterization Including Circular Dichroism and In Vitro Testing on Bovine GPx-1. Arch. Pharm. Chem. Life Sci. 2014, 347, 153–160. [Google Scholar] [CrossRef]
- Rajendran, J.G.; Mankoff, D.A.; O’Sullivan, F.; Peterson, L.M.; Schwartz, D.L.; Conrad, E.U.; Spence, A.M.; Muzi, M.; Farwell, D.G.; Krohn, K.A. Hypoxia and Glucose Metabolism in Malignant Tumors: Evaluation by [18F]Fluoromisonidazole and [18F]Fluorodeoxyglucose Positron Emission Tomography Imaging. Clin. Cancer Res. 2004, 10, 2245–2252. [Google Scholar] [CrossRef]
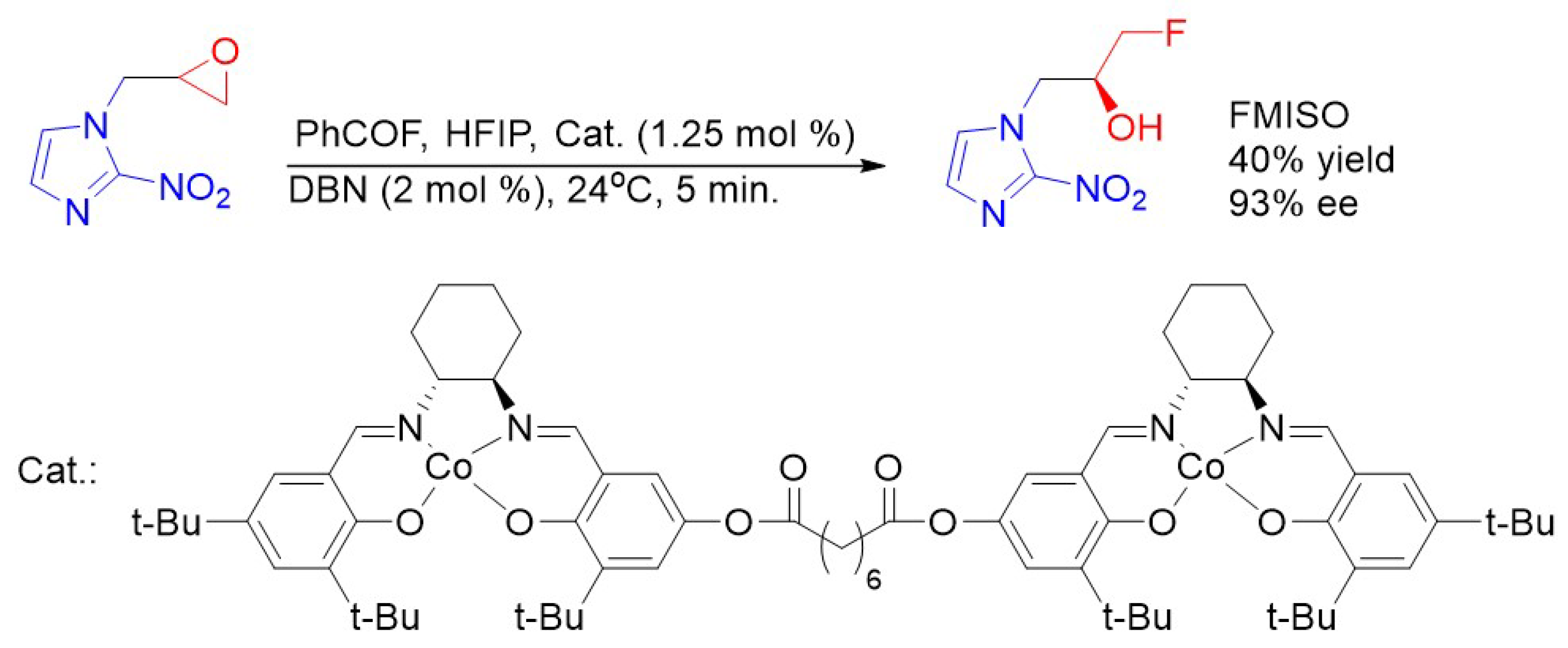
- Nieto, E.; Alajarin, R.; Alvarez-Builla, J.; Larranaga, I.; Gorospe, E.; Pozo, M.A. A New and Improved Synthesis of the Hypoxia Marker [18F]-FMISO. Synthesis 2010, 21, 3700–3704. [Google Scholar]
- Kwon, Y.-D.; Seol, E.; Lim, S.-T.; Sohn, M.-H.; Kim, H.-K.; Jung, Y.; Lee, S.J. Practical Synthesis of 1-(2-nitro-1H-imidazol-1-yl)-3-(tosyloxy)propan-2-yl acetate for the Radiosynthesis of [18F]-FMISO. Bull. Korean Chem. Soc. 2015, 36, 559–563. [Google Scholar]
- Kwon, Y.-D.; Jung, Y.; Lim, S.T.; Sohn, M.-H.; Kim, H.-K. Facile and Efficient Synthesis of [18F]Flouromisonidazole Using Novel 2-Nitroimidazole Derivatives. J. Braz. Chem. Soc. 2016, 27, 1150–1156. [Google Scholar]
- Revunov, E.; Zhuravlev, F. Co(salen)-mediated Enantioselective Radiofluorination of Epoxides. Radiosynthesis of Enantiomerically Enriched [18F]F-MISO via Kinetic Resolution. J. Fluor. Chem. 2013, 156, 130–135. [Google Scholar] [CrossRef]
- Graham, T.J.A.; Lambert, R.F.; Ploessl, K.; Kung, H.F.; Doyle, A.G. Enantioselective Radiosynthesis of Positron Emission Tomography (PET) Tracers Containing [18F]Fluorohydrins. J. Am. Chem. Soc. 2014, 136, 5291–5294. [Google Scholar] [CrossRef]
- Kalow, J.A.; Doyle, A.G. Mechanistic Investigations of Cooperative Catalysis in the Enantioselective Fluorination of Epoxides. J. Am. Chem. Soc. 2011, 133, 16001–16012. [Google Scholar] [CrossRef]
- Borzecka, W.; Lavandera, I.; Gotor, V. Biocatalyzed Synthesisof Both Enantiopure Fluoromisonidazole Antipodes. Tetrahedron Lett. 2013, 54, 5022–5025. [Google Scholar] [CrossRef]
- Zhang, W.; Fan, W.; Zhou, Z.; Garrison, J. Synthesis and Evaluation of a Radiolabeled Phosphoramide Mustard with Selectivity for Hypoxic Cancer Cells. ACS Med. Chem. Lett. 2017, 8, 1269–1274. [Google Scholar] [CrossRef]
- Duan, J.-X.; Matteucci, M.; Davar, N.; Andersen, D. TH-302 Solid Forms and Method Related Thereto. WO Patent 2016011195 A1, 21 January 2016. [Google Scholar]
- Murthy, S.S.M.; Bhilare, S.; Cardozo, J.; Chrysochos, N.; Schulzke, C.; Sanghvi, Y.S.; Gunturu, K.C.; Kapdi, A.R. Pd/PTABS: Low-Temperature Thioetherification of Chloro(hetero)arenes. J. Org. Chem. 2019, 84, 8921–8940. [Google Scholar]
- Sharma, S.; Anand, R.; Cham, P.S.; Raina, S.; Vishwakarma, R.A.; Singh, P.P. A Concise and Sequential Synthesis of the Nitroimidazooxazole Based Drug, Delamanid and Related Compounds. RSC Adv. 2020, 10, 17085–17093. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Ahmed, R.; Raina, S.; Vishwakarama, R.A.; Singh, P.P. Process for the Preparation of Derivatives of 1,1-dialkylethane-1,2-diols as Useful Intermediates. WO Patent 2020202205 A1, 8 October 2020. [Google Scholar]
- Fairlamb, A.; Patterson, S.; Wylie, S.; Read, K. Treatment of Parasitic Disease. WO Patent 2017072523 A1, 4 May 2017. [Google Scholar]
- Chen, G.J.; Zhu, M.L.; Chen, Y.X.; Miao, X.Q.; Guo, M.; Jiang, N.; Zhai, X. An Efficient and Practical Protocol for the Production of Pretomanid (PA-824) via a Novel Synthetic Strategy. Chem. Pap. 2020, 74, 3937–3945. [Google Scholar] [CrossRef]
- Patterson, S.; Wyllie, S.; Stoyanovski, L.; Perry, M.R.; Simeons, F.R.C.; Norval, S.; Osuna-Cabello, M.; De Rycker, M.; Read, K.D.; Fairlamb, A.H. The R Enantiomer of the Antitubercular Drud PA-824 as a Potential Oral Treatment for Visceral Leishmaniasis. Antimicrob. Agents Chemother. 2013, 57, 4699–4706. [Google Scholar] [CrossRef]
- Strassfeld, D.A.; Wickens, Z.K.; Picazo, E.; Jacobsen, E.N. Highly Enantioselective, Hydrogen-Bond-Donor Catalyzed Additions to Oxetanes. J. Am. Chem. Soc. 2020, 142, 9175–9180. [Google Scholar] [CrossRef]
- Marsini, M.A.; Reider, P.J.; Sorensen, E.J. A Concise and Convergent Synthesis of PA-824. J. Org. Chem. 2010, 75, 7479–7482. [Google Scholar] [CrossRef]
- Thompson, A.M.; O’Connor, P.D.; Marshall, A.J.; Blaser, A.; Yardley, V.; Maes, L.; Gupta, S.; Launay, D.; Braillard, S.; Chatelain, E.; et al. Development of (6R)-2-Nitro-6-[4-(trifluoromethoxy)phenoxy]-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (DNDI-8219): A New Lead for Visceral Leishmaniasis. J. Med. Chem. 2018, 61, 2329–2352. [Google Scholar] [CrossRef]
- Rami, M.; Dubois, L.; Parvathaneni, N.-K.; Alterio, V.; van Kujik, S.J.A.; Monti, S.M.; Lambin, P.; De Simone, G.; Supuran, C.T.; Winum, J.-Y. Hypoxia-Targeting Carbonic Anhydrase IX Inhibitors by a New Series of Nitroimidazole-Sulfonamides/Sulfamides/Sulfamates. J. Med. Chem. 2013, 56, 8512–8520. [Google Scholar] [CrossRef]
- Samant, B.S.; Sukhthankar, M.G. Compounds Containing 2-Substituted Imidazole Ring for Treatment Against Human African Trypanosomiasis. Bioorg. Med. Chem. Lett. 2011, 21, 1015–1018. [Google Scholar] [CrossRef]
- Natarajan, P.; Chaudhary, R.; Venugopalan, P. Silver(I)-Promoted ipso-Nitrtion of Carboxylic Acids by Nitronium Tetrafluoroborate. J. Org. Chem. 2015, 80, 10498–10504. [Google Scholar] [CrossRef]
- Zeb, A.; Malik, I.; Rasheed, S.; Choudhary, I.M.; Basha, F.Z. Metronidazole Esters: A New Class of Antiglycation Agents. Med. Chem. 2012, 8, 846–852. [Google Scholar] [CrossRef]
- Pi, J.; Zhao, T.; Yang, S.; Dong, J.; Rao, K.; Li, B.; Deng, J.; Zhang, Q. Environment-friendly Method for Metronidazole Synthesis. CN Patent 105348200, 24 February 2016. [Google Scholar]
- Zhao, T.; Li, B.; Zhang, W.; Ai, J.; Pi, J.; Zhang, Q.; Xie, G.; Wu, M. Method for Synthesizing Metronidazole Under Catalysis of Solid Acids. CN Patent 110172039, 27 August 2019. [Google Scholar]
- Mandalapu, D.; Kushwaha, B.; Gupta, S.; Singh, N.; Shukla, M.; Kumar, J.; Tanpula, D.K.; Sankhwar, S.N.; Maikhuri, J.P.; Siddiqi, M.I.; et al. 2-Methyl-4/5-nitroimidazole Derivatives Potentiated Against Sexually Transmitted Trichomonas: Design, Synthesis, Biology and 3D-QSAR Study. Eur. J. Med. Chem. 2016, 124, 820–839. [Google Scholar] [CrossRef]
- Ansari, M.A.; Saad, S.M.; Khan, K.M.; Salar, U.; Taslimi, P.; Taskin-Tok, T.; Saleem, F.; Jahangir, A. Biology-Oriented Drug Synthesis and Evaluation of Secnidazole Esters as Novel Enzyme Inhibitors. Arch. Pharm. 2021, 355, e2100376. [Google Scholar] [CrossRef]
- Pantala, R.C.M.; Khagga, M.; Bhavani, R.; Bhavani, V. Novel Salt of Tinidazole with Improved Solubility and Antibacterial Activity. Orient. J. Chem. 2017, 33, 490–499. [Google Scholar]
- Pantala, R.C.M.; Khagga, M.; Bhavani, R.; Bhavani, V. Synthesis, Characterization and Biological Activity of Novel Salt/Molecular Salts of Tinidazole. Orient. J. Chem. 2017, 33, 859–872. [Google Scholar]
- Li, Y.M.; Rizvi, S.; Hu, D.Q.; Sun, D.W.; Gao, A.H.; Zhou, Y.B.; Li, J.; Jiang, X.F. Selective Late-Stage Oxygenation of Sulfides with Ground-State Oxygen by Uranyl Photocatalysis. Angew. Chem. Int. Ed. 2019, 58, 13499–13506. [Google Scholar] [CrossRef]
- Shin, Y.; Noel, R.; Banerjee, S.; Kojetin, D.; Song, X.; He, Y.; Lin, L.; Cameron, M.D.; Burris, T.P.; Kamenecka, T.M. Small Molecule Tertiary Amines as Agonists of the Nuclear Hormone Receptor Rev-erbα. Bioorg. Med. Chem. Lett. 2012, 22, 4413–4417. [Google Scholar] [CrossRef]
- De, S.K.; Stebbins, J.L.; Chen, L.-H.; Riel-Mehan, M.; Machleidt, T.; Dahl, R.; Yuan, H.; Emdadi, A.; Barile, E.; Chen, V.; et al. Design, Synthesis, and Structure−Activity Relationship of Substrate Competitive, Selective, and in Vivo Active Triazole and Thiadiazole Inhibitors of the c-Jun N-Terminal Kinase. J. Med. Chem. 2009, 52, 1943–1952. [Google Scholar] [CrossRef]
- Stokes, J.M.; Yang, K.; Swanson, K.; Jin, W.; Cubillos-Ruiz, A.; Donghia, N.M.; MacNair, C.R.; French, S.; Carfrae, L.A.; Bloom-Ackermann, Z.; et al. A Deep Learning Approach to Antibiotic Discovery. Cell 2020, 180, 688–702. [Google Scholar] [CrossRef]
- Karpenko, I.; Deev, S.; Kiselev, O.; Charushin, V.; Rusinov, V.; Ulomsky, E.; Deeva, E.; Yanvarev, D.; Ivanov, A.; Smirnova, O.; et al. Antiviral Properties, Metabolism, and Pharmacokinetics of a Novel Azolo-1,2,4-Triazine-Derived Inhibitor of Influenza A and B Virus Replication. Antimicrob. Agents Chemother. 2010, 54, 2017–2022. [Google Scholar] [CrossRef]
- Sabitov, A.U.; Belousov, V.V.; Edin, A.S.; Oleinichenko, E.V.; Gladunova, E.P.; Tikhonova, E.P.; Kuzmina, T.Y.; Kalinina, Y.S.; Sorokin, P.V. Practical Experience of Using Riamilovir in Treatment of Patients with Moderate COVID-19. Antibiot. Chemother. 2020, 65, 27–30. [Google Scholar] [CrossRef]
- Voinkov, E.K.; Drokin, R.A.; Ulomskiy, E.N.; Slepukhin, P.A.; Rusinov, V.L.; Chupakhin, O.N. Crystal Structure of Medicinal Product Triazavirin. J. Chem. Crystallogr. 2019, 49, 213. [Google Scholar] [CrossRef]
- Shestakova, T.S.; Khalymbadzha, I.A.; Deev, S.L.; Eltsov, O.S.; Rusinov, V.L.; Shenkarev, Z.O.; Arseniev, A.S.; Chupakhin, O.N. Synthesis of the [2H,15N]-labeled Antiviral Drug “Triazavirine”. Russ. Chem. Bull. Int. Ed. 2011, 60, 729–732. [Google Scholar] [CrossRef]
- Shestakova, T.S.; Deev, S.L.; Khalymbadzha, I.A.; Rusinov, V.L.; Paramonov, A.S.; Arseniev, A.S.; Shenkarev, Z.O.; Charushin, V.N.; Chupakhin, O.N. Antiviral Drug Triazavirin, Selectively Labeled with 2H, 13C and 15N Stable Isotopes. Synthesis and Properties. Chem. Heterocycl. Compd. 2021, 57, 479–482. [Google Scholar] [CrossRef] [PubMed]
















































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
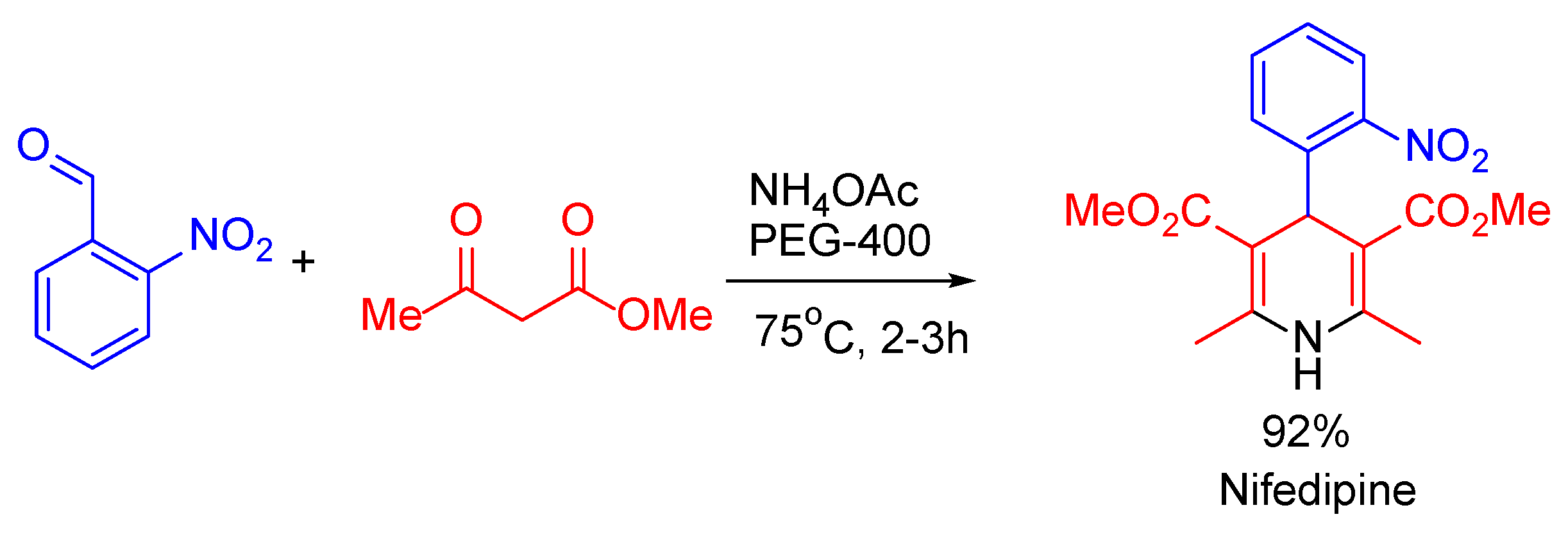{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
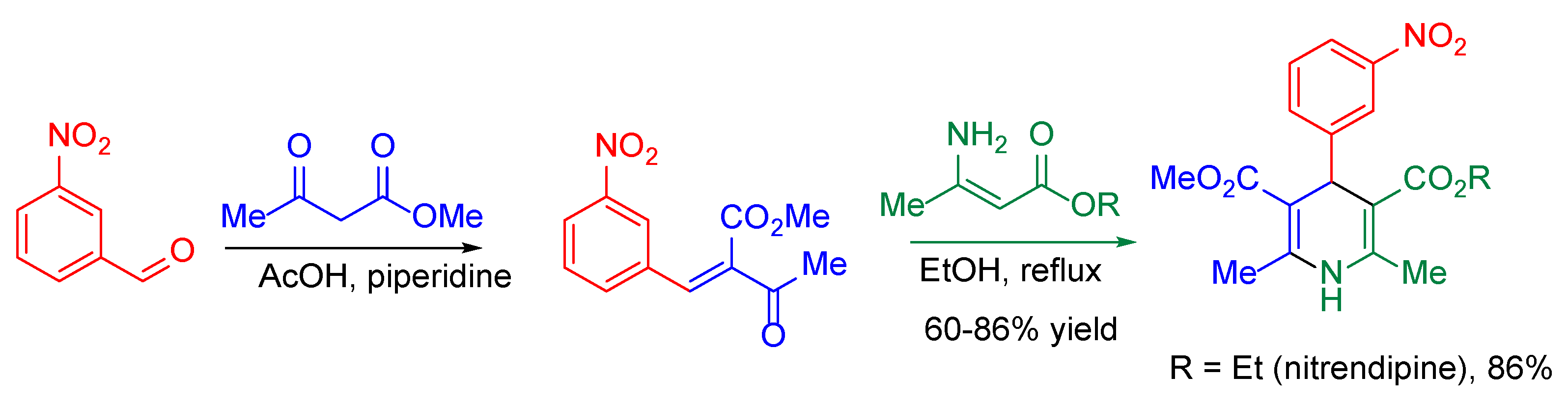{kind=link}
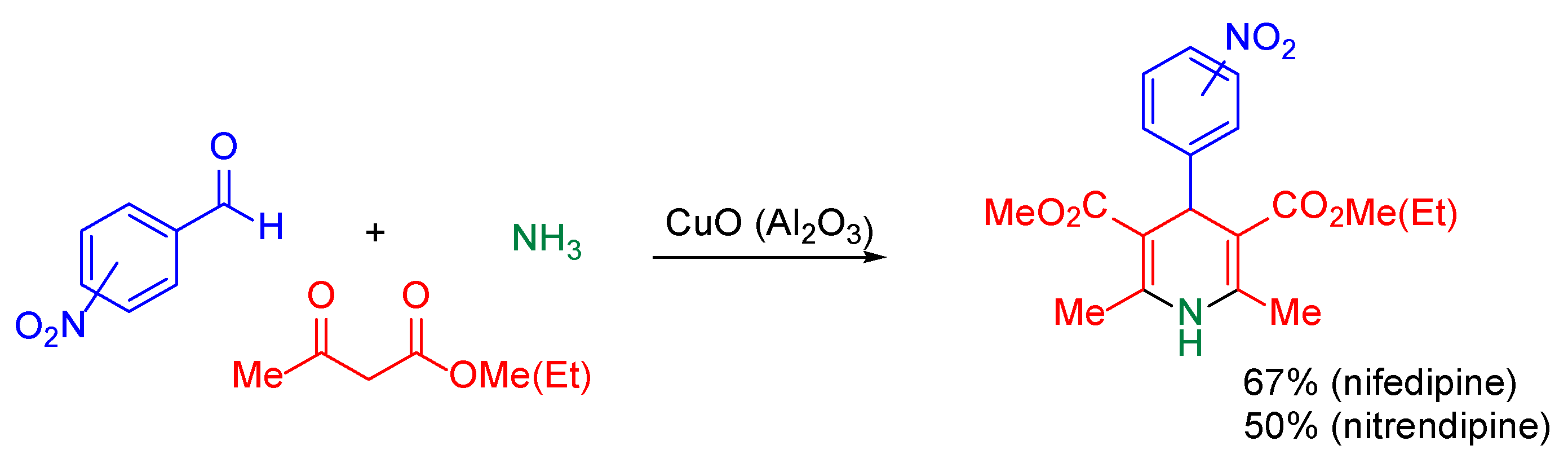{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
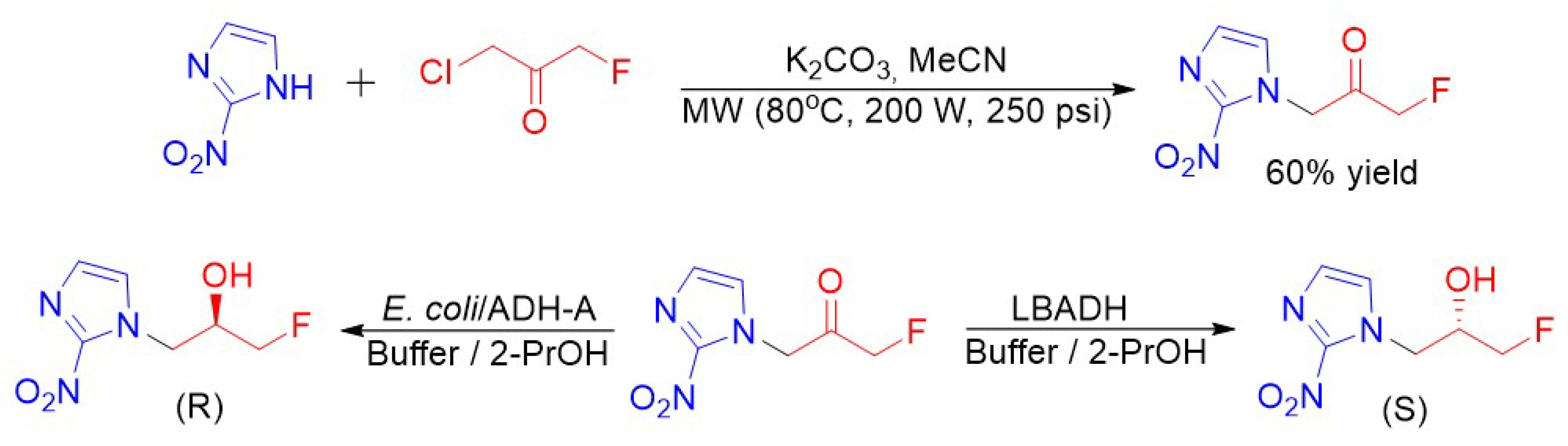{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
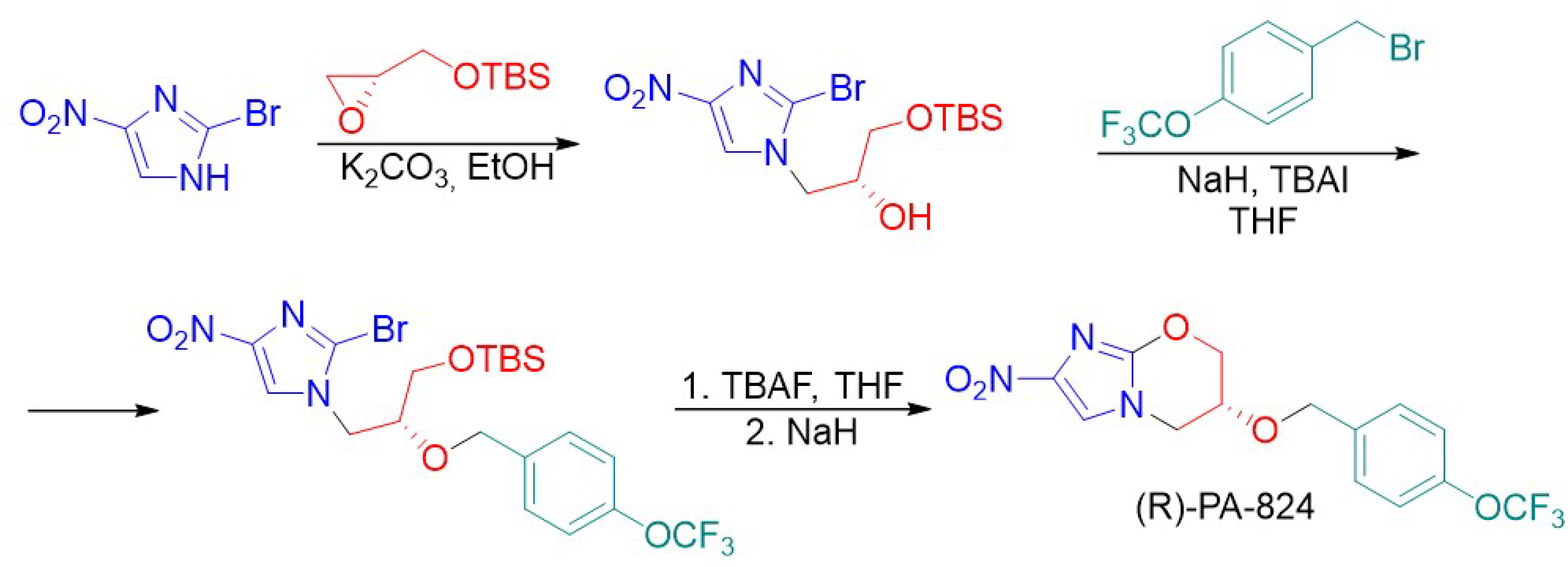{kind=link}
{kind=link}
{kind=link}
{kind=link}
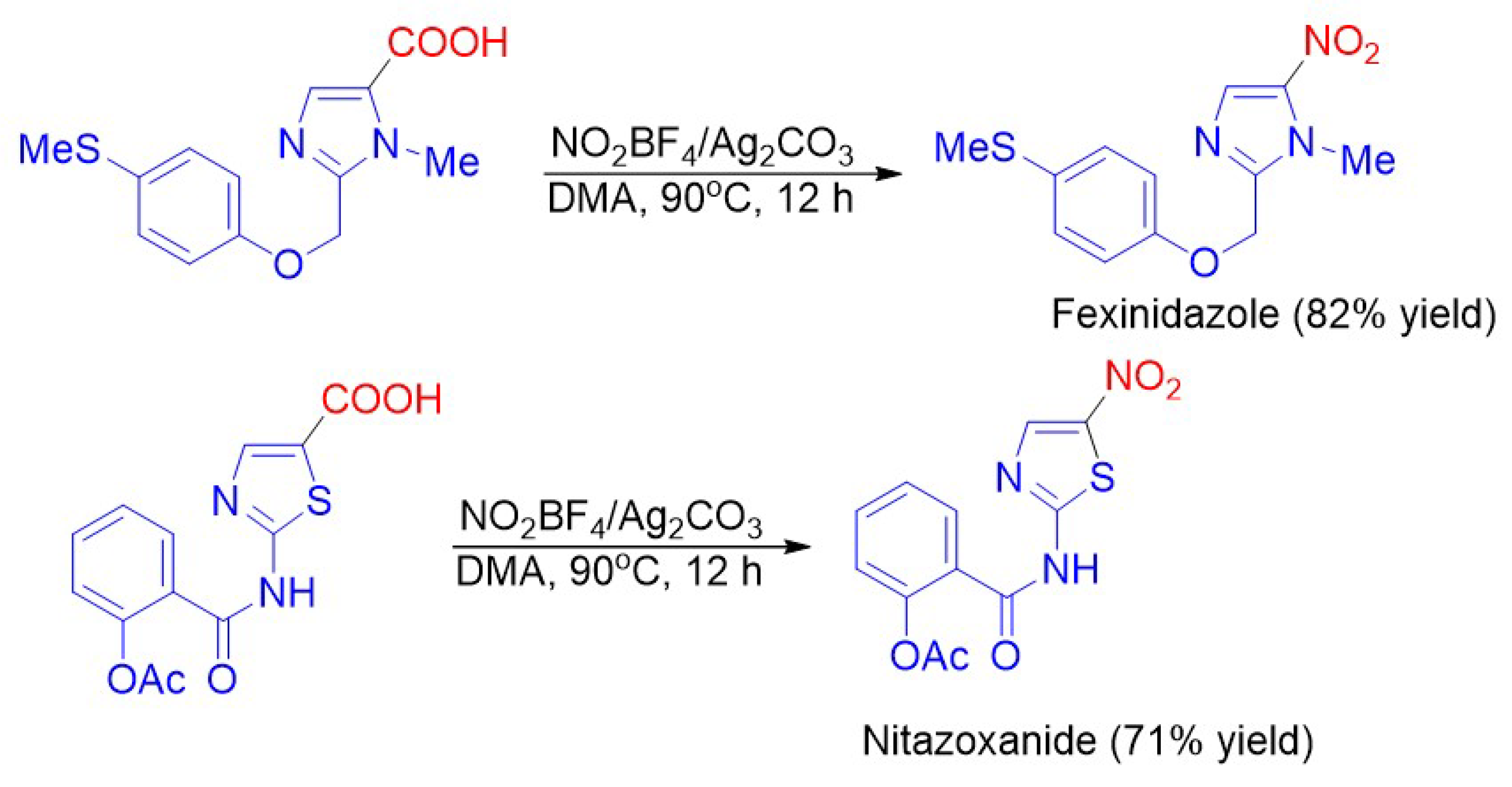{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
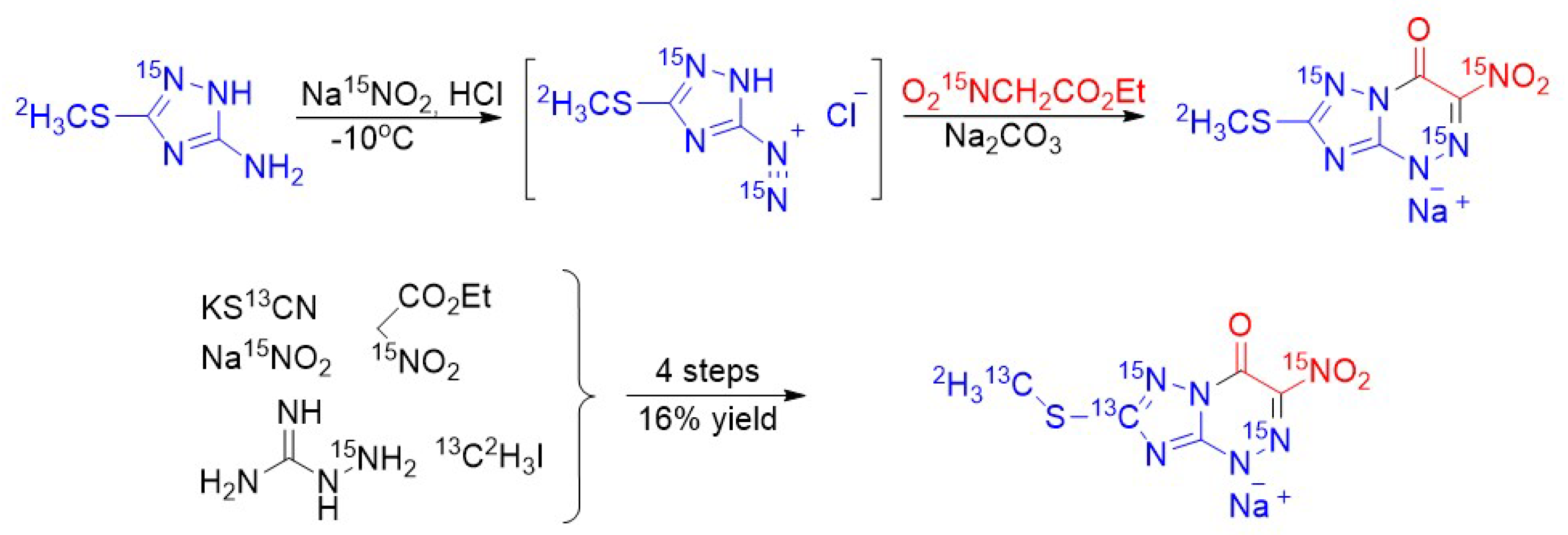{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bastrakov, M.; Starosotnikov, A. Recent Progress in the Synthesis of Drugs and Bioactive Molecules Incorporating Nitro(het)arene Core. Pharmaceuticals 2022, 15, 705. https://doi.org/10.3390/ph15060705
Bastrakov M, Starosotnikov A. Recent Progress in the Synthesis of Drugs and Bioactive Molecules Incorporating Nitro(het)arene Core. Pharmaceuticals. 2022; 15(6):705. https://doi.org/10.3390/ph15060705
Chicago/Turabian StyleBastrakov, Maxim, and Alexey Starosotnikov. 2022. "Recent Progress in the Synthesis of Drugs and Bioactive Molecules Incorporating Nitro(het)arene Core" Pharmaceuticals 15, no. 6: 705. https://doi.org/10.3390/ph15060705
APA StyleBastrakov, M., & Starosotnikov, A. (2022). Recent Progress in the Synthesis of Drugs and Bioactive Molecules Incorporating Nitro(het)arene Core. Pharmaceuticals, 15(6), 705. https://doi.org/10.3390/ph15060705