Abstract
Chagas and leishmaniasis are two neglected diseases considered as public health problems worldwide, for which there is no effective, low-cost, and low-toxicity treatment for the host. Naphthoquinones are ligands with redox properties involved in oxidative biological processes with a wide variety of activities, including antiparasitic. In this work, in silico methods of quantitative structure–activity relationship (QSAR), molecular docking, and calculation of ADME (absorption, distribution, metabolism, and excretion) properties were used to evaluate naphthoquinone derivatives with unknown antiprotozoal activity. QSAR models were developed for predicting antiparasitic activity against Trypanosoma cruzi, Leishmania amazonensis, and Leishmania infatum, as well as the QSAR model for toxicity activity. Most of the evaluated ligands presented high antiparasitic activity. According to the docking results, the family of triazole derivatives presented the best affinity with the different macromolecular targets. The ADME results showed that most of the evaluated compounds present adequate conditions to be administered orally. Naphthoquinone derivatives show good biological activity results, depending on the substituents attached to the quinone ring, and perhaps the potential to be converted into drugs or starting molecules.
Keywords:
chagas; leishmaniasis; naphthoquinones; antiprotozoal evaluation; QSAR; molecular docking; ADME 1. Introduction
Chagas and leishmaniasis are two parasitic infectious diseases endemic to Latin America, considered by the World Health Organization (WHO) as neglected tropical diseases, for which there is currently no effective, safe, and economical chemotherapy treatment. For chagas, the WHO estimates that between 7 and 8 million people are infected, with 12,000 deaths and 70 million people at risk of contracting it per year [1,2], while for leishmaniasis, there is an estimated figure of 12 million infected people, 1.6 million new cases each year, and 350 million people at risk of acquiring it [3,4,5]. In response to this problem, a growing number of investigations, mainly in Latin America, are being carried out to find new powerful anti-chagas and anti-leishmaniasis agents with low toxicity.
Chagas disease is a zoonosis caused by the protozoan Trypanosoma cruzi, which affects around 150 species of mammals, including humans, and is mainly vector-borne by hematophagous insects of the Tritominidae subfamily (popularly known as kissing bugs, bed bugs, and whistles, among other names). Since 1970, the treatment against this pathology has been based mainly on the antiparasitic drugs Nifurtimox (NFX) and Benznidazole (BNZ). These medicines have an effectiveness of 70% during the acute phase, where the prevailing parasitic form is blood trypomastigotes, and decreases to 20% in the chronic phase, where the predominant form corresponds to the intracellular phase of amastigotes [6,7]. It has been proven that NFX and BNZ have a high toxicity for mammalian cells, with serious side effects, such as peripheral neuropathies, anorexia, nausea, and vomiting, as well as neurological reactions such as anxiety and disorientation, among others [8]. On the other hand, leishmaniasis encompasses a group of infectious diseases caused by at least 20 species of the Leishmania genus, including L. braziliensis, L. amazonensis, L. infantum, L. guyanensis, L. panamensis, and L. mexicana, which are the species that affect humans. Its transmission is mainly through vectors via insect bites of the Psychodidae family. This disease presents three different clinical forms in humans, canines, and several wild vertebrates: cutaneous, mucocutaneous, and visceral [3]. Current treatment against leishmaniasis is based on the use of pentavalent antimonials, such as Miltefosine, Amphotericin B (AmB), Paromomycin, and Pentamidine; however, there are restrictions on the use of these drugs due to their side effects, which include hepatic, cardiac, and renal toxicity [5]. Additionally, they present limitations, such as high production cost and increased resistance by the parasite [9].
Naphthoquinones stand out among the most studied natural compounds and synthetic derivatives for their anti-chagas and anti-leishmaniasis (mainly against L. infantum and L. amazonensis) activity. β-lapachone derivatives have exhibited potential anticancer, antiviral, antiparasitic (including anti-chagas and anti-leishmanial activity), antimicrobial, anti-inflammatory, anti-obesity, antioxidant, and neuroprotective activity, while showing low levels of toxicity to normal cells [10]. Naphthoimidazoles derived from β-lapachone have been prepared, and their trypanocidal activity has been evaluated using electron microscopy, flow cytometry, and biochemical techniques, indicating that some compounds lead to an oxidative imbalance, which generates the production of ROS and the death of the parasite [10,11,12,13]. De Silva et al. carried out molecular docking studies on two cysteine proteases: cruzin and rhodesain, which are fundamental in the metabolism of the T. cruzi parasite [14]. This allowed for the identification of 14 naphthoquinone derivatives with potential antiprotozoal activity, which were synthesized and tested in vitro. On the other hand, 2,3-diphenyl-1,4-naphthoquinone (DPNQ) is considered a potential chemotherapeutic agent against T. cruzi due to its high trypanocidal activity in phenotypic screening and experimental murine infection by T. cruzi. Treatment with DPNQ in infected female mice promoted a halving of the parasite load, and ensured a 60% survival rate of the animal [15]. In 2021, Becerra et al. used pharmacophoric models based on different studies to design and subsequently synthesize nine phenolic derivatives, which were tested against T. cruzi strains, most of which showed good activity compared to BNZ, and the best prospects showed low toxicity [16]. Plumbagin is a plant-derived naphthoquinone metabolite (5-hydroxy-2-methyl-1,4-naphthaquinone) that inhibits trypanothione reductase, and has been validated as a drug which is responsible for promoting oxidative stress in Leishmania [9]. Similarly, naphtherocarpanquinone (LQB-118), which has been evaluated against cutaneous leishmaniasis, showed activity against intracellular amastigotes of L. amazonensis. It is proposed, through an anti-leishmanial model, that a good level of administration could counteract this clinical form of leishmaniasis [17]. The leishmanicidal activity of a series of substituted bis-2-hydroxy-1,4-naphthoquinones prepared from lawsone with aliphatic and aromatic aldehydes has also been evaluated [18]. Overall, four of the bis-lawsone analogs showed results similar to Pentamidine, but without cytotoxicity to host cells. Other studies demonstrated the high activity and low toxicity for host cells of derivatives of nor-α-lapachone and α-lapachone fused with triazole; despite them, however, resistance similar to that presented by the reference drug (Pentamidine) was observed [19]. Likewise, α-lapachone has shown a similar inhibition to sodium stibogluconate (Pentostam) [20]. Acetylisolapachol showed greater activity against L. amazonensis and does not exhibit a risk to the host [21].
Continuing with our search for novel structures with high antiparasitic activity and low toxicity [22], in this work, we have carried out an extensive in silico study of substituted derivatives of 1,4-naphthoquinone as potential anti-chagas and anti-leishmanial candidates. QSAR modeling of anti-T. cruzi in trypomastigotes, anti-L. amazonensis, anti-L. infantum, and toxicity has been carried out, based on the antiparasitic derivatives of naphthoquinone and other related structures reported in the literature. The best QSAR models were used to evaluate the predictive antiparasitic and toxicity activity of 68 1,4-naphthoquinone derivatives with hitherto unknown biological activity. This study was complemented by the evaluation of the 68 candidates against 5 macromolecular targets related to vital processes for the survival of these protozoa. In the case of T. cruzi, trypanothione reductase (TcTR) and lanosterol α-demethylase (TcLαD) proteins were selected. For the Leishmania genus in general, trypanothione reductase (LTR) protein was selected, while for the L. amazonensis and L. infantum strains, arginase (LaA) and tyrosine aminotransferase (LiTA) were selected, respectively. Additionally, the ADME (absorption, distribution, metabolism, and excretion) properties of the 68 naphthoquinone derivatives were evaluated. The best anti-chagas and anti-leishmanial candidates derived from this in silico study are currently in the organic synthesis stage at our lab for their subsequent in vitro evaluation.
2. Results and Discussion
2.1. QSAR Modeling
In total, four QSAR models were performed based on in vitro reports of naphthoquinone derivatives and related structures with anti-T. cruzi (in trypomastigotes) [23,24,25,26,27,28,29,30,31], anti-L. amazonensis [18,32,33,34,35,36,37,38,39], and anti-L. infantum [40,41,42,43,44,45] activities, as well as toxicity [30,46,47,48,49,50,51] evaluation. For each QSAR study, the top seven models were generated, which are shown in Tables S1 and S2 in the Supplementary Materials. In turn, the best model (in bold) among the seven was selected, which was performed considering high homogeneity (fitting, R2) between the calibration and validation groups with the lowest root mean square error (RMSE), thereby avoiding over-adjusted models. For selecting the best model, the Ockham’s principle of parsimony was also considered, which states that if there are models with statistical parameters of equal quality, the simplest one should be selected [52].
2.1.1. QSAR Model for Anti-chagas Activity
Equation (1) shows the best QSAR model for the anti-chagas activity predicted as LogIC50, which was developed based on 153 derivatives of 1,2-naphthoquinone and 1,4-naphthoquinone (Figure A1) evaluating in vitro blood trypomastigotes of strain Y of T. cruzi. A description of the molecular descriptors that define this model is listed in Table 1.

Table 1.
Molecular descriptors of the best anti-chagas QSAR model as defined by Equation (1).
The descriptors and are fingerprints calculated in Fragmentor [53], and refer to the presence of oxygen followed by an unsaturation; while the second was calculated in PaDeL [54], and belongs to the MACCS keys, a group of 166 free access fragments [55], which involves an oxygen–oxygen arrangement at a distance equal to three bonds and two intermediate atoms of any type. According to this QSAR model (Equation (1)), these molecular descriptors contribute negatively to the evaluated activity (i.e., they decrease the activity). The other descriptors of the model were calculated in the program QUBILS-MAS [56]. The descriptors , , and are calculated from the Kurtosis, which is a statistical invariant, while belongs to the Minkowski indicators. is a quadratic index, and are bilinear, and is linear. The descriptor is a global index that contributes to the biological activity. This is related to molar refractivity, which can sometimes be used to model London dispersion forces or attractive van der Waals interactions; these are factors related to the presence of strong interactions between a ligand and the active sites of a given macromolecular receptor. However, refractivity is the consequence of repulsive nonbonding interactions, and is highly dependent on the flexibility of the ligand [57]. The descriptor describes the electronegativity and polarizability of the fragment of carbon atoms in aliphatic chains, while is associated with the refractivity and charge of the subset of heteroatoms in the molecule [4]. Both descriptors contribute negatively to the anti-chagas activity. Considering the nature of descriptors and , it is hypothesized that the anti-chagas activity could be related to the ability of the ligands to dock and inhibit essential macromolecules in the metabolism of T. cruzi. For its part, the descriptor provides a positive contribution to the biological activity, and is a function of the mass of aromatic carbon atoms, which suggests that the presence of aromatic systems improves activity.
Table S3 in the Supplementary Materials shows the intercorrelation relationship between each pair of descriptors included in the anti-chagas QSAR model, while Table S4 lists the values of each descriptor for each molecule. Table S5 shows the experimental values together with those predicted by the anti-chagas activity model as log IC50.
2.1.2. QSAR Model for Anti-L. amazonensis Activity
For the anti-L. amazonensis (cutaneous and mucocutaneous leishmaniasis) activity (expressed as LogIC50), the QSAR model with four descriptors was selected (Table S1), which is described according to Equation (2). This model was developed based on 60 ligands (Figure A2), with in vitro anti-leishmanial activity reported as IC50:
where ,, , and are described as shown in Table 2.
Table 2.
Molecular descriptors in the anti-L. amazonensis QSAR model, as described by Equation (2).
The descriptor refers to the Klekota–Roth fingerprint KRFP2, and it comprises part of six of the seven anti-L. amazonensis QSAR models considered (Table S1). This descriptor provides a measure of chemical similarity related to a bond between two carbon atoms, where each carbon atom has, as its substituents, a hydrogen atom and two nonhydrogen atoms [54,58]. On the other hand, , , and refer to 2D type descriptors. The minHBint9 () descriptor describes the topological state of the atom’s environment, such as the electronic interactions present in the atoms of the molecule at a topological distance of nine with each atom. According to Equation (2), this characteristic decreases the IC50, that is, the less topological distance there is between the atoms that make up the structure, the better its antiprotozoal activity [59]. The IC3 descriptor () is based on the three-order neighborhood symmetry. This means that molecules with lower symmetry will have a better predictive activity [60]. Finally, the descriptor MDEC-23 () refers to the information regarding the edge of the molecular distance among all secondary and tertiary carbons. The presence and value of enhance the anti-leishmaniasis activity predicted by the model, indicating that the presence of secondary and tertiary carbon atoms in these molecules is related to their biological activity [61,62,63].
2.1.3. QSAR Model for anti-L. infantum Activity
For predicting anti-L. infantum (visceral leishmaniasis) activity such as LogIC50, the model with six descriptors was selected (Table S1), which is represented according to Equation (3). The molecules used for constructing this QSAR model are shown in Figure A3:
where are 2D descriptors described as shown in Table 3.
Table 3.
Molecular descriptors in the anti-L. infantum QSAR model, as described by Equation (3).
The MATS3c () descriptor refers to Moran’s autocorrelation, which expresses the partial charge values of atoms separated by three distances, that is, it estimates the correlation of charges divided into three bonds [64,65]. The descriptor nHBint7 () counts resistance E-state descriptors for hydrogen bonds with a path length of seven. The – descriptors are part of the QuBiLS-MAS program [66]. The descriptor refers to the partition algorithm, which is used to calculate estimates of most neutral organic compounds that have C, H, O, N, S, Se, P, B, Si, and halogen atoms. This descriptor is based on AlogP, which can also estimate local hydrophobicity, visualize molecular hydrophobicity maps, and evaluate hydrophobic interactions when protein–ligand complexes are formed [67,68]. The descriptor considers bilinear indices and aliphatic chain carbon atoms, and correlates the topological area of the polar surface and the van der Waals volume. The polar surface area (PSA) is a molecular descriptor widely used in the study of drug transport properties, related to the penetration of the blood–brain barrier and its intestinal absorption. Additionally, the descriptor alludes to the sum of the contributions of polar atoms such as oxygen, nitrogen, and hydrogen to the molecular surface area [69,70]. The descriptor correlates the carbon atoms of aliphatic chains with the van der Waals volume and charge. Molecular volume is defined as a measure of the space around electron-filled atomic nuclei, and is geometrically interpreted as the combined volume of the superimposed spheres centered on the nuclei, similar to a space-filling molecular model [71,72]. Finally, the descriptor presents a nonstochastic matrix of order 15; in this case, the descriptor considers the heteroatoms different from C and H, correlating them with the partition algorithm and estimating the different atoms. This descriptor includes most of the zwitterionic compounds that have amine, carboxylic acids, and ammonium halide salts. The descriptor is also based on an intrinsically atomistic model, which is useful for drug design, since it makes estimates of the local or general hydrophobicity of a molecule [66,68].
Table S7 in the Supporting Materials shows the intercorrelation relationship between each pair of descriptors included in the anti-leishmanial QSAR models (Equations (2) and (3)), while Table S8 (L. amazonensis) and Table S7 (L. infantum) show the values of each descriptor calculated for each molecule used in the development of these QSAR models. Tables S10 and S11 list the experimental and model-predicted values of anti-leishmanial activity as log IC50.
2.1.4. QSAR Model for Toxicity
From the QSAR study for toxicity (expressed as LogIC50), a model with five descriptors was selected (Table S2), which is represented in Equation (4). This model was developed based on 76 naphthoquinone derivatives, as shown in Figure A4.
where are 2D descriptors described as shown in Table 4.
Table 4.
Molecular descriptors in the QSAR model of toxicity as described by Equation (4).
The toxicity model descriptors were calculated using the QUBILS-MAS program [66]. The descriptors , , , and are calculated from the Kurtosis, which is a statistical invariant of distribution. The descriptor correlates with physicochemical properties, polarized topological surface area, and refractivity. As indicated above, the topological polar surface area of a molecule depends on the sum of the surface area of polar atoms, such as oxygen, nitrogen, and hydrogen, and facilitates the ability of a molecule to penetrate cells. According to this, the greater the value of the polar topological surface in a molecule, the greater its probability of being transported [73]. Meanwhile, refractivity is a measure of the volume occupied by an atom or a group of atoms [69]. These last two properties allow for a theoretical prediction of the pharmacological potential of a molecule in a biological environment [74]. The descriptor correlates the polarization with the charge of aromatic carbon atoms because it facilitates the distortion of the atomic or molecular charge in electromagnetic fields. This descriptor refers to an electronic parameter, which impacts chemical–biological interactions [75]. The descriptor correlates the polar surface area with the polarization of the carbon atoms of the aromatic moiety and the heteroatoms attached to this moiety. The descriptor is associated with the partition algorithm, heteroatoms, and electronegativity. This descriptor is based on the tendency of a heteroatom or functional group to attract electrons and estimate the local or general hydrophobicity of a molecule [67,68,69,70,71,72,73,74,76,77]. Finally, the descriptor refers to the charge and the carbon atoms in the aliphatic chains.
Table S13 in Supporting Materials shows the intercorrelation relationship between each pair of descriptors included in the QSAR model of toxicity, while Table S15 shows the experimental and predicted values in log IC50 used for constructing of the QSAR model of toxicity.
2.1.5. Validation of QSAR Models
Table 5 contains a compilation of the statistical parameters used for the internal and external validation of the four QSAR models developed. From Y-randomization, it was found that RMSEcal < RMSErand, thus indicating that all QSAR models are robust due to the absence of random correlation [78] Internal validation by the Leave-One-Out (LOO) method yielded a value of the squared correlation coefficient () ≥ 0.5 [78,79], which ensures the statistical stability of each model. In addition to presenting good internal validation parameters, all of the QSAR models are predictive, since they meet the following requirements: the slopes k and k’ of the plots of observed and predicted values are in the range 0.85–1.15 (with k corresponding to the case when the predicted values are plotted on the x-axis and the experimental values on the y-axis, while k’ is the inverse graph). The CCC statistically evaluates the models, this parameter verifies the difference between the experimental and predicted values. The squared correlation coefficients have values greater than 0.5, which validate the models [80].
Table 5.
Internal and external validation parameters for each best QSAR model.
Figure 1 shows the dispersion diagram of the experimental values of anti-chagas and anti-Leishmania (L. amazonensis and L. infantum) activity and toxicity expressed as Log(IC50) as a function of the values predicted by each model. In all cases, it is observed how points adopt a linear trend around the line of perfect fit (in green), which confirms a multivariate linear relationship.
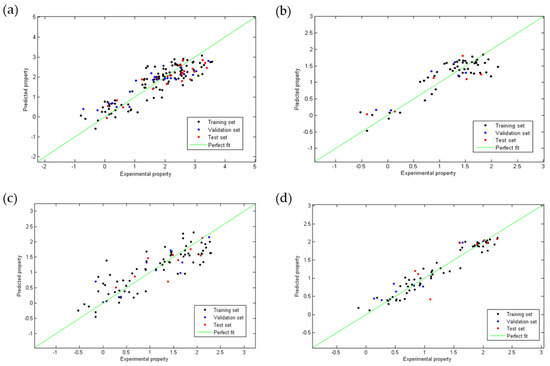
Figure 1.
Linear correlation plots between the experimental and the predicted values obtained using the QSAR Equations (1) (a), (2) (b), (3) (c) and (4) (d).
Figure 2 shows the plots of the residuals for the four QSAR models developed. It was observed that for anti-chagas (Figure 2a) and anti-leishmaniasis (Figure 2b,c) activity, no residual is greater than three times the standard deviation (3S) of the model (outliers), while the toxicity model (Figure 2d) presented one outlier of the test group. In all QSAR models, the points follow a random distribution around the line at , which suggests that these properties are modeled using of multiple linear regression.
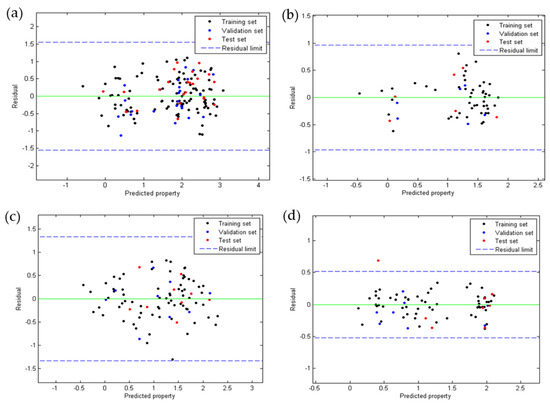
Figure 2.
Dispersion of residues according to QSAR Equations (1) (a), (2) (b), (3) (c), and (4) (d).
Due to the low diversity of molecules considered in the development of the QSAR models, the acceptable predictions are restricted to structural analogs derived from naphthoquinone, whose influence value is less than the critical influence value (h*) in each model (Figure 3). As shown in Figure 3a,c, for anti-chagas and anti-L. infantum activity, all molecules of the validation group and of the test group are within the domain of applicability, and molecules of the calibration group are considered outside the value of h*, a fact that reinforces the predictive capacity of these models. For the anti-L. amazonensis activity model, Figure 3b shows how a molecule of the test group is rejected, thus demonstrating its ability to reject molecules with large differences. For its part, the toxicity model shows that all molecules considered for its development and validation are within the domain of applicability.
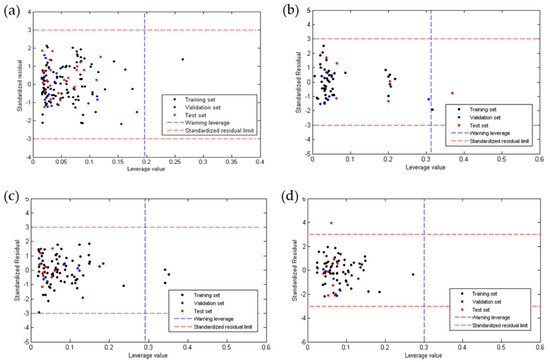
2.1.6. Molecular Design and Applicability of QSAR Models
Both ortho-naphthoquinone (1,2-substituted) and para-naphthoquinone (1,4-substituted) are recognized as highly active cores due to the synergy between its acid base and oxidation reduction properties [81], whose derivatives have exhibited several tunable antiparasitic effects according to the substitutions made in their fused rings [82]. As can be verified from the structural compilation used for constructing our QSAR antiprotozoal models (Figure A1, Figure A2 and Figure A3 in Appendix A), a high in vitro anti-chagas or anti-leishmanial effect is achieved when substitutions with heterocyclic, aromatic, or aliphatic groups are made at the 2 and 3 positions, for the case of the para-naphthoquinone, or at the 3 and 4 positions, for the case of the ortho-naphthoquinone nucleus.
The molecular design of our selection group was based on the following principles: (i) restriction to only derivatives of the 1,4-naphthoquinone nucleus at positions 2 and 3; (ii) use of open-chain and heterocyclic substituents at these positions; (iii) use of nitrogenous derivatives in position 2; (iv) variation in position 3 with electro-withdrawing and electro-donor groups; and (v) substitution in the amino nitrogen, located in position 2, with both highly functionalized aromatic and heterocycle rings. Due to this strategy, four families of 1,4-naphthoquinone derivatives (Figure 4) were selected for in silico evaluation: (a) 2-chloro-3-arylamino family (NQ–Chlorine); (b) 2-amino-3-arylamino family (NQ–Amine); (c) 2-amino-3-triazolamino family (NQ–Triazole); and (d) phenazine family (NQ–Phenazine). In particular, the use of triazoles and phenazines as substituents was established based on the high intrinsic activity of these groups, as well as their enhancing effect when incorporated into other active nucleus [23,82,83,84].
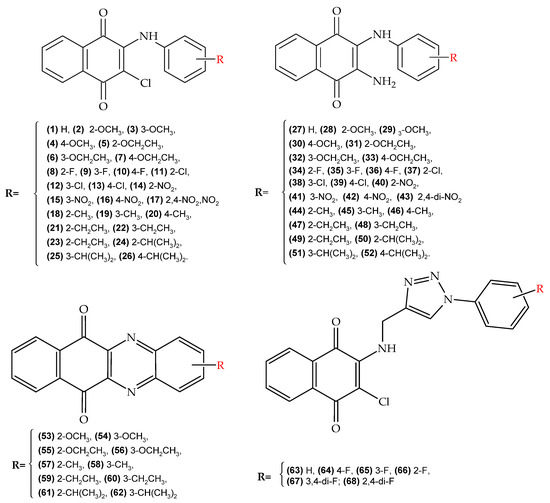
Figure 4.
Molecular design of 1,4-naphthoquinone derivatives with potential antiparasitic activity (chagas and leishmaniasis). (top-left) (NQ–Chlorine family), (top-right) (NQ–Amine family), (bottom-left) (NQ–Phenazine), and (bottom-right) (NQ–Triazole family).
QSAR models established according to Equations (1)–(4) were used to predict anti-chagas (IC50 in trypomastigotes), anti-L. amazonensis (cutaneous and mucocutaneous leishmaniasis), and anti-L. infantum (visceral leishmaniasis) activity, as well as toxicity, of the 68 derivatives of 1,4-naphthoquinone shown in Figure 4, which are structures that do not present experimental anti-chagas or anti-leishmanial activity (in vitro or in vivo) reported so far.
Figure 5a shows the prediction values of anti-chagas activity (IC50) for the 68 1,4-naphthoquinone derivatives evaluated using QSAR Equation (1). As a result, 47 (69%) of the 68 evaluated molecules had a better predicted IC50 than the experimental value of the reference drug BNZ. Overall, 10 of these derivatives (structures 17, 43, 53–60) were not within the applicability domain of the model, so their predicted activity should be considered unreliable. High anti-chagasic activity was found for the NQ–Phenazine derivatives substituted with the isopropyl group (structures 61 and 62), as well as for the NQ–Chlorine and NQ–Amine derivatives containing the same substituent (structures 24–26 and 51–52, respectively). Additionally, the derivatives coupled to the triazole ring (NQ–Triazole family) show a slightly better predicted activity than BNZ. According to this QSAR model, those amino derivatives with electron-withdrawing substituents (nitro and fluorine, structures 34–36 and 40–42, respectively) present the less parasitic activity toward T. cruzi trypomastigotes.
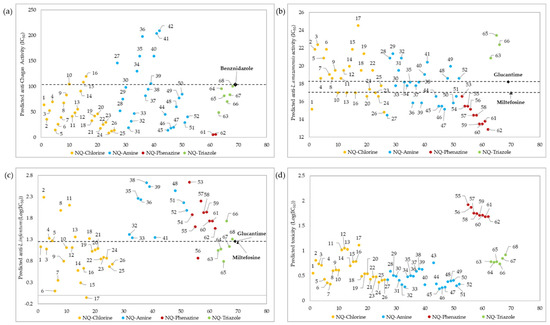
Figure 5.
Prediction of anti-chagas (IC50) (Table S6) (a), anti-L. amazonensis (IC50) (b), and anti-L. infantum (LogIC50) (Table S12) (c) activity, as well as toxicity (LogIC50) (Table S16) (d), for the 68 1,4-naphthoquinone derivatives (x-axis) evaluated using QSAR Equations (1)–(4), respectively. Only structures with reliable activity, as determined by the influence value (h*), are presented. The predicted values for anti-L. infantum and toxicity are presented in LogIC50 because of the wide dispersion of the data.
The predicted IC50 values of anti-Leishmania activity (L. amazonensis and L. infantum) for the 68 naphthoquinone derivatives evaluated with QSAR Equations (2) and (3) are presented in Figure 5b,c respectively. According to the influence value (h*), 63 (93%) of the 68 naphthoquinone derivatives showed reliable anti-Leishmania activity, and five molecules (42, 43, 64, 67 and 68) did not show reliable activity. It was found that all molecules belonging to NQ–Phenazine family, as well as some derivatives of the NQ–Chloro (structures 1, 20, 24, and 26), and NQ–Amine families (structures 27, 36, 39, 44–47, 50–51), presented a better anti-L. amazonensis effect than the reference drugs Miltefosine and Glucantime (see Figure 5b), unlike the all of NQ–Triazole derivatives that had lower results than the reference drugs. According to Equation (3), 51 derivatives presented a reliable predictive anti-L. infantum activity, while the other 17 molecules were not found within the influence parameter (0.24). Among the molecules that presented a reliable activity, the best activity was that of molecule 6 together with several other NQ–Chloro derivatives (structures 1, 3, 6–7, 9–10, 12, 14–17, and 19–26), one NQ–Phenazine derivative (structure 56), and four of the NQ–Triazole derivatives (structures 63–65 and 67), all of which presented better activity than the reference drug. All of the NQ–Amine derivatives presented lower anti-L. infantum activity than the two reference drugs.
The toxicity values (LogIC50) predicted by QSAR Equation (4) for the 68 naphthoquinone derivatives are shown in Figure 5d. A total of 64 molecules presented a reliable predicted activity, while 4 of them (structures 41–42, 53 and 54) were outside of the influence parameter (h*). Among the 68 molecules evaluated, all of the structures belonging to the NQ–Phenazine family presented the lowest toxicity values.
2.2. Molecular Docking
Ligand–protein docking simulations were carried out to determine the most effective binding mode of each of the 68 1,4-naphthoquinone derivatives within the catalytic sites of the 5 chosen molecular targets (TcTR, TcLαD, LTR, LaR, and LiTA). These macromolecular receptors were selected due to their relevance in the processes of survival, metabolism, reproduction, and proliferation of the parasites T. cruzi, L. amazonensis, and L. infantum. Conformational flexibility was allowed in all rotational bonds of the ligand, while the protein was used as a rigid structure. The best poses were selected according to the MVD scoring function, which helped to elucidate the electronic and structural aspects of the binding mode of the ligands in the active site of each protein.
2.2.1. Docking in Trypanothione Reductase and Lanosterol α-Demethylase Proteins of T. Cruzi
For the TcTR protein a 496 Å3 cavity was used, while for the TcLαD protein, a 481 Å3 cavity was used (Figure S1). The results of the ligand–protein coupling are shown in Figure 6a,b, where the 68 1,4-naphthoquinone derivatives show a good interaction (from −128.75 to −79.68 kcal/mol) with the two selected molecular targets. In this study, as the interaction energy decreases, the affinity of the compounds with the enzyme improves.
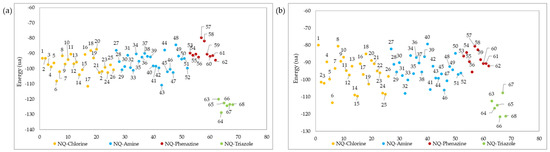
Figure 6.
Ligand–protein docking energies of 1,4-naphthoquinone derivatives against (a) TcTR and (b) TcLαD receptors.
As shown in Figure 6a, the triazole-fused naphthoquinone derivatives had better interaction energy (from −128.75 to −120.03 kcal/mol) with the active site of the TcTR. Among these, NQ–Triazole structure 64 had the best affinity, presenting 4 hydrogen bonds: 1 with the Asn 340 A residue, another with the Arg 355 A residue, and 1 with the Gly 459 B residue (Figure 7a), with the latter 2 not present in its triazole analogs; as well as steric interactions with the amino acids His 461, Thr 335, Ile 339, Glu 466, Glu 19, Pro 336, and Ser 470. Chacón et al. [85] also reported that the hydrogen bond interaction with the Gly 459 residue provided the best affinity energy to the quinoxaline derivative of the group they evaluated; and reported for this compound the same hydrophobic interactions. Similarly, the natural substrate co-crystallized in the active site of the TcTR protein presented interactions with residues Glu 19 A, Gly 459 B, Pro 336 A, Ile 339 A, and His 461 B [86]. NQ–Triazole 66 presented an energy close to that of molecule 64, and in addition to the same hydrophobic interactions, it presented unions with the amino acids Leu 18 A, Tyr 111 A, and Val 54, which are also part of the TcTR-trypanothione union (Figure 7b). The only hydrogen bond that this derivative presents together with the other NQ–triazole molecules is with the Ser 15 A residue, which is a solvent-mediated hydrogen bond interaction [86]. Note that this interaction occurs only in this series of compounds. Table S17 in the Supporting Materials shows the interaction energy of each ligand, as well as the hydrogen bonds with the different amino acid residues of the active site of the TcTR and TcLαD proteins.
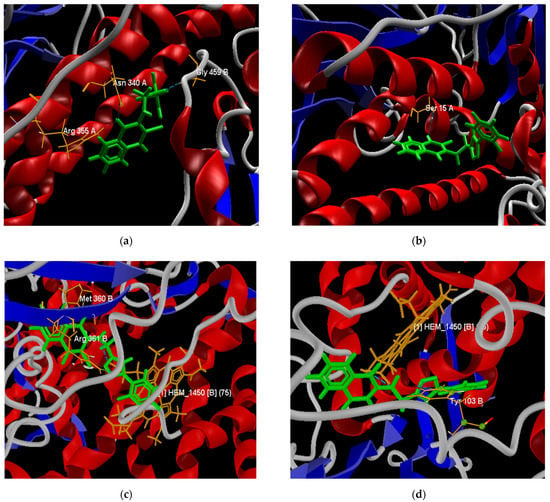
Figure 7.
Best poses of derivatives 64 (a) and 66 (b) in the active site of the TcTR protein. Best poses of derivatives 66 (c) and 68 (d) in the active site of the TcLαD protein.
NQ–Chlorine derivatives 6 (−108.09 kcal/mol), and 17 (−111.59 kcal/mol), as well as NQ–Amine derivative 43 (−110.87 kcal/mol), also presented high ligand–protein interaction energies. Although these compounds have hydrogen bonds in the TcTR active site (Figure 7c), the strength of these hydrogen bonds is comparable to those in compounds that have the least favored interaction energies, i.e., NQ–Phenazine structures 57 (−79.68 kcal/mol) and 58 (−81.93 kcal/mol). From the above, it follows that the compounds 6, 17, and 43 achieve their highest affinity and stability through other types of interactions.
The molecular docking evaluation against the TcLαD protein showed that molecule 66 was again the ligand with the best affinity (−121.60 kcal/mol), followed by molecule 68 (−121.18 kcal/mol). Both structures belong to the family of triazoles substituted with fluorine atoms in the terminal aromatic ring, in this case, in the meta and ortho-para positions, respectively. Derivative 66 presents three hydrogen bonds between the three nitrogen atoms of the triazole ring with the donor oxygen atom of the Tyr 103 (B) residue; this type of interaction was also found in the binding site of Fluconazole and Posaconazole with a single hydrogen bond [87]. Ligand 66 also exhibits hydrophobic interactions with residues Tyr 116 (B), Ala 291 (B), Ala 287 (B), Met 460 (B), Phe 290 (B), Met 106 (B), and Met 40 (B). A capture the pose of compound 66 at the active site of the TcLαD target is presented in Figure 7c. Structure 68 presents two hydrogen bonds, the strongest with the iron protoporphyrin IX enzymatic cofactor (HEM_1450 or Heme) co-crystallized in the amino acid chain B, and the other with the Tyr 103 residue (B) (Figure 7d), which is consistent with the interactions reported for this receptor with Fluconazole and Posaconazole [87]. The steric interactions that stabilize ligand 68, also present in the two azoles mentioned [87], occur with residues Ala 291 (B), Ala 287 (B), Tyr 116 (B), and Phe 110 (B). Cardoso et al. reported a hydrogen bond between their furan–naphthoquinones with the Tyr 116 residue in the binding site of this protein [88]; however, for molecules 66 and 68, the interactions with this residue are of the steric type. These same authors report cation-π interactions with the Fe of the Heme group, which were not observed in any 1,4-naphthoquinone derivative tested here.
Other structures that presented favorable interaction energies with the active site of the TcLαD protein were the NQ–Amine derivatives 32 (−107.97 kcal/mol), 41 (−105.71 kcal/mol), and 46 (−106.12 kcal/mol), and the NQ–Chlorine derivatives 6 (−113.43 kcal/mol), 14 (−108.92 kcal/mol), 15 (−109.63 kcal/mol), 24 (−107.88 kcal/mol), and 25 (−108.50 kcal/mol). Among these, the lowest energy was found for the derivative substituted with an ethoxide group in the meta position of the phenylamino aromatic ring (structure 6), which did not present hydrogen bond interactions. The other 30 molecules did not present formation of hydrogen bonds with any amino acid residue of the active site.
2.2.2. Docking in Trypanothione Reductase, Arginase, and Aminotransferase Proteins of Leismania Genus
The docking evaluation of the 68 1,4-naphptoquinone derivatives in the catalytic sites of the proteins trypanothione reductase (LTr), arginase (LaA), and aminotransfera (LiAT) was carried out using cavities of 705, 305, and 1171 Å3, respectively (Figure S2).
The ligand–protein interaction energies for the best pose of each of the 68 derivatives evaluated are shown in Figure 8. In all cases, the most effective interactions against the three macromolecular receptors of the Leishmania genus were obtained for derivatives belonging to the NQ–triazoles. Molecules 65, 66, and 67 showed the best interaction energies (−158,024, −121,516, and −121,426 kcal/mol) against LTr, LaA, and LiAt active sites, respectively, while derivative 64 showed the second-best interaction energy (−155,221, −116,708, and −119,504 kcal/mol) in all three docking studies. Only in the case of the docking evaluation against the LaA site (Figure 8b) did derivative 17, belonging to the NQ–Chlorine family, and derivatives 32, 33, 40, 41, and 43 of the NQ–amino family, show a better affinity interaction than member 63 of the NQ–triazole family.
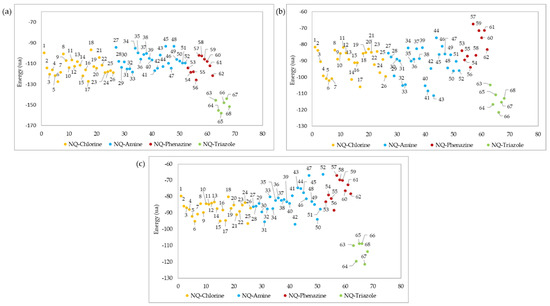
These results conform with some studies of drugs used to combat parasitic diseases based on triazoles or azoles. These compounds lead to alterations in the mitochondria and accumulation of lipid bodies, thus interfering with the biosynthesis of the cell membrane [89], and leading to cell death of the parasite [90,91]. Thus, triazole-substituted naphthoquinone derivatives have potential antiparasitic activity against Leishmania, since they have also been shown to be a type of compound tolerable by patients [89].
The two best poses adopted within the active site for each docking evaluation are presented in Figure 9. In the case of the poses of derivatives 64 and 65 against the LTr protein (Figure 9a,b), both present interactions by hydrogen bonds with the amino acid residues Ser 14, Thr 335, Cys 52, and Lys 60 of LTr. The two cysteine residues (Cys52 and Cys57) present in the active site form the disulfide bond in the oxidized form of the protein [92], which is a bond critical in the parasite’s defense mechanism, while the interactions with the residues Thr 335 and Lys 60 are destined exclusively to the binding domain of FAD [93]. In this way, the binding of the ligands to these last residues prevents the binding of FAD and its orientation toward the active site during the reduction, which inhibits the LTr enzyme [93].
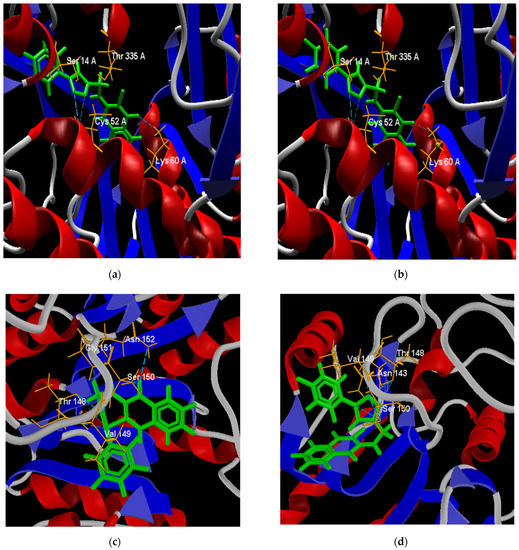
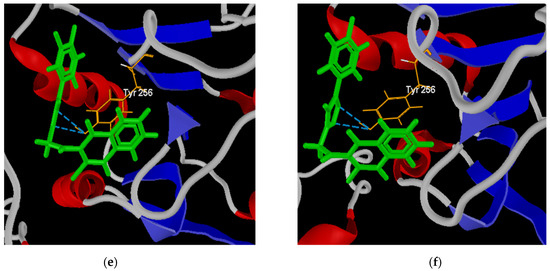
Figure 9.
Best poses of derivatives: 64 (a) and 65 (b) in the active site of the LTr protein; 64 (c) and 66 (d) in the active site of the LaA protein; and 64 (e) and 67 (f) in the active site of the LiAt protein.
The best positions of structures 64 and 66 in the active site of the LaA protein (Figure 9c,d) presented both interactions by hydrogen bonds with residues Ser 150, Thr 148, and Val 149. Structure 64 presented hydrogen bonds with residues Gly151 and Asn 152, while structure 66 formed an interaction with residue Asn 143. In similar studies [94,95], some flavonoid ligands showed interactions with amino acid residues Ser150, Asn 152, and Asn153 within the active site, exhibited high in vitro activity against L. amazonensis cultures, and low toxicity in mammalian cells. Residues Ser150 and Asn143 are involved in the Mn(II) metal bridge in coordination with the active site of arginase, which is required to conduct its catalytic activity [95,96]. According to this, molecules 64 and 66 inhibit the coupling of the metallic bridge, and would be good inhibitors of the arginase protein.
The poses of structures 64 and 67 facing the LiAt receptor are presented in Figure 9e,f. Both structures present interaction by hydrogen bonds with the residue Tyr 256. It has been shown that this residue prevents the rotation of the pyridine ring of the pyridoxal phosphate cofactor until the entrance of the amino acid, avoiding its stability [97].
2.3. ADME Analysis
Theoretical early estimation of ADME properties for the series of 68 1,4-naphthoquinone derivatives was performed in the free SwissADME web tool [98]. The data of lipophilicity, solubility, pharmacokinetic properties, and skin permeability parameters are collected in Table S21.
Figure 10 shows the estimated lipophilicity values of the 68 ligands, expressed as the logarithm of the octanol–water partition coefficient (Log Po/w). This parameter makes it possible to determine hydrophobicity, which is a parameter considered in the initial phases of drug development, and which allows inferring how the compound will behave in biological fluids and its possible diffusion through biological membranes [77,99]. The lipophilic results show that the 68 derivatives have a Log p < 5 [100], so according to the Lipinski Rules, they can be administered orally [101]. NQ–Amine derivative 43 presented the lowest value (log Po/w = 0.49), that is, it is more hydrophilic than the rest.
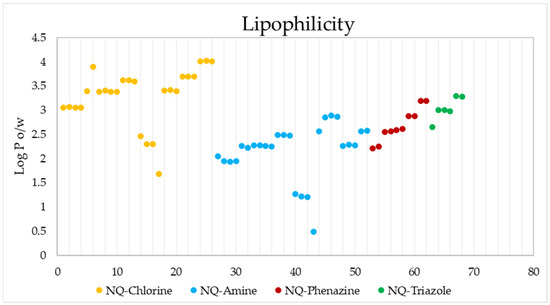
Figure 10.
Lipophilicity values of the 68 naphthoquinone derivatives.
Figure 11 shows the SWISSADME solubility values (expressed as log S) of the 68 derivatives according to the ESOL (Estimated SOLubility), solubility adapted by Ali et.al., and SILICO-IT [102] methods. The ESOL method estimates the solubility in water directly from the molecular structure, followed by its weight [103]; the Ali method incorporates the effect of the topological polar surface area [104]; and the SILICO-IT method is calculated using a fragmented procedure [102]. The solubility (Log S) scales used are insoluble < −10, poor < −6, moderate < −4, soluble < −2, and high < 0. The calculated solubility data presented ESOL values of −5.42 < Log S < −3.57, Ali values of −7.04 < Log S < −3.9 and SILICO-IT values of −8.33 < Log S < −4.28, indicating that all 68 compounds have moderate to poor water solubility.
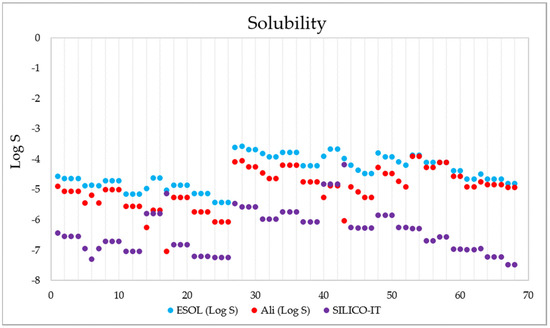
Figure 11.
Solubility data (in Log S) calculated for the 68 1,4-naphthoquinone derivatives using the ESOL (blue), Ali-adapted (orange), and SILICO-IT (magenta) methods.
The evaluation of pharmacokinetic parameters of gastrointestinal (GI) absorption, P-gp substrate, and cytochrome P450 (CYP) inhibition or interaction for all 68 derivatives is presented in Figure 12.
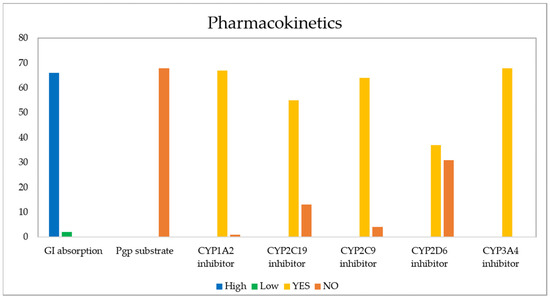
Figure 12.
Pharmacokinetic properties calculated for the 68 naphthoquinone derivatives.
The GI barrier has a complex structure given the characteristics of a semi-permeable membrane, which allows fat-soluble molecules to penetrate it through a diffusion process, as is the case with most drugs [105]. As Figure 13 shows, passive human gastrointestinal (GI) absorption data estimate that 97% of the 68 compounds present high absorption in the digestive tract, while the remaining 3%, corresponding to structures 17 and 43, would present low intestinal absorption. For its part, P-glycoprotein (PGP) is a permeability protein that pumps substances out of the body and prevents their absorption, which causes drug resistance to form [105,106]. It was found that none of the compounds behaved as a substrate for P-gp; therefore, they could conduct the function without any resistance [107].
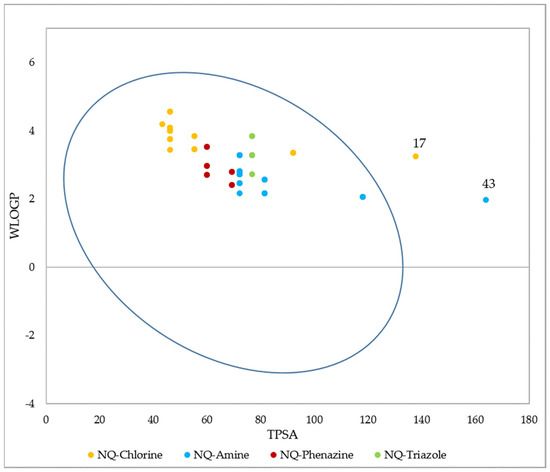
Figure 13.
BOILED-Egg (adapted with permission from Ref. [108]) evaluation of gastrointestinal absorption for the 68 1,4-naphthoquinone derivatives. Molecules inside the oval indicate that they have GI permeability.
Another form of drug administration is through the skin (transdermal distribution), which allows the transport of substances through the epidermis. This parameter is related to the skin permeability coefficient (Kp), whose values indicate that the more negative the log Kp (with Kp in cm/s), the less permeant the molecule is [109,110]. Figure 14 shows the skin permeability of the 68 evaluated compounds, where it is evident that the best candidates to be administered transdermally are NQ–Chloro derivatives; among them, the best are 25, 26, and 27, while the ligand with the lowest permeability is 43.
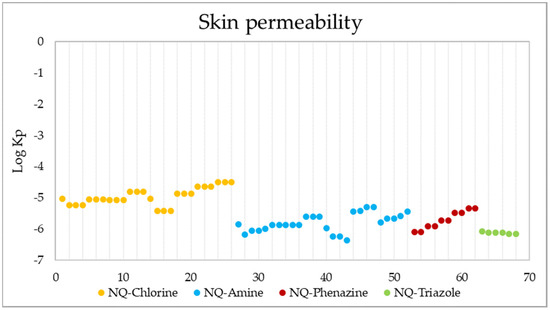
Figure 14.
Skin permeability values in Log Kp (Kp in cm/s) of the 68 derivatives.
The main enzyme isoforms CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4 [98] are involved in drug metabolism, and inhibitors block their metabolic activity in a dose-dependent manner. Structure 43 was found to be the only derivative unable to inhibit CYP1A2, while 13 derivatives (36–38, 40–43, and 54–58), 4 derivatives (35–37, and 57), and 31 derivatives (11–16, 24–30, 40–43, 55–68) projected a negative inhibitory response of CYP2C19, CYP2C9, and CYP2D6, respectively. In each of these cases, the activity of the remaining molecules would be affected by their metabolism, due to their ability to bind to these enzymes, which may generate adverse effects such as high toxicity [111,112,113]. None of the 68 molecules reported the inhibition of CYP3A4.
Based on a similarity to drugs, filters based on Lipinski (Pfizer) [101], Ghose (Amgen) [114], Veber (GSK) [115], Egan (Pharmacia) [116], and Muegge (Bayer) [117] rules were also evaluated, as well as the bioavailability score. These filters qualitatively define the feasibility of a compound to become an oral drug candidate. The results show that 100% of the 1,4-naphthoquinone derivatives comply with the Lipinski and Ghose rules, and 98.5% comply with the Veber and Egan rules. For these last two cases, molecule 43 presented a violation, caused by a value of TPSA = 163.83 Å2 that is above the established ranges (TPSA ≤ 140 and TPSA ≤ 131.6, respectively). Finally, the Muegge filter indicated that 90% of the derivatives present valid conditions to become oral drugs. The remaining 10% presented a violation since their XLOGP value is outside of the allowed range (−2 ≤ XLOGP ≤ 5) (structures 21–26), while the TPSA value of structure 43 is again above the range settled down. All compounds had a bioavailability value of 0.55, indicating oral bioavailability based on Lipinski’s rules. Additionally, it was established that an AB score of 0.55 is required to be considered a sufficiently absorbable molecule orally [118].
3. Materials and Methods
3.1. QSAR Modelling
3.1.1. In Vitro Anti-Chagas and Anti-Leishmaniasis Data
All of the in vitro data used for constructing of the antiparasitic and toxicity QSAR models were taken from the literature. For constructing the anti-chagasic activity model, 153 structures derived from naphthoquinone were used, some of which were fused with triazoles and oxanes, among other heterocycles (Figure A1). All of these derivatives showed anti-T. cruzi activity, evaluated in vitro under the following experimental conditions. The stock solutions of the compounds were prepared in DMSO. Strain Y trypomastigotes were obtained at the peak of parasitemia from albino mice, then isolated through differential centrifugation, and resuspended in Dulbecco’s Modified Eagle Medium (DME), with a concentration of 107 parasite cells/mL in the presence of 10% mouse blood. A total of 100 µL of this solution was added to 100 µL of the compound solutions. The cell count was determined in a Neubauer chamber, and the trypanocidal activity was expressed as IC50, which corresponds to a concentration that allows the lysis of 50% of the parasites [23,24,25,26,27,28,29,30,31].
For the QSAR modeling of cutaneous anti-leishmaniasis (L. amazonensis) activity, 60 molecules (Figure A2) containing functional groups such as quinone, triazole, indole, and amine with high trypanocidal activity reported as IC50 were used. The anti-leishmanial activity of these structures was evaluated by tests with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT) bromide. In these bioassays, promastigotes were seeded in RPMI medium supplemented with 10% FBS, and cultured in a 96-well plate with a concentration of 10 5 cells/plate at 37 °C. After the seeding period, the MTT solution was added, the formed complex was dissolved in DMSO, the supernatant was removed, and the cells were incubated under the same seeding conditions in the presence of various concentrations of the compounds. The concentration that inhibited 50% of parasite growth was determined as IC50 by linear regression [18,32,33,34,35,36,37,38,39].
For constructing the QSAR model of anti-visceral leishmaniasis (L. infantum) activity, 90 molecules (Figure A3) derived from quinone, triazole, and indole with activity reported as IC50 were used. The anti-leishmanial activity evaluation for these molecules was conducted using incubation in RPMI medium with 12% fetal calf serum (FBS), at a concentration of 105 cells/mL, for 48 h at 25 °C. After the incubation process, promastigote growth was estimated by counting the parasites with a Neubauer hemocytometer. The 50% inhibitory concentration (IC50) was defined as the drug concentration required to inhibit 50% of the parasite growth [40,41,42,43,44,45].
The QSAR model of toxicity was built from 76 structures (Figure A4) derived from naphthoquinone, some fused with triazoles, pyrazoles, imidazoles, and aromatic rings, with experimental data measured according to the following parameters. Mouse fibroblasts (L929) were used and determined by the reduction of 3-(4,5-dimethyl-2-thiazol)-2,5-diphenyl-2H-tetrazolium bromide (MTT), expressed as IC50 [30,46,47,48,49,50,51].
3.1.2. Development of Antiprotozoal QSAR Models
All molecules were processed in MDL mol (V2000) format using the ACD/I-Lab software version 11.0 13 [119]. The calculation of 83,180 fingerprint-type, one-dimensional, and two-dimensional molecular descriptors was performed in the programs PaDEL-Descriptor [54], Mold2 [120], QuBiLS-MAS [56], and Fragmentor [121] (all free access). By using the balanced subsets method, the data sets were divided into calibration, validation, and test groups, using 70, 15, and 15%, for the anti-chagas activity model, 80%, 10%, and 10% for the anti-leishmanial activity models, and 70, 15, and 15% for toxicity, respectively. Subsequently, the Replacement Method (RM) [122], which is available in the MatLab programming language, was applied to explore the set of descriptors. RM is an unequivocal algorithm based on multivariable linear regression (MLR), which searches for the best subsets of descriptors in large databases. By using the RM algorithm, multivariable regression models of up to seven descriptors were generated for each model, which was carried out in the MATLAB software version 7.12.0 [123].
3.1.3. Validation of the QSAR Models
Validation of all QSAR models was performed through both internal and external validation processes. Internal validation was performed using the LOO (Leave-One-Out) validation method. For the internal validation of each QSAR model, the statistical parameters R2 LOO (square correlation coefficient of LOO) and SLOO (standard deviation of LOO) were determined. Additionally, the statistical robustness of the models was determined using randomization Y [124,125]. For its part, the external validation verified the predictive capacity, applying the methodology proposed by Golbraikh and Tropsha [78], where the correlation coefficients between the predicted and experimental properties of the compounds in the test set are estimated.
3.2. Molecular Docking
The structures of the macromolecular targets used for molecular docking were extracted from the Protein Data Bank (PDB). In the case of T. cruzi, trypanothione reductase (TcTR) [86] and lanosterol α-demethylase (TcLαD) [87] proteins were selected, with 1BZL and 2WUZ coding and a resolution of 2.40 Å and 2.45 Å, respectively. For the Leishmania genus in general, the protein trypanothione reductase (LTR) [126,127] with code 2JK6 and a resolution of 2.95 Å was selected, while for the strains L. amazonensis and L. infantum arginase (LaA) [128] and tyrosine aminotransferase (LiTA) [129] were selected, with codes 4IU0 and 4IX8 and resolutions of 1.77 Å and 2.35 Å, respectively.
Before the molecular docking simulations, the proteins were prepared in the Molegro Virtual Docker (MVD) program [130], where bond assignment, bond order, hybridization, missing hydrogens, charge assignment, and atom tripos were performed. To validate the software and optimize the molecular docking parameters, a re-docking procedure of the co-crystallized ligands was carried out. For each protein, a cavity was created by delimiting the coupling space of the co-crystallized ligand, and the best pose (Figure 15) was selected based on the lowest RMSD value (<2) [131].
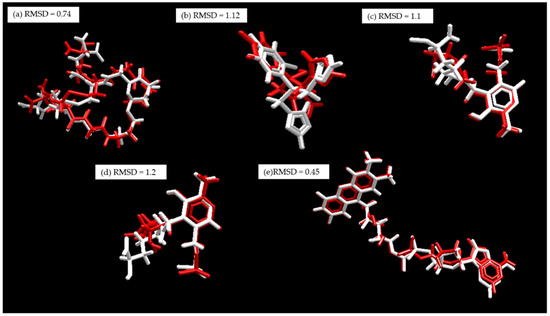
Figure 15.
Re-coupling, white: co-crystallized substrate. Red: Substrate coupled using MVD. (a) TcTR; (b) TcLαD, (c) LiTA, (d) LaA, and (e) LTR.
For the preparation of the ligands, the structures of the naphthoquinone derivatives (68) were built in the ACD-Labs program, and saved as .mol files. These structures were then optimized in the Gaussian 09W program [132] by simultaneously relaxing all geometric parameters to the theoretical level B3LYP/6–31 + G(d).
For the molecular docking simulations, 5 poses (conformation and orientation) were generated for each of the 68 ligands evaluated, using the search algorithm MolDock Optimizer, which was executed with 10 repetitions. The data analysis was performed in the Molegro Virtual Modeller (MVM) program, where the best pose in each case was selected, and the data were plotted based on the energy calculated using the scoring function (Equation (5)).
where is the intermolecular interaction energy of the ligand–protein, and the is the internal energy of the ligand. The is shown in Equation (6):
where the EPLP term represents the PLP (piecewise linear potential) energy, which consists of the use of two different parameter sets, described as follows: one for approximation of the steric term (van der Waals) among atoms, and the other potential for the hydrogen binding. The second term () is related to the electrostatic interactions among overloaded atoms. It is a Coulomb potential with a dielectric constant dependent on the distance (D(r) = 4r). The numerical value of 332.0 is responsible for the electrostatic energy unit to be given in kilocalories per mol. The qi and qj terms represent the charges of the atoms i and j, respectively. The rij term indicates the interatomic distance between the atoms i and j [133].
The is shown in Equation (7).
The first part of the equation (double summation) is among all pairs of atoms in the ligand, taking off those which are connected by two bonds. The second term characterizes the torsional energy, where θ is the torsional angle of the bond and θ0 is its corresponding value in the equilibrium. The average of the torsional energy bond contribution is used if several torsions could be determined. The last term, Eclash, assigns a penalty of 1.000 if the distance between two heavy atoms (more than two bonds apart) is shorter than 2.0 Å, not considering infeasible ligand conformations [134]. The docking search algorithm that is applied in the MVD program considers an evolutionary algorithm, the interactive optimization techniques which are inspired by Darwinian evolution theory, and a new hybrid search algorithm called guided differential evolution. This hybrid combines the differential evolution optimization technique with a cavity prediction algorithm during the search process, thereby allowing for fast and accurate identification of potential binding modes (poses) [133,134,135].
3.3. ADME Analysis
The ADME study was conducted using the SWISSADME web tool of the Swiss Institute of Bioinformatics [98]. This free web tool allows you to calculate properties, such as lipophilicity, water solubility, pharmacokinetics, and drug similarity, based on the structure or SMILE of the molecule.
4. Conclusions
A total of four QSAR models based on naphthoquinone derivative structures were developed and validated, three of them for antiprotozoal activity against T. cruzi, L. amazonensis, and L. infantum, and one for toxicity prediction. All QSAR models were built using in vitro inhibitory concentrations (IC50) which were previously reported. The anti-T. cruzi model was developed based on 153 molecules, the anti-L. amazonensis used 60 molecules, the anti-L. infantum model used 90 structures, and the toxicity model was built with 76. According to the anti-T. cruzi QSAR model, the anti-chagasic activity is favored by the refractivity, electronegativity, and polarizability of the molecules. These characteristics possibly give them a better binding capacity and inhibition of essential macromolecules in the metabolism of the parasite. Additionally, it was found that the presence of aromatic fragments in these structures increases their antiparasitic activity. Of the proposed molecules, structures 61 and 62, belonging to the derivatives fused with phenazine, and 24, 25, and 54, which are phenylamino derivatives, presented a better activity against T. cruzi predicted by the QSAR model compared to that of the reference drug. The QSAR anti-L. amazonensis predicts that molecules with less symmetry and with the presence of secondary and tertiary carbons will be more active. Thus, phenazines 62, 60, 59, 61, 58, and 57 showed a predicted potential activity against L. amazonensis, as well as derivatives 56, 27, and 26 of the phenylamine family. A total of 22 structures showed better predicted activity than Miltefosine, and 37 demonstrated better results than Glucantime. According to the anti-L. infantum QSAR model, the activity is a function of LogP, which is an essential characteristic for ligands to be considered drugs, since it gives them the ability to permeate biological membranes. Similarly, the activity increases with PSA, which is related to the penetration of the blood–brain barrier and its intestinal absorption. The derivatives with the best predicted anti-L. infantum activity were 6, 15, 7, 14, and 16, which belong to the phenylamino family. Additionally, it is highlighted that 23 molecules were more active than the reference drugs (Miltefosine and Glucantime).
According to the results of molecular docking, all the evaluated compounds present very favorable interaction energies with the selected receptors, which are essential in the metabolism of both T. cruzi and Leishmania. In the TcTR protein, the derivatives fused with triazole stand out. The best of these was structure 64, which, unlike its analogs, features two hydrogen bonds between the triazole ring and the Gly 459 residue. Similarly, the triazole derivatives presented the best interaction energies for the TcLαD protein. The best of them was structure 66. This derivative presents three interactions of hydrogen bonds between the triazole ring and the Tyr 103 residue, which is a fundamental residue in the active site of said receptor. The triazole derivatives 65, 66, and 67 presented the best interaction affinities against the catalytic sites of all leishmania proteins evaluated (Trypanothione reductase, Arginase, and Tyrosine Aminotransferase). These types of derivatives have a wide variety of biological activities, and according to our results, they are considered potent inhibitors of macromolecular targets. Ligands belonging to this type of molecules are currently used as active principles of antiparasitic drugs because they generate cell deformation and alterations in the mitochondria, thus interfering with the biosynthesis of the parasite’s cell membrane, causing cell death.
The results of ADME showed that the 68 evaluated compounds met the requirements to be administered orally. ADME calculations included lipophilicity, solubility, pharmacokinetics, and drug-likeness.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ph15060687/s1, Figure S1: Cavities in the active site of TcTR (a) and TcLαD (b) proteins; Figure S2: Cavities in the active site of LTR (a). LaA (b). and LiTA (c) proteins; Table S1: Antiparasitic QSAR models for chagas and leishmaniasis protozoa based on naphthoquinone derivatives obtained by the replacement method; Table S2: Toxicity QSAR models based on naphthoquinone derivatives obtained by the replacement method; Table S3: Intercorrelation matrix of the QSAR model descriptors for anti-chagas activity; Table S4: Numerical values of the QSAR anti-chagas model descriptors; Table S5: Experimental and predicted values of anti-chagas activity. Residuals and influence values h (h^lim = 0.196) are reported. ^ validation group and * test group; Table S6: Values predicted by the QSAR model of anti-chagas activity of the 68 naphthoquinone derivatives (prediction group); Table S7: Intercorrelation matrix of the QSAR model descriptors for anti-leishmanial activity; Table S8: Numerical values of the QSAR anti-L. amazonensis model descriptors; Table S9: Numerical values of the QSAR anti-L. infantum model descriptors; Table S10: Experimental and predicted values of anti-L. amazonensis activity. Residuals and influence values h (h^lim = 0.3125) are reported. ^ validation group and * test group; Table S11: Experimental and predicted values of anti-L. infantum activity. Residuals and influence values h (h^lim = 0.24) are reported. ^ validation group and * test group; Table S12: Values predicted by the QSAR models of anti-leishmanial activity of the 68 naphthoquinone derivatives (prediction group); Table S13: Intercorrelation matrix of the QSAR model descriptors for toxicity; Table S12: Numerical values of the QSAR toxicity model descriptors Table S14. Numerical values of the QSAR toxicity model descriptors; Table S15: Experimental and predicted values of toxicity. Residuals and influence values h (h^lim = 0.3125) are reported. ^ validation group and * test group; Table S16: Values predicted by the QSAR model of toxicity of the 68 naphthoquinone derivatives (prediction group); Table S17: Results of the coupling of naphthoquinone derivatives in the active site of the TcTR and TcLαD proteins. Table S18: Results of the docking of naphthoquinone derivatives in the active site of the LTR protein; Table S19: Results of the docking of naphthoquinone derivatives in the active site of the LaA protein; Table S20: Results of the docking of naphthoquinone derivatives in the active site of the LiTA protein; Table S21: ADME parameters.
Author Contributions
Conceptualization, J.A.G.C., D.L.N.G. and A.C.-C.; methodology, L.S.P.C., K.A.A.S., J.A.G.C. and D.L.N.G.; software, L.S.P.C., K.A.A.S., J.A.G.C., D.L.N.G. and P.R.D.; validation, L.S.P.C., K.A.A.S., D.L.N.G. and P.R.D.; formal analysis, L.S.P.C., K.A.A.S., J.A.G.C., D.L.N.G. and P.R.D.; investigation, L.S.P.C., K.A.A.S. and D.L.N.G.; resources, J.A.G.C.; data curation, L.S.P.C., K.A.A.S. and D.L.N.G.; writing—original draft preparation, L.S.P.C., K.A.A.S. and J.A.G.C.; writing—review and editing, L.S.P.C., K.A.A.S., J.A.G.C., D.L.N.G., P.R.D., A.C.-C. and W.E.R.N.; visualization, L.S.P.C., K.A.A.S. and J.A.G.C.; supervision, J.A.G.C.; project administration, J.A.G.C.; funding acquisition, J.A.G.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Universidad Pedagógica y Tecnológica de Colombia (UPTC) through projects SGI2033, SGI2949, SGI3038, and SGI3188 of the Dirección de Investigaciones (DIN).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article and Supplementary Material.
Acknowledgments
The authors greatly acknowledge the financial support provided by the Universidad Pedagógica y Tecnológica de Colombia (UPTC) and Ministerio de Ciencia, Tecnología e Innovación de Colombia (Minciencias). K.A.A.S. especially thanks the Ministerio de Ciencia, Tecnología e Innovación de Colombia (Minciencias) for awarding a Young Researcher Scholarship through call 891 of 2020. P.R.D. gives thanks to Ministerio de Ciencia, Tecnología e Innovacion Productiva de Argentina for the electronic library facilities. P.R.D. is a member of the scientific researcher career of CONICET.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Appendix A

Figure A1.
Molecules with reported biological activity (IC50) used for the development of the QSAR anti-T. cruzi.

Figure A2.
Molecules with reported biological activity used for the development of the anti-L. amazonensis QSAR model.

Figure A3.
Molecules with reported biological activity used for the development of the anti-L. infantum QSAR model.

Figure A4.
Molecules with reported toxicity in mouse fibroblasts (L929) used for the development of the QSAR model of toxicity.
References
- Enfermedad de Chagas—OPS/OMS. Available online: https://www.paho.org/es/temas/enfermedad-chagas (accessed on 5 April 2022).
- Dos Santos Naujorks, A.A.; Da Silva, A.O.; Da Silva Lopez, R.; De Albuquerque, S.; Beatriz, A.; Marquez, M.R.; Pires De Lima, D. Novel naphthoquinone derivatives and evaluation of trypanocidal and leishmanicidal activities. J. Mater. Chem. B 2018, 6, 1–3. [Google Scholar] [CrossRef]
- Alonso-Padilla, J.; Rodríguez, A. High Throughput Screening for Anti–Trypanosoma cruzi Drug Discovery. PLoS Negl. Trop. Dis. 2014, 8, e3259. [Google Scholar] [CrossRef] [PubMed]
- Coura, J.R. Present situation and new strategies for Chagas disease chemotherapy: A proposal. Mem. Inst. Oswaldo Cruz 2009, 104, 549–554. [Google Scholar] [CrossRef]
- Schmunis, G.A.; Yadon, Z.E. Chagas disease: A new worldwide challenge. Acta Trop. 2010, 115, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Arias, D.G.; Garay, A.S.; Rodrigues, D.; Forastieri, P.S.; Luna, L.E.; Bürgui, M.D.L.M.; Prieta, C.; Iglesia, A.A.; Cravero, R.M.; Guerrero, S.A. Rational design of nitrofuran derivatives: Synthesis and valuation as inhibitors of Trypanosoma cruzi trypanothione reductase. Eur. J. Med. Chem. 2017, 125, 1088–1097. [Google Scholar] [CrossRef] [PubMed]
- Urbina, J.A. Ergosterol biosynthesis and drug development for Chagas disease. Mem. Inst. Oswaldo Cruz 2009, 104, 311–318. [Google Scholar] [CrossRef]
- Valdez, R.H.; Düsman Tonin, L.T.; Ueda-Nakamura, T.; Dias Filho, B.P.; Monrgado-Diaz, J.A.; Sarragitto, M.H.; Vataru Nakamura, C. Biological activity of 1,2,3,4-tetrahydro-β-carboline-3-carboxamides against Trypanosoma cruzi. Acta Trop. 2009, 110, 7–14. [Google Scholar] [CrossRef]
- De Souza, E.M.; Lansiaux, A.; Bailly, C.; Wilson, W.D.; Hu, Q.; Boykin, D.W.; Batista, M.M.; Araújo-Jorge, T.C.; Soerio, M.N.C. Phenyl substitution of furamidine markedly potentiates its anti-parasitic activity against Trypanosoma cruzi and Leishmania amazonensis. Biochem. Pharmacol. 2004, 68, 593–600. [Google Scholar] [CrossRef]
- Rolim Neto, P.J.; Luiz Gomes, C.; Wanderley Sales, V.D.A.; de Melo, C.G.; Ferreira da Silva, R.M.; Vicente Nishimura, R.H.; Araújo, R. Beta-lapachone: Natural occurrence, physicochemical properties, biological activities, toxicity and synthesis. Phytochemistry 2021, 186, 112713. [Google Scholar] [CrossRef]
- Menna-Barreto, R.F.S.; Henriques-Pons, A.; Pinto, A.V.; Morgado-Diaz, J.A.; Soares, M.J.; De Castro, S.L. Effect of a β-lapachone-derived naphthoimidazole on Trypanosoma cruzi: Identification of target organelles. J. Antimicrob. Chemother. 2005, 56, 1034–1041. [Google Scholar] [CrossRef]
- Menna-Barreto, R.F.S.; Corrêa, J.R.; Pinto, A.V.; Soares, M.J.; De Castro, S.L. Mitochondrial disruption and DNA fragmentation in Trypanosoma cruzi induced by naphthoimidazoles synthesized from β-lapachone. Parasitol. Res. 2007, 101, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Menna-Barreto, R.F.S.; Correa, J.R.; Cascabulho, C.M.; Fernandes, M.C.; Pinto, A.V.; Soares, M.J.; De Castro, S.L. Naphthoimidazoles promote different death phenotypes in Trypanosoma cruzi. Parasitology 2009, 136, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.R.; Souza Guimaraes, A.; Do Nascimento, J.; Do Santo Nascimento, I.J.; Barbosa da Silva, E.; McKerrow, J.H.; Cardoso, S.H.; da Silva-Júnior, E.F. Computer-aided design of 1,4-naphthoquinone-based inhibitors targeting cruzain and rhodesain cysteine proteases. Bioorg. Med. Chem. 2021, 41, 116213. [Google Scholar] [CrossRef] [PubMed]
- Ramos, E.I.; Garza, K.M.; Krauth-Siegel, R.L.; Bader, J.; Martinez, L.E.; Maldonado, R.A. 2,3-Diphenyl-1,4-Naphthoquinone: A potential chemotherapeutic agent against trypanosoma cruzi. J. Parasitol. 2009, 95, 461–466. [Google Scholar] [CrossRef]
- Becerra, N.A.; Espinosa-Bustos, C.; Vázquez, K.; Rivera, G.; Paulino, M.; Cantero, J.; Nogueda, B.; Chacón-Vargas, F.; Castillo-Velazquez, U.; Rodríguez, A.F.E.; et al. Expanding the chemical space of aryloxy-naphthoquinones as potential anti-Chagasic agents: Synthesis and trypanosomicidal activity. Med. Chem. Res. 2021, 30, 2256–2265. [Google Scholar] [CrossRef]
- Da Cunha-Júnior, E.F.; Pacienza-Lima, W.; Alves Ribeiro, G.; Daher Netto, C.; Do Canto-Cavalheiro, M.M.; Monteiro da Silva, A.J.; Ribeiro Costa, P.R.; Rossi-Bergamann, B.; Torres-Santo, E.C. Effectiveness of the local or oral delivery of the novel naphthopterocarpanquinone LQB-118 against cutaneous leishmaniasis. J. Antimicrob. Chemother. 2011, 66, 1555–1559. [Google Scholar] [CrossRef]
- De Araújo, M.V.; De Souza, P.S.O.; De Queiroz, A.C.; Da Matta, B.; Brandao Leite, A.; Da Silva, A.E.; de Franca, J.A.A.; Silva, T.M.S.; Camara, C.A.; Alexandre-Moreira, M.S. Synthesis, leishmanicidal activity and theoretical evaluations of a series of substituted bis-2-Hydroxy-1,4-Naphthoquinones. Molecules 2014, 19, 15180–15195. [Google Scholar] [CrossRef]
- Guimarães, T.T.; Pinto, M.C.F.R.; Lanza, J.S.; Melo, M.N.; Do Monte-Neto, R.L.; De Melo, I.M.M.; Diogo, E.B.T.; Ferreira, V.F.; Camara, C.A.; Valenca, W.O.; et al. Potent naphthoquinones against antimony-sensitive and -resistant Leishmania parasites: Synthesis of novel α- And nor-α-lapachone-based 1,2,3-triazoles by copper-catalyzed azide-alkyne cycloaddition. Eur. J. Med. Chem. 2013, 63, 523–530. [Google Scholar] [CrossRef]
- Teixeira, M.J.; De Almeida, Y.M.; Viana, J.R.; Holanda Filha, J.G.; Rodrigues, T.P.; Prata, J.R., Jr.; Coelho, I.C.B.; Rao, V.S.; Pompeu, M.M.L. In vitro and in vivo leishmanicidal activity of 2-hydroxy-3-(3-methyl-2-butenyl)-1,4-naphthoquinone (lapachol). Phytother. Res. 2001, 15, 44–48. [Google Scholar] [CrossRef]
- Lima, N.M.F.; Corraia, C.S.; Leon, L.L.; Machado, G.M.C.; De Fátima Madeir, A.M.; Santana, G.A.E.; Goulart, M.O.F. Antileishmanial activity of lapachol analogues. Mem. Inst. Oswaldo Cruz 2004, 99, 757–761. [Google Scholar] [CrossRef]
- Pandey, R.K.; Kumbhar, B.V.; Sundar, S.; Kunwar, A.; Prajapati, V.K. Structure-based virtual screening, molecular docking, ADMET and molecular simulations to develop benzoxaborole analogs as potential inhibitor against Leishmania donovani trypanothione reductase. J. Recept. Signal Transduct. 2017, 37, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Park, S.Y.; Lee, H.J.; Suh, M.E.; Schollmeyer, D.; Lee, C.O. Synthesis and cytotoxicity of 6,11-Dihydro-pyrido- and 6,11-Dihydro-benzo [2,3-b]phenazine-6,11-dione derivatives. Bioorg. Med. Chem. 2003, 11, 1709–1714. [Google Scholar] [CrossRef]
- Da Silva Júnior, E.N.; Diogo, E.B.T.; Dias, G.G.; Rodrigues, B.L.; Guimarães, T.T.; Valenca, W.O.; Camara, C.A.; De Oliveira, M.R.N.; Camara, C.A.; De Oliveira, R.A.; et al. Synthesis and anti-Trypanosoma cruzi activity of Naphthoquinone-containing Triazoles: Electrochemical studies on the effects of the quinoidal moiety. Bioorg. Med. Chem. 2013, 21, 6337–6348. [Google Scholar] [CrossRef]
- Bahia, S.B.B.B.; Reis, W.J.; Jardim, G.A.M.; Souto, F.T.; De Simone, C.A.; Gatto, C.C.; Menna-Barreto, R.F.S.; De Castro, S.L.; Cavalvanti, B.C.; Pessoa, C.; et al. Molecular hybridization as a powerful tool towards multitarget quinoidal systems: Synthesis, trypanocidal and antitumor activities of naphthoquinone-based 5-iodo-1,4-disubstituted-, 1,4- and 1,5-disubstituted-1,2,3-triazoles. Med. Chem. Comm. 2016, 7, 1555–1563. [Google Scholar] [CrossRef]
- Pinto, A.V.; Pinto, C.N.; Pinto, M.D.C.F.R.; Rita, R.S.; Pezzella, C.A.C. Trypanocidal Activity of Synthetic Heterocyclic Derivatives of Active Quinones from Tabebuia sp. Arzneimittelforschung 1997, 47, 21045. [Google Scholar] [CrossRef]
- Jardim, G.A.M.; Silva, T.L.; Goulart, M.O.F.; De Simone, C.A.; Barbosa, J.M.C.; Salomão, K.; De Castro, S.L.; Bower, J.F.; Da Silva Júnio, R.E.N. Rhodium-catalyzed C-H bond activation for the synthesis of quinonoid compounds: Significant Anti-Trypanosoma cruzi activities and electrochemical studies of functionalized quinones. Eur. J. Med. Chem. 2017, 136, 406–419. [Google Scholar] [CrossRef]
- Ferreira, S.B.; Salomão, K.; De Carvalho da Silva, F.; Ventura Pinto, A.; Kaiser, C.R.; Pinto, A.C.; Ferreira, V.F.; De Castro, S.L. Synthesis and anti-Trypanosoma cruzi activity of β-lapachone analogues. Eur. J. Med. Chem. 2011, 46, 3071–3077. [Google Scholar] [CrossRef]
- Da Silva Júnior, E.N.; De Mel, I.M.M.; Diogo, E.B.T.; Costa, V.A.; De Souza Felho, J.D.; Valencia, W.O.; Camara, C.A.; De Oliveira, R.N.; De Araujo, A.S.; Emery, F.S.; et al. On the search for potential anti-Trypanosoma cruzi drugs: Synthesis and biological evaluation of 2-hydroxy-3-methylamino and 1, 2, 3-triazolic naphthoquinoidal compounds obtained by click chemistry reactions. Eur. J. Med. Chem. 2012, 52, 304–312. [Google Scholar] [CrossRef]
- Da Silva, E.N.; Menna-Barreto, R.F.S.; Pinto, M.C.F.R.; Silva, R.S.F.; Teixeira, D.V.; De Souza, C.B.V.; De Simone, C.A.; De Castro, S.L.; Ferreira, V.L.; Pinto, A.V. Naphthoquinoidal [1,2,3]-triazole, a new structural moiety active against Trypanosoma cruzi. Eur. J. Med. Chem. 2008, 43, 1774–1780. [Google Scholar] [CrossRef]
- Da Silva Júnior, E.N.; De Souza, M.C.; Fernandes, M.C.; Menna-Barreto, R.F.S.; Pinto, M.C.F.R.; De Assis Lopes, F.; de Simone, C.A.; Andrade, C.K.; Pinto, A.V.; Ferreira, V.F.; et al. Synthesis and anti-Trypanosoma cruzi activity of derivatives from nor-lapachones and lapachones. Bioorg. Med. Chem. 2008, 16, 5030–5038. [Google Scholar] [CrossRef]
- Ryu, C.K.; Kim, D.H. The antimicrobial activities of some 1,4-naphthalenediones (III). Arch. Pharmacal Res. 1993, 16, 161–163. [Google Scholar] [CrossRef]
- Salomão, K.; De Santana, N.A.; Molina, M.T.; De Castro, S.L.; Menna-Barreto, R.F.S. Trypanosoma cruzi mitochondrial swelling and membrane potential collapse as primary evidence of the mode of action of naphthoquinone analogues. BMC Microbiol. 2013, 13, 196. [Google Scholar] [CrossRef] [PubMed]
- Valderrama, J.A.; Benites, J.; Cortés, M.; Pessoa-Mahana, H.; Prina, E.; Fournet, A. Studies on quinones. Part 38: Synthesis and leishmanicidal activity of sesquiterpene 1,4-quinones. Bioorg. Med. Chem. 2003, 13, 4713–4718. [Google Scholar] [CrossRef] [PubMed]
- Valderrama, J.A.; Zamorano, C.; González, M.F.; Prina, E.; Fournet, A. Studies on quinones. Part 39: Synthesis and leishmanicidal activity of acylchloroquinones and hydroquinones. Bioorg. Med. Chem. 2005, 13, 4153–4159. [Google Scholar] [CrossRef]
- Da Silva, A.J.M.; Netto, C.D.; Pacienza-Lima, W.; Torres-Santo, E.C.; Rosii-Bergmann, B.; Maurel, S.; Valentin, A.; Costa, P.R.R. Antitumoral, Antileishmanial and Antimalarial Activity of Pentacyclic 1,4-Naphthoquinone Derivatives. J. Braz. Chem. Soc. 2009, 20, 176–182. [Google Scholar] [CrossRef]
- De Araújo, M.V.; David, C.C.; Netto, J.C.; De Oliveira, L.A.P.L.; Da Silva, K.C.J.; Dos Santos, J.M.; Da Silva, J.K.S.; De A Brandão, V.B.C.; Silva, T.M.S.; Camara, C.A.; et al. Evaluation on the leishmanicidal activity of 2-N,N′-dialkylamino-1,4-naphthoquinone derivatives. Exp. Parasitol. 2017, 176, 46–51. [Google Scholar] [CrossRef]
- Da Silva, P.R.; De Oliveira, J.F.; Da Silva, A.L.; Marques Queiroz, C.; de Lima, M.D.C.A.; Sampaio Feitosa, A.P.; Araújo Duarte, D.M.; Da Silva, A.C.; Brelaz de Castro, M.C.A.; Alves Pereira, V.R.; et al. Novel indol-3-yl-thiosemicarbazone derivatives: Obtaining, evaluation of in vitro leishmanicidal activity and ultrastructural studies. Chem.-Biol. Interact. 2020, 315, 108899. [Google Scholar] [CrossRef]
- Scarim, C.B.; Jornada, D.H.; Machado, M.G.M.; Ferreira, C.M.R.; Dos Santos, J.L.; Chung, M.C. Thiazole, thio and semicarbazone derivatives against tropical infective diseases: Chagas disease, human African trypanosomiasis (HAT), leishmaniasis, and malaria. Eur. J. Med. Chem. 2019, 162, 378–395. [Google Scholar] [CrossRef]
- Pinto, E.G.; Santo, I.O.; Schmidt, T.J.; Borborema, S.E.T.; Ferreira, V.F.; Rocha, D.R.; Templone, A.G. Potential of 2-hydroxy-3-phenylsulfanylmethyl-[1,4]-naphthoquinones against Leishmania (L.) infantum: Biological activity and structure-activity relationships. PLoS ONE 2014, 9, e105127. [Google Scholar] [CrossRef]
- Tapia, R.A.; Tapia, R.A.; Prieto, Y.; Pautet, M.; Domard, M.; Domard, M.; Sarciron, M.E.; Walchshofer, N.; Walchshofer, N.; Fillion, H. Synthesis and Antileishmanial Activity of Indoloquinones Containing a Fused Benzothiazole Ring. Eur. J. Org. Chem 2002, 2002, 4005–4010. [Google Scholar] [CrossRef]
- Di Giorgio, C.; Delmas, F.; Filloux, N.; Robin, M.; Seferian, L.; Azas, N.; Gasquet, M.; Costa, M.; Timon-David, P.M.; Galy, J.P. In vitro activities of 7-substituted 9-chloro and 9-amino-2-methoxyacridines and their bis- and tetra-acridine complexes against Leishmania infantum. Antimicrob. Agents Chemother. 2003, 47, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Cunha Araújo, I.A.; De Paula, R.C.; Alves, C.L.; Ferraz Faria, K.; De Oliveira, M.M.; Gonçalves Mendes, G.; Ferreira Abdia Dias, E.M.; Ribeiro, R.R.; De Oliveira, A.B.; Da Silva, S.M. Efficacy of lapachol on treatment of cutaneous and visceral leishmaniasis. Exp. Parasitol. 2019, 199, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos Faiões, V.; Da Frota, L.C.R.; Cunha-Junior, E.F.; Barcellos, J.C.F.; Da Silva, T.; Netto, C.D.; Gonçalves Da Silva, S.A.; Da Silva, A.J.M.; Costa, P.R.R.; Torres-Santos, E.C. Second-generation pterocarpanquinones: Synthesis and antileishmanial activity. J. Venom. Anim. Toxins Incl. Trop. Dis. 2018, 24, 35. [Google Scholar] [CrossRef] [PubMed]
- Jentzsch, J.; Koto, W.S.; Al Nasr, I.S.; Schobert, R.; Ersfeld, K.; Biersack, B. New Antiparasitic Bis-Naphthoquinone Derivatives. Chem. Biodivers. 2020, 17, e1900597. [Google Scholar] [CrossRef]
- Ramírez-Macías, I.; Marín, C.; Es-Samti, H.; Fernandéz, A.; Guardia, J.J.; Zentar, H.; Agil, a.; Chamboun, R.; Alvarez-Manzaneda, E.; Sánchez-Moreno, M. Taiwaniaquinoid and abietane quinone derivatives with trypanocidal activity against T. cruzi and Leishmania spp. Parasitol. Int. 2012, 61, 405–413. [Google Scholar] [CrossRef]
- Da Cruz, E.H.G.; Hussene, C.M.B.; Diasa, G.G.; Diogo, E.B.T.; De Melo, I.M.M.; Rodrigues, B.L.; Da Silva Júnior, E.N.; Da Silva, M.G.; Valencia, W.O.; Camara, C.A. 1,2,3-Triazole-, arylamino- and thio-substituted 1,4-naphthoquinones: Potent antitumor activity, electrochemical aspects, and bioisosteric replacement of C-ring-modified lapachones. Bioorg. Med. Chem. 2014, 22, 1608–1619. [Google Scholar] [CrossRef]
- Ramirez, O.; Motta-Mena, L.B.; Cordova, A.; Garza, K.M. A small library of synthetic Di-substituted 1, 4-naphthoquinones induces ROS-mediated cell death in murine fibroblasts. PLoS ONE 2014, 9, e106828. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, Z.; Zhang, W.; Yan, D. 2-Substituted-1-(2-morpholinoethyl)-1H-naphtho [2,3-d]imidazole-4,9-diones: Design, synthesis and antiproliferative activity. Bioorg. Med. Chem. Lett. 2018, 28, 2454–2458. [Google Scholar] [CrossRef]
- Baiju, T.V.; Almeida, R.G.; Sivanandam, S.T.; De Simone, C.A.; Brito, L.M.; Cavalcanti, B.C.; Pessoa, C.; Namboothiti, I.N.N.; Da Silva Júnior, E.N. Quinonoid compounds via reactions of lawsone and 2-aminonaphthoquinone with α-bromonitroalkenes and nitroallylic acetates: Structural diversity by C-ring modification and cytotoxic evaluation against cancer cells. Eur. J. Med. Chem. 2018, 151, 686–704. [Google Scholar] [CrossRef]
- Da Silva Júnior, E.N.; De Souza, M.C.B.V.; Pinto, A.V.; Pinto, C.C.; Goulart, M.O.F.; Barros, F.W.A.; Pessoa, C.; Costa-Lotufo, L.V.; Montenegro, R.C.; de Moraes, M.O.; et al. Synthesis and potent antitumor activity of new arylamino derivatives of nor-β-lapachone and nor-α-lapachone. Bioorg. Med. Chem. 2007, 15, 7035–7041. [Google Scholar] [CrossRef]
- Hoffmann, R.; Minkin, V.I.; Carpenter, B.K. Ockham’s Razor and chemistry. Int. J. Philos. Chem. 1997, 3, 3–28. [Google Scholar] [CrossRef]
- Laboratoire de Chemoinformatique. Available online: http://infochim.u-strasbg.fr/spip.php?rubrique41 (accessed on 29 August 2021).
- Wei Yap, C. PaDEL-Descriptor: An Open Source Software to Calculate Molecular Descriptors and Fingerprints. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Durant, J.L.; Leland, B.A.; Henry, D.R.; Nourse, J.G. Reoptimization of MDL keys for use in drug discovery. J. Chem. Inf. Model. 2002, 42, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Martiní, J.R.V.; Ponce, Y.M.; Jacas, C.R.G.; Mayorga, K.M. QuBiLS—MAS, open source multi—platform software for atom-and bond-based topological descriptors computations. J. Cheminform. 2017, 9, 35. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Crippen, G.M. Atomic Physicochemical Parameters for Three-Dimensional-Structure-Directed Quantitative Structure-Activity Relationships. 2. Modeling Dispersive and Hydrophobic Interactions. J. Chem. Inf. Comput. Sci. 1987, 27, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Klekota, J.; Roth, F.P. Chemical substructures that enrich for biological activity. Bioinformatics 2008, 24, 2518–2525. [Google Scholar] [CrossRef]
- Martínez, M.J.; Ponzoni, I.; Díaz, M.F.; Vazquez, G.E.; Soto, A.J. Visual analytics in cheminformatics: User—supervised descriptor selection for QSAR methods. J. Cheminform. 2015, 7, 39. [Google Scholar] [CrossRef]
- Garro Martinez, J.C.; Andrada, M.F.; Vega-Hissi, E.G.; Garibotto, F.M.; Nogueras, M.; Rodríguez, R.; Cobo, J.; Enriz, R.D.; Estrada, M.R. Dihydrofolate reductase inhibitors: A quantitative structure—activity relationship study using 2D-QSAR and 3D-QSAR methods. Med. Chem. Res. 2016, 26, 247–261. [Google Scholar] [CrossRef]
- Insubria QSAR PaDEL-Descriptor Model for Prediction of Endocrine Disruptors Chemicals (EDC) Estrogen Receptor (ER)-Binding Affinity. Available online: http://padel.nus.edu.sg/software/padeldescriptor/index.html (accessed on 5 October 2021).
- Samuelson, J. Why Metronidazole Is Active against both Bacteria and Parasites. ASM 1999, 43, 1533–1541. [Google Scholar] [CrossRef]
- Cardoso-Silva, J.; Papadatos, G.; Papageorgiou, L.G.; Tsoka, S. Optimal Piecewise Linear Regression Algorithm for QSAR Modelling. Mol. Inform. 2019, 38, 1800028. [Google Scholar] [CrossRef]
- Thurston, B.A.; Ferguson, A.L. Machine learning and molecular design of self-assembling-conjugated oligopeptides. Mol. Simul. 2018, 44, 930–945. [Google Scholar] [CrossRef]
- Ibrahum, Z.Y.; Uzairu, A.; Shallangwa, G.; Abechi, S. Theoretical design of novel antimalarial agents against P. falciparum strain, Dd2 through the QSAR modeling of synthesized 2′-substituted triclosan derivatives. Heliyon 2020, 6, e05032. [Google Scholar] [CrossRef] [PubMed]
- García-Jacas, C.R.; Barigye, S.J.; Marrero-Ponce, Y.; Acevedo-Martínez, L.; Valdés-Martiní, J.R.; Contreras-Torres, E. QuBiLS-MIDAS: A Parallel Free-Software for Molecular Descriptors Computation Based on Multilinear Algebraic Maps. J. Comput. Chem. 2014, 35, 1395–1409. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. Prediction of Hydrophobic (Lipophilic) Properties of Small Organic Molecules Using Fragmental Methods: An Analysis of ALOGP and CLOGP Methods. J. Phys. Chem. 1998, 5639, 3762–3772. [Google Scholar] [CrossRef]
- Lanez, T.; Ahmedi, R. AlogP calculation of octanol-water partition coefficient of ferrocene derivatives. Int. J. Pharm.Tech. Res. 2015, 8, 408–414. [Google Scholar] [CrossRef][Green Version]
- Prasanna, S.; Doerksen, R.J. Topological Polar Surface Area: A Useful Descriptor in 2D-QSAR. Curr. Med. Chem. 2009, 16, 21–41. [Google Scholar] [CrossRef]
- Organisation de Coopération et de Développement Economiques. Guidance Document on the Validation of (Quantitative)Structure-Activity Relationships [(Q)SAR] Models; OECD: Paris, France, 2007. [Google Scholar]
- Ciubotariu, D.; Medeleanu, M.; Vlaia, V.; Olariu, T.; Ciubotariu, C.; Dragos, D.; Corina, S. Molecular van der Waals Space and Topological Indices from the Distance Matrix. Molecules 2004, 9, 1053–1078. [Google Scholar] [CrossRef]
- Andrew, G.; Pablo, R. Chemometrics Applocations and Research QSAR in Medicinal Chemistry, 1st ed.; Appe Academic Press: Burlington, ON, Canada; Cambridge, MA, USA, 2016; pp. 366–381. [Google Scholar]
- Fernandes, J.; Gattass, C.R. Topological polar surface area defines substrate transport by multidrug resistance associated protein 1 (MRP1/ABCC1). J. Med. Chem 2009, 52, 1214–1218. [Google Scholar] [CrossRef]
- Gozalbes, R.; Doucet, J.P.; Derouin, F. Application of Topological Descriptors in QSAR and Drug Design: History and New Trends. Curr. Drug Targets Infect. Disord. 2002, 2, 93–102. [Google Scholar] [CrossRef]
- Tandon, H.; Ranjan, P.; Chakraborty, T.; Suhag, V. Polarizability: A promising descriptor to study chemical–biological interactions. Mol. Divers. 2021, 25, 249–262. [Google Scholar] [CrossRef]
- Putz, M.V.; Ionaşcu, C.; Putz, A.M.; Ostafe, V. Alert-QSAR. Implications for electrophilic theory of chemical carcinogenesis. Int. J. Mol. Sci. 2011, 12, 5098–5134. [Google Scholar] [CrossRef] [PubMed]
- Bhal, S.K.; Kassam, K.; Peirson, I.G.; Pearl, G.M. The rule of five revisited: Applying log D in place of log P in drug-likeness filters. Mol. Pharm. 2007, 4, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Golbraikh, A.; Tropsha, A. Beware of q 2! J. Mol. Graph. Model. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Nossa, D.L.; Rozo Núñez, W.E.; Gómez Castaño, J.A.; Duchowicz, P.R. Antiprotozoal QSAR modelling for trypanosomiasis (Chagas disease) based on thiosemicarbazone and thiazole derivatives. J. Mol. Graph. Model. 2021, 103, 107821. [Google Scholar] [CrossRef] [PubMed]
- Roy, K. On some aspects of validation of predictive quantitative structure—Activity relationship models. Drug Discov. 2007, 2, 1567–1578. [Google Scholar] [CrossRef] [PubMed]
- López López, L.I.; Nery Flores, S.D.; Silva Belmares, S.Y.; Sáenz Galindo, A. Naphthoquinones: Biological properties and synthesis of lawsone and derivatives—A structured review. Vitae 2014, 21, 248–258. [Google Scholar]
- Rodrigues Coura, J.; De Castro, S.L. A critical review on chagas disease chemotherapy. Mem. Inst. Oswaldo Cruz 2002, 97, 3–24. [Google Scholar] [CrossRef]
- Silva, R.S.F.; De Amorim, M.B.C.; Pinto, F.R.; Emery, F.S.; Goulart, M.O.F.; Pinto, A.V. Chemoselective Oxidation of Benzophenazines by m-CPBA: N-Oxidation vs. Oxidative Cleavage. J. Braz. Chem. Soc. 2007, 18, 759–764. [Google Scholar] [CrossRef][Green Version]
- Pacheco, P.A.F.; Galvão, R.M.S.; Faria, A.F.M.; Von Ranke, N.I.; Rangel, M.S.; Ribeiro, T.M.; Bello, M.I.; Rodrigues, C.R.; Ferreira, V.F.; Da Rocha, D.R.; et al. 8-Hydroxy-2-(1H-1,2,3-triazol-1-yl)-1,4-naphtoquinone derivatives inhibited P2X7 Receptor-Induced dye uptake into murine Macrophages. Bioorg. Med. Chem. 2019, 27, 1449–1455. [Google Scholar] [CrossRef]
- Chacón-Vargas, K.F.; Nogueda-Torres, B.; Sánchez-Torres, L.E.; Suarez-Contreras, E.; Villalobos-Rocha, J.C.; Torres-Martinez, Y.; Lara-Ramirez, E.E.; Fiorani, G.; Krauth-Siegel, R.L.; Bolognesi, M.L.; et al. Trypanocidal activity of quinoxaline 1,4 Di-N-oxide derivatives as trypanothione reductase inhibitors. Molecules 2017, 22, 220. [Google Scholar] [CrossRef]
- Bond, C.S.; Zhang, Y.; Berriman, M.; Cunningham, M.L.; Fairlamb, A.H.; Hunter, W.N. Crystal structure of Trypanosoma cruzi trypanothione reductase in complex with trypanothione, and the structure-based discovery of new natural product inhibitors. Structure 1999, 7, 81–89. [Google Scholar] [CrossRef]
- Chen, C.K.; Leung, S.F.; Guilbert, C.; Jacobson, M.P.; Mckerrow, J.H.; Podust, L.M. Structural characterization of CYP51 from Trypanosoma cruzi and Trypanosoma brucei bound to the antifungal drugs posaconazole and fluconazole. PLoS Negl. Trop. Dis. 2010, 4, e651. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, M.F.C.; Forezi, L.S.M.; De Souza, A.S.; Faria, A.F.M.; Galvão, R.M.S.; Bello, M.L.; Da Silva, F.C.; Faria, R.X.; Ferreira, V.F. Tandem Synthesis of Furanaphthoquinones via Enamines and Evaluation of Their Antiparasitic Effects against Trypanosoma cruzi. J. Braz. Chem. Soc. 2022, 33, 238–250. [Google Scholar] [CrossRef]
- Inacio Filho, J.D. Estudo do Efeito da EGCG In Vitro e In Vivo, Suas Associações e Mecanismo Molecular de Ação em Leishmania Infantum. Ph.D. Thesis, Fundação Oswaldo Cruz, Rios de Janeiro, Brazil, 2018. [Google Scholar]
- Reynolds, K.A. Design and Synthesis of Quinoline, Cinchona Alkaloids and Other Potential Inhibitors or Leishmaniasis. Ph.D. Thesis, Griffith University, Brisbane, Australia, 2012. [Google Scholar]
- Braga, S.S. Multi-target drugs active against leishmaniasis: A paradigm of drug repurposing. Eur. J. Med. Chem. 2019, 183, 111660. [Google Scholar] [CrossRef] [PubMed]
- Ilari, A.; Baiocco, P.; Messori, L.; Fiorillo, A.; Boffi, A.; Gramiccia, M.; Di Muccio, T.; Colotti, G. A gold-containing drug against parasitic polyamine metabolism:The X-ray structure of trypanothione reductase from Leishmania infantum in complex with auranofin reveals a dual mechanism of enzyme inhibition. Amino Acids 2012, 42, 803–811. [Google Scholar] [CrossRef]
- Venkatesan, S.K.; Shukla, A.K.; Dubey, V.K. Molecular Docking Studies of Selected Tricyclic and Quinone Derivatives on Trypanothione Reductase of Leishmania infantum. J. Comp. Chem. 2010, 32, 174–182. [Google Scholar] [CrossRef]
- Manjolin, L.C.; Dos Reis, M.B.G.; Do Carmo Maquiaveli, C.; Santos-Filho, O.A.; Da Silva, E.R. Dietary flavonoids fisetin, luteolin and their derived compounds inhibit arginase, a central enzyme in Leishmania (Leishmania) amazonensis infection. Food Chem. 2013, 141, 2253–2262. [Google Scholar] [CrossRef]
- De Lima, E.C.; Castelo-Branco, F.S.; Maquiaveli, C.C.; Farias, A.B.; Rennó, M.N.; Boechat, N.; Silva, E.R. Phenylhydrazides as inhibitors of Leishmania amazonensis arginase and antileishmanial activity. Bioorg. Med. Chem. 2019, 27, 3853–3859. [Google Scholar] [CrossRef]
- Kanyo, Z.F.; Scolnick, L.R.; Ash, D.E.; Christianson, D.W. Structure of unique binuclear manganese cluster in arginase. Lett. Nat. 1996, 383, 554–557. [Google Scholar] [CrossRef]
- Estrada, E.; Molina, E. Novel local (fragment-based) topological molecular descriptors for QSPR / QSAR and molecular design. J. Mol. Graph. Model. 2001, 20, 54–64. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Zafar, F.; Gupta, A.; Thangavel, K.; Khayana, K.; Sani, A.A.; Ghosal, A.; Tandon, P.; Nishat, N. Physicochemical and Pharmacokinetic Analysis of Anacardic Acid Derivatives. ACS Omega 2020, 5, 6021–6030. [Google Scholar] [CrossRef] [PubMed]
- Bhal, S.K. Advanced Chemistry Development (ACD/LABS). Log P—Making Sense of the Value; Advanced Chemistry Development: Toronto, ON, Canada, 2007. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Mahanthesh, M.T.; Ranjith, D.; Raghavendra, Y.; Jyothi, R.; Narappa, G.; Ravi, M.V. Swiss ADME prediction of phytochemicals present in Butea monosperma (Lam.) Taub. J. Pharmacogn. Phytochem. 2020, 9, 1799–1809. [Google Scholar]
- Delaney, J.S. ESOL: Estimating aqueous solubility directly from molecular structure. J. Chem. Inf. Comput. Sci. 2004, 44, 1000–1005. [Google Scholar] [CrossRef]
- Ali, P.; Camilleri, P.; Brown, M.B.; Hutt, A.J.; Kirton, S.B. Revisiting the general solubility equation: In silico prediction of aqueous solubility incorporating the effect of topographical polar surface area. J. Chem. Inf. Model. 2012, 52, 420–428. [Google Scholar] [CrossRef]
- Löbenberg, R.; Amidon, G.L.; Ferraz, H.G.; Bou-Chacra, N. Mechanism of gastrointestinal drug absorption and application in therapeutic drug delivery. In Therapeutic Delivery Methods: A Concise Overview of Emerging Areas; Future Science Ltd.: London, UK, 2013; pp. 8–22. [Google Scholar] [CrossRef]
- Osorio, E.J.; Robledo, S.M.; Arango, G.J.; Muskus, C.E. Leishmania: Papel de la glicoproteína P en la mediación de resistencia a medicamentos y estrategias de reversión. Biomédica 2005, 25, 242. [Google Scholar] [CrossRef]
- Finch, A.; Pillans, P. P-glycoprotein and its role in drug-drug interactions. Aust. Prescr. 2014, 37, 137–139. [Google Scholar] [CrossRef]
- Daina, A.; Zoete, V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. Chem. Pub. Soc. Eur. 2016, 11, 1117–1121. [Google Scholar] [CrossRef]
- Potts, R.O.; Guy, R.H. Predicting Skin Permeability. Pharm. Res. 1992, 9, 663–669. [Google Scholar] [CrossRef]
- Ranjith, D.; Ravikumar, C. SwissADME predictions of pharmacokinetics and drug-likeness properties of small molecules present in Ipomoea mauritiana Jacq. J. Pharmacogn. Phytochem. 2019, 8, 2063–2073. Available online: http://www.swissadme.ch/index.php (accessed on 30 April 2022).
- Jaimes-Santoyo, J.; De Montesinos-Sampedro, A.; Barbosa-Cobos, R.E.; Moreno-Mutio, S.G.; Rodriguez-Ballesteros, D.; Ramos-Cervantes, T.; Ocharán-Hernández, M.E.; Toscano-Garibay, J.; Beltrán-Ramírez, O. El Citocromo P-450. Rev. Hosp. Juárez Mex. 2014, 81, 250–256. [Google Scholar]
- Deodhar, M.; Al Rihani, S.B.; Arwood, M.J.; Drakjian, L.; Dow, P.; Turgeon, J.; Michaud, V. Mechanisms of cyp450 inhibition: Understanding drug-drug interactions due to mechanism-based inhibition in clinical practice. Pharmaceutics 2020, 12, 846. [Google Scholar] [CrossRef] [PubMed]
- Ogu, C.C.; Maxa, J.L. Drug Interactions Due to Cytochrome P450. Bayl. Univ. Med. Cent. 2017, 8280, 11–14. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.J.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple Selection Criteria for Drug-like Chemical Matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef]
- Martin, Y.C. A Bioavailability Score. J. Med. Chem. 2005, 48, 3164–3170. [Google Scholar] [CrossRef]
- ACD/ChemSketch; Versión 2021.1.3; Advanced Chemistry Development, Inc.: Toronto, ON, Canada, 2021; Available online: www.acdlabs.com (accessed on 30 April 2022).
- Mold2_FDA. Available online: https://www.fda.gov/science-research/bioinformatics-tools/mold2 (accessed on 29 August 2021).
- Laboratory of Chemoinformatique. 2013. Available online: http://infochim.u-strasbg.fr/ (accessed on 30 April 2022).
- Mercader, A.G.; Duchowicz, P.; Ferna, F. Replacement Method and Enhanced Replacement Method Versus the Genetic Algorithm Approach for the Selection of Molecular Descriptors in QSPR/QSAR Theories. J. Chem. Inf. Model. 2010, 50, 1542–1548. [Google Scholar] [CrossRef]
- Mathworks, MATLAB—Mathworks—MATLAB & Simulink. 2016. Available online: https://nl.mathworks.com/products/matlab.html%0Ahttp://www.mathworks.com/products/matlab/ (accessed on 29 August 2021).
- Gramatica, P. Principles of QSAR models validation: Internal and external. QSAR Comb. Sci. 2007, 26, 694–701. [Google Scholar] [CrossRef]
- Veerasamy, R.; Rajak, H.; Jain, A.; Sivadasan, S.; Varghese, C.P.; Agrawal, R.K. Validation of QSAR Models—Strategies and Importance. Int. J. Drug Des. Discov. 2011, 2, 511–519. [Google Scholar]
- Singh, B.K.; Sarkar, N.; Jagannadham, M.V.; Dubey, V.K. Modeled structure of trypanothione reductase of Leishmania infantum. BMB Rep. 2008, 41, 444–447. [Google Scholar] [CrossRef] [PubMed]
- Baiocco, P.; Colotti, G.; Franceschini, S.; Ilari, A. Molecular Basis of Antimony Treatment in Leishmaniasis. J. Med. Chem. 2009, 52, 2603–2612. [Google Scholar] [CrossRef]
- Antonio, E.L.D.; Ullman, B.; Roberts, S.C.; Dixit, U.P.; Wilson, M.E.; Hai, Y.; Christianson, D.W. Crystal structure of arginase from Leishmania mexicana and implications for the inhibition of polyamine biosynthesis in parasitic infections. Arch. Biochem. Biophys. 2013, 535, 163–176. [Google Scholar] [CrossRef]
- Moreno, M.A.; Abramov, A.; Abendroth, J.; Alonso, A.; Zhang, S.; Alcolea, P.J.; Edwards, T.; Lorimer, D.; Myley, P.J.; Larraga, V. Structure of tyrosine aminotransferase from Leishmania infantum. Acta Crystallogr. 2014, 70, 583–587. [Google Scholar] [CrossRef]
- Molexus, Molegro Virtual Docker-Manual, Copyrigh, M. 2019. Available online: http://molexus.io/molegro-virtual-docker/ (accessed on 30 April 2022).
- Filgueira De Azevedo, W. Docking Screens for Drug Discovery; Humana Press: Ria Grande Do Sul, Brazil, 2019. [Google Scholar]
- Frisch, J.P.M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzewski, V.G.; Montgomery, J.A., Jr.; Stratmann, R.E.; Burant, J.C.; et al. Gaussian 98, Revision A. 7; Gaussian Inc.: Pittsburgh, PA, USA, 1998; p. 49. [Google Scholar]
- Thomsen, R.; Christensen, M.H. MolDock: A New Technique for High-Accuracy Molecular Docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef]
- De Azevedo, W.F., Jr. MolDock Applied to Structure-Based Virtual Screening. Curr. Drug Targets 2010, 11, 327–334. [Google Scholar] [CrossRef]
- Da Cunha, E.F.F.; Azevedo Martins, R.C.; Girão Albuquerque, M.; de Alencastro, R.B. LIV-3D-QSAR model for estrogen receptor ligands. J. Mol. Model. 2004, 10, 297–304. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).