
Structural Elucidation of Rift Valley Fever Virus L Protein towards the Discovery of Its Potential Inhibitors

 ,
,  , , ,
, , ,  ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Protein Structural Analysis
2.2. Binding Site Determination
2.3. Molecular Docking
2.4. ADME and Toxicity Prediction Analysis
2.5. MD Simulation
2.5.1. Root Mean Square Deviation (RMSD)
2.5.2. Root Mean Square Fluctuation (RMSF) Analysis
2.5.3. Radius of Gyration (Rg)
2.5.4. B-Factor Analysis
2.6. MMGBA/PBSA Analysis
3. Materials and Methods
3.1. Homology Modeling
3.2. Structure Validation
3.3. Target Protein Preparation
3.4. Compound Preparation
3.5. Structure-Based Virtual Screening
3.6. ADME and Toxicity Prediction Analysis
3.7. MD Simulations
3.8. MMGBA/PBSA Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pepin, M.; Bouloy, M.; Bird, B.H.; Kemp, A.; Paweska, J. Rift Valley fever virus (Bunyaviridae: Phlebovirus): An update on pathogenesis, molecular epidemiology, vectors, diagnostics and prevention. Vet. Res. 2010, 41, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bird, B.H.; Ksiazek, T.G.; Nichol, S.T.; MacLachlan, N.J. Rift Valley fever virus. J. Am. Vet. Med. Assoc. 2009, 234, 883–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartman, A. Rift valley fever. Clin. Lab. Med. 2017, 37, 285–301. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, T.; Makino, S. The pathogenesis of Rift Valley fever. Viruses 2011, 3, 493–519. [Google Scholar] [CrossRef] [Green Version]
- Drake, J.M.; Hassan, A.N.; Beier, J.C. A statistical model of Rift Valley fever activity in Egypt. J. Vector Ecol. 2013, 38, 251–259. [Google Scholar] [CrossRef] [Green Version]
- Helmy, Y.A.; El-Adawy, H.; Abdelwhab, E.M. A comprehensive review of common bacterial, parasitic and viral zoonoses at the human-animal interface in Egypt. Pathogens 2017, 6, 33. [Google Scholar] [CrossRef]
- Gaudreault, N.N.; Indran, S.V.; Bryant, P.; Richt, J.A.; Wilson, W.C. Comparison of Rift Valley fever virus replication in North American livestock and wildlife cell lines. Front. Microbiol. 2015, 6, 664. [Google Scholar] [CrossRef]
- McElroy, A.K.; Albariño, C.G.; Nichol, S.T. Development of a RVFV ELISA that can distinguish infected from vaccinated animals. Virol. J. 2009, 6, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Lumley, S.; Horton, D.L.; Hernandez-Triana, L.L.; Johnson, N.; Fooks, A.R.; Hewson, R. Rift Valley fever virus: Strategies for maintenance, survival and vertical transmission in mosquitoes. J. Gen. Virol. 2017, 98, 875–887. [Google Scholar] [CrossRef] [Green Version]
- Ikegami, T. Molecular biology and genetic diversity of Rift Valley fever virus. Antiviral Res. 2012, 95, 293–310. [Google Scholar] [CrossRef] [Green Version]
- Gerrard, S.R.; Nichol, S.T. Synthesis, proteolytic processing and complex formation of N-terminally nested precursor proteins of the Rift Valley fever virus glycoproteins. Virology 2007, 357, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Müller, R.; Poch, O.; Delarue, M.; Bishop, D.; Bouloy, M. Rift Valley fever virus L segment: Correction of the sequence and possible functional role of newly identified regions conserved in RNA-dependent polymerases. J. Gen. Virol. 1994, 75, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Poch, O.; Sauvaget, I.; Delarue, M.; Tordo, N. Identification of four conserved motifs among the RNA-dependent polymerase encoding elements. EMBO J. 1989, 8, 3867–3874. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, P.; Malet, H.; Cusack, S.; Reguera, J. Structural insights into bunyavirus replication and its regulation by the vRNA promoter. Cell 2015, 161, 1267–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinsella, E.; Martin, S.G.; Grolla, A.; Czub, M.; Feldmann, H.; Flick, R. Sequence determination of the Crimean–Congo hemorrhagic fever virus L segment. Virology 2004, 321, 23–28. [Google Scholar] [CrossRef] [Green Version]
- Bouloy, M.; Weber, F. Molecular biology of Rift Valley fever virus. Open Virol. J. 2010, 4, 8. [Google Scholar] [CrossRef]
- Morin, B.; Coutard, B.; Lelke, M.; Ferron, F.; Kerber, R.; Jamal, S.; Frangeul, A.; Baronti, C.; Charrel, R.; De Lamballerie, X. The N-terminal domain of the arenavirus L protein is an RNA endonuclease essential in mRNA transcription. PLoS Pathog. 2010, 6, e1001038. [Google Scholar] [CrossRef]
- Patterson, J.; Holloway, B.; Kolakofsky, D. La Crosse virions contain a primer-stimulated RNA polymerase and a methylated cap-dependent endonuclease. J. Virol. 1984, 52, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Gogrefe, N.; Reindl, S.; Günther, S.; Rosenthal, M. Structure of a functional cap-binding domain in Rift Valley fever virus L protein. PLoS Pathog. 2019, 15, e1007829. [Google Scholar] [CrossRef] [Green Version]
- Sarwar, S.; Abdul Qadir, M.; Alharthy, R.D.; Ahmed, M.; Ahmad, S.; Vanmeert, M.; Mirza, M.U.; Hameed, A. Folate Conjugated Polyethylene Glycol Probe for Tumor-Targeted Drug Delivery of 5-Fluorouracil. Molecules 2022, 27, 1780. [Google Scholar] [CrossRef]
- Salo-Ahen, O.M.H.; Alanko, I.; Bhadane, R.; Bonvin, A.M.J.J.; Honorato, R.V.; Hossain, S.; Juffer, A.H.; Kabedev, A.; Lahtela-Kakkonen, M.; Larsen, A.S. Molecular Dynamics Simulations in Drug Discovery and Pharmaceutical Development. Processes 2021, 9, 71. [Google Scholar] [CrossRef]
- Ahmad, S.; Lee, Y.K.; Nazir, M.; Abdul Rahman, N.; Trant, J.F.; Abdullah, I. Fragment-based in silico design of SARS CoV-2 main protease inhibitors. Chem. Biol. Drug Des. 2021, 98, 604–619. [Google Scholar] [CrossRef] [PubMed]
- Durrani, F.G.; Gul, R.; Mirza, M.U.; Kaderbhai, N.N.; Froeyen, M.; Saleem, M. Mutagenesis of DsbAss is Crucial for the Signal Recognition Particle Mechanism in Escherichia coli: Insights from Molecular Dynamics Simulations. Biomolecules 2019, 9, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalid, H.; Landry, K.B.; Ijaz, B.; Ashfaq, U.A.; Ahmed, M.; Kanwal, A.; Froeyen, M.; Mirza, M.U. Discovery of novel Hepatitis C virus inhibitor targeting multiple allosteric sites of NS5B polymerase. Infect. Genet. Evol. 2020, 84, 104371. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.U.; Ahmad, S.; Abdullah, I.; Froeyen, M. Identification of novel human USP2 inhibitor and its putative role in treatment of COVID-19 by inhibiting SARS-CoV-2 papain-like (PLpro) protease. Comput. Biol. Chem. 2020, 89, 107376. [Google Scholar] [CrossRef]
- Mirza, M.U.; Vanmeert, M.; Ali, A.; Iman, K.; Froeyen, M.; Idrees, M. Perspectives towards antiviral drug discovery against Ebola virus. J. Med. Virol. 2019, 91, 2029–2048. [Google Scholar] [CrossRef]
- ul Qamar, M.T.; Ahmad, S.; Khan, A.; Mirza, M.U.; Ahmad, S.; Abro, A.; Chen, L.-L.; Almatroudi, A.; Wei, D.-Q. Structural probing of HapR to identify potent phytochemicals to control Vibrio cholera through integrated computational approaches. Comput. Biol. Med. 2021, 138, 104929. [Google Scholar] [CrossRef]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational methods in drug discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [Green Version]
- Jácome, R.; Becerra, A.; de León, S.P.; Lazcano, A. Structural analysis of monomeric RNA-dependent polymerases: Evolutionary and therapeutic implications. PLoS ONE 2015, 10, e0139001. [Google Scholar] [CrossRef] [Green Version]
- Van Der Linden, L.; Vives-Adrián, L.; Selisko, B.; Ferrer-Orta, C.; Liu, X.; Lanke, K.; Ulferts, R.; De Palma, A.M.; Tanchis, F.; Goris, N. The RNA template channel of the RNA-dependent RNA polymerase as a target for development of antiviral therapy of multiple genera within a virus family. PLoS Pathog. 2015, 11, e1004733. [Google Scholar] [CrossRef] [Green Version]
- Ferrer-Orta, C.; Arias, A.; Escarmís, C.; Verdaguer, N. A comparison of viral RNA-dependent RNA polymerases. Curr. Opin. Struct. Biol. 2006, 16, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.U.; Vanmeert, M.; Froeyen, M.; Ali, A.; Rafique, S.; Idrees, M. In silico structural elucidation of RNA-dependent RNA polymerase towards the identification of potential Crimean-Congo Hemorrhagic Fever Virus inhibitors. Sci. Rep. 2019, 9, 6809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godoy, A.S.; Lima, G.M.; Oliveira, K.I.; Torres, N.U.; Maluf, F.V.; Guido, R.V.; Oliva, G. Crystal structure of Zika virus NS5 RNA-dependent RNA polymerase. Nat. Commun. 2017, 8, 14764. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Feng, T.; Cheng, J.; Li, Y.; Yin, X.; Zeng, W.; Jin, X.; Li, Y.; Guo, F.; Jin, T. Structure of the NS5 methyltransferase from Zika virus and implications in inhibitor design. Biochem. Biophys. Res. Commun. 2017, 492, 624–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hercík, K.; Kozak, J.; Šála, M.; Dejmek, M.; Hřebabecký, H.; Zborníková, E.; Smola, M.; Ruzek, D.; Nencka, R.; Boura, E. Adenosine triphosphate analogs can efficiently inhibit the Zika virus RNA-dependent RNA polymerase. Antivir. Res. 2017, 137, 131–133. [Google Scholar] [CrossRef]
- Pattnaik, A.; Palermo, N.; Sahoo, B.R.; Yuan, Z.; Hu, D.; Annamalai, A.S.; Vu, H.L.; Correas, I.; Prathipati, P.K.; Destache, C.J. Discovery of a non-nucleoside RNA polymerase inhibitor for blocking Zika virus replication through in silico screening. Antivir. Res. 2018, 151, 78–86. [Google Scholar] [CrossRef]
- Lu, G.; Gong, P. Crystal structure of the full-length Japanese encephalitis virus NS5 reveals a conserved methyltransferase-polymerase interface. PLoS Pathog. 2013, 9, e1003549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malet, H.; Egloff, M.-P.; Selisko, B.; Butcher, R.E.; Wright, P.J.; Roberts, M.; Gruez, A.; Sulzenbacher, G.; Vonrhein, C.; Bricogne, G. Crystal structure of the RNA polymerase domain of the West Nile virus non-structural protein 5. J. Biol. Chem. 2007, 282, 10678–10689. [Google Scholar] [CrossRef] [Green Version]
- Noble, C.G.; Lim, S.P.; Chen, Y.-L.; Liew, C.W.; Yap, L.; Lescar, J.; Shi, P.-Y. Conformational flexibility of the Dengue virus RNA-dependent RNA polymerase revealed by a complex with an inhibitor. J. Virol. 2013, 87, 5291–5295. [Google Scholar] [CrossRef] [Green Version]
- Noble, C.G.; Chen, Y.-L.; Dong, H.; Gu, F.; Lim, S.P.; Schul, W.; Wang, Q.-Y.; Shi, P.-Y. Strategies for development of dengue virus inhibitors. Antivir. Res. 2010, 85, 450–462. [Google Scholar] [CrossRef] [PubMed]
- El Sahili, A.; Lescar, J. Dengue virus non-structural protein 5. Viruses 2017, 9, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Francesco, R.; Tomei, L.; Altamura, S.; Summa, V.; Migliaccio, G. Approaching a new era for hepatitis C virus therapy: Inhibitors of the NS3-4A serine protease and the NS5B RNA-dependent RNA polymerase. Antivir. Res. 2003, 58, 1–16. [Google Scholar] [CrossRef]
- Dhanak, D.; Duffy, K.J.; Johnston, V.K.; Lin-Goerke, J.; Darcy, M.; Shaw, A.N.; Gu, B.; Silverman, C.; Gates, A.T.; Nonnemacher, M.R. Identification and biological characterization of heterocyclic inhibitors of the hepatitis C virus RNA-dependent RNA polymerase. J. Biol. Chem. 2002, 277, 38322–38327. [Google Scholar] [CrossRef] [Green Version]
- Gemma, S.; Brogi, S.; Novellino, E.; Campiani, G.; Maga, G.; Brindisi, M.; Butini, S. HCV-targeted antivirals: Current status and future challenges. Curr. Pharm. Des. 2014, 20, 3445–3464. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.U.; Saadabadi, A.; Vanmeert, M.; Salo-Ahen, O.M.H.; Abdullah, I.; Claes, S.; De Jonghe, S.; Schols, D.; Ahmad, S.; Froeyen, M. Discovery of HIV entry inhibitors via a hybrid CXCR4 and CCR5 receptor pharmacophore-based virtual screening approach. Eur. J. Pharm. Sci. 2020, 155, 105537. [Google Scholar] [CrossRef] [PubMed]
- Cavasotto, C.N.; Phatak, S.S. Homology modeling in drug discovery: Current trends and applications. Drug Discov. Today 2009, 14, 676–683. [Google Scholar] [CrossRef]
- Hillisch, A.; Pineda, L.F.; Hilgenfeld, R. Utility of homology models in the drug discovery process. Drug Discov. Today 2004, 9, 659–669. [Google Scholar] [CrossRef]
- Oshiro, C.; Bradley, E.K.; Eksterowicz, J.; Evensen, E.; Lamb, M.L.; Lanctot, J.K.; Putta, S.; Stanton, R.; Grootenhuis, P.D.J. Performance of 3D-database molecular docking studies into homology models. J. Med. Chem. 2004, 47, 764–767. [Google Scholar] [CrossRef]
- Kairys, V.; Fernandes, M.X.; Gilson, M.K. Screening drug-like compounds by docking to homology models: A systematic study. J. Chem. Inf. Model 2006, 46, 365–379. [Google Scholar] [CrossRef]
- Fernandes, M.X.; Kairys, V.; Gilson, M.K. Comparing ligand interactions with multiple receptors via serial docking. J. Chem. Inf. Comput. Sci. 2004, 44, 1961–1970. [Google Scholar] [CrossRef]
- McGovern, S.L.; Shoichet, B.K. Information decay in molecular docking screens against holo, apo, and modeled conformations of enzymes. J. Med. Chem. 2003, 46, 2895–2907. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.U.; Rafique, S.; Ali, A.; Munir, M.; Ikram, N.; Manan, A.; Salo-Ahen, O.M.; Idrees, M. Towards peptide vaccines against Zika virus: Immunoinformatics combined with molecular dynamics simulations to predict antigenic epitopes of Zika viral proteins. Sci. Rep. 2016, 6, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Liu, L.; Liu, A.; Yan, L.; He, Y.; Shen, S.; Hu, M.; Guo, Y.; Liu, H.; Liu, C. Structure of severe fever with thrombocytopenia syndrome virus L protein elucidates the mechanisms of viral transcription initiation. Nat. Microbiol. 2020, 5, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Vogel, D.; Thorkelsson, S.R.; Quemin, E.R.; Meier, K.; Kouba, T.; Gogrefe, N.; Busch, C.; Reindl, S.; Günther, S.; Cusack, S. Structural and functional characterization of the severe fever with thrombocytopenia syndrome virus L protein. Nucleic Acids Res. 2020, 48, 5749–5765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenberg, D. The discovery of the α-helix and β-sheet, the principal structural features of proteins. Proc. Natl. Acad. Sci. USA 2003, 100, 11207–11210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef]
- Sidorova, A.E.; Malyshko, E.V.; Lutsenko, A.O.; Shpigun, D.K.; Bagrova, O.E. Protein Helical Structures: Defining Handedness and Localization Features. Symmetry 2021, 13, 879. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Skolnick, J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Watson, J.D.; Thornton, J.M. ProFunc: A server for predicting protein function from 3D structure. Nucleic Acids Res. 2005, 33, W89–W93. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Li, J.; Gao, G.F.; Tien, P.; Liu, W. Bunyavirales ribonucleoproteins: The viral replication and transcription machinery. Crit. Rev. Microbiol. 2018, 44, 522–540. [Google Scholar] [CrossRef] [PubMed]
- Pflug, A.; Guilligay, D.; Reich, S.; Cusack, S. Structure of influenza A polymerase bound to the viral RNA promoter. Nature 2014, 516, 355. [Google Scholar] [CrossRef] [PubMed]
- Arnold, J.J.; Ghosh, S.K.B.; Cameron, C.E. Poliovirus RNA-dependent RNA polymerase (3Dpol): Divalent cation modulation of primer, template, and nucleotide selection. J. Biol. Chem. 1999, 274, 37060–37069. [Google Scholar] [CrossRef] [Green Version]
- Ferrer-Orta, C.; Arias, A.; Pérez-Luque, R.; Escarmís, C.; Domingo, E.; Verdaguer, N. Sequential structures provide insights into the fidelity of RNA replication. Proc. Natl. Acad. Sci. USA 2007, 104, 9463–9468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amroun, A.; Priet, S.; de Lamballerie, X.; Quérat, G. Bunyaviridae RdRps: Structure, motifs, and RNA synthesis machinery. Crit. Rev. Microbiol. 2017, 43, 753–778. [Google Scholar] [CrossRef]
- Rothwell, P.J.; Waksman, G. Structure and mechanism of DNA polymerases. Adv. Protein Chem. 2005, 71, 401–440. [Google Scholar]
- Biswas, S.K.; Nayak, D.P. Mutational analysis of the conserved motifs of influenza A virus polymerase basic protein 1. J. Virol. 1994, 68, 1819–1826. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, A.B.; de la Torre, J.C. Genetic and biochemical evidence for an oligomeric structure of the functional L polymerase of the prototypic arenavirus lymphocytic choriomeningitis virus. J. Virol. 2005, 79, 7262–7268. [Google Scholar] [CrossRef] [Green Version]
- Vázquez, A.L.; Alonso, J.M.M.; Parra, F. Mutation analysis of the GDD sequence motif of a calicivirus RNA-dependent RNA polymerase. J. Virol. 2000, 74, 3888–3891. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zheng, H.; Gao, F.; Tian, D.; Yuan, S. Mutational analysis of the SDD sequence motif of a PRRSV RNA-dependent RNA polymerase. Sci. China Life Sci. 2011, 54, 870–879. [Google Scholar] [CrossRef] [Green Version]
- Abdelrheem, D.A.; Rahman, A.A.; Elsayed, K.N.; Abd El-Mageed, H.; Mohamed, H.S.; Ahmed, S.A. Isolation, characterization, in vitro anticancer activity, dft calculations, molecular docking, bioactivity score, drug-likeness and admet studies of eight phytoconstituents from brown alga sargassum platycarpum. J. Mol. Struct. 2021, 1225, 129245. [Google Scholar] [CrossRef]
- Bergeron, É.; Albariño, C.G.; Khristova, M.L.; Nichol, S.T. Crimean-Congo hemorrhagic fever virus-encoded ovarian tumor protease activity is dispensable for virus RNA polymerase function. J. Virol. 2010, 84, 216–226. [Google Scholar] [CrossRef] [Green Version]
- Dunn, E.F.; Pritlove, D.C.; Jin, H.; Elliott, R.M. Transcription of a recombinant bunyavirus RNA template by transiently expressed bunyavirus proteins. Virology 1995, 211, 133–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beerens, N.; Selisko, B.; Ricagno, S.; Imbert, I.; Van Der Zanden, L.; Snijder, E.J.; Canard, B. De novo initiation of RNA synthesis by the arterivirus RNA-dependent RNA polymerase. J. Virol. 2007, 81, 8384–8395. [Google Scholar] [CrossRef] [Green Version]
- Boonrod, K.; Chotewutmontri, S.; Galetzka, D.; Krczal, G. Analysis of tombusvirus revertants to identify essential amino acid residues within RNA-dependent RNA polymerase motifs. J. Gen. Virol. 2005, 86, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, A.O.; Spaan, W.J.M.; Snijder, E.J. Nidovirus transcription: How to make sense? J. Gen. Virol. 2006, 87, 1403–1421. [Google Scholar] [CrossRef] [PubMed]
- Qamar, M.T.; Ashfaq, U.A.; Tusleem, K.; Mumtaz, A.; Tariq, Q.; Goheer, A.; Ahmed, B. In-silico identification and evaluation of plant flavonoids as dengue NS2B/NS3 protease inhibitors using molecular docking and simulation approach. Pak. J. Pharm. Sci. 2017, 30, 2119–2137. [Google Scholar] [PubMed]
- Muhseen, Z.T.; Hameed, A.R.; Al-Hasani, H.M.; Ahmad, S.; Li, G. Computational Determination of Potential Multiprotein Targeting Natural Compounds for Rational Drug Design Against SARS-COV-2. Molecules 2021, 26, 674. [Google Scholar] [CrossRef]
- Piper, M.E.; Gerrard, S.R. A novel system for identification of inhibitors of Rift Valley fever virus replication. Viruses 2010, 2, 731–747. [Google Scholar] [CrossRef] [Green Version]
- Ausubel, F.M. Current Protocols in Molecular Biology; Greene Pub. Associates and Wiley-Interscience: New York, NY, USA, 1987; Volume 1. [Google Scholar]
- Graci, J.D.; Cameron, C.E. Mechanisms of action of ribavirin against distinct viruses. Rev. Med. Virol. 2006, 16, 37–48. [Google Scholar] [CrossRef]
- Allison, A.C.; Eugui, E.M. Mycophenolate mofetil and its mechanisms of action. Immunopharmacology 2000, 47, 85–118. [Google Scholar] [CrossRef]
- Seley-Radtke, K.L.; Yates, M.K. The evolution of nucleoside analogue antivirals: A review for chemists and non-chemists. Part 1: Early structural modifications to the nucleoside scaffold. Antivir. Res. 2018, 154, 66–86. [Google Scholar] [CrossRef] [PubMed]
- Kuchta, R.D. Nucleotide Analogues as Probes for DNA and RNA Polymerases. Curr. Protoc. Chem. 2010, 2, 111–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, J.; Kumar, S.; Li, X.; Jockusch, S.; Russo, J.J. Nucleotide analogues as inhibitors of viral polymerases. BioRxiv 2020. [Google Scholar]
- Ghazwani, M.Y.; Bakheit, A.H.; Hakami, A.R.; Alkahtani, H.M.; Almehizia, A.A. Virtual Screening and Molecular Docking Studies for Discovery of Potential RNA-Dependent RNA Polymerase Inhibitors. Crystals 2021, 11, 471. [Google Scholar] [CrossRef]
- Gajjar, N.D.; Dhameliya, T.M.; Shah, G.B. In search of RdRp and Mpro inhibitors against SARS CoV-2: Molecular docking, molecular dynamic simulations and ADMET analysis. J. Mol. Struct. 2021, 1239, 130488. [Google Scholar] [CrossRef]
- Lin, J.H.; Lu, A.Y. Role of pharmacokinetics and metabolism in drug discovery and development. Pharmacol. Rev. 1997, 49, 403–449. [Google Scholar]
- Tsaioun, K.; Bottlaender, M.; Mabondzo, A. ADDME–Avoiding Drug Development Mistakes Early: Central nervous system drug discovery perspective. BMC Neurol. 2009, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Alavijeh, M.S.; Chishty, M.; Qaiser, M.Z.; Palmer, A.M. Drug metabolism and pharmacokinetics, the blood-brain barrier, and central nervous system drug discovery. NeuroRx 2005, 2, 554–571. [Google Scholar] [CrossRef] [Green Version]
- Thomas, V.H.; Bhattachar, S.; Hitchingham, L.; Zocharski, P.; Naath, M.; Surendran, N.; Stoner, C.L.; El-Kattan, A. The road map to oral bioavailability: An industrial perspective. Expert Opin. Drug Metab. Toxicol. 2006, 2, 591–608. [Google Scholar] [CrossRef]
- Das, P.; Majumder, R.; Mandal, M.; Basak, P. In-Silico approach for identification of effective and stable inhibitors for COVID-19 main protease (Mpro) from flavonoid based phytochemical constituents of Calendula officinalis. J. Biomol. Struct. Dyn. 2020, 39, 6265–6280. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, I. Virtual screening of Microalgal compounds as potential inhibitors of Type 2 Human Transmembrane serine protease (TMPRSS2). arXiv 2021, arXiv:2108.13764. [Google Scholar]
- Anwar, F.; Saleem, U.; Ahmad, B.; Ashraf, M.; Rehman, A.U.; Froeyen, M.; Kee, L.Y.; Abdullah, I.; Mirza, M.U.; Ahmad, S. New naphthalene derivative for cost-effective AChE inhibitors for Alzheimer’s treatment: In silico identification, in vitro and in vivo validation. Comput. Biol. Chem. 2020, 89, 107378. [Google Scholar] [CrossRef] [PubMed]
- Guterres, H.; Im, W. Improving protein-ligand docking results with high-throughput molecular dynamics simulations. J. Chem. Inf. Model 2020, 60, 2189–2198. [Google Scholar] [CrossRef]
- Vanmeert, M.; Razzokov, J.; Mirza, M.U.; Weeks, S.D.; Schepers, G.; Bogaerts, A.; Rozenski, J.; Froeyen, M.; Herdewijn, P.; Pinheiro, V.B. Rational design of an XNA ligase through docking of unbound nucleic acids to toroidal proteins. Nucleic Acids Res. 2019, 47, 7130–7142. [Google Scholar] [CrossRef] [Green Version]
- Rehman, H.M.; Mirza, M.U.; Ahmad, M.A.; Saleem, M.; Froeyen, M.; Ahmad, S.; Gul, R.; Alghamdi, H.A.; Aslam, M.S.; Sajjad, M. A putative prophylactic solution for COVID-19: Development of novel multiepitope vaccine candidate against SARS-COV-2 by comprehensive immunoinformatic and molecular modelling approach. Biology 2020, 9, 296. [Google Scholar] [CrossRef]
- Koulgi, S.; Jani, V.; Uppuladinne, M.V.; Sonavane, U.; Joshi, R. Remdesivir-bound and ligand-free simulations reveal the probable mechanism of inhibiting the RNA dependent RNA polymerase of severe acute respiratory syndrome coronavirus 2. RSC Adv. 2020, 10, 26792–26803. [Google Scholar] [CrossRef]
- Hosseini, M.; Chen, W.; Xiao, D.; Wang, C. Computational molecular docking and virtual screening revealed promising SARS-CoV-2 drugs. Precis. Clin. Med. 2021, 4, 1–16. [Google Scholar] [CrossRef]
- Rudrapal, M.; Gogoi, N.; Chetia, D.; Khan, J.; Banwas, S.; Alshehri, B.; Alaidarous, M.A.; Laddha, U.D.; Khairnar, S.J.; Walode, S.G. Repurposing of phytomedicine-derived bioactive compounds with promising anti-SARS-CoV-2 potential: Molecular docking, MD simulation and drug-likeness/ADMET studies. Saudi J. Biol. Sci. 2021, 29, 2432–2446. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-point binding free energy calculation with MM/PBSA and MM/GBSA: Strategies and applications in drug design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef] [PubMed]
- Fiser, A.; Sali, A. Modeller: Generation and refinement of homology-based protein structure models. Methods Enzym. 2003, 374, 461–491. [Google Scholar] [CrossRef]
- Fan, H.; Irwin, J.J.; Webb, B.M.; Klebe, G.; Shoichet, B.K.; Sali, A. Molecular Docking Screens Using Comparative Models of Proteins. J. Chem. Inf. Model 2009, 49, 2512–2527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariani, V.; Kiefer, F.; Schmidt, T.; Haas, J.; Schwede, T. Assessment of template based protein structure predictions in CASP9. Proteins 2011, 79 (Suppl. 10), 37–58. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.J.; Mao, B.; Aramini, J.M.; Montelione, G.T. Assessment of template-based protein structure predictions in CASP10. Proteins 2014, 82 (Suppl. 2), 43–56. [Google Scholar] [CrossRef] [Green Version]
- Webb, B.; Sali, A. Protein Structure Modeling with MODELLER. Methods Mol. Biol. 2014, 1137, 1–15. [Google Scholar] [CrossRef]
- Consortium, U. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Sussman, J.L.; Lin, D.; Jiang, J.; Manning, N.O.; Prilusky, J.; Ritter, O.; Abola, E.E. Protein Data Bank (PDB): Database of three-dimensional structural information of biological macromolecules. Acta Crystallogr. Sect. D Biol. Crystallogr. 1998, 54, 1078–1084. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Eswar, N.; Eramian, D.; Webb, B.; Shen, M.-Y.; Sali, A. Protein structure modeling with MODELLER. In Structural Proteomics; Springer: Berlin/Heidelberg, Germany, 2008; pp. 145–159. [Google Scholar]
- Querin, O.; Young, V.; Steven, G.; Xie, Y. Computational efficiency and validation of bi-directional evolutionary structural optimisation. Comput. Methods Appl. Mech. Eng. 2000, 189, 559–573. [Google Scholar] [CrossRef]
- Tahir ul Qamar, M.; Mirza, M.U.; Song, J.-M.; Rao, M.J.; Zhu, X.; Chen, L.-L. Probing the structural basis of Citrus phytochrome B using computational modelling and molecular dynamics simulation approaches. J. Mol. Liq. 2021, 340, 116895. [Google Scholar] [CrossRef]
- Gopalakrishnan, K.; Sowmiya, G.; Sheik, S.; Sekar, K. Ramachandran plot on the web (2.0). Protein Pept. Lett. 2007, 14, 669–671. [Google Scholar] [CrossRef] [PubMed]
- Case, D.; Darden, T.; Cheatham, T., III; Simmerling, C.; Wang, J.; Duke, R.; Luo, R.; Crowley, M.; Walker, R.; Zhang, W. AMBER 10; University of California: San Francisco, CA, USA, 2008. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Dallakyan, S.; Olson, A.J. Small-Molecule Library Screening by Docking with PyRx. In Chemical Biology; Springer: Berlin/Heidelberg, Germany, 2015; pp. 243–250. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Jejurikar, B.L.; Rohane, S.H. Drug Designing in Discovery Studio. Asian J. Res. Chem. 2021, 14, 135–138. [Google Scholar]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, A.; Arooj, M.; Butt, T.T.; Zahid, S.; Zahid, F.; Jafar, T.H.; Waquar, S.; Gan, S.H.; Ahmad, S.; Mirza, M.U. In silico and in vivo characterization of cabralealactone, solasodin and salvadorin in a rat model: Potential anti-inflammatory agents. Drug Des. Dev. Ther. 2018, 12, 1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naheed, N.; Maher, S.; Saleem, F.; Khan, A.; Wadood, A.; Rasheed, S.; Choudhary, M.I.; Froeyen, M.; Abdullah, I.; Mirza, M.U. New isolate from Salvinia molesta with antioxidant and urease inhibitory activity. Drug Dev. Res. 2021, 82, 1169–1181. [Google Scholar] [CrossRef] [PubMed]
- Alamri, M.A.; Tahir ul Qamar, M.; Mirza, M.U.; Bhadane, R.; Alqahtani, S.M.; Muneer, I.; Froeyen, M.; Salo-Ahen, O.M.H. Pharmacoinformatics and molecular dynamics simulation studies reveal potential covalent and FDA-approved inhibitors of SARS-CoV-2 main protease 3CLpro. J. Biomol. Struct. Dyn. 2020, 39, 4936–4948. [Google Scholar] [CrossRef] [PubMed]
- Man, V.H.; Wu, X.; He, X.; Xie, X.-Q.; Brooks, B.R.; Wang, J. Determination of van der Waals Parameters Using a Double Exponential Potential for Nonbonded Divalent Metal Cations in TIP3P Solvent. J. Chem. Theory Comput. 2021, 17, 1086–1097. [Google Scholar] [CrossRef]
- Amin, S.A.; Ghosh, K.; Gayen, S.; Jha, T. Chemical-informatics approach to COVID-19 drug discovery: Monte Carlo based QSAR, virtual screening and molecular docking study of some in-house molecules as papain-like protease (PLpro) inhibitors. J. Biomol. Struct. Dyn. 2021, 39, 4764–4773. [Google Scholar] [CrossRef]
- Ngo, S.T.; Quynh Anh Pham, N.; Thi Le, L.; Pham, D.-H.; Vu, V.V. Computational determination of potential inhibitors of SARS-CoV-2 main protease. J. Chem. Inf. Model 2020, 60, 5771–5780. [Google Scholar] [CrossRef]
- Kräutler, V.; Van Gunsteren, W.F.; Hünenberger, P.H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 2001, 22, 501–508. [Google Scholar] [CrossRef]
- Bussi, G.; Parrinello, M. Accurate sampling using Langevin dynamics. Phys. Rev. E 2007, 75, 056707. [Google Scholar] [CrossRef] [Green Version]
- Bhowmik, D.; Nandi, R.; Jagadeesan, R.; Kumar, N.; Prakash, A.; Kumar, D. Identification of potential inhibitors against SARS-CoV-2 by targeting proteins responsible for envelope formation and virion assembly using docking based virtual screening, and pharmacokinetics approaches. Infect. Genet. Evol. 2020, 84, 104451. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham III, T.E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Kouznetsova, V.L.; Zhang, A.; Tatineni, M.; Miller, M.A.; Tsigelny, I.F. Potential COVID-19 papain-like protease PLpro inhibitors: Repurposing FDA-approved drugs. PeerJ 2020, 8, e9965. [Google Scholar] [CrossRef] [PubMed]
- Alamri, M.A.; ul Qamar, M.T.; Afzal, O.; Alabbas, A.B.; Riadi, Y.; Alqahtani, S.M. Discovery of anti-MERS-CoV small covalent inhibitors through pharmacophore modeling, covalent docking and molecular dynamics simulation. J. Mol. Liq. 2021, 330, 115699. [Google Scholar] [CrossRef]
- Kuzmanic, A.; Zagrovic, B. Determination of ensemble-average pairwise root mean-square deviation from experimental B-factors. Biophys. J. 2010, 98, 861–871. [Google Scholar] [CrossRef] [Green Version]
- Homeyer, N.; Gohlke, H. Free energy calculations by the molecular mechanics Poisson— Boltzmann surface area method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Duan, L.; Chen, F.; Liu, H.; Wang, Z.; Pan, P.; Zhu, F.; Zhang, J.Z.H.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 7. Entropy effects on the performance of end-point binding free energy calculation approaches. Phys. Chem. Chem. Phys. 2018, 20, 14450–14460. [Google Scholar] [CrossRef] [PubMed]
- Tuccinardi, T. What is the current value of MM/PBSA and MM/GBSA methods in drug discovery? Expert Opin. Drug Discov. 2021, 16, 1233–1237. [Google Scholar] [CrossRef] [PubMed]
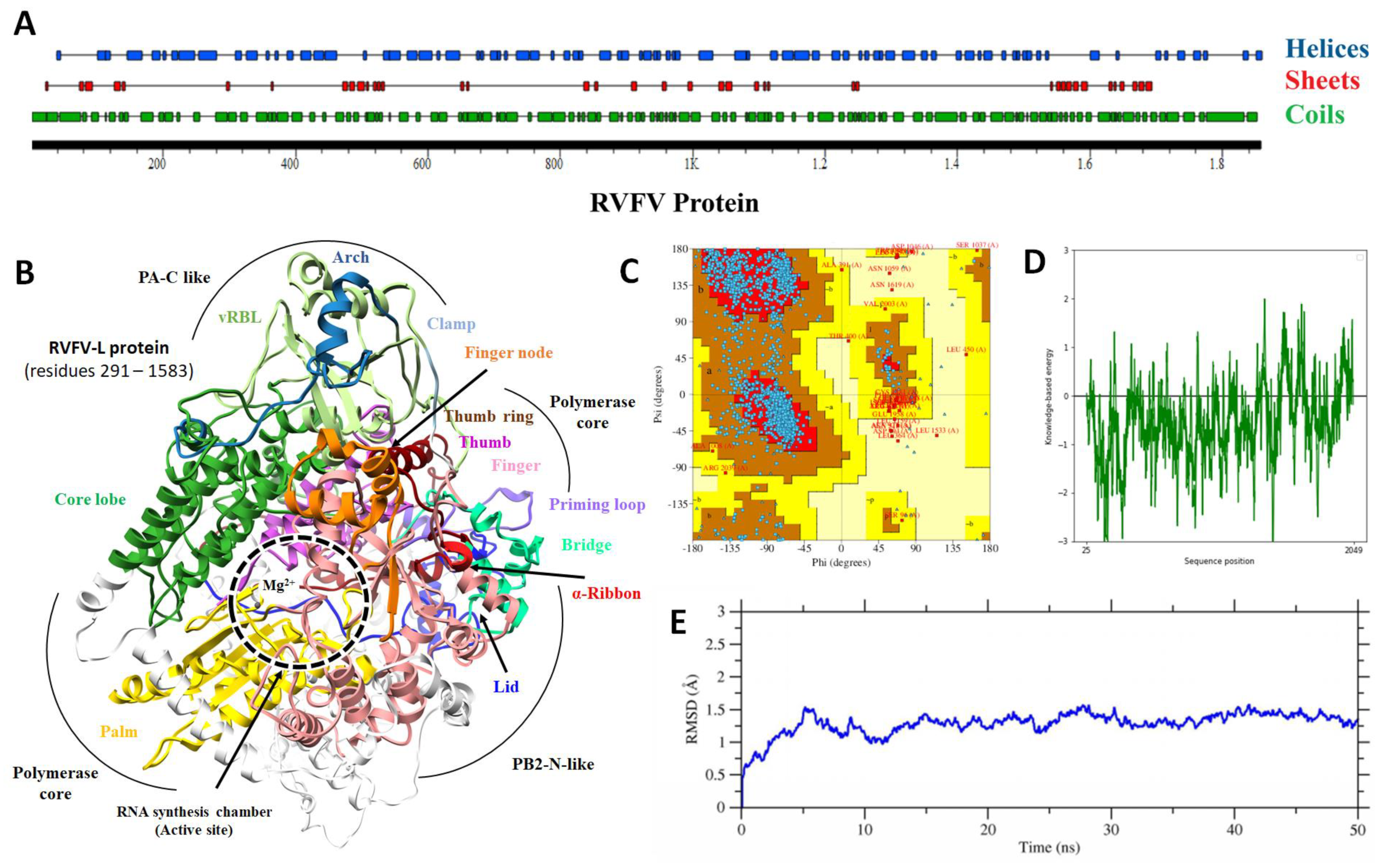




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Docked Complex | Chemical Structure | Binding Energy kcal/mol | Binding Residues (Amino Acid ID) | |
|---|---|---|---|---|
| Van der Waals | Hydrogen Bonds | |||
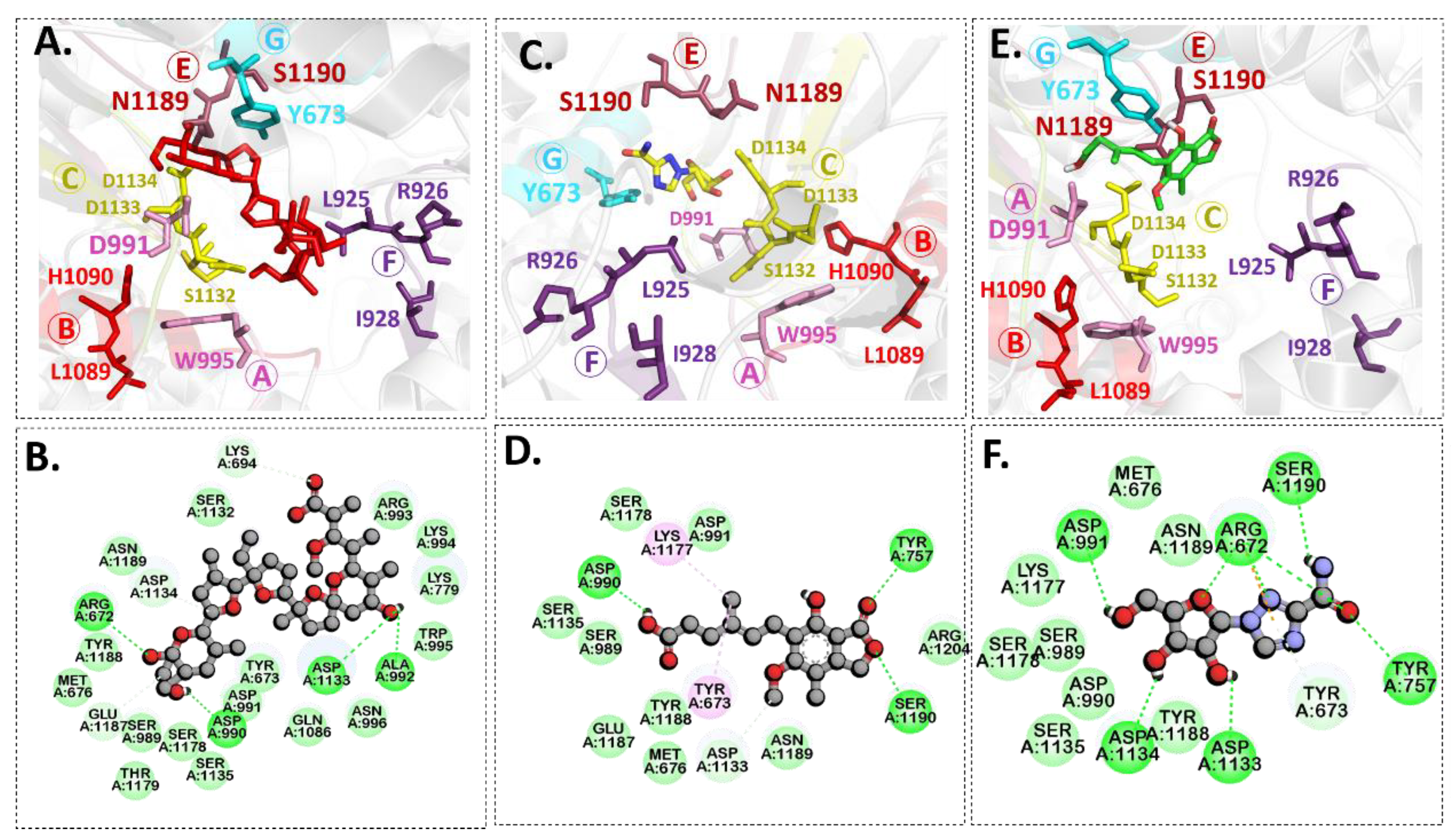
| A-317491 |  | −8.7 | 673, 1132, 1133 | 1090, 995, 1134, 925, 1086, 924, 996, 779, 992, 1177, 993, 991, 676, 672, 1188, 1189 |
| Monensin | 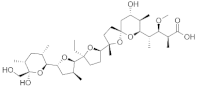 | −8.5 | 676, 1187, 989, 1179, 1135, 991, 673, 995, 694, 1134, 1189 | 672, 990, 1133, 992 |
| VER155008 | 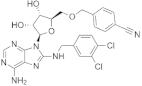 | −7.8 | 925, 1189, 1187, 676, 989, 1179, 1178, 673, 1133, 1086 | 779, 1190, 1188, 672, 991, 1177 |
| Khasianine |  | −7.1 | 1189, 1204, 924, 1133, 925, 993, 995, 996, 994, 779, 694, 1177, 670, 669, 673, 1134 | 997 |
| Mycophenolic acid | 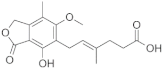 | −7.0 | 1181, 1180, 1127, 1126, 1183, 751, 667, 985, 1171, 1128, 668, 1170 | 1197, 984 |
| Ribavirin |  | −6.7 | 1135, 1188, 990, 989, 1178, 1177, 676, 1189 | 1134, 757, 1190, 672, 991, 1133 |
| Parameters | Compounds | ||
|---|---|---|---|
| A-317491 | Khasianine | VER155008 | |
| Absorption | |||
| BBB | No | No | No |
| GI absorption | Low | Low | Low |
| Caco-2 permeability | −6.019 | −5.356 | −5.727 |
| Human oral bioavailability | 0.56 | 0.17 | 0.17 |
| Log P | 5.296 | 2.723 | 2.136 |
| TPSA (Å2) | 141.44 | 179.56 | 166.21 |
| Metabolism | |||
| P-glycoprotein substrate | No | Yes | No |
| P-glycoprotein inhibitor | No | No | No |
| CYP450 2C9 substrate | No | No | No |
| CYP450 2D6 substrate | No | No | No |
| CYP450 3A4 substrate | No | No | No |
| CYP450 1A2 inhibitor | No | No | No |
| CYP450 2C9 inhibitor | Yes | No | No |
| CYP450 2D6 inhibitor | No | No | No |
| CYP450 2C19 inhibitor | No | No | No |
| CYP450 3A4 inhibitor | No | No | Yes |
| Toxicity | |||
| AMES Toxicity | Non-toxic | Non-toxic | Non-toxic |
| Carcinogens | Non-carcinogenic | Non-carcinogenic | Non-carcinogenic |
| Acute oral toxicity | 2500 mg/kg | 500 mg/kg | 7000 mg/kg |
| Energy Component | Average | Standard Error of Mean | Average | Standard Error of Mean | Average | Standard Error of Mean | Average | Standard Error of Mean | Average | Standard Error of Mean | Average | Standard Error of Mean |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A-1317491 | Khasianine | Monensin | Mycophenolic Acid | Ribavirin | VER155008 | |||||||
| MM-GBSA | ||||||||||||
| ΔEvdw | −65.37 | 2.72 | −54.45 | 4.89 | −53.83 | 3.23 | −48.81 | 5.00 | −26.20 | 3.57 | −51.64 | 3.87 |
| ΔEele | −56.86 | 6.92 | −257.10 | 17.11 | −80.72 | 5.55 | −30.21 | 4.83 | −155.72 | 7.14 | −87.45 | 11.03 |
| ΔGp | 74.46 | 6.80 | 275.40 | 16.39 | 100.29 | 4.76 | 42.30 | 4.30 | 158.69 | 5.83 | 98.08 | 8.68 |
| ΔGnp | −7.10 | 0.19 | −6.51 | 0.31 | −6.92 | 0.39 | −5.04 | 0.18 | −3.75 | 0.26 | −5.72 | 0.29 |
| ΔEMM | −122.23 | 7.68 | −311.55 | 18.54 | −134.56 | 6.12 | −79.03 | 8.37 | −181.93 | 7.33 | −139.09 | 9.68 |
| ΔGsol | 67.36 | 6.68 | 268.88 | 16.23 | 93.36 | 4.68 | 37.26 | 4.21 | 154.93 | 5.86 | 92.35 | 8.74 |
| ΔGtotal | −54.87 | 2.75 | −42.66 | 4.16 | −41.19 | 2.89 | −41.76 | 4.96 | −26.99 | 4.216 | −46.73 | 3.41 |
| MM-PBSA | ||||||||||||
| ΔEvdw | −65.37 | 2.72 | −54.45 | 4.89 | −53.83 | 3.23 | −48.81 | 5.00 | −26.20 | 3.57 | −51.64 | 3.87 |
| ΔEele | −56.86 | 6.92 | −257.10 | 17.1 | −80.72 | 5.55 | −30.21 | 4.83 | −155.72 | 7.14 | −87.45 | 11.03 |
| ΔGp | 95.85 | 7.89 | 288.12 | 16.80 | 114.98 | 8.24 | 57.57 | 5.53 | 166.11 | 6.22 | 118.11 | 7.49 |
| ΔGnp | −4.73 | 0.10 | −4.99 | 0.13 | −5.52 | 0.16 | −3.18 | 0.08 | −2.34 | 0.07 | −4.44 | 0.12 |
| ΔEMM | −122.2 | 7.68 | −311.55 | 18.54 | −134.56 | 6.12 | −79.03 | 8.37 | −181.93 | 7.33 | −139.09 | 9.68 |
| ΔGsol | 91.12 | 7.84 | 283.12 | 16.76 | 109.45 | 8.19 | 54.39 | 5.53 | 163.77 | 6.20 | 113.66 | 7.49 |
| ΔGtotal | −31.11 | 4.89 | −28.42 | 4.30 | −25.10 | 5.39 | −24.64 | 6.16 | −18.16 | 5.23 | −25.42 | 5.87 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alamri, M.A.; Mirza, M.U.; Adeel, M.M.; Ashfaq, U.A.; Tahir ul Qamar, M.; Shahid, F.; Ahmad, S.; Alatawi, E.A.; Albalawi, G.M.; Allemailem, K.S.; et al. Structural Elucidation of Rift Valley Fever Virus L Protein towards the Discovery of Its Potential Inhibitors. Pharmaceuticals 2022, 15, 659. https://doi.org/10.3390/ph15060659
Alamri MA, Mirza MU, Adeel MM, Ashfaq UA, Tahir ul Qamar M, Shahid F, Ahmad S, Alatawi EA, Albalawi GM, Allemailem KS, et al. Structural Elucidation of Rift Valley Fever Virus L Protein towards the Discovery of Its Potential Inhibitors. Pharmaceuticals. 2022; 15(6):659. https://doi.org/10.3390/ph15060659
Chicago/Turabian StyleAlamri, Mubarak A., Muhammad Usman Mirza, Muhammad Muzammal Adeel, Usman Ali Ashfaq, Muhammad Tahir ul Qamar, Farah Shahid, Sajjad Ahmad, Eid A. Alatawi, Ghadah M. Albalawi, Khaled S. Allemailem, and et al. 2022. "Structural Elucidation of Rift Valley Fever Virus L Protein towards the Discovery of Its Potential Inhibitors" Pharmaceuticals 15, no. 6: 659. https://doi.org/10.3390/ph15060659
APA StyleAlamri, M. A., Mirza, M. U., Adeel, M. M., Ashfaq, U. A., Tahir ul Qamar, M., Shahid, F., Ahmad, S., Alatawi, E. A., Albalawi, G. M., Allemailem, K. S., & Almatroudi, A. (2022). Structural Elucidation of Rift Valley Fever Virus L Protein towards the Discovery of Its Potential Inhibitors. Pharmaceuticals, 15(6), 659. https://doi.org/10.3390/ph15060659