Oncostatin M Receptor as a Therapeutic Target for Radioimmune Therapy in Synovial Sarcoma

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
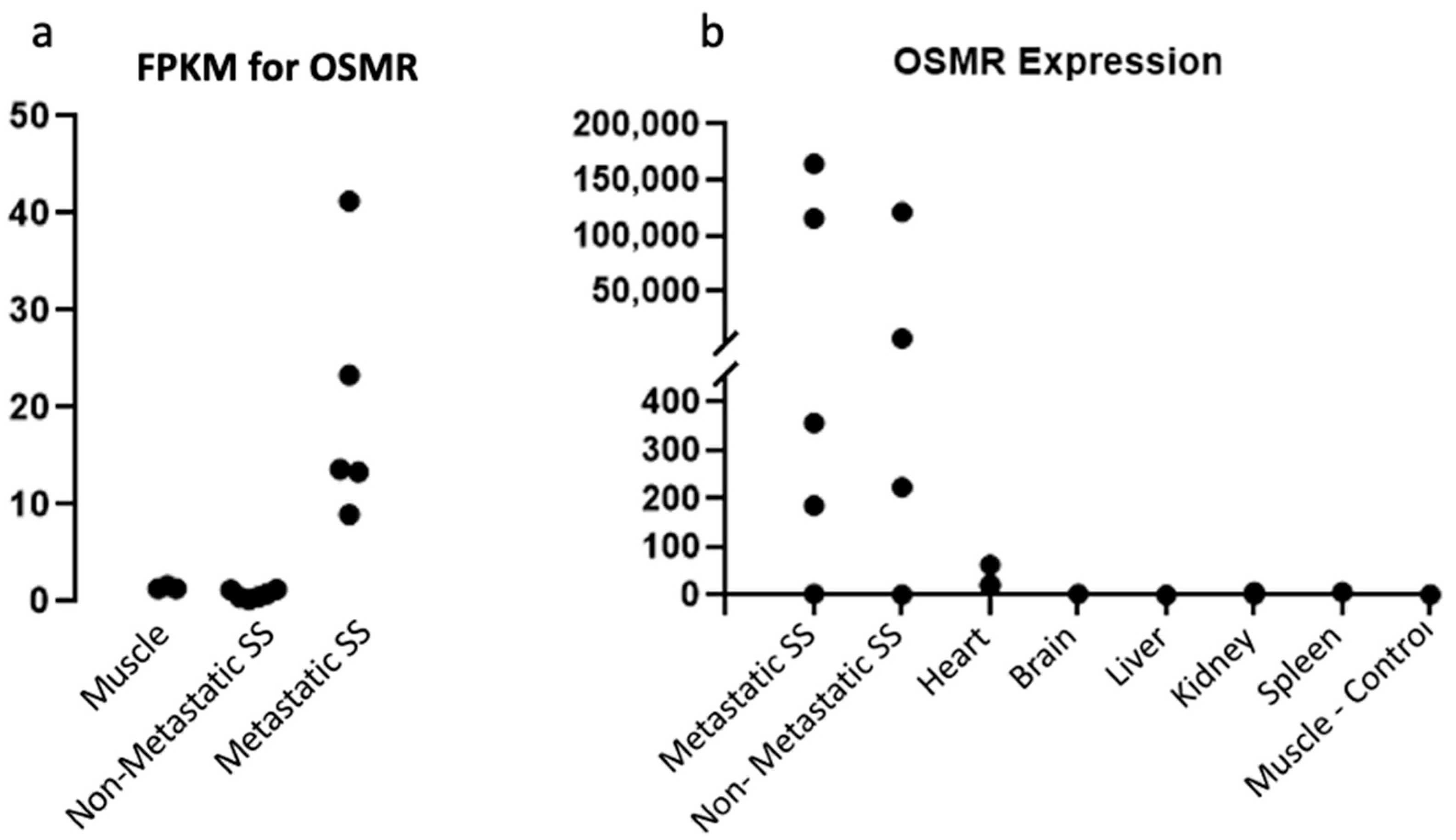
2.1. OSMR Is Present in Sufficient Quantities for Therapeutic Targeting
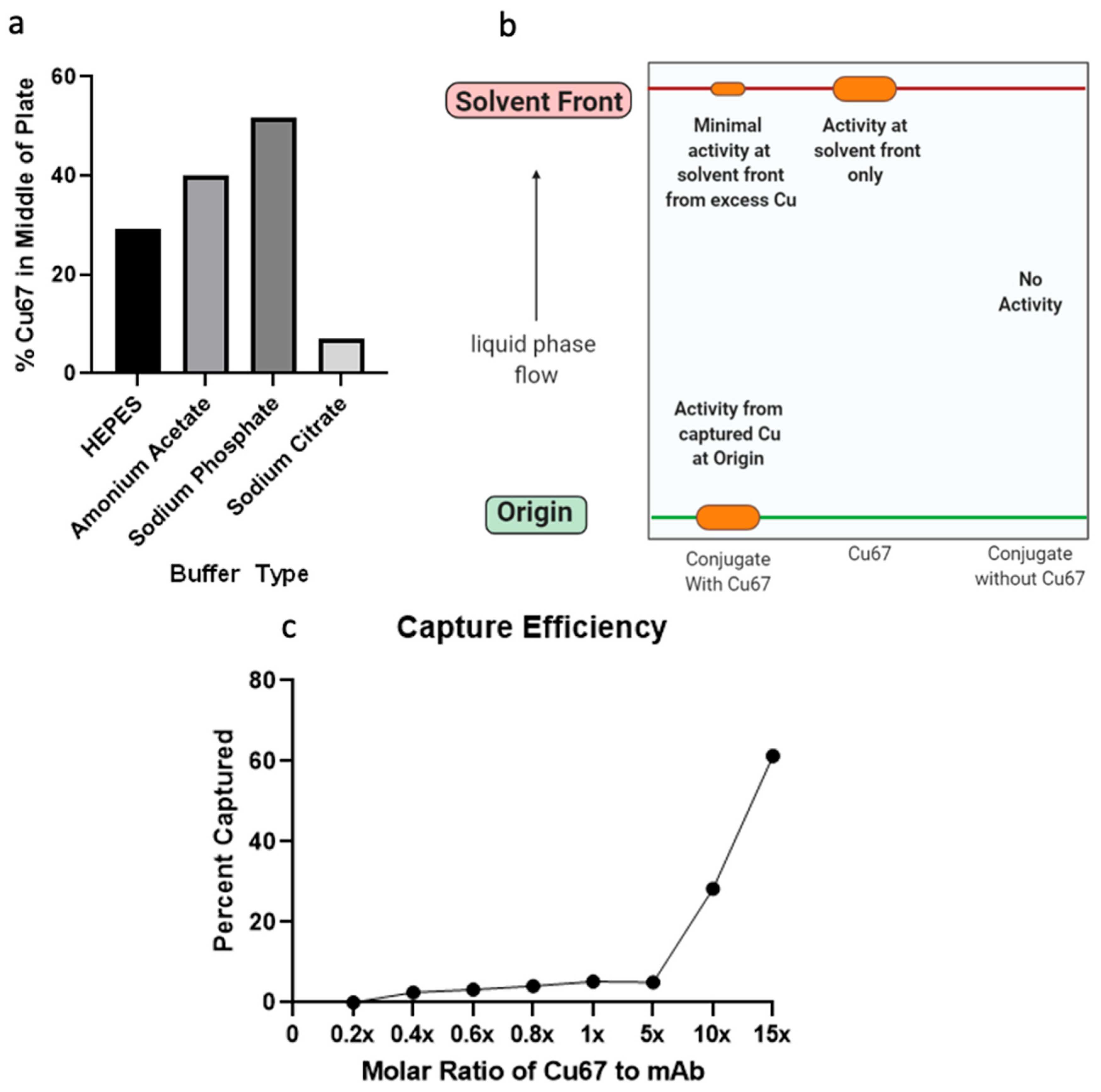
2.2. Anti-OSMR Drug Design
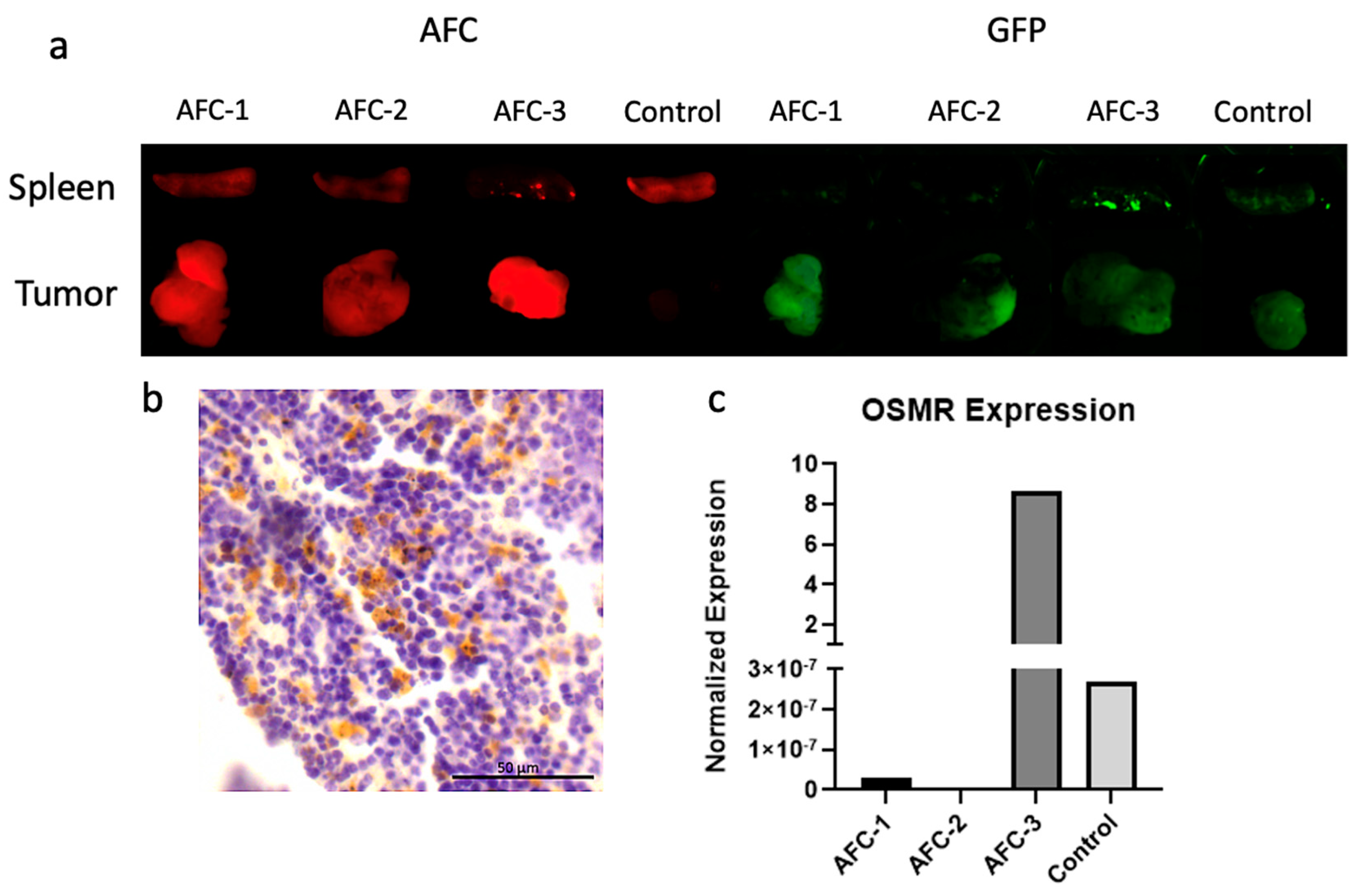
2.3. Pharmacokinetic Studies of Antibody-Fluorescent Conjugate
3. Discussion
4. Materials and Methods
4.1. Mouse Models
4.2. RNAseq
4.3. Slide Preparation
4.4. Immunohistochemistry
4.5. Western Blot
4.6. RTqPCR
4.7. ADC Preparation
4.7.1. Antibody Purification
4.7.2. Chelator Conjugation
4.7.3. Radiolabeling of ADC
4.7.4. AFC Preparation
4.8. Murine Pharmacokinetic Studies
4.9. Sandwich ELISA Binding Assay
4.10. Thin Layer Chromatography
4.11. Thermal Shift Stability Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Surveillance, Epidemiology, and End Results Program (SEER). Cancer of Soft Tissue Including Heart—Cancer Stat Facts. Available online: https://seer.cancer.gov/statfacts/html/soft.html (accessed on 5 October 2020).
- National Cancer Institute. Synovial Sarcoma. 2019. Available online: https://www.cancer.gov/pediatric-adult-rare-tumor/rare-tumors/rare-soft-tissue-tumors/synovial-sarcoma (accessed on 18 August 2020).
- Ladenstein, R.; Treuner, J.; Koscielniak, E.; d’Oleire, F.; Keim, M.; Gadner, H.; Niethammer, D.; Jürgens, H.; Ritter, J.; Schmidt, D. Synovial sarcoma of childhood and adolescence. Report of the German CWS-81 study. Cancer 1993, 71, 3647–3655. [Google Scholar] [CrossRef]
- Eilber, F.C.; Brennan, M.F.; Eilber, F.R.; Eckardt, J.J.; Grobmyer, S.R.; Riedel, E.; Forscher, C.; Maki, R.G.; Singer, S. Chemotherapy Is Associated with Improved Survival in Adult Patients with Primary Extremity Synovial Sarcoma. Ann. Surg. 2007, 246, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Cancer Research UK. Survival for Soft Tissue Sarcomas. Available online: https://www.cancerresearchuk.org/about-cancer/soft-tissue-sarcoma/survival (accessed on 9 May 2022).
- Deshmukh, R.; Mankin, H.J.; Singer, S. Synovial Sarcoma: The Importance of Size and Location for Survival. Clin. Orthop. Relat. Res. 2004, 419, 155–161. [Google Scholar] [CrossRef]
- Krieg, A.H.; Hefti, F.; Speth, B.M.; Jundt, G.; Guillou, L.; Exner, U.G.; von Hochstetter, A.R.; Cserhati, M.D.; Fuchs, B.; Mouhsine, E.; et al. Synovial sarcomas usually metastasize after >5 years: A multicenter retrospective analysis with minimum follow-up of 10 years for survivors. Ann. Oncol. 2011, 22, 458–467. [Google Scholar] [CrossRef]
- Turc-Carel, C.; Dal Cin, P.; Limon, J.; Rao, U.; Li, F.P.; Corson, J.M.; Zimmerman, R.; Parry, D.M.; Cowan, J.M.; Sandberg, A.A. Involvement of chromosome X in primary cytogenetic change in human neoplasia: Nonrandom translocation in synovial sarcoma. Proc. Natl. Acad. Sci. USA 1987, 84, 1981–1985. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Abro, B.; Kaushal, M.; Chen, L.; Chen, T.; Gondim, M.; Yan, W.; Neidich, J.; Dehner, L.P.; Pfeifer, J.D. Tumor mutation burden and checkpoint immunotherapy markers in primary and metastatic synovial sarcoma. Hum. Pathol. 2020, 100, 15–23. [Google Scholar] [CrossRef]
- Potter, J.W.; Jones, K.B.; Barrott, J.J. Sarcoma—The standard-bearer in cancer discovery. Crit. Rev. Oncol. Hematol. 2018, 126, 1–5. [Google Scholar] [CrossRef]
- Richards, C.D. The Enigmatic Cytokine Oncostatin M and Roles in Disease. ISRN Inflamm. 2013, 2013, 512103. [Google Scholar] [CrossRef] [Green Version]
- Mahony, C.B.; Pasche, C.; Bertrand, J.Y. Oncostatin M and Kit-Ligand Control Hematopoietic Stem Cell Fate during Zebrafish Embryogenesis. Stem Cell Rep. 2018, 10, 1920–1934. [Google Scholar] [CrossRef]
- Tanaka, M.; Hirabayashi, Y.; Sekiguchi, T.; Inoue, T.; Katsuki, M.; Miyajima, A. Targeted disruption of oncostatin M receptor results in altered hematopoiesis. Blood 2003, 102, 3154–3162. [Google Scholar] [CrossRef] [Green Version]
- Mosley, B.; De Imus, C.; Friend, D.; Boiani, N.; Thoma, B.; Park, L.S.; Cosman, D. Dual Oncostatin M (OSM) Receptors: Cloning and characterization of an alternative signaling subunit conferring osm-specific receptor activation. J. Biol. Chem. 1996, 271, 32635–32643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Cancer Institute. Radiopharmaceuticals Emerging as New Cancer Therapy. 2020. Available online: https://www.cancer.gov/news-events/cancer-currents-blog/2020/radiopharmaceuticals-cancer-radiation-therapy (accessed on 5 April 2022).
- White, J.M.; Escorcia, F.E.; Viola, N.T. Perspectives on metals-based radioimmunotherapy (RIT): Moving forward. Theranostics 2021, 11, 6293–6314. [Google Scholar] [CrossRef] [PubMed]
- Gudkov, S.V.; Shilyagina, N.Y.; Vodeneev, V.A.; Zvyagin, A.V. Targeted Radionuclide Therapy of Human Tumors. Int. J. Mol. Sci. 2015, 17, 33. [Google Scholar] [CrossRef] [PubMed]
- Buchmann, I.; Meyer, R.G.; Mier, W.; Haberkorn, U. Myeloablative radioimmunotherapy in conditioning prior to haematological stem cell transplantation: Closing the gap between benefit and toxicity? Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 484–498. [Google Scholar] [CrossRef]
- Lhuillier, C.; Rudqvist, N.-P.; Elemento, O.; Formenti, S.C.; Demaria, S. Radiation therapy and anti-tumor immunity: Exposing immunogenic mutations to the immune system. Genome Med. 2019, 11, 40. [Google Scholar] [CrossRef] [Green Version]
- Barrott, J.J.; Kafchinski, L.A.; Jin, H.; Potter, J.W.; Kannan, S.D.; Kennedy, R.; Mosbruger, T.; Wang, W.-L.; Tsai, J.-W.; Araujo, D.M.; et al. Modeling synovial sarcoma metastasis in the mouse: PI3′-lipid signaling and inflammation. J. Exp. Med. 2016, 213, 2989–3005. [Google Scholar] [CrossRef]
- Yap, T.A.; Carden, C.P.; Kaye, S.B. Beyond chemotherapy: Targeted therapies in ovarian cancer. Nat. Rev. Cancer 2009, 9, 167–181. [Google Scholar] [CrossRef]
- Xie, Y.-H.; Chen, Y.-X.; Fang, J.-Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef]
- Baudino, T.A. Targeted Cancer Therapy: The Next Generation of Cancer Treatment. Curr. Drug Discov. Technol. 2015, 12, 3–20. [Google Scholar] [CrossRef]
- Targeted Drug Therapy for Soft Tissue Sarcoma. Available online: https://www.cancer.org/cancer/soft-tissue-sarcoma/treating/targeted-therapy.html (accessed on 5 April 2022).
- Qiao, Z.; Shiozawa, K.; Kondo, T. Proteomic approach toward determining the molecular background of pazopanib resistance in synovial sarcoma. Oncotarget 2017, 8, 109587–109595. [Google Scholar] [CrossRef] [Green Version]
- Desar, I.M.E.; Fleuren, E.D.G.; van der Graaf, W.T.A. Systemic Treatment for Adults with Synovial Sarcoma. Curr. Treat. Opt. Oncol. 2018, 19, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deller, M.C.; Hudson, K.R.; Ikemizu, S.; Bravo, J.; Jones, E.Y.; Heath, J.K. Crystal structure and functional dissection of the cytostatic cytokine oncostatin M. Structure 2000, 8, 863–874. [Google Scholar] [CrossRef]
- Cooper, M.S.; Ma, M.T.; Sunassee, K.; Shaw, K.P.; Williams, J.D.; Paul, R.L.; Donelly, P.S.; Blower, P.J. Comparison of 64Cu-complexing bifunctional chelators for radioimmunoconjugation: Labeling efficiency, specific activity and in vitro/in vivo stability. Bioconjug. Chem. 2012, 23, 1029–1039. [Google Scholar] [CrossRef] [Green Version]
- Hao, G.; Mastren, T.; Silvers, W.; Hassan, G.; Öz, O.K.; Sun, X. Copper-67 radioimmunotheranostics for simultaneous immunotherapy and immuno-SPECT. Sci. Rep. 2021, 11, 3622. [Google Scholar] [CrossRef] [PubMed]
- Poot, A.J.; Lam, M.G.E.H.; van Noesel, M.M. The Current Status and Future Potential of Theranostics to Diagnose and Treat Childhood Cancer. Front. Oncol. 2020, 10, 578286. [Google Scholar] [CrossRef] [PubMed]
- Larson, S.M.; Carrasquillo, J.A.; Cheung, N.-K.V.; Press, O.W. Radioimmunotherapy of human tumours. Nat. Rev. Cancer 2015, 15, 347–360. [Google Scholar] [CrossRef]
- Fisher, R.; Pusztai, L.; Swanton, C. Cancer heterogeneity: Implications for targeted therapeutics. Br. J. Cancer 2013, 108, 479–485. [Google Scholar] [CrossRef] [Green Version]
- The Human Protein Atlas. Expression of OSMR in Cancer—Summary. Available online: https://www.proteinatlas.org/ENSG00000145623-OSMR/pathology (accessed on 26 October 2021).
- Yang, Q.; Parker, C.L.; McCallen, J.D.; Lai, S.K. Addressing challenges of heterogeneous tumor treatment through bispecific protein-mediated pretargeted drug delivery. J. Control. Release 2015, 220, 715–726. [Google Scholar] [CrossRef] [Green Version]
- Fleuren, E.D.G.; Versleijen-Jonkers, Y.M.H.; Heskamp, S.; van Herpen, C.M.L.; Oyen, W.J.G.; van der Graaf, W.T.A.; Boerman, O.C. Theranostic applications of antibodies in oncology. Mol. Oncol. 2014, 8, 799–812. [Google Scholar] [CrossRef]
- Parker, C.; Nilsson, S.; Heinrich, D.; Helle, S.I.; O’Sullivan, J.M.; Fosså, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; Seke, M.; et al. Alpha Emitter Radium-223 and Survival in Metastatic Prostate Cancer. N. Engl. J. Med. 2013, 369, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Haldar, M.; Hancock, J.D.; Coffin, C.M.; Lessnick, S.L.; Capecchi, M.R. A Conditional Mouse Model of Synovial Sarcoma: Insights into a Myogenic Origin. Cancer Cell 2007, 11, 375–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geethadevi, A.; Nair, A.; Parashar, D.; Ku, Z.; Xiong, W.; Deng, H.; Li, Y.; George, J.; McAllister, D.M.; Sun, Y.; et al. Oncostatin M Receptor–Targeted Antibodies Suppress STAT3 Signaling and Inhibit Ovarian Cancer Growth. Cancer Res. 2021, 81, 5336–5352. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.Y.; Liu, Q.; Yang, H.; Tan, Q.; Gan, L.Q.; Ren, F.L.; Wang, H. OSMR gene effect on the pathogenesis of chronic autoimmune Urticaria via the JAK/STAT3 pathway. Mol. Med. 2018, 24, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCollum, S.; Kalivas, A.; Kirkham, M.; Kunz, K.; Okojie, J.; Pavek, A.; Barrott, J. Oncostatin M Receptor as a Therapeutic Target for Radioimmune Therapy in Synovial Sarcoma. Pharmaceuticals 2022, 15, 650. https://doi.org/10.3390/ph15060650
McCollum S, Kalivas A, Kirkham M, Kunz K, Okojie J, Pavek A, Barrott J. Oncostatin M Receptor as a Therapeutic Target for Radioimmune Therapy in Synovial Sarcoma. Pharmaceuticals. 2022; 15(6):650. https://doi.org/10.3390/ph15060650
Chicago/Turabian StyleMcCollum, Sarah, Austen Kalivas, Matthew Kirkham, Kaden Kunz, Jeffrey Okojie, Adriene Pavek, and Jared Barrott. 2022. "Oncostatin M Receptor as a Therapeutic Target for Radioimmune Therapy in Synovial Sarcoma" Pharmaceuticals 15, no. 6: 650. https://doi.org/10.3390/ph15060650
APA StyleMcCollum, S., Kalivas, A., Kirkham, M., Kunz, K., Okojie, J., Pavek, A., & Barrott, J. (2022). Oncostatin M Receptor as a Therapeutic Target for Radioimmune Therapy in Synovial Sarcoma. Pharmaceuticals, 15(6), 650. https://doi.org/10.3390/ph15060650