
Drug Design by Pharmacophore and Virtual Screening Approach


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
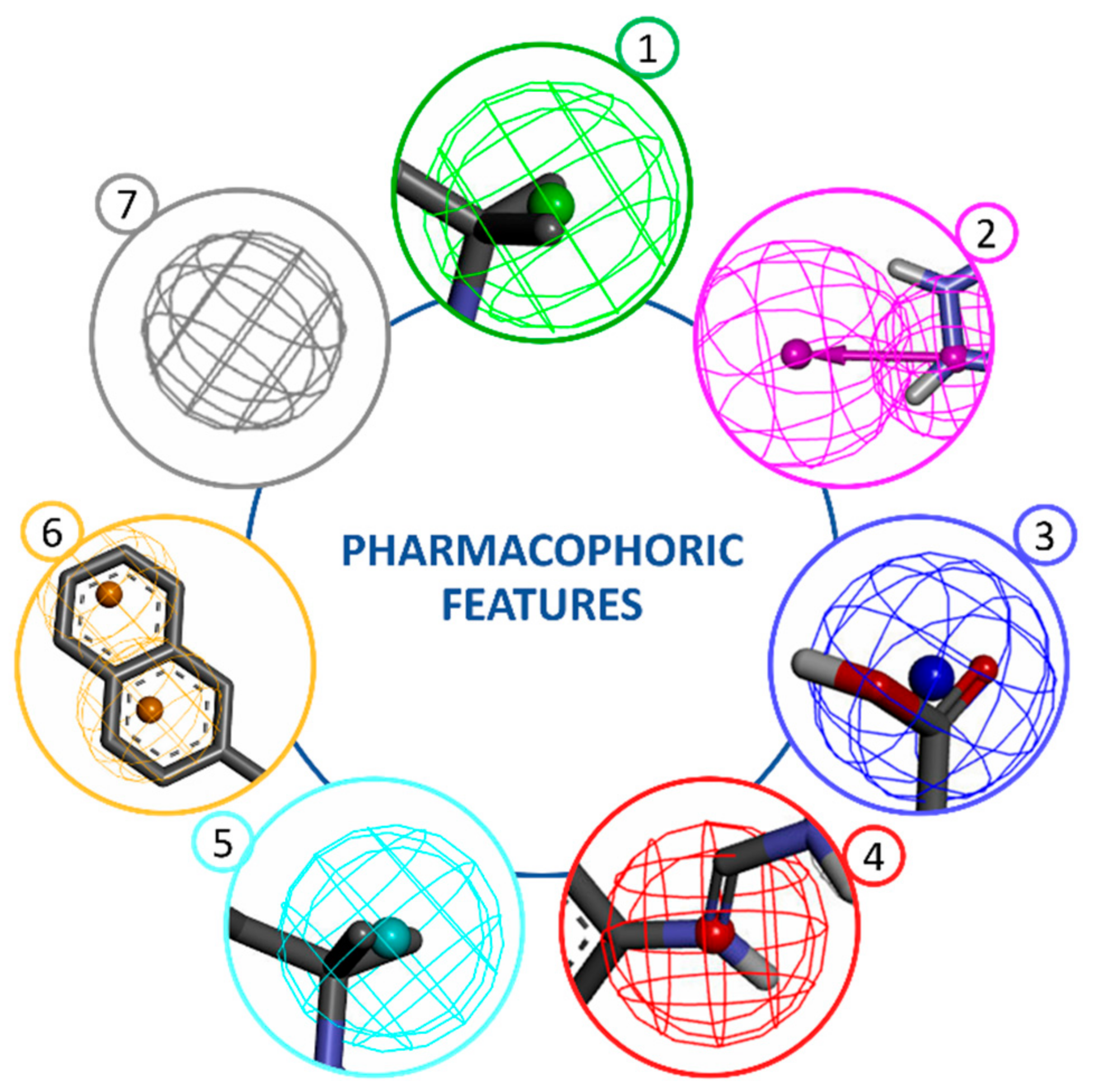
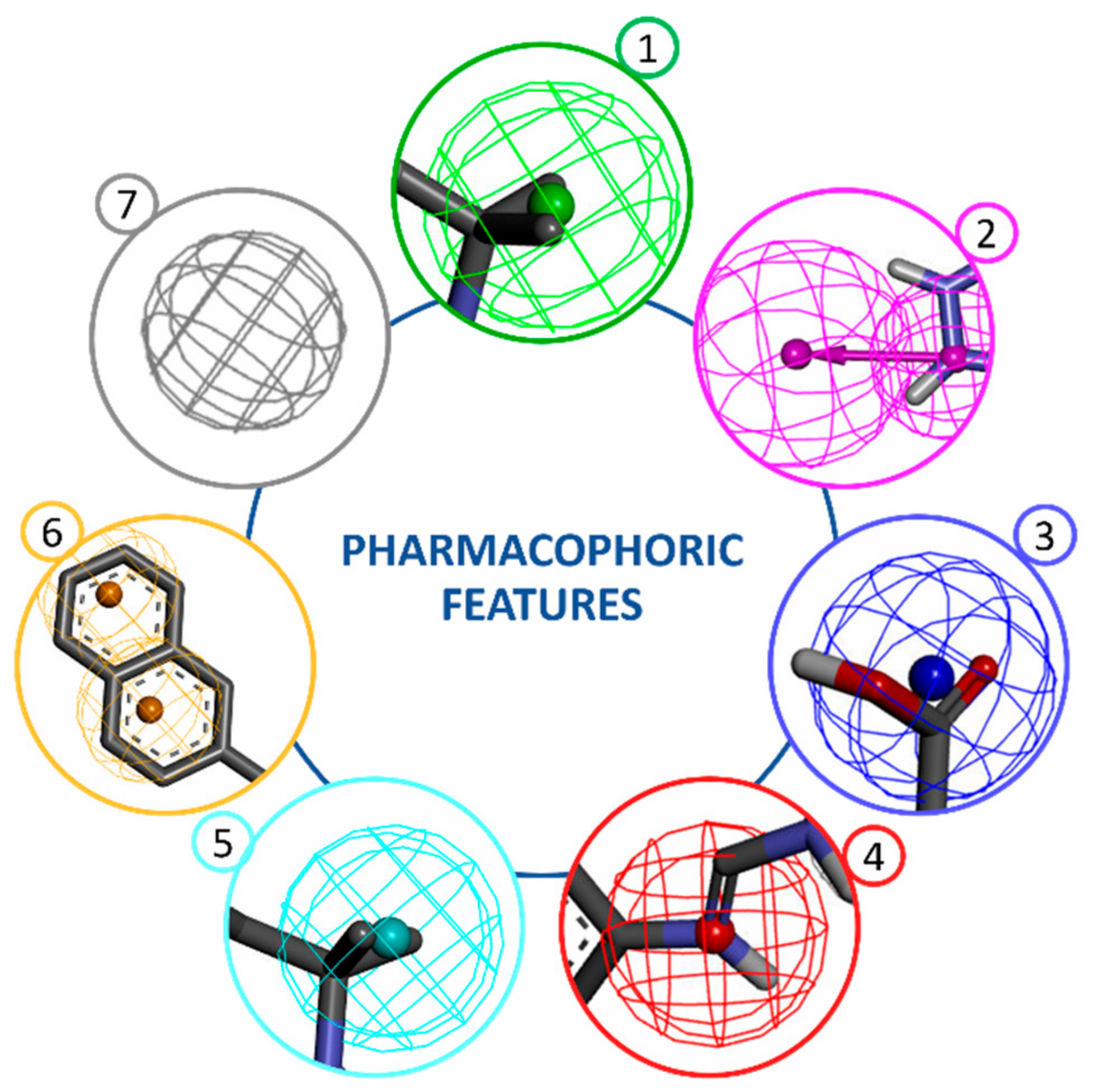
:1. Introduction
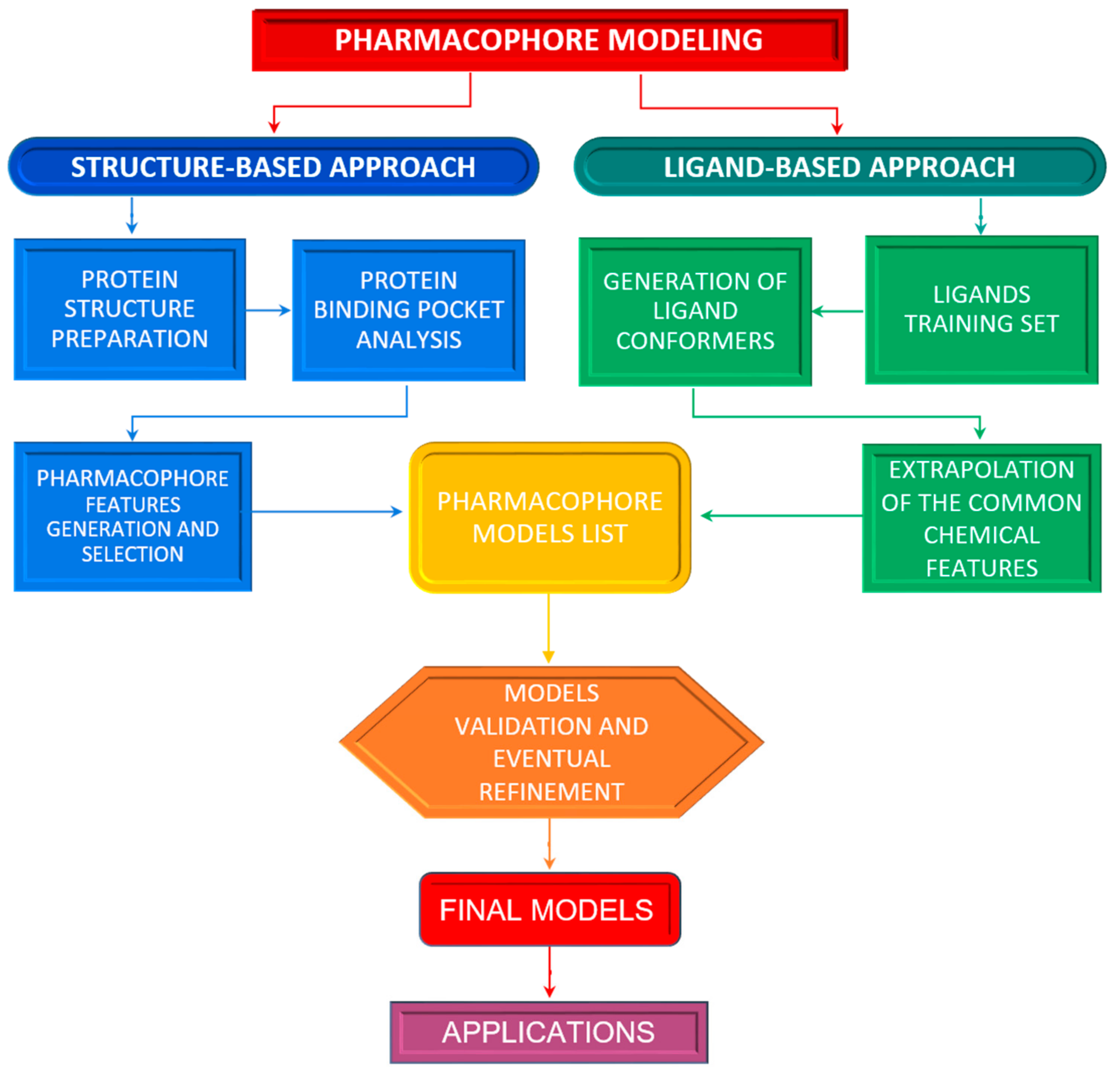
2. Structure-Based Pharmacophore Modelling
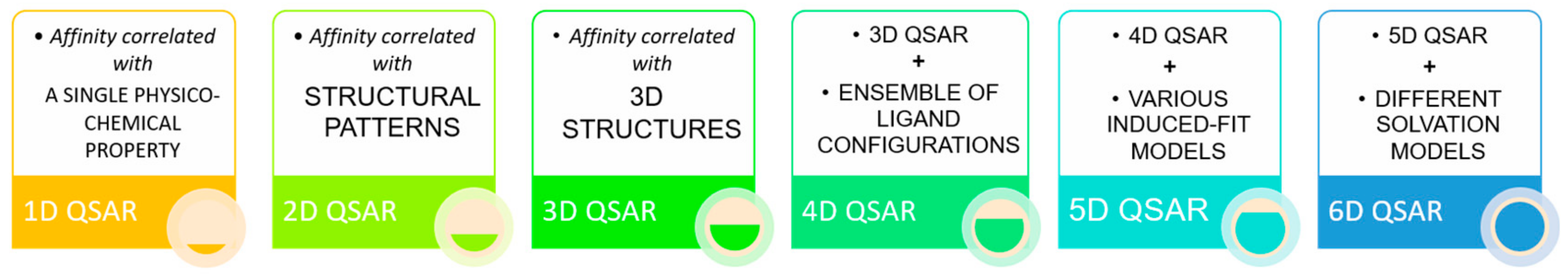
3. Ligand-Based Pharmacophore Modelling
Pharmacophore Models Validation
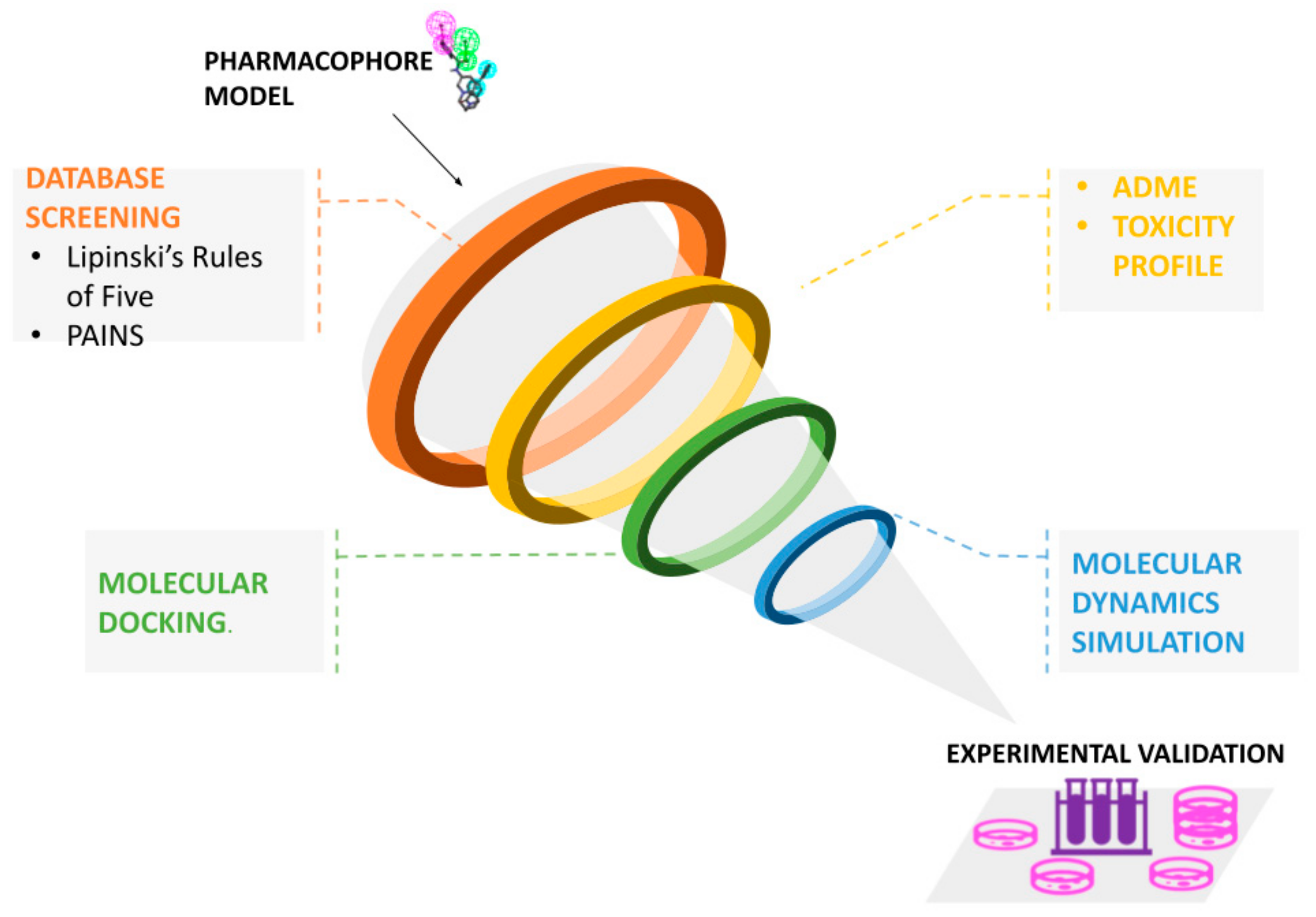
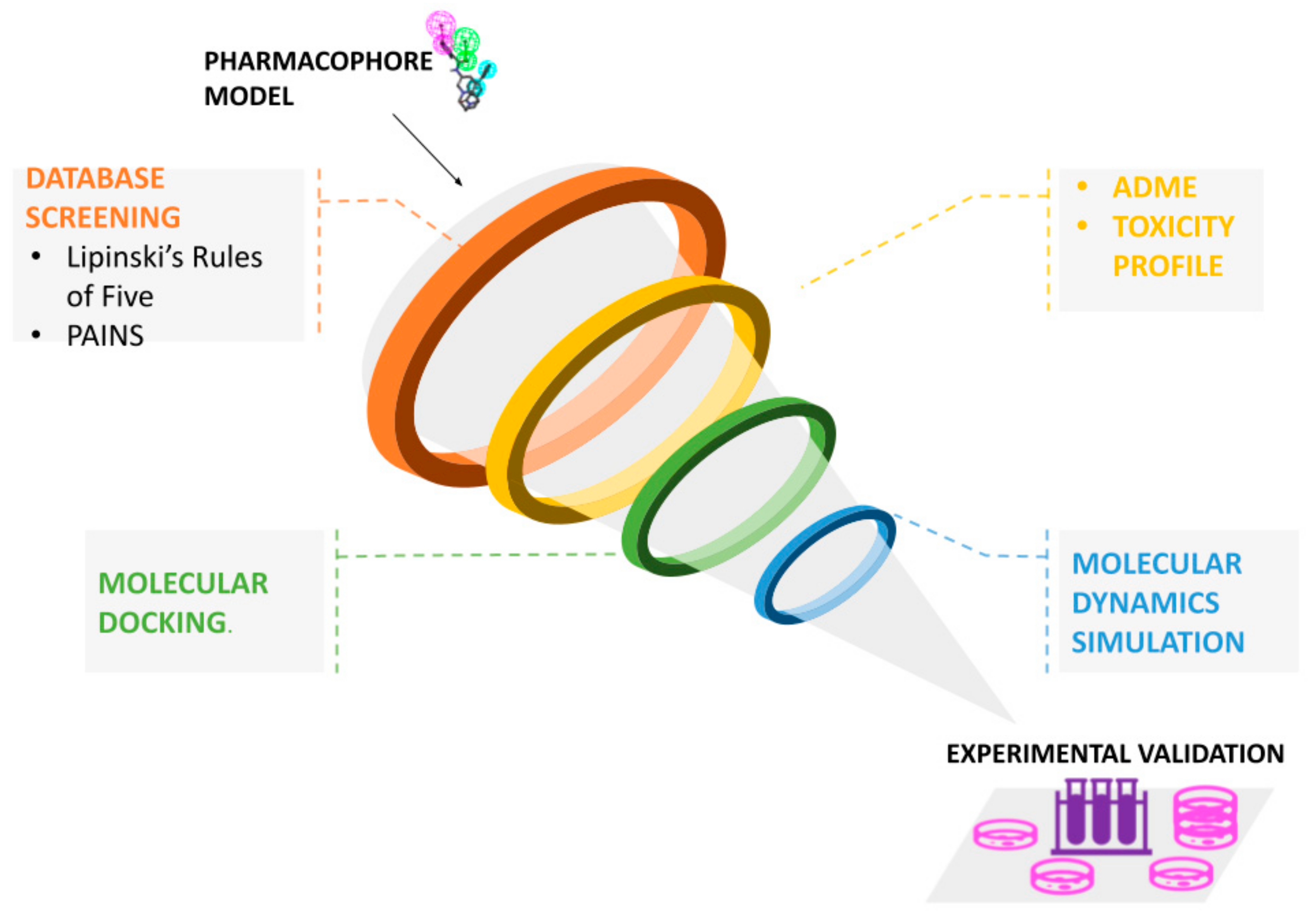
4. Pharmacophore-Based Virtual Screening
5. Limitations and Possible Solutions
6. Software for Pharmacophore Modelling
7. Examples and Case Studies
8. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Song, C.M.; Lim, S.J.; Tong, J.C. Recent Advances in Computer-Aided Drug Design. Brief. Bioinform. 2009, 10, 579–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voet, A.; Banwell, E.F.; Sahu, K.K.; Heddle, J.G.; Zhang, K.Y.J. Protein Interface Pharmacophore Mapping Tools for Small Molecule Protein: Protein Interaction Inhibitor Discovery. Curr. Top. Med. Chem. 2013, 3, 989–1001. [Google Scholar] [CrossRef] [PubMed]
- Seidel, T.; Schuetz, D.A.; Garon, A.; Langer, T. The Pharmacophore Concept and Its Applications in Computer-Aided Drug Design. Prog. Chem. Org. Nat. Prod. 2019, 110, 99–141. [Google Scholar] [PubMed]
- Seidel, T.; Wieder, O.; Garon, A.; Langer, T. Applications of the Pharmacophore Concept in Natural Product Inspired Drug Design. Mol. Inform. 2020, 39, 1–11. [Google Scholar] [CrossRef]
- Wermuth, C.G.; Ganellin, C.R.; Lindberg, P.; Mitscher, L.A. Glossary of terms used in medicinal chemistry. Pure Appl. Chem. 1998, 70, 1129–1142. [Google Scholar] [CrossRef]
- Kaserer, T.; Beck, K.R.; Akram, M.; Odermatt, A.; Schuster, D.; Willett, P. Pharmacophore Models and Pharmacophore-Based Virtual Screening: Concepts and Applications Exemplified on Hydroxysteroid Dehydrogenases. Molecules 2015, 20, 22799–22832. [Google Scholar] [CrossRef] [Green Version]
- Vuorinen, A.; Schuster, D. Methods for generating and applying pharmacophore models as virtual screening filters and for bioactivity profiling. Methods 2015, 71, 113–134. [Google Scholar] [CrossRef]
- Sanders, M.P.A.; McGuire, R.; Roumen, L.; de Esch, I.J.P.; de Vlieg, J.; Klomp, J.P.G.; de Graaf, C. From the Protein’s Perspective: The Benefits and Challenges of Protein Structure-Based Pharmacophore Modeling. MedChemComm 2012, 3, 28–38. [Google Scholar] [CrossRef]
- Chandrasekaran, B.; Agrawal, N.; Kaushik, S. Pharmacophore Development. In Encyclopedia of Bioinformatics and Computational Biology: ABC of Bioinformatics; Elsevier: Amsterdam, The Netherlands, 2018; Volume 1–3, pp. 677–687. [Google Scholar] [CrossRef]
- Muhammed, M.T.; Aki-Yalcin, E. Homology modeling in drug discovery: Overview, current applications, and future perspectives. Chem. Biol. Drug Des. 2019, 93, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Protein-ligand docking: Current status and future challenges. Proteins 2006, 65, 15–26. [Google Scholar] [CrossRef]
- Xie, Z.R.; Hwang, M.J. Methods for predicting protein-ligand binding sites. Methods Mol. Biol. 2015, 1215, 383–398. [Google Scholar]
- Goodford, P.J. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 1985, 28, 849–857. [Google Scholar] [CrossRef]
- Böhm, H.J. The computer program LUDI: A new method for the de novo design of enzyme inhibitors. J. Comput. Aided Mol. Des. 1992, 6, 61–78. [Google Scholar] [CrossRef]
- Choudhury, C.; Narahari Sastry, G. Pharmacophore Modelling and Screening: Concepts, Recent Developments and Applications in Rational Drug Design. Chall. Adv. Comput. Chem. Phys. 2019, 27, 25–53. [Google Scholar]
- Yang, S.Y. Pharmacophore modeling and applications in drug discovery: Challenges and recent advances. Drug Discov. Today 2010, 15, 444–450. [Google Scholar] [CrossRef]
- Huang, N.; Shoichet, B.K.; Irwin, J.J. Benchmarking Sets for Molecular Docking. J. Med. Chem. 2006, 49, 6789–6801. [Google Scholar] [CrossRef] [Green Version]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Chichester, C.; Digles, D.; Siebes, R.; Loizou, A.; Groth, P.; Harland, L. Drug discovery FAQs: Workflows for answering multidomain drug discovery questions. Drug Discov. Today 2015, 20, 399–405. [Google Scholar] [CrossRef] [Green Version]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL Database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Cheng, D.; Shrivastava, S.; Tzur, D.; Gautam, B.; Hassanali, M. DrugBank: A knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008, 36, D901–D906. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bryant, S.H.; Cheng, T.; Wang, J.; Gindulyte, A.; Shoemaker, B.A.; Thiessen, P.A.; He, S.; Zhang, J. PubChem BioAssay: 2017 update. Nucleic Acids Res. 2017, 45, D955–D963. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.S.; Paules, R.S.; Simeonov, A.; Fitzpatrick, S.C.; Crofton, K.M.; Casey, W.M.; Mendrick, D.L. The US Federal Tox21 Program: A strategic and operational plan for continued leadership. ALTEX 2018, 35, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Gurung, A.B.; Ali, M.A.; Lee, J.; Farah, M.A.; Al-Anazi, K.M. An Updated Review of Computer-Aided Drug Design and Its Application to COVID-19. Biomed. Res. Int. 2021, 2021, 8853056. [Google Scholar] [CrossRef]
- Seidel, T.; Ibis, G.; Bendix, F.; Wolber, G. Strategies for 3D pharmacophore-based virtual screening. Drug Discov. Today Technol. 2010, 7, e221–e228. [Google Scholar] [CrossRef]
- Wolber, G.; Dornhofer, A.A.; Langer, T. Efficient overlay of small organic molecules using 3D pharmacophores. J. Comput. Aided Mol. Des. 2006, 20, 773–788. [Google Scholar] [CrossRef]
- Tyagi, R.; Singh, A.; Chaudhary, K.K. Pharmacophore modeling and its application. In Bioinformatics: Methods and Applications; Singh, D.B., Pathak, R.K., Eds.; Academic Press: Cambridge, MA, USA, 2021. [Google Scholar]
- Barnum, D.; Greene, J.; Smellie, A.; Sprague, P. Identification of common functional configurations among molecules. J. Chem. Inf. Comput. Sci. 1996, 36, 563–571. [Google Scholar] [CrossRef]
- Prieto-Martínez, F.D.; Norinder, U.; Medina-Franco, J.L. Cheminformatics Explorations of Natural Products. In Progress in the Chemistry of Organic Natural Products; Kinghorn, A.D., Gibbons, H.F.S., Kobayashi, J., Asakawa, Y., Liu, J.K., Eds.; Springer: Cham, Switzerland, 2019. [Google Scholar]
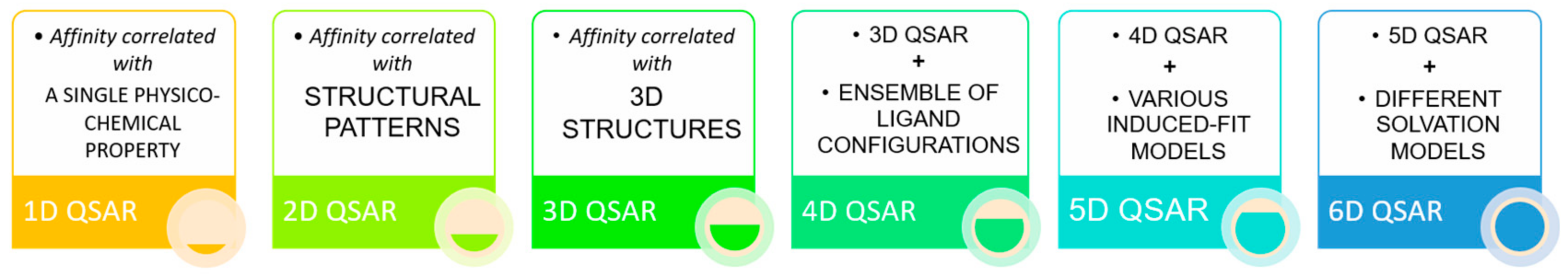
- Lill, M.A. Multi-dimensional QSAR in drug discovery. Drug Discov. Today 2007, 12, 1013–1017. [Google Scholar] [CrossRef]
- Lu, S.-H.; Wu, J.W.; Liu, H.-L.; Zhao, J.-H.; Liu, K.-T.; Chuang, C.-K.; Lin, H.-Y.; Tsai, W.-B.; Ho, Y. The discovery of potential acetylcholinesterase inhibitors: A combination of pharmacophore modeling, virtual screening, and molecular docking studies. J. Biomed. Sci. 2011, 18, 8. [Google Scholar] [CrossRef] [Green Version]
- Triballeau, N.; Acher, F.; Brabet, I. Virtual Screening Workflow Development Guided by the “Receiver Operating Characteristic” Curve Approach. Application to High-Throughput Docking on Metabotropic Glutamate Receptor Subtype 4. J. Med. Chem. 2005, 48, 2534–2547. [Google Scholar] [CrossRef]
- Mitra, I.; Saha, A.; Roy, K. Pharmacophore mapping of arylamino-substituted benzo[b]thiophenes as free radical scavengers. J. Mol. Model. 2010, 16, 1585–1596. [Google Scholar] [CrossRef]
- Maia, E.H.B.; Assis, L.C.; de Oliveira, T.A.; da Silva, A.M.; Taranto, A.G. Structure-Based Virtual Screening: From Classical to Artificial Intelligence. Front. Chem. 2020, 8, 343. [Google Scholar] [CrossRef]
- Sunseri, J.; Koes, D.R. Pharmit: Interactive exploration of chemical space. Nucleic Acids Res. 2016, 44, W442–W448. [Google Scholar] [CrossRef] [Green Version]
- Koes, D.R.; Camacho, C.J. ZINCPharmer: Pharmacophore search of the ZINC database. Nucleic Acids Res. 2012, 40, W409–W414. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021, 49, D437–D451. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. ZINC—A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, K.A.; Huang, X. Machine Learning-Based Virtual Screening and Its Applications to Alzheimer’s Drug Discovery: A Review. Curr. Pharm. Des. 2018, 24, 3347–3358. [Google Scholar] [CrossRef]
- Lionta, E.; Spyrou, G.; Vassilatis, D.K.; Cournia, Z. Structure-based virtual screening for drug discovery: Principles, applications and recent advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef]
- Tyagi, M.; Begnini, F.; Poongavanam, V.; Doak, B.C.; Kihlberg, J. Drug Syntheses Beyond the Rule of 5. Chemistry 2020, 26, 49–88. [Google Scholar] [CrossRef] [Green Version]
- Allen, W.J.; Balius, T.E.; Mukherjee, S.; Brozell, S.R.; Moustakas, D.T.; Lang, P.T.; Case, D.A.; Kuntz, I.D.; Rizzo, R.C. DOCK 6: Impact of new features and current docking performance. J. Comput. Chem. 2015, 36, 1132–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Huey, R.; Olson, A.J. Using AutoDock for ligand-receptor docking. In Current Protocols in Bioinformatics; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008; Chapter 8, Unit 8.14. [Google Scholar]
- Pal, S.; Kumar, V.; Kundu, B.; Bhattacharya, D.; Preethy, N.; Reddy, M.P.; Talukdar, A. Ligand-based Pharmacophore Modeling, Virtual Screening and Molecular Docking Studies for Discovery of Potential Topoisomerase I Inhibitors. Comput. Struct. Biotechnol. J. 2019, 17, 291–310. [Google Scholar] [CrossRef] [PubMed]
- Boz, E.; Stein, M. Accurate Receptor-Ligand Binding Free Energies from Fast QM Conformational Chemical Space Sampling. Int. J. Mol. Sci. 2021, 22, 3078. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Huai, Z.; He, Q.; Liu, Z. A General Picture of Cucurbit[8]uril Host-Guest Binding. J. Chem. Inf. Model. 2021, 61, 6107–6134. [Google Scholar] [CrossRef]
- Case, D.A. Normal mode analysis of protein dynamics. Curr. Opin. Struct. Biol. 1994, 4, 285–290. [Google Scholar] [CrossRef]
- Gallicchio, E.; Levy, R.M. Recent theoretical and computational advances for modeling pro-tein-ligand binding affinities. Adv. Protein Chem. Struct Biol. 2011, 85, 27–80. [Google Scholar]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Qiu, L.; Yan, Y.; Sun, Z.; Song, J.; Zhang, J.Z.H. Interaction entropy for computational alanine scanning in protein-protein binding. Wires Comput. Mol. Sci. 2018, 8, e1342. [Google Scholar] [CrossRef]
- Whitesides, G.M.; Krishnamurthy, V.M. Designing ligands to bind proteins. Q. Rev. Biophys. 2005, 38, 385–395. [Google Scholar] [CrossRef]
- Polishchuk, P.; Kutlushina, A.; Bashirova, D.; Mokshyna, O.; Madzhidov, T. Virtual Screening Using Pharmacophore Models Retrieved from Molecular Dynamic Simulations. Int. J. Mol. Sci. 2019, 20, 5834. [Google Scholar] [CrossRef] [Green Version]
- Wieder, M.; Garon, A.; Perricone, U.; Boresch, S.; Seidel, T.; Almerico, A.M.; Langer, T. Common Hits Approach: Combining Pharmacophore Modeling and Molecular Dynamics Simulations. J. Chem. Inf. Model. 2017, 57, 365–385. [Google Scholar] [CrossRef]
- Lombino, J.; Gulotta, M.R.; De Simone, G.; Mekni, N.; De Rosa, M.; Carbone, D.; Parrino, B.; Cascioferro, S.M.; Diana, P.; Padova, A.; et al. Dynamic-shared Pharmacophore Approach as Tool to Design New Allosteric PRC2 Inhibitors, Targeting EED Binding Pocket. Mol. Inform. 2021, 40, e2000148. [Google Scholar] [CrossRef]
- Caporuscio, F.; Tafi, A. Pharmacophore modelling: A forty year old approach and its modern synergies. Curr. Med. Chem. 2011, 18, 2543–2553. [Google Scholar] [CrossRef]
- Prathipati, P.; Dixit, A.; Saxena, A.K. Computer-aided drug design: Integration of structure-based and ligand-based approaches in drug design. Curr. Comput. Aided Drug Des. 2007, 3, 133–148. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Wolber, G.; Langer, T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef]
- Salam, N.K.; Nuti, R.; Sherman, W. Novel method for generating structure-based pharmacophores using energetic analysis. J. Chem. Inf. Model. 2009, 49, 2356–2368. [Google Scholar] [CrossRef]
- Sutter, J.; Li, J.; Maynard, A.J.; Goupil, A.; Luu, T.; Nadassy, K. New features that improve the pharmacophore tools from Accelrys. Curr. Comput. Aided Drug Des. 2011, 7, 173–180. [Google Scholar] [CrossRef]
- Li, H.; Sutter, J.; Hoffmann, R. HypoGen: An Automated System for Generating Predictive 3D Pharmacophore Models. In Pharmacophore Perception, Development, and Use in Drug Design; Güner, O.F., Ed.; International University Line: La Jolla, CA, USA, 2000. [Google Scholar]
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. PHASE: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Aided Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988, 10, 5959–5967. [Google Scholar] [CrossRef]
- Klebe, G.; Abraham, U.; Mietzner, T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J. Med. Chem. 1994, 37, 4130–4146. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lai, L. Pocket v.2: Further Developments on Receptor-Based Pharmacophore Modeling. J. Chem. Inf. Model. 2006, 46, 2684–2691. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Dror, O.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PharmaGist: A webserver for ligand-based pharmacophore detection. Nucleic Acids Res. 2008, 36, W223–W228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragno, R. www.3d-qsar.com: A web portal that brings 3-D QSAR to all electronic devices-the Py-CoMFA web application as tool to build models from pre-aligned datasets. J. Comput. Aided Mol. Des. 2019, 33, 855–864. [Google Scholar] [CrossRef]
- Reddy, A.S.; Pati, S.P.; Kumar, P.P.; Pradeep, H.N.; Sastry, G.N. Virtual screening in drug discovery—A computational perspective. Curr. Protein Pept. Sci. 2007, 8, 329–351. [Google Scholar] [CrossRef]
- Kar, S.; Roy, K. How far can virtual screening take us in drug discovery? Expert Opin. Drug Discov. 2013, 8, 245–261. [Google Scholar] [CrossRef]
- Danishuddin, M.; Khan, A.U. Structure based virtual screening to discover putative drug candidates: Necessary considerations and successful case studies. Methods 2015, 71, 135–145. [Google Scholar] [CrossRef]
- Damm-Ganamet, K.L.; Arora, N.; Becart, S.; Edwards, J.P.; Lebsack, A.D.; McAllister, H.M.; Nelen, M.I.; Rao, N.L.; Westover, L.; Wiener, J.J.M.; et al. Accelerating Lead Identification by High Throughput Virtual Screening: Prospective Case Studies from the Pharmaceutical Industry. J. Chem. Inf. Model. 2019, 59, 2046–2062. [Google Scholar] [CrossRef]
- Temml, V.; Kutil, Z. Structure-based molecular modeling in SAR analysis and lead optimization. Comput. Struct. Biotechnol. J. 2021, 19, 1431–1444. [Google Scholar] [CrossRef]
- Schuster, D.; Maurer, E.M.; Laggner, C.; Nashev, L.G.; Wilckens, T.; Langer, T.; Odermatt, A. The Discovery of New 11β-Hydroxysteroid Dehydrogenase Type 1 Inhibitors by Common Feature Pharmacophore Modeling and Virtual Screening. J. Med. Chem. 2006, 49, 3454–3466. [Google Scholar] [CrossRef]
- Dotolo, S.; Cervellera, C.; Russo, M.; Russo, G.L.; Facchiano, A. Virtual Screening of Natural Compounds as Potential PI3K-AKT1 Signaling Pathway Inhibitors and Experimental Validation. Molecules 2021, 26, 492. [Google Scholar] [CrossRef]
- Cervellera, C.; Russo, M.; Dotolo, S.; Facchiano, A.; Russo, G.L. STL1, a New AKT Inhibitor, Synergizes with Flavonoid Quercetin in Enhancing Cell Death in A Chronic Lymphocytic Leukemia Cell Line. Molecules 2021, 26, 5810. [Google Scholar] [CrossRef]
- Rahman, N.; Basharat, Z.; Yousuf, M.; Castaldo, G.; Rastrelli, L.; Khan, H. Virtual Screening of Natural Products against Type II Transmembrane Serine Protease (TMPRSS2), the Priming Agent of Coronavirus 2 (SARS-CoV-2). Molecules 2020, 25, 2271. [Google Scholar] [CrossRef]
- Rossi Sebastiano, M.; Ermondi, G.; Hadano, S.; Caron, G. AI-based protein structure databases have the potential to accelerate rare diseases research: AlphaFoldDB and the case of IAHSP/Alsin. Drug Discov. Today 2021, 27, 1652–1660. [Google Scholar] [CrossRef]
- Song, T.; Zhang, X.; Ding, M.; Rodriguez-Paton, A.; Wang, S.; Wang, G. DeepFusion: A Deep Learning Based Multi-Scale Feature Fusion Method for Predicting Drug-Target Interactions. Methods 2022, in press. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giordano, D.; Biancaniello, C.; Argenio, M.A.; Facchiano, A. Drug Design by Pharmacophore and Virtual Screening Approach. Pharmaceuticals 2022, 15, 646. https://doi.org/10.3390/ph15050646
Giordano D, Biancaniello C, Argenio MA, Facchiano A. Drug Design by Pharmacophore and Virtual Screening Approach. Pharmaceuticals. 2022; 15(5):646. https://doi.org/10.3390/ph15050646
Chicago/Turabian StyleGiordano, Deborah, Carmen Biancaniello, Maria Antonia Argenio, and Angelo Facchiano. 2022. "Drug Design by Pharmacophore and Virtual Screening Approach" Pharmaceuticals 15, no. 5: 646. https://doi.org/10.3390/ph15050646
APA StyleGiordano, D., Biancaniello, C., Argenio, M. A., & Facchiano, A. (2022). Drug Design by Pharmacophore and Virtual Screening Approach. Pharmaceuticals, 15(5), 646. https://doi.org/10.3390/ph15050646