
Nitro-Group-Containing Thiopeptide Derivatives as Promising Agents to Target Clostridioides difficile

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
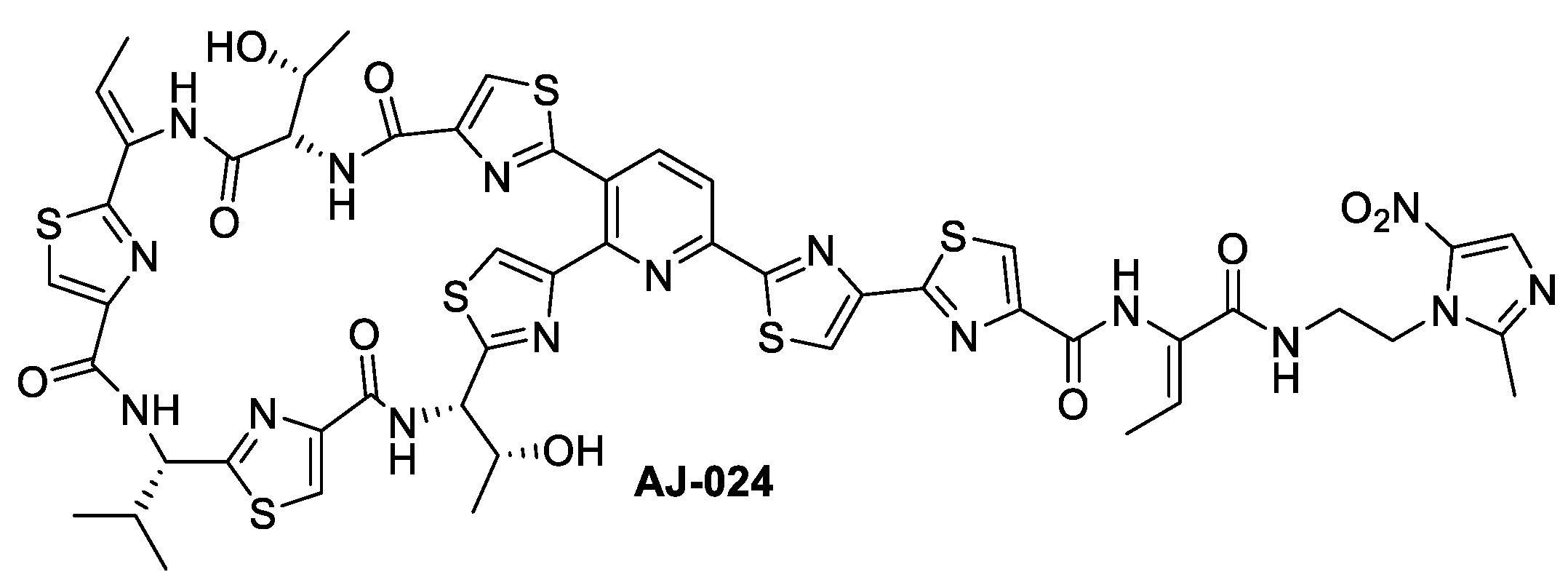
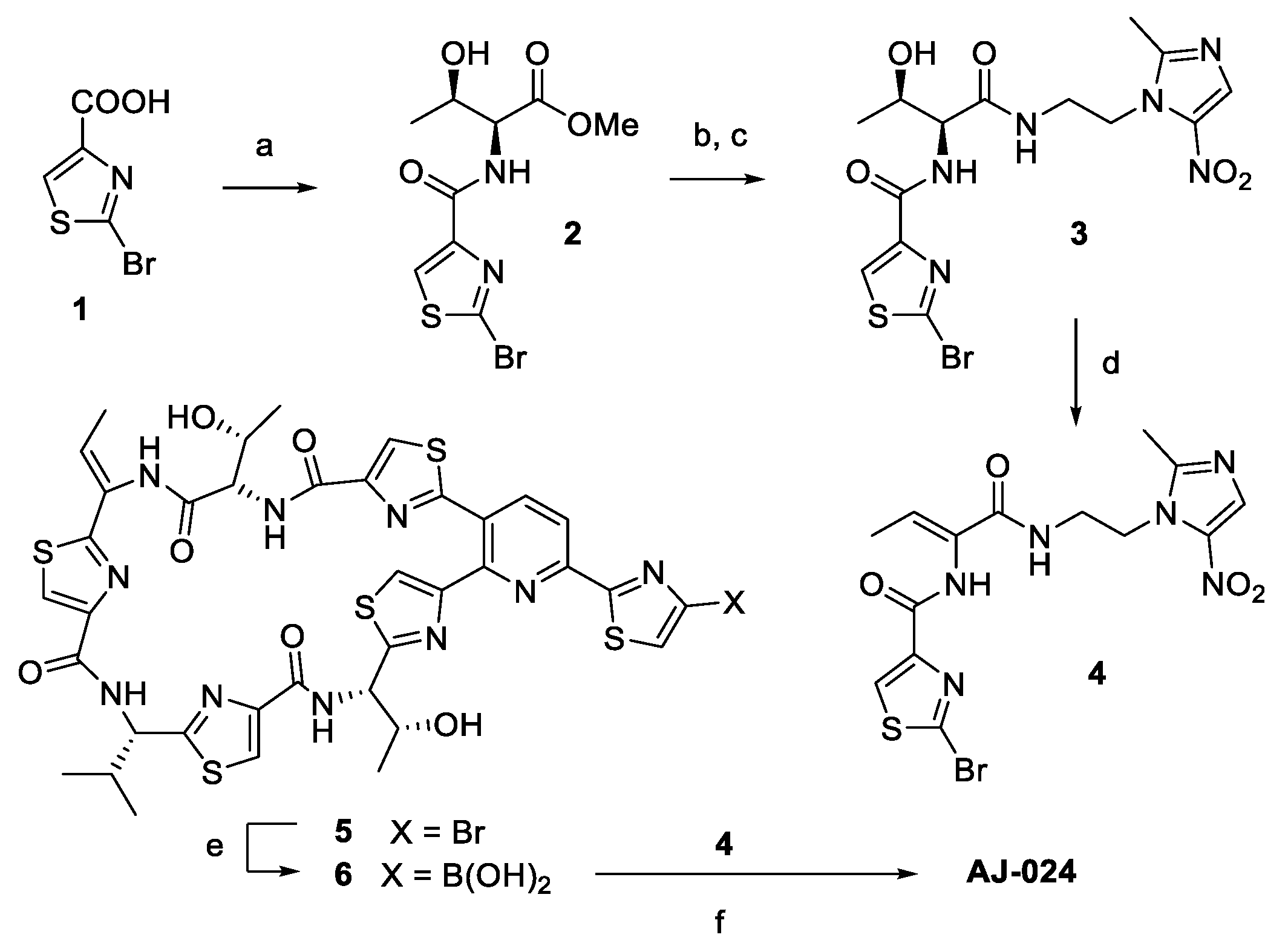
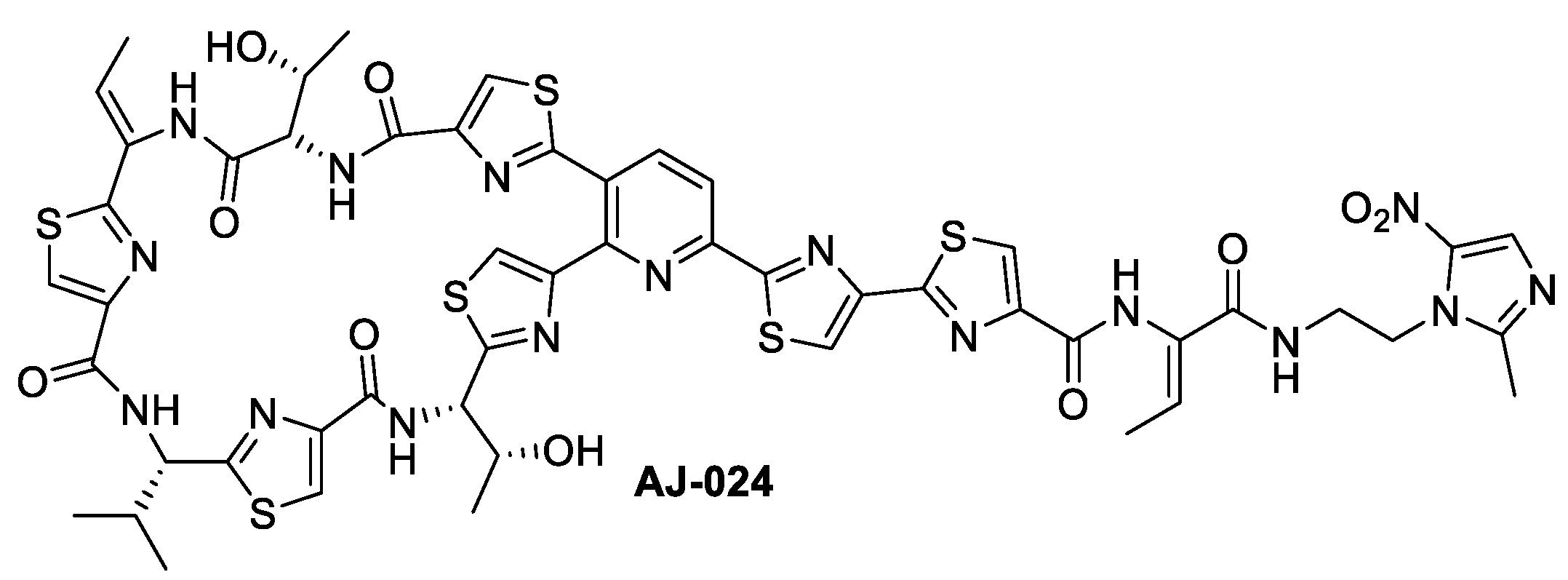
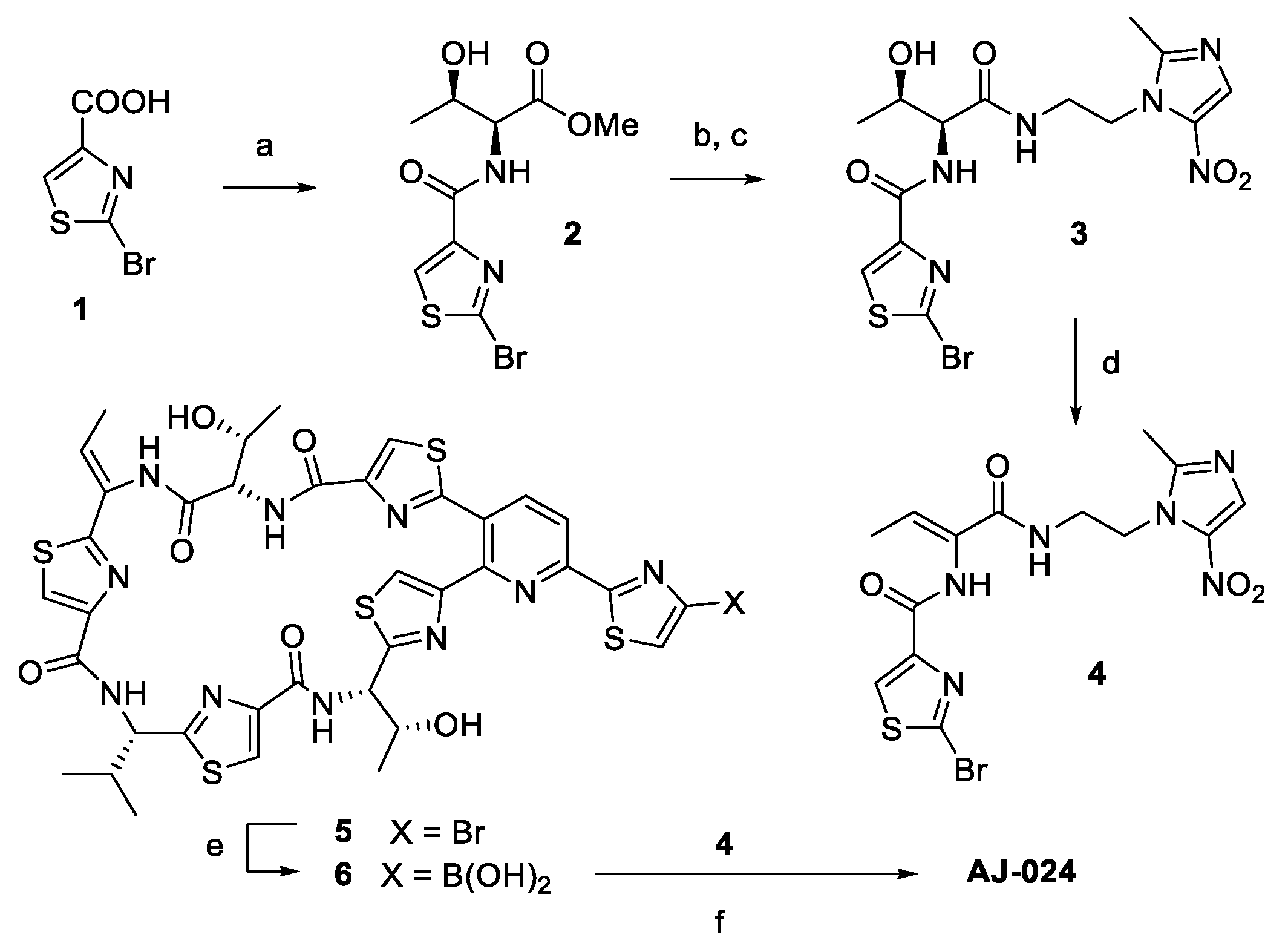
2.1. Chemical Synthesis of AJ-024 and Antibacterial Activity Thereof against Representative C. difficile Strains
2.2. Superior In Vitro Activity of AJ-024 against Extensively Classified C. difficile Clinical Isolates
2.3. Antimicrobial Activities of AJ-024 against Other Bacterial Species
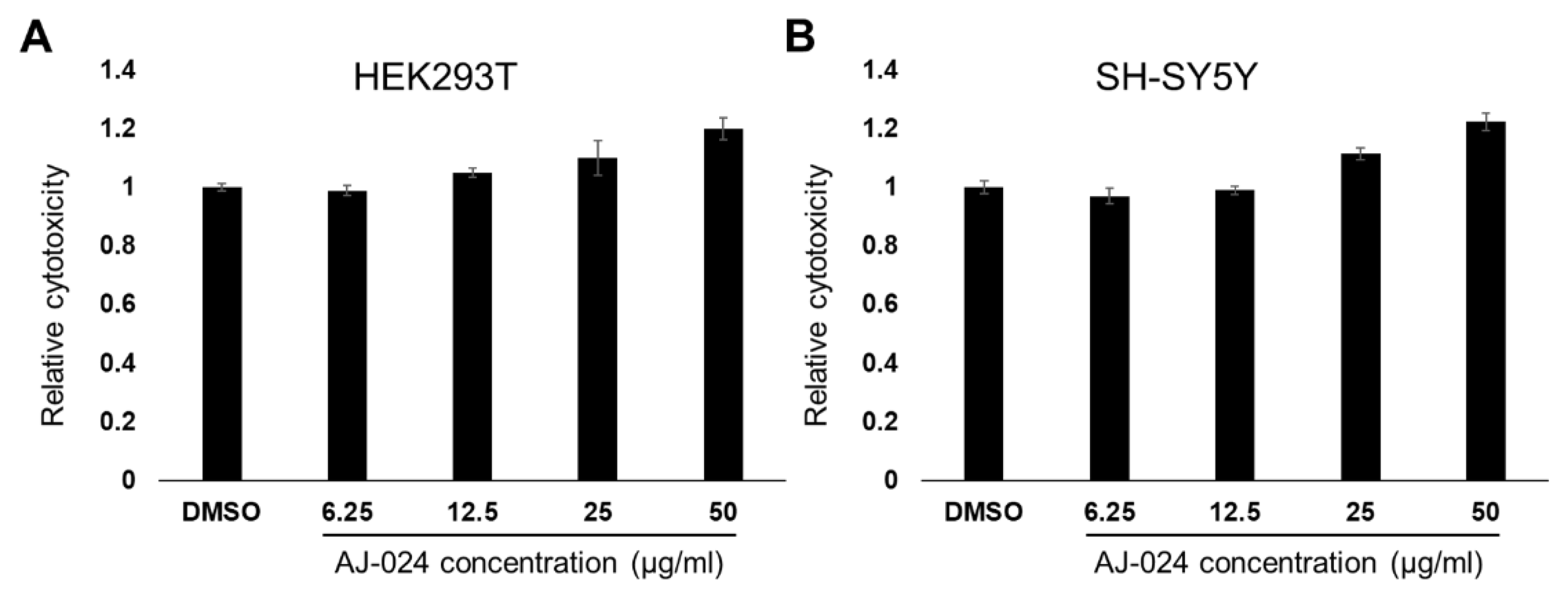
2.4. AJ-024 Has No Appreciable Human Cellular Toxicity
2.5. Favorable ADME Properties of AJ-024
2.6. Time-Kill Assay of AJ-024
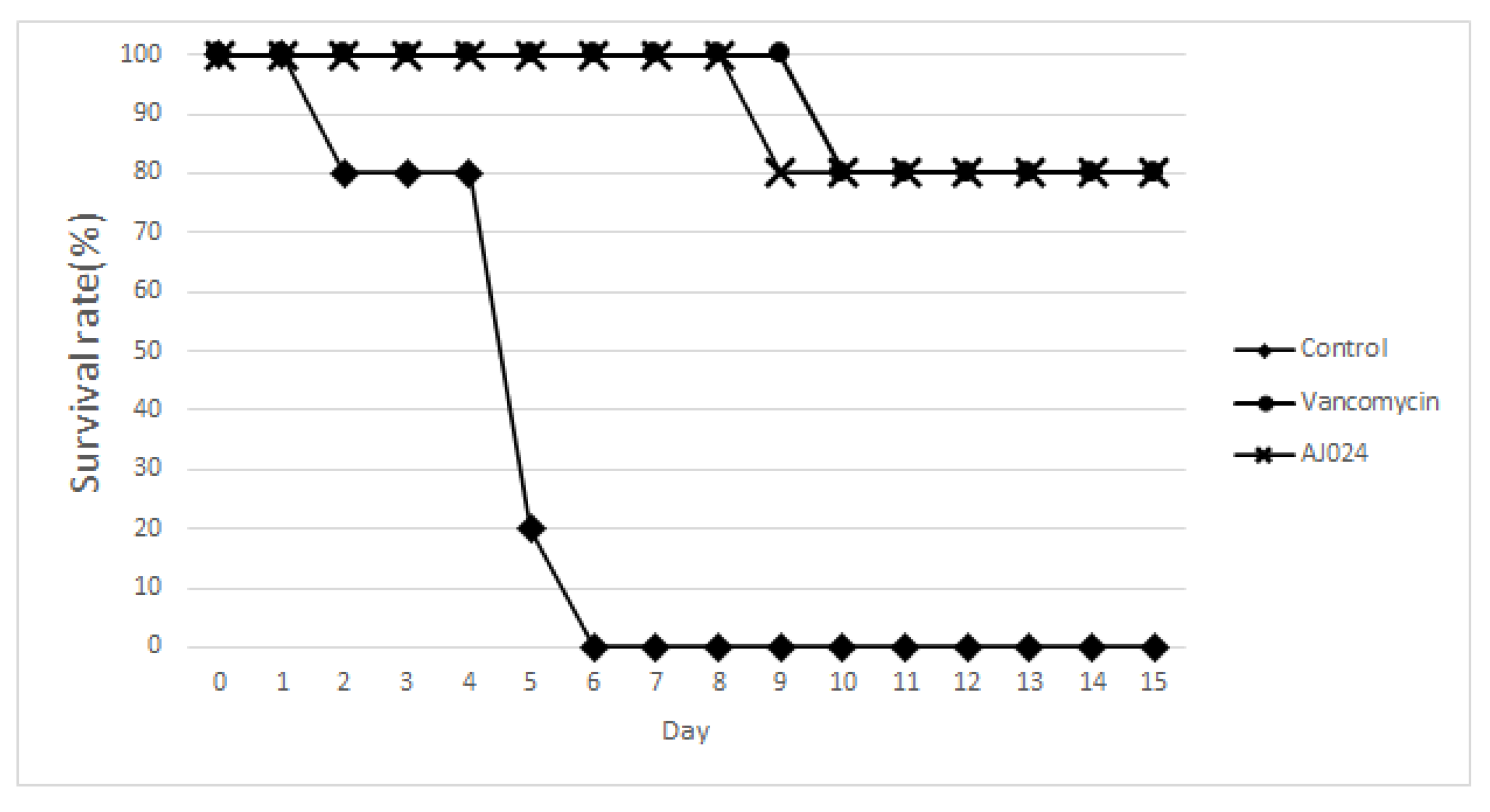
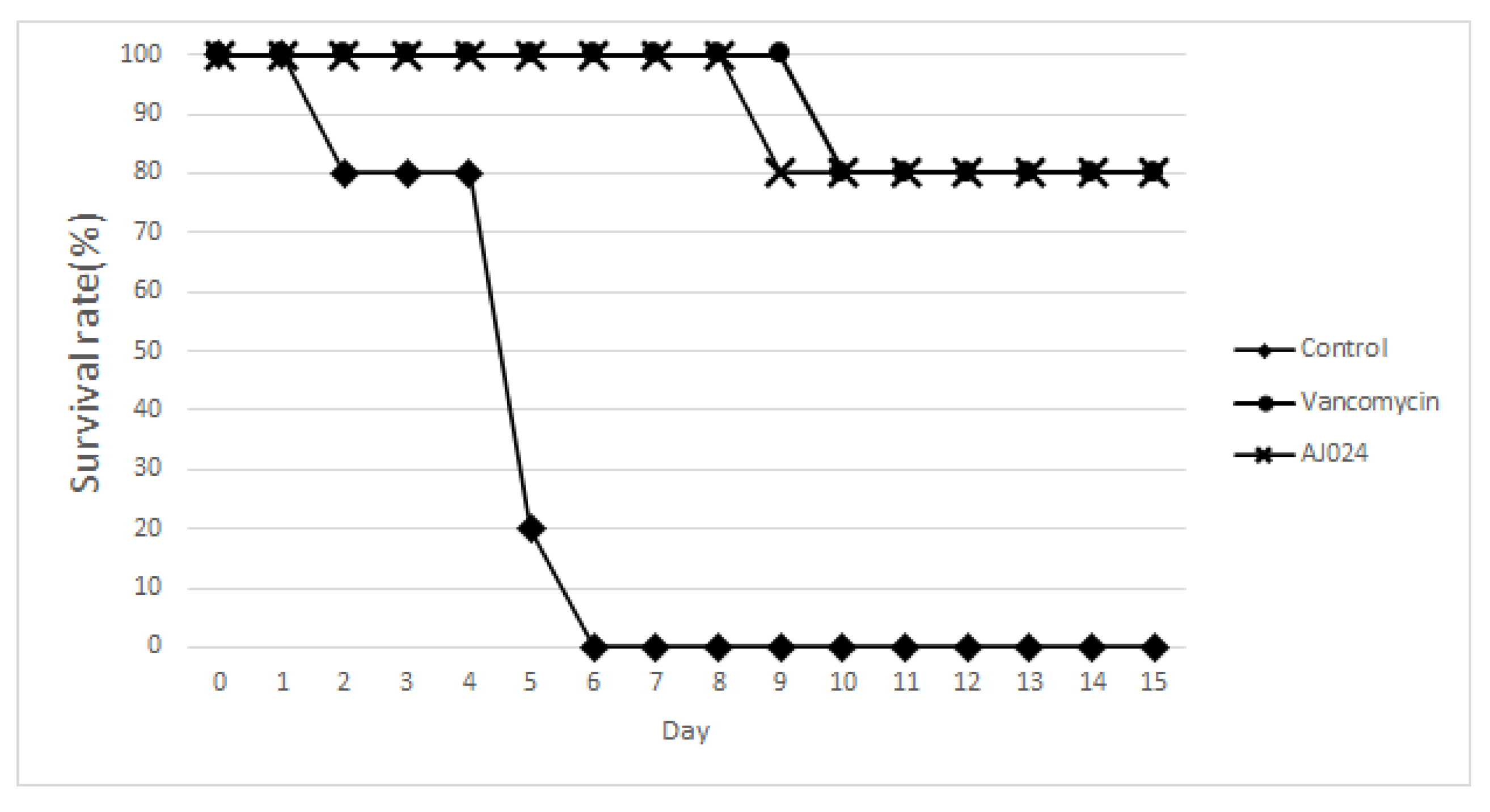
2.7. Effect of AJ-024 on Acute CDI In Vivo Infection Model
3. Materials and Methods
3.1. Chemistry
Synthesis of AJ-024
Synthesis of Methyl (2-bromothiazole-4-carbonyl)-L-threoninate (Compound 2)
Synthesis of 2-bromo-N-((2S,3R)-3-hydroxy-1-((2-(2-methyl-5-nitro-1H-imidazol-1-yl)ethyl)amino)-1-oxobutan-2-yl)thiazole-4-carboxamide (Compound 3)
Synthesis of (Z)-2-bromo-N-(1-((2-(2-methyl-5-nitro-1H-imidazol-1-yl)ethyl)amino)-1-oxobut-2-en-2-yl)thiazole-4-carboxamide (Compound 4)
Synthesis of ((12Z,32Z,72Z,112Z,4S,8S,12Z,15S)-26-(4-bromothiazol-2-yl)-12-ethylidene-4,15-bis((R)-1-hydroxyethyl)-8-isopropyl-5,9,13,16-tetraaza-1(2,4),3,7,11(4,2) -tetrathiazola-2(3,2)-pyridinacycloheptadecaphane-6,10,14,17-tetraone (AJ-024)
3.2. Cellular Toxicity
3.3. Time-Kill Assays
3.4. In Vivo Bacteria Inoculation
3.5. Mouse IBD-C. difficile Comorbidity Infection Model
3.6. Inhibition of CYP Enzymes Activity by AJ-024
3.7. In Vitro Metabolic Stability of AJ-024
3.8. Plasma Protein Binding Study of AJ-024
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fletcher, J.R.; Pike, C.M.; Parsons, R.J.; Rivera, A.J.; Foley, M.H.; McLaren, M.R.; Montgomery, S.A.; Theriot, C.M. Clostridioides difficile Exploits Toxin-Mediated Inflammation to Alter the Host Nutritional Landscape and Exclude Competitors from the Gut Microbiota. Nat. Commun. 2021, 12, 462. [Google Scholar] [CrossRef] [PubMed]
- Czepiel, J.; Dróżdż, M.; Pituch, H.; Kuijper, E.J.; Perucki, W.; Mielimonka, A.; Goldman, S.; Wultańska, D.; Garlicki, A.; Biesiada, G. Clostridium difficile Infection: Review. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 1211–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayedy, L.; Kothari, D.; Richards, R.J. Toxic Megacolon Associated Clostridium difficile Colitis. World J. Gastrointest. Endosc. 2010, 2, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Rao, K.; Erb-Downward, J.R.; Walk, S.T.; Micic, D.; Falkowski, N.; Santhosh, K.; Mogle, J.A.; Ring, C.; Young, V.B.; Huffnagle, G.B.; et al. The Systematic Inflammatory Response to Clostridium difficile Infection. PLoS ONE 2014, 9, e92578. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC). Clostridiodides Difficile Infection. 2019. Available online: https://www.cdc.gov/hai/organisms/cdiff/cdiff_infect.html (accessed on 25 March 2022).
- Valiente, E.; Cairns, M.D.; Wren, B.W. The Clostridium difficile PCR Ribotype 027 Lineage: A Pathogen on the Move. Clin. Microbiol. Infect. 2014, 20, 396–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tay, H.L.; Chow, A.; Ng, T.M.; Lye, D.C. Risk Factors and Treatment Outcomes of Severe Clostridioides difficile Infection in Singapore. Sci. Rep. 2019, 9, 13440. [Google Scholar] [CrossRef] [Green Version]
- Centers for Disease Control and Prevention (CDC). Antibiotic/Antimicrobial Resistance (AR/AMR), Biggest Threats and Data. Available online: https://www.cdc.gov/drugresistance/biggest-threats.html (accessed on 25 March 2022).
- Blanco, N.; Robinson, G.L.; Heil, E.L.; Perlmutter, R.; Wilson, L.E.; Brown, C.H.; Heavner, M.S.; Nadimpalli, G.; Lemkin, D.; Morgan, D.J.; et al. Impact of a C. difficile Infection (CDI) Reduction Bundle and Its Components on CDI Diagnosis and Prevention. Am. J. Infect. Control. 2021, 49, 319–326. [Google Scholar] [CrossRef]
- Mergenhagen, K.A.; Wojciechowski, A.L.; Paladino, J.A. A Review of the Economics of Treating Clostridium difficile Infection. Pharmacoeconomics 2014, 32, 639–650. [Google Scholar] [CrossRef]
- Jiang, Y.; Sarpong, E.M.; Sears, P.; Obi, E.N. Budget Impact Analysis of Fidaxomicin Versus Vancomycin for the Treatment of Clostridioides difficile Infection in the United States. Infect. Dis. Ther. 2022, 11, 111–126. [Google Scholar] [CrossRef]
- Fatima, R.; Aziz, M. The Hypervirulent Strain of Clostridium difficile: NAP1/B1/027-a Brief Overview. Cureus 2019, 11, e3977. [Google Scholar] [CrossRef] [Green Version]
- Kullar, R.; Tran, M.N.; Goldstein, E. Investigational Treatment Agents for Recurrent Clostridioides difficile Infection (rCDI). J. Exp. Pharmacol. 2020, 12, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Jarrad, A.M.; Karoli, T.; Blaskovich, M.A.; Lyras, D.; Cooper, M.A. Clostridium difficile Drug Pipeline: Challenges in Discovery and Development of New Agents. J. Med. Chem. 2015, 58, 5164–5185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miethke, M.; Pieroni, M.; Weber, T.; Brönstrup, M.; Hammann, P.; Halby, L.; Arimondo, P.B.; Glaser, P.; Aigle, B.; Bode, H.B.; et al. Towards the Sustainable Discovery and Development of New Antibiotics. Nat. Rev. Chem. 2021, 5, 726–749. [Google Scholar] [CrossRef] [PubMed]
- Dingsdag, S.A.; Hunter, N. Metronidazole: An Update on Metabolism, Structure–Cytotoxicity and Resistance Mechanisms. J. Antimicrob. Chemother. 2018, 73, 265–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, S.F.; Peng, L.P.; Zhang, H.Z.; Rasheed, S.; Kumar, K.V.; Zhou, C.H. Novel Hybrids of Metronidazole and Quinolones: Synthesis, Bioactive Evaluation, Cytotoxicity, Preliminary Antimicrobial Mechanism and Effect of Metal Ions on Their Transportation by Human Serum Albumin. Eur. J. Med. Chem. 2014, 86, 318–334. [Google Scholar] [CrossRef] [PubMed]
- Bagley, M.C.; Dale, J.W.; Merritt, E.A.; Xiong, X. Thiopeptide Antibiotics. Chem. Rev. 2005, 105, 685–714. [Google Scholar] [CrossRef]
- Harms, J.M.; Wilson, D.N.; Schluenzen, F.; Connell, S.R.; Stachelhaus, T.; Zaborowska, Z.; Spahn, C.M.T.; Fucini, P. Translational Regulation via L11: Molecular Switches on the Ribosome Turned on and off by Thiostrepton and Micrococcin. Mol. Cell. 2008, 30, 26–38. [Google Scholar] [CrossRef]
- Chan, D.C.K.; Burrows, L.L. Thiopeptide: Antibiotics with Unique Chemical Structures and Diverse Biological Activities. J. Antibiot. 2021, 74, 161–175. [Google Scholar] [CrossRef]
- Zheng, Q.; Wang, Q.; Wang, S.; Wu, J.; Gao, Q.; Liu, W. Thiopeptide Antibiotics Exhibit a Dual Mode of Action Against Intracellular Pathogens by Affecting Both Host and Microbe. Chem. Biol. 2015, 22, 1002–1007. [Google Scholar] [CrossRef] [Green Version]
- Hwang, H.-J.; Son, Y.-J.; Kim, D.; Lee, J.; Shin, Y.-J.; Kwon, Y.; Ciufolini, M.A. Diversity-Oriented Routes to Thiopeptide Antibiotics: Total Synthesis and Biological Evaluation of Micrococcin P2. Org. Biomol. Chem. 2022, 20, 1893–1899. [Google Scholar] [CrossRef]
- Miyaura, N.; Suzuki, A. Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, D.; Fawley, W.; Kachrimanidou, M.; Bowden, R.; Crook, D.W.; Fung, R.; Golubchik, T.; Harding, R.M.; Jeffery, K.J.; Jolley, K.A.; et al. Multilocus Sequence Typing of Clostridium difficile. J. Clin. Microbiol. 2010, 48, 770–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacCannell, D.R.; Louie, T.J.; Gregson, D.B.; Laverdiere, M.; Labbe, A.C.; Laing, F.; Henwick, S. Molecular Analysis of Clostridium difficile PCR Ribotype 027 Isolates from Eastern and Western Canada. J. Clin. Microbiol. 2006, 44, 2147–2152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujitani, S.; George, W.L.; Morgan, M.A.; Nichols, S.; Murthy, A.R. Implications for Vancomycin-Resistant Enterococcus Colonization Associated with Clostridium difficile Infections. Am. J. Infect.Control. 2011, 39, 188–193. [Google Scholar] [CrossRef]
- Zhou, F.; Hamza, T.; Fluer, A.S.; Zhang, Y.; Yu, H.; Chen, K.; Heath, J.E.; Chen, Y.; Huang, H.; Feng, H. Mice with Inflammatory Bowel Disease are Susceptible to Clostridium difficile Infection with Severe Disease Outcomes. Inflamm. Bowel. Dis. 2018, 24, 573–582. [Google Scholar] [CrossRef] [Green Version]
- Saleh, M.M.; Frisbee, A.L.; Leslie, J.L.; Buonomo, E.L.; Cowardin, C.A.; Ma, J.Z.; Simpson, M.E.; Scully, K.W.; Abhyankar, M.M.; Petri, W.A. Colitis-Induced Th17 Cells Increase the Risk for Severe Subsequent Clostiridium difficile infection. Cell Host Microbe 2019, 25, 756–765. [Google Scholar] [CrossRef]
- Solomon, K. The Host Immune Response to Clostridium difficile Infection. Ther. Adv. Infect. Dis. 2013, 1, 19–35. [Google Scholar] [CrossRef] [Green Version]
- Issa, M.; Ananthakrishnan, A.N.; Binion, D.G. Clostridium difficile and Inflammatory Bowel Disease. Inflamm. Bowel. Dis. 2008, 14, 1432–1442. [Google Scholar] [CrossRef]
- Clinical and Laboratory Standards Institute. Methods for Antimicrobial Susceptibility Testing of Anaerobic Bacteria, 9th ed.; M11; CLSI: Wayne, PA, USA, 2018. [Google Scholar]
- Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing, 29th ed.; M100; CLSI: Wayne, PA, USA, 2019. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial Strains | Ribotype | AJ-024 | Vancomycin |
|---|---|---|---|
| C. difficile ATCC 9689 | 001 | 0.25 | 1 |
| C. difficile ATCC 43255 | 087 | 0.25 | 1 |
| C. difficile ATCC 51695 | 001 | 0.25 | 1 |
| C. difficile ATCC-BAA-1871 | 001 | 0.25 | 1 |
| C. difficile ATCC 17858 | 054 | 0.5 | 1 |
| Clade | Ribotype | n | MLST | Toxin | VAN | AJ-024 |
|---|---|---|---|---|---|---|
| 1 | RT001 | 3 | ST3 | A+B+ | 2 | 0.5 |
| RT002 | 4 | ST8 | A+B+ | 1–2 | 0.25–1 | |
| RT012 | 3 | ST54 | A+B+ | 1–2 | 0.25 | |
| RT014 | 3 | ST14 | A+B+ | 1 | 0.5 | |
| RT015 | 3 | ST35 | A+B+ | 1 | 0.5 | |
| RT018 | 10 | ST17 | A+B+ | 0.5–2 | 0.25–1 | |
| RT293 | 2 | ST129 | A+B+ | 1 | 0.5–1 | |
| 2 | RT027 | 3 | ST1 | A+B+CDT+ | 1 | 0.5 |
| nt | 1 | ST67 | A+B+CDT+ | 2 | 0.5 | |
| 3 | RT130 | 5 | ST5 | A+B+CDT+ | 1 | 0.25–1 |
| nt | 3 | ST221 | A+B+CDT+ | 1 | 0.25 | |
| 4 | RT017 | 10 | ST37 | A−B+ | 0.5–1 | 0.25–0.5 |
| 5 | RT078 | 3 | ST11 | A+B+CDT+ | 1 | 0.25–0.5 |
| MIC range | 0.5–2 | 0.25–1 | ||||
| MIC50 | 1 | 0.5 |
| MIC, μg/mL | |||||
|---|---|---|---|---|---|
| Gram Positive | Strain | AJ-024 | VAN | LIZ | CIP |
| S. aureus | ATCC 25923 | 0.25 | 0.5 | 2 | 0.5 |
| S. aureus | ATCC 43300 | 0.25 | 0.5 | 2 | 1 |
| S. aureus | Clinical isolate | 0.5 | >32 | 1 | >32 |
| E. faecalis | ATCC 29212 | 0.25 | 0.5 | 2 | 1 |
| E. faecalis | Clinical isolate | 0.25 | >32 | 4 | >32 |
| E. faecium | ATCC 19434 | 0.25 | 1 | 2 | 1 |
| E. faecium | Clinical isolate | 0.5 | >32 | 2 | 32 |
| Gram Negative | |||||
| E. coli | ATCC 25922 | >32 | >32 | >32 | 0.06 |
| K. pneumoniae | ATCC 10031 | >32 | >32 | >32 | 0.06 |
| P. aeruginosa | ATCC 27853 | >32 | >32 | >32 | 0.5 |
| A. baumannii | ATCC 17904 | >32 | >32 | >32 | 0.5 |
| CYP Inhibition | PPB | MS | PS | |||||
|---|---|---|---|---|---|---|---|---|
| 1A2 | 2C9 | 2C19 | 2D6 | 3A4 | % Bound | % Remaining (30 min) | % Remaining (30 min) | |
| AJ-024 | 68.9 | 73.3 | 57.2 | 77.6 | 62.8 | 99.9 | 63.8 | 95.9 |
| Ketoconazole | 94.3 | >100 | 97.6 | >100 | 27.9 | |||
| Dexamethasone | 78.8 | |||||||
| Warfarin | 98.9 | |||||||
| Verapamil | 16.3 | |||||||
| Procaine | 1.4 | |||||||
| Enalapril | 95.2 | |||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.; Kim, Y.-R.; Hwang, H.-J.; Ciufolini, M.A.; Lee, J.; Lee, H.; Clovis, S.; Jung, S.; Oh, S.-H.; Son, Y.-J.; et al. Nitro-Group-Containing Thiopeptide Derivatives as Promising Agents to Target Clostridioides difficile. Pharmaceuticals 2022, 15, 623. https://doi.org/10.3390/ph15050623
Kim D, Kim Y-R, Hwang H-J, Ciufolini MA, Lee J, Lee H, Clovis S, Jung S, Oh S-H, Son Y-J, et al. Nitro-Group-Containing Thiopeptide Derivatives as Promising Agents to Target Clostridioides difficile. Pharmaceuticals. 2022; 15(5):623. https://doi.org/10.3390/ph15050623
Chicago/Turabian StyleKim, Dahyun, Young-Rok Kim, Hee-Jong Hwang, Marco A. Ciufolini, Jusuk Lee, Hakyeong Lee, Shyaka Clovis, Sungji Jung, Sang-Hun Oh, Young-Jin Son, and et al. 2022. "Nitro-Group-Containing Thiopeptide Derivatives as Promising Agents to Target Clostridioides difficile" Pharmaceuticals 15, no. 5: 623. https://doi.org/10.3390/ph15050623
APA StyleKim, D., Kim, Y.-R., Hwang, H.-J., Ciufolini, M. A., Lee, J., Lee, H., Clovis, S., Jung, S., Oh, S.-H., Son, Y.-J., & Kwak, J.-H. (2022). Nitro-Group-Containing Thiopeptide Derivatives as Promising Agents to Target Clostridioides difficile. Pharmaceuticals, 15(5), 623. https://doi.org/10.3390/ph15050623