
Immunosuppressant Therapies in COVID-19: Is the TNF Axis an Alternative?



Abstract
1. Introduction
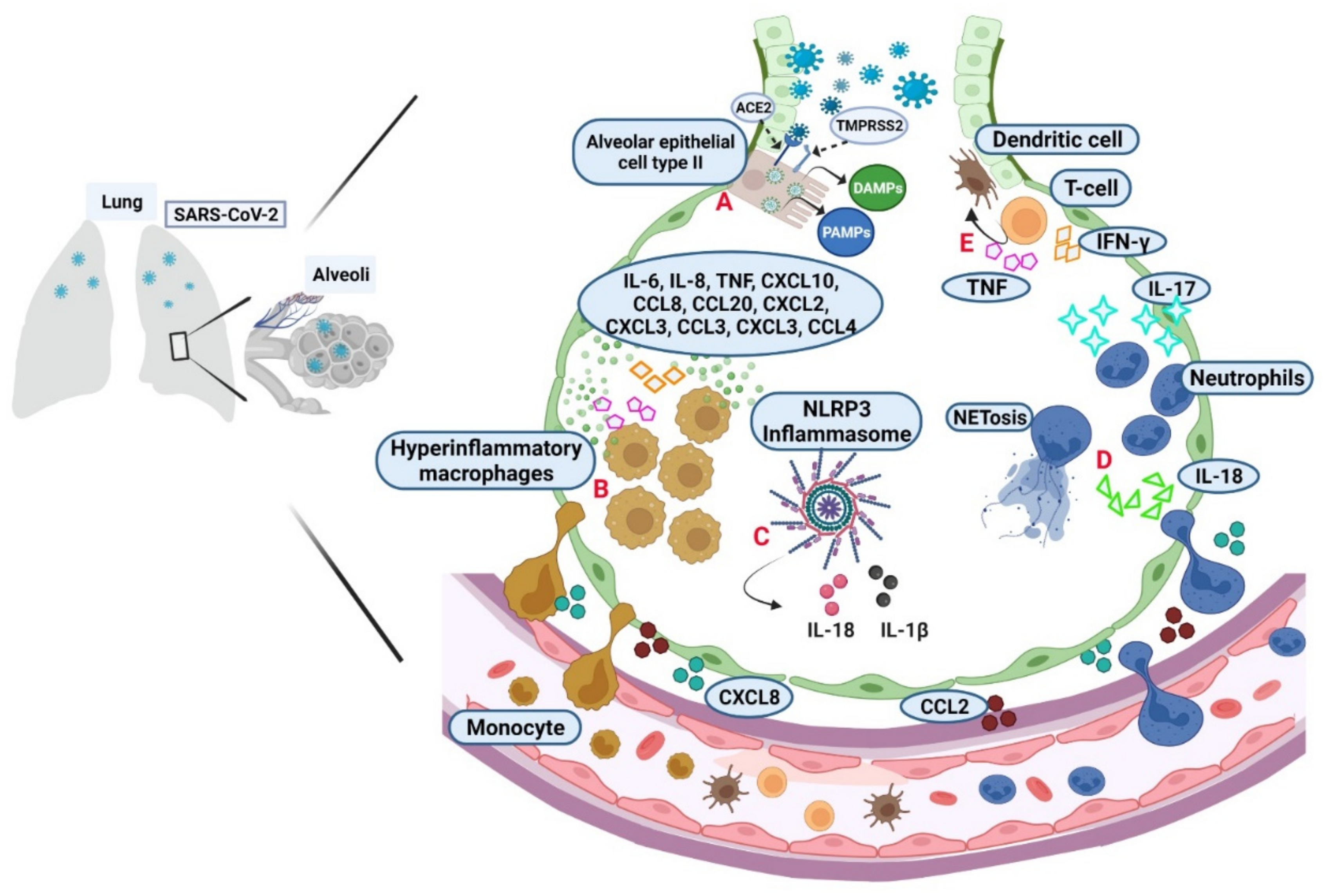
2. More than One Rout Favor the Cytokine Storm in COVID-19
3. Relevant Targets of the Inflammatory Response for Immunotherapy in COVID-19
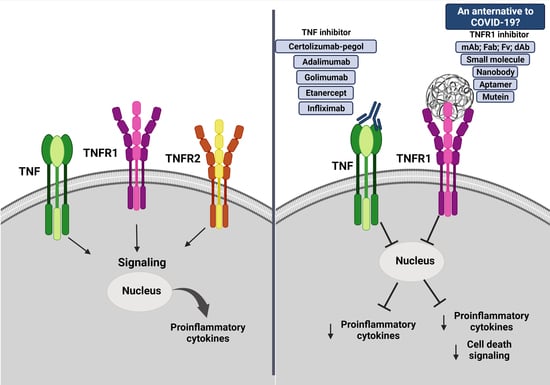
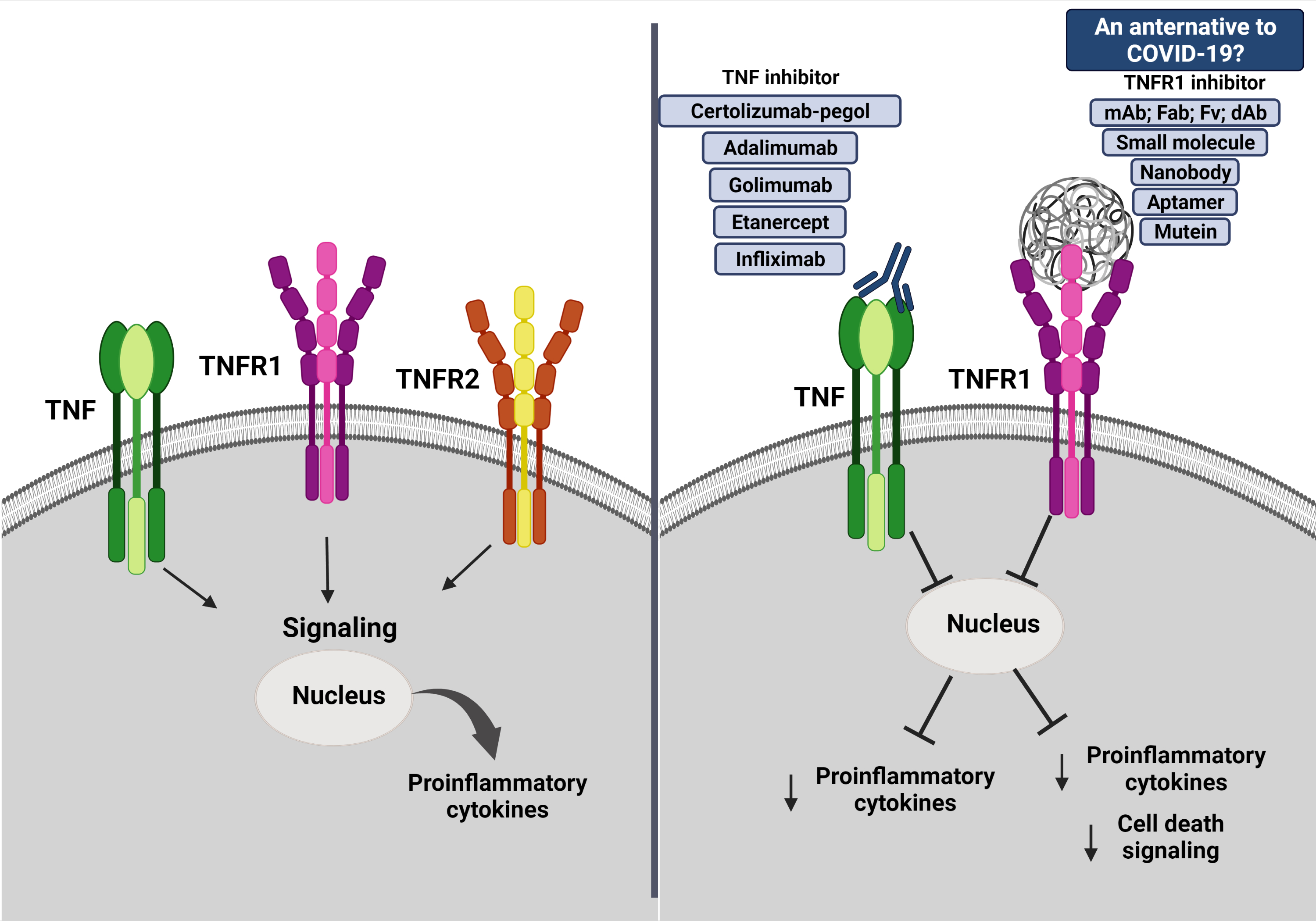
4. What Is TNF and Its Inhibitors?
5. Could TNF Be Considered a Target in COVID-19?
6. Could Be sTNFR1 a Treatment Target for COVID-19?
7. What We Know from Preliminary Results about TNFR1-Selective Inhibitors under Progress?
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADAM17 | A Disintegrin and Metalloprotease 17 |
| ARDS | Acute Respiratory Distress Syndrome |
| ACE2 | Angiotensin-Converting Enzyme 2 |
| BAL | Bronchoalveolar lavage |
| DAMPs | Danger-Associated Molecular Patterns |
| IL-6R | Interleukin 6 Receptor |
| IBD | Inflammatory Bowel Disease |
| iNOS | Inducible Nitric Oxide Synthase |
| IRF1 | Inducible nitric oxide synthase |
| JAK | Janus kinase |
| mAbs | Monoclonal antibodies |
| MAP | Mycobacterium avium subspecies paratuberculosis |
| MAS | Macrophage Activation Syndrome |
| NET | Neutrophil Extracellular Trap |
| NF-κB | Nuclear Factor-kappa B |
| NK | Natural killer |
| NO | Nitric Oxide |
| NRP1 | Neuropilin 1 |
| PAMPs | Pathogen-Associated Molecular Patterns |
| PANoptosis | Pyroptosis, Necroptosis, and Apoptosis |
| PLAD | Pre-ligand-binding assembly domain |
| PRRs | Pattern Recognition Receptors |
| SARS-CoV-2 | Severe Acute Respiratory Syndrome coronavirus-2 |
| sTNFR1 | soluble TNFR1 |
| sTNF | soluble TNF |
| SNPs | Single nucleotide polymorphisms |
| TLR | Toll-like receptors |
| TNF | Tumor Necrosis Factor |
| tmTNF | Transmembrane-TNF |
| TMPRSS2 | Type II Transmembrane Serine Protease |
| TNFR1 | TNF receptor 1 |
| TNFR2 | TNF receptor 2 |
| TNFRs | TNF receptors |
| TNFRSF | TNF receptor superfamily |
| VOC | Variants of Concern |
References
- Yokota, S.; Miyamae, T.; Kuroiwa, Y.; Nishioka, K. Novel Coronavirus Disease 2019 (COVID-19) and Cytokine Storms for More Effective Treatments from an Inflammatory Pathophysiology. J. Clin. Med. 2021, 10, 801. [Google Scholar] [CrossRef] [PubMed]
- Acuti Martellucci, C.; Flacco, M.E.; Cappadona, R.; Bravi, F.; Mantovani, L.; Manzoli, L. SARS-CoV-2 Pandemic: An Overview. Adv. Biol. Regul. 2020, 77, 100736. [Google Scholar] [CrossRef] [PubMed]
- Cucinotta, D.; Vanelli, M. WHO Declares COVID-19 a Pandemic. Acta Bio Med. Atenei Parm. 2020, 91, 157–160. [Google Scholar] [CrossRef]
- WHO Coronavirus Disease (COVID-19) Dashboard. Available online: https://covid19.who.int (accessed on 18 March 2022).
- World Health Organization. COVID-19 Strategic Preparedness and Response Plan: Monitoring and Evaluation Framework. 2021. Available online: https://www.who.int/publications/i/item/WHO-WHE-2021.07-eng (accessed on 18 March 2022).
- Kannan, S.; Shaik Syed Ali, P.; Sheeza, A. Omicron (B.1.1.529)-Variant of Concern-Molecular Profile and Epidemiology: A Mini Review. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 8019–8022. [Google Scholar] [CrossRef]
- Nealon, J.; Cowling, B.J. Omicron Severity: Milder but Not Mild. Lancet Lond. Engl. 2022, 399, 412–413. [Google Scholar] [CrossRef]
- Hsu, J.Y.; Mao, Y.C.; Liu, P.Y.; Lai, K.L. Pharmacology and Adverse Events of Emergency-Use Authorized Medication in Moderate to Severe COVID-19. Pharm. Basel Switz. 2021, 14, 955. [Google Scholar] [CrossRef]
- Buszko, M.; Nita-Lazar, A.; Park, J.-H.; Schwartzberg, P.L.; Verthelyi, D.; Young, H.A.; Rosenberg, A.S. Lessons Learned: New Insights on the Role of Cytokines in COVID-19. Nat. Immunol. 2021, 22, 404–411. [Google Scholar] [CrossRef]
- Sinha, P.; Matthay, M.A.; Calfee, C.S. Is a “Cytokine Storm” Relevant to COVID-19? JAMA Intern. Med. 2020, 180, 1152–1154. [Google Scholar] [CrossRef]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the Eye of the Cytokine Storm. Microbiol. Mol. Biol. Rev. MMBR 2012, 76, 16–32. [Google Scholar] [CrossRef]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-α and IFN-γ Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2020, 184, 149–168.e17. [Google Scholar] [CrossRef]
- Fara, A.; Mitrev, Z.; Rosalia, R.A.; Assas, B.M. Cytokine Storm and COVID-19: A Chronicle of pro-Inflammatory Cytokines. Open Biol. 2020, 10, 200160. [Google Scholar] [CrossRef]
- Júnior, M.L.; de Souza, L.M.; Dutra, R.E.; de Melo Valente, R.G.; Melo, T.S. Review on Therapeutic Targets for COVID-19: Insights from Cytokine Storm. Postgrad. Med. J. 2021, 97, 391–398. [Google Scholar] [CrossRef]
- Steeland, S.; Libert, C.; Vandenbroucke, R.E. A New Venue of TNF Targeting. Int. J. Mol. Sci. 2018, 19, 1442. [Google Scholar] [CrossRef]
- Mootoo, A.; Stylianou, E.; Arias, M.A.; Reljic, R. TNF-Alpha in Tuberculosis: A Cytokine with a Split Personality. Inflamm. Allergy Drug Targets 2009, 8, 53–62. [Google Scholar] [CrossRef]
- Kollias, G.; Douni, E.; Kassiotis, G.; Kontoyiannis, D. The Function of Tumour Necrosis Factor and Receptors in Models of Multi-Organ Inflammation, Rheumatoid Arthritis, Multiple Sclerosis and Inflammatory Bowel Disease. Ann. Rheum. Dis. 1999, 58 (Suppl. S1), I32–I39. [Google Scholar] [CrossRef]
- Feldmann, M.; Maini, R.N.; Woody, J.N.; Holgate, S.T.; Winter, G.; Rowland, M.; Richards, D.; Hussell, T. Trials of Anti-Tumour Necrosis Factor Therapy for COVID-19 Are Urgently Needed. Lancet 2020, 395, 1407–1409. [Google Scholar] [CrossRef]
- Robinson, P.C.; Liew, D.F.L.; Liew, J.W.; Monaco, C.; Richards, D.; Shivakumar, S.; Tanner, H.L.; Feldmann, M. The Potential for Repurposing Anti-TNF as a Therapy for the Treatment of COVID-19. Med 2020, 1, 90–102. [Google Scholar] [CrossRef]
- Ravelli, A.; Minoia, F.; Davì, S.; Horne, A.; Bovis, F.; Pistorio, A.; Aricò, M.; Avcin, T.; Behrens, E.M.; De Benedetti, F.; et al. 2016 Classification Criteria for Macrophage Activation Syndrome Complicating Systemic Juvenile Idiopathic Arthritis: A European League Against Rheumatism/American College of Rheumatology/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Arthritis Rheumatol. 2016, 68, 566–576. [Google Scholar] [CrossRef]
- Karki, R.; Kanneganti, T.-D. The “Cytokine Storm”: Molecular Mechanisms and Therapeutic Prospects. Trends Immunol. 2021, 42, 681–705. [Google Scholar] [CrossRef]
- Potdar, A.A.; Dube, S.; Naito, T.; Li, K.; Botwin, G.; Haritunians, T.; Li, D.; Casero, D.; Yang, S.; Bilsborough, J.; et al. Altered Intestinal ACE2 Levels Are Associated With Inflammation, Severe Disease, and Response to Anti-Cytokine Therapy in Inflammatory Bowel Disease. Gastroenterology 2021, 160, 809–822.e7. [Google Scholar] [CrossRef]
- Angeli, F.; Zappa, M.; Reboldi, G.; Trapasso, M.; Cavallini, C.; Spanevello, A.; Verdecchia, P. The Pivotal Link between ACE2 Deficiency and SARS-CoV-2 Infection: One Year Later. Eur. J. Intern. Med. 2021, 93, 28–34. [Google Scholar] [CrossRef]
- Li, X.Z.; Qiu, Y.; Jeffery, L.; Liu, F.; Feng, R.; He, J.S.; Tan, J.Y.; Ye, Z.Y.; Lin, S.N.; Ghosh, S.; et al. Down-Regulation of Colonic ACE2 Expression in Patients With Inflammatory Bowel Disease Responding to Anti-TNF Therapy: Implications for COVID-19. Front. Med. 2021, 7, 613475. [Google Scholar] [CrossRef]
- Aleksova, A.; Gagno, G.; Sinagra, G.; Beltrami, A.P.; Janjusevic, M.; Ippolito, G.; Zumla, A.; Fluca, A.L.; Ferro, F. Effects of SARS-CoV-2 on Cardiovascular System: The Dual Role of Angiotensin-Converting Enzyme 2 (ACE2) as the Virus Receptor and Homeostasis Regulator-Review. Int. J. Mol. Sci. 2021, 22, 4526. [Google Scholar] [CrossRef]
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the Receptor-Binding Domain (RBD) of 2019 Novel Coronavirus: Implication for Development of RBD Protein as a Viral Attachment Inhibitor and Vaccine. Cell. Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef]
- Schultze, J.L.; Aschenbrenner, A.C. COVID-19 and the Human Innate Immune System. Cell 2021, 184, 1671–1692. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 Facilitates SARS-CoV-2 Cell Entry and Infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.-C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System: Celebrating the 20th Anniversary of the Discovery of ACE2. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef]
- Ambrocio-Ortiz, E.; Pérez-Rubio, G.; Del Ángel-Pablo, A.D.; Buendía-Roldán, I.; Chávez-Galán, L.; Hernández-Zenteno, R.D.; Ramírez-Venegas, A.; Rojas-Serrano, J.; Mejía, M.; Pérez-Padilla, R.; et al. Angiotensin-Converting Enzyme 2 (ACE2) in the Context of Respiratory Diseases and Its Importance in Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Infection. Pharm. Basel Switz. 2021, 14, 805. [Google Scholar] [CrossRef]
- Calligaris, M.; Cuffaro, D.; Bonelli, S.; Spanò, D.P.; Rossello, A.; Nuti, E.; Scilabra, S.D. Strategies to Target ADAM17 in Disease: From Its Discovery to the IRhom Revolution. Mol. Basel Switz. 2021, 26, 944. [Google Scholar] [CrossRef]
- Xiao, L.; Sakagami, H.; Miwa, N. ACE2: The Key Molecule for Understanding the Pathophysiology of Severe and Critical Conditions of COVID-19: Demon or Angel? Viruses 2020, 12, 491. [Google Scholar] [CrossRef]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pöhlmann, S. TMPRSS2 and ADAM17 Cleave ACE2 Differentially and Only Proteolysis by TMPRSS2 Augments Entry Driven by the Severe Acute Respiratory Syndrome Coronavirus Spike Protein. J. Virol. 2014, 88, 1293–1307. [Google Scholar] [CrossRef]
- Matsuyama, S.; Nagata, N.; Shirato, K.; Kawase, M.; Takeda, M.; Taguchi, F. Efficient Activation of the Severe Acute Respiratory Syndrome Coronavirus Spike Protein by the Transmembrane Protease TMPRSS2. J. Virol. 2010, 84, 12658–12664. [Google Scholar] [CrossRef]
- Shulla, A.; Heald-Sargent, T.; Subramanya, G.; Zhao, J.; Perlman, S.; Gallagher, T. A Transmembrane Serine Protease Is Linked to the Severe Acute Respiratory Syndrome Coronavirus Receptor and Activates Virus Entry. J. Virol. 2011, 85, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Channappanavar, R.; Kanneganti, T.-D. Coronaviruses: Innate Immunity, Inflammasome Activation, Inflammatory Cell Death, and Cytokines. Trends Immunol. 2020, 41, 1083–1099. [Google Scholar] [CrossRef]
- Medina-Quero, K.; Barreto-Rodriguez, O.; Mendez-Rodriguez, V.; Sanchez-Moncivais, A.; Buendia-Roldan, I.; Chavez-Galan, L. SARS-CoV-2 Infection: Understanding the Immune System Abnormalities to Get an Adequate Diagnosis. Bosn. J. Basic Med. Sci. 2021, 21, 503–514. [Google Scholar] [CrossRef]
- Paludan, S.R.; Mogensen, T.H. Innate Immunological Pathways in COVID-19 Pathogenesis. Sci. Immunol. 2022, 7, eabm5505. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Wen, W.; Fan, X.; Hou, W.; Su, B.; Cai, P.; Li, J.; Liu, Y.; Tang, F.; Zhang, F.; et al. COVID-19 Immune Features Revealed by a Large-Scale Single-Cell Transcriptome Atlas. Cell 2021, 184, 1895–1913.e19. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.; Aktar, S.; Rahman, M.M.; Chowdhury, M.M.H. NLRP3 Inflammasome Activation in COVID-19: An Interlink between Risk Factors and Disease Severity. Microbes Infect. 2022, 24, 104913. [Google Scholar] [CrossRef]
- Rodrigues, T.S.; de Sá, K.S.G.; Ishimoto, A.Y.; Becerra, A.; Oliveira, S.; Almeida, L.; Gonçalves, A.V.; Perucello, D.B.; Andrade, W.A.; Castro, R.; et al. Inflammasomes Are Activated in Response to SARS-CoV-2 Infection and Are Associated with COVID-19 Severity in Patients. J. Exp. Med. 2021, 218, e20201707. [Google Scholar] [CrossRef]
- Ackermann, M.; Anders, H.-J.; Bilyy, R.; Bowlin, G.L.; Daniel, C.; De Lorenzo, R.; Egeblad, M.; Henneck, T.; Hidalgo, A.; Hoffmann, M.; et al. Patients with COVID-19: In the Dark-NETs of Neutrophils. Cell Death Differ. 2021, 28, 3125–3139. [Google Scholar] [CrossRef]
- Tomar, B.; Anders, H.-J.; Desai, J.; Mulay, S.R. Neutrophils and Neutrophil Extracellular Traps Drive Necroinflammation in COVID-19. Cells 2020, 9, 1383. [Google Scholar] [CrossRef]
- Del Valle, D.M.; Kim-Schulze, S.; Huang, H.-H.; Beckmann, N.D.; Nirenberg, S.; Wang, B.; Lavin, Y.; Swartz, T.H.; Madduri, D.; Stock, A.; et al. An Inflammatory Cytokine Signature Predicts COVID-19 Severity and Survival. Nat. Med. 2020, 26, 1636–1643. [Google Scholar] [CrossRef]
- Kox, M.; Waalders, N.J.B.; Kooistra, E.J.; Gerretsen, J.; Pickkers, P. Cytokine Levels in Critically Ill Patients With COVID-19 and Other Conditions. JAMA 2020, 324, 1565–1567. [Google Scholar] [CrossRef]
- Wilson, J.G.; Simpson, L.J.; Ferreira, A.-M.; Rustagi, A.; Roque, J.; Asuni, A.; Ranganath, T.; Grant, P.M.; Subramanian, A.; Rosenberg-Hasson, Y.; et al. Cytokine Profile in Plasma of Severe COVID-19 Does Not Differ from ARDS and Sepsis. JCI Insight 2020, 5, e140289. [Google Scholar] [CrossRef]
- Cortegiani, A.; Ippolito, M.; Greco, M.; Granone, V.; Protti, A.; Gregoretti, C.; Giarratano, A.; Einav, S.; Cecconi, M. Rationale and Evidence on the Use of Tocilizumab in COVID-19: A Systematic Review. Pulmonology 2021, 27, 52–66. [Google Scholar] [CrossRef]
- Mahase, E. COVID-19: Anti-TNF Drug Adalimumab to Be Trialled for Patients in the Community. BMJ 2020, 371, m3847. [Google Scholar] [CrossRef]
- Aouba, A.; Baldolli, A.; Geffray, L.; Verdon, R.; Bergot, E.; Martin-Silva, N.; Justet, A. Targeting the Inflammatory Cascade with Anakinra in Moderate to Severe COVID-19 Pneumonia: Case Series. Ann. Rheum. Dis. 2020, 79, 1381–1382. [Google Scholar] [CrossRef]
- Atal, S.; Fatima, Z. IL-6 Inhibitors in the Treatment of Serious COVID-19: A Promising Therapy? Pharm. Med. 2020, 34, 223–231. [Google Scholar] [CrossRef]
- Huet, T.; Beaussier, H.; Voisin, O.; Jouveshomme, S.; Dauriat, G.; Lazareth, I.; Sacco, E.; Naccache, J.M.; Bézie, Y.; Laplanche, S.; et al. Anakinra for Severe Forms of COVID-19: A Cohort Study. Lancet Rheumatol. 2020, 2, e393–e400. [Google Scholar] [CrossRef]
- Lythgoe, M.P.; Middleton, P. Ongoing Clinical Trials for the Management of the COVID-19 Pandemic. Trends Pharmacol. Sci. 2020, 41, 363–382. [Google Scholar] [CrossRef]
- Lu, R.-M.; Hwang, Y.-C.; Liu, I.-J.; Lee, C.-C.; Tsai, H.-Z.; Li, H.-J.; Wu, H.-C. Development of Therapeutic Antibodies for the Treatment of Diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef]
- Rodrugues, E.B.; Farah, M.E.; Maia, M.; Penha, F.M.; Regatieri, C.; Melo, G.B.; Pinheiro, M.M.; Zanetti, C.r. Therapeutic monoclonal antibodies in ophthalmology. Prog. Retin. Eye Res. 2009, 28, 117–144. [Google Scholar] [CrossRef]
- Gautret, P.; Million, M.; Jarrot, P.-A.; Camoin-Jau, L.; Colson, P.; Fenollar, F.; Leone, M.; La Scola, B.; Devaux, C.; Gaubert, J.Y.; et al. Natural History of COVID-19 and Therapeutic Options. Expert Rev. Clin. Immunol. 2020, 16, 1159–1184. [Google Scholar] [CrossRef]
- Liu, B.; Li, M.; Zhou, Z.; Guan, X.; Xiang, Y. Can We Use Interleukin-6 (IL-6) Blockade for Coronavirus Disease 2019 (COVID-19)-Induced Cytokine Release Syndrome (CRS)? J. Autoimmun. 2020, 111, 102452. [Google Scholar] [CrossRef]
- Le, T.T.; Karmouty-Quintana, H.; Melicoff, E.; Le, T.T.; Weng, T.; Chen, N.-Y.; Pedroza, M.; Zhou, Y.; Davies, J.; Philip, K.; et al. Blockade of IL-6 Trans Signaling Attenuates Pulmonary Fibrosis. J. Immunol. Baltim. Md. 1950 2014, 193, 3755–3768. [Google Scholar] [CrossRef]
- NIH COVID-19 Treatment Guidelines. Available online: https://www.covid19treatmentguidelines.nih.gov/about-the-guidelines/whats-new/ (accessed on 1 February 2022).
- Vela, D.; Vela-Gaxha, Z.; Rexhepi, M.; Olloni, R.; Hyseni, V.; Nallbani, R. Efficacy and Safety of Tocilizumab versus Standard Care/Placebo in Patients with COVID-19; a Systematic Review and Meta-analysis of Randomized Clinical Trials. Br. J. Clin. Pharmacol. 2022, 88, 1955–1963. [Google Scholar] [CrossRef]
- Kaplon, H.; Chenoweth, A.; Crescioli, S.; Reichert, J.M. Antibodies to Watch in 2022. mAbs 2022, 14, 2014296. [Google Scholar] [CrossRef]
- Dougan, M.; Nirula, A.; Azizad, M.; Mocherla, B.; Gottlieb, R.L.; Chen, P.; Hebert, C.; Perry, R.; Boscia, J.; Heller, B.; et al. Bamlanivimab plus Etesevimab in Mild or Moderate COVID-19. N. Engl. J. Med. 2021, 385, 1382–1392. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Gonzalez-Rojas, Y.; Juarez, E.; Crespo Casal, M.; Moya, J.; Falci, D.R.; Sarkis, E.; Solis, J.; Zheng, H.; Scott, N.; et al. COMET-ICE Investigators. Early Treatment for COVID-19 with SARS-CoV-2 Neutralizing Antibody Sotrovimab. N. Engl. J. Med. 2021, 385, 1941–1950. [Google Scholar] [CrossRef] [PubMed]
- Therapeutic Goods Administration. Australian Public Assessment Report for Sotrovimab, Proprietary Product Name: Xevudy; Sponsor: GlaxoSmithKline Australia Pty Ltd. Available online: https://www.tga.gov.au/sites/default/files/auspar-sotrovimab-210820.pdf (accessed on 13 February 2022).
- Syed, Y.Y. Regdanvimab: First Approval. Drugs 2021, 81, 2133–2137. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Casirivimab/Imdevimab: First Approval. Drugs 2021, 81, 2047–2055. [Google Scholar] [CrossRef]
- Bachove, I.; Chang, C. Anakinra and Related Drugs Targeting Interleukin-1 in the Treatment of Cryopyrin-Associated Periodic Syndromes. Open Access Rheumatol. Res. Rev. 2014, 6, 15–25. [Google Scholar] [CrossRef]
- Maes, B.; Bosteels, C.; De Leeuw, E.; Declercq, J.; Van Damme, K.; Delporte, A.; Demeyere, B.; Vermeersch, S.; Vuylsteke, M.; Willaert, J.; et al. Treatment of Severely Ill COVID-19 Patients with Anti-Interleukin Drugs (COV-AID): A Structured Summary of a Study Protocol for a Randomised Controlled Trial. Trials 2020, 21, 468. [Google Scholar] [CrossRef]
- Hwang, Y.-C.; Lu, R.-M.; Su, S.-C.; Chiang, P.-Y.; Ko, S.-H.; Ke, F.-Y.; Liang, K.-H.; Hsieh, T.-Y.; Wu, H.-C. Monoclonal Antibodies for COVID-19 Therapy and SARS-CoV-2 Detection. J. Biomed. Sci. 2022, 29, 1. [Google Scholar] [CrossRef]
- Kedor, C.; Listing, J.; Zernicke, J.; Weiß, A.; Behrens, F.; Blank, N.; Henes, J.C.; Kekow, J.; Rubbert-Roth, A.; Schulze-Koops, H.; et al. Canakinumab for Treatment of Adult-Onset Still’s Disease to Achieve Reduction of Arthritic Manifestation (CONSIDER): Phase II, Randomised, Double-Blind, Placebo-Controlled, Multicentre, Investigator-Initiated Trial. Ann. Rheum. Dis. 2020, 79, 1090–1097. [Google Scholar] [CrossRef]
- Sfriso, P.; Bindoli, S.; Doria, A.; Feist, E.; Galozzi, P. Canakinumab for the Treatment of Adult-Onset Still’s Disease. Expert Rev. Clin. Immunol. 2020, 16, 129–138. [Google Scholar] [CrossRef]
- Caricchio, R.; Abbate, A.; Gordeev, I.; Meng, J.; Hsue, P.Y.; Neogi, T.; Arduino, R.; Fomina, D.; Bogdanov, R.; Stepanenko, T.; et al. Effect of Canakinumab vs Placebo on Survival Without Invasive Mechanical Ventilation in Patients Hospitalized With Severe COVID-19: A Randomized Clinical Trial. JAMA 2021, 326, 230–239. [Google Scholar] [CrossRef]
- Jorgensen, S.C.J.; Tse, C.L.Y.; Burry, L.; Dresser, L.D. Baricitinib: A Review of Pharmacology, Safety, and Emerging Clinical Experience in COVID-19. Pharmacotherapy 2020, 40, 843–856. [Google Scholar] [CrossRef]
- Kalil, A.C.; Patterson, T.F.; Mehta, A.K.; Tomashek, K.M.; Wolfe, C.R.; Ghazaryan, V.; Marconi, V.C.; Ruiz-Palacios, G.M.; Hsieh, L.; Kline, S.; et al. ACTT-2 Study Group Members. Baricitinib plus Remdesivir for Hospitalized Adults with COVID-19. N. Engl. J. Med. 2021, 384, 795–807. [Google Scholar] [CrossRef]
- Biddle, K.; White, J.; Sofat, N. What Is the Full Potential of Baricitinib in Treating Patients with COVID-19? Expert Rev. Clin. Immunol. 2022, 1–5. [Google Scholar] [CrossRef]
- Holbrook, J.; Lara-Reyna, S.; Jarosz-Griffiths, H.; McDermott, M. Tumour Necrosis Factor Signalling in Health and Disease. F1000Research 2019, 8. F1000 Faculty Rev-111. [Google Scholar] [CrossRef]
- Monaco, C.; Nanchahal, J.; Taylor, P.; Feldmann, M. Anti-TNF Therapy: Past, Present and Future. Int. Immunol. 2015, 27, 55–62. [Google Scholar] [CrossRef]
- Horiuchi, T.; Mitoma, H.; Harashima, S.; Tsukamoto, H.; Shimoda, T. Transmembrane TNF-Alpha: Structure, Function and Interaction with Anti-TNF Agents. Rheumatol. Oxf. Engl. 2010, 49, 1215–1228. [Google Scholar] [CrossRef]
- Parameswaran, N.; Patial, S. Tumor Necrosis Factor-α Signaling in Macrophages. Crit. Rev. Eukaryot. Gene Expr. 2010, 20, 87–103. [Google Scholar] [CrossRef]
- Ruiz, A.; Palacios, Y.; Garcia, I.; Chavez-Galan, L. Transmembrane TNF and Its Receptors TNFR1 and TNFR2 in Mycobacterial Infections. Int. J. Mol. Sci. 2021, 22, 5461. [Google Scholar] [CrossRef]
- Pimentel-Muiños, F.X.; Seed, B. Regulated Commitment of TNF Receptor Signaling: A Molecular Switch for Death or Activation. Immunity 1999, 11, 783–793. [Google Scholar] [CrossRef]
- Lainez, B.; Fernandez-Real, J.M.; Romero, X.; Esplugues, E.; Cañete, J.D.; Ricart, W.; Engel, P. Identification and Characterization of a Novel Spliced Variant That Encodes Human Soluble Tumor Necrosis Factor Receptor 2. Int. Immunol. 2004, 16, 169–177. [Google Scholar] [CrossRef]
- Hernaez, B.; Alcamí, A. Virus-Encoded Cytokine and Chemokine Decoy Receptors. Curr. Opin. Immunol. 2020, 66, 50–56. [Google Scholar] [CrossRef]
- Levine, S.J. Mechanisms of Soluble Cytokine Receptor Generation. J. Immunol. Baltim. Md 1950 2004, 173, 5343–5348. [Google Scholar] [CrossRef]
- Sultana, S.; Bishayi, B. Neutralization of TNFR-1 and TNFR-2 Modulates S. Aureus Induced Septic Arthritis by Regulating the Levels of pro Inflammatory and Anti Inflammatory Cytokines during the Progression of the Disease. Immunol. Lett. 2018, 196, 33–51. [Google Scholar] [CrossRef]
- US Food and Drug Administration. FDA Drug Safety Communication: Drug Labels for the Tumor Necrosis Factor-Alpha (TNFα) Blockers Now Include Warnings about Infection with Legionella and Listeria Bacteria. FDA 2011. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-drug-labels-tumor-necrosis-factor-alpha-tnfa-blockers-now-include (accessed on 12 March 2022).
- Lim, H.; Lee, S.H.; Lee, H.T.; Lee, J.U.; Son, J.Y.; Shin, W.; Heo, Y.-S. Structural Biology of the TNFα Antagonists Used in the Treatment of Rheumatoid Arthritis. Int. J. Mol. Sci. 2018, 19, 768. [Google Scholar] [CrossRef]
- Caporali, R.; Allanore, Y.; Alten, R.; Combe, B.; Durez, P.; Iannone, F.; Nurmohamed, M.T.; Lee, S.J.; Kwon, T.S.; Choi, J.S.; et al. Efficacy and Safety of Subcutaneous Infliximab versus Adalimumab, Etanercept and Intravenous Infliximab in Patients with Rheumatoid Arthritis: A Systematic Literature Review and Meta-Analysis. Expert Rev. Clin. Immunol. 2021, 17, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Wiens, A.; Correr, C.J.; Pontarolo, R.; Venson, R.; Quinalha, J.V.; Otuki, M.F. A Systematic Review and Meta-Analysis of the Efficacy and Safety of Etanercept for Treating Rheumatoid Arthritis. Scand. J. Immunol. 2009, 70, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Mitoma, H.; Horiuchi, T.; Tsukamoto, H.; Ueda, N. Molecular Mechanisms of Action of Anti-TNF-α Agents-Comparison among Therapeutic TNF-α Antagonists. Cytokine 2018, 101, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zheng, Y.; Chen, X. Drugs for Autoimmune Inflammatory Diseases: From Small Molecule Compounds to Anti-TNF Biologics. Front. Pharmacol. 2017, 8, 460. [Google Scholar] [CrossRef] [PubMed]
- Kaymakcalan, Z.; Sakorafas, P.; Bose, S.; Scesney, S.; Xiong, L.; Hanzatian, D.K.; Salfeld, J.; Sasso, E.H. Comparisons of Affinities, Avidities, and Complement Activation of Adalimumab, Infliximab, and Etanercept in Binding to Soluble and Membrane Tumor Necrosis Factor. Clin. Immunol. Orlando Fla 2009, 131, 308–316. [Google Scholar] [CrossRef]
- Sherman, M.; Tsynman, D.N.; Kim, A.; Arora, J.; Pietras, T.; Messing, S.; St Hilaire, L.; Yoon, S.; Decross, A.; Shah, A.; et al. Sustained Improvement in Health-Related Quality of Life Measures in Patients with Inflammatory Bowel Disease Receiving Prolonged Anti-Tumor Necrosis Factor Therapy. J. Dig. Dis. 2014, 15, 174–179. [Google Scholar] [CrossRef]
- van Schouwenburg, P.A.; Rispens, T.; Wolbink, G.J. Immunogenicity of Anti-TNF Biologic Therapies for Rheumatoid Arthritis. Nat. Rev. Rheumatol. 2013, 9, 164–172. [Google Scholar] [CrossRef]
- Patel, S.; Wadhwa, M. Therapeutic Use of Specific Tumour Necrosis Factor Inhibitors in Inflammatory Diseases Including COVID-19. Biomed. Pharmacother. Biomed. Pharmacother. 2021, 140, 111785. [Google Scholar] [CrossRef]
- Liuzzo, G.; Patrono, C. COVID 19: In the Eye of the Cytokine Storm. Eur. Heart J. 2021, 42, 150–151. [Google Scholar] [CrossRef]
- Leisman, D.E.; Ronner, L.; Pinotti, R.; Taylor, M.D.; Sinha, P.; Calfee, C.S.; Hirayama, A.V.; Mastroiani, F.; Turtle, C.J.; Harhay, M.O.; et al. Cytokine Elevation in Severe and Critical COVID-19: A Rapid Systematic Review, Meta-Analysis, and Comparison with Other Inflammatory Syndromes. Lancet Respir. Med. 2020, 8, 1233–1244. [Google Scholar] [CrossRef]
- Palacios, Y.; Ruiz, A.; Ramón-Luing, L.A.; Ocaña-Guzman, R.; Barreto-Rodriguez, O.; Sánchez-Monciváis, A.; Tecuatzi-Cadena, B.; Regalado-García, A.G.; Pineda-Gudiño, R.D.; García-Martínez, A.; et al. Severe COVID-19 Patients Show an Increase in Soluble TNFR1 and ADAM17, with a Relationship to Mortality. Int. J. Mol. Sci. 2021, 22, 8423. [Google Scholar] [CrossRef]
- Baslılar, S.; Pehlivan, O. Evaluation of Factors Affecting the Frequency and Clinical Course of COVID-19 in Patients Using Anti-TNF-Alpha Agents. Rev. Assoc. Med. Bras. 1992 2021, 67, 1286–1292. [Google Scholar] [CrossRef]
- Izadi, Z.; Brenner, E.J.; Mahil, S.K.; Dand, N.; Yiu, Z.Z.N.; Yates, M.; Ungaro, R.C.; Zhang, X.; Agrawal, M.; Colombel, J.-F.; et al. Association Between Tumor Necrosis Factor Inhibitors and the Risk of Hospitalization or Death Among Patients With Immune-Mediated Inflammatory Disease and COVID-19. JAMA Netw. Open 2021, 4, e2129639. [Google Scholar] [CrossRef]
- Kokkotis, G.; Kitsou, K.; Xynogalas, I.; Spoulou, V.; Magiorkinis, G.; Trontzas, I.; Trontzas, P.; Poulakou, G.; Syrigos, K.; Bamias, G. Systematic Review with Meta-Analysis: COVID-19 Outcomes in Patients Receiving Anti-TNF Treatments. Aliment. Pharmacol. Ther. 2022, 55, 154–167. [Google Scholar] [CrossRef]
- Foundation CsaC. Resources for IBD Healthcare Professionals: 2019 Novel Coronavirus (COVID-19) (2020). Available online: https://www.crohnscolitisfoundation.org/coronavirus/ibd-medication (accessed on 18 March 2022).
- Crohn´s and Colitis Foundation. Crohn’s and Colitis Foundation. National Scientific Advisory Committee (NSAC). Available online: https://www.crohnscolitisfoundation.org/about/national-scientific-advisory-committee (accessed on 18 March 2022).
- Rubin, D.T.; Abreu, M.T.; Rai, V.; Siegel, C.A. Management of Patients With Crohn’s Disease and Ulcerative Colitis During the Coronavirus Disease-2019 Pandemic: Results of an International Meeting. Gastroenterology 2020, 159, 6–13.e6. [Google Scholar] [CrossRef]
- Rubin, D.T.; Feuerstein, J.D.; Wang, A.Y.; Cohen, R.D. AGA Clinical Practice Update on Management of Inflammatory Bowel Disease During the COVID-19 Pandemic: Expert Commentary. Gastroenterology 2020, 159, 350–357. [Google Scholar] [CrossRef]
- Keewan, E.; Beg, S.; Naser, S.A. Anti-TNF-α Agents Modulate SARS-CoV-2 Receptors and Increase the Risk of Infection Through Notch-1 Signaling. Front. Immunol. 2021, 12, 1662. [Google Scholar] [CrossRef]
- Kopan, R. Notch Signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a011213. [Google Scholar] [CrossRef]
- Li, W.; Wang, D.; Sun, X.; Zhang, Y.; Wang, L.; Suo, J. ADAM17 Promotes Lymph Node Metastasis in Gastric Cancer via Activation of the Notch and Wnt Signaling Pathways. Int. J. Mol. Med. 2019, 43, 914–926. [Google Scholar] [CrossRef]
- Minozzi, S.; Bonovas, S.; Lytras, T.; Pecoraro, V.; González-Lorenzo, M.; Bastiampillai, A.J.; Gabrielli, E.M.; Lonati, A.C.; Moja, L.; Cinquini, M.; et al. Risk of Infections Using Anti-TNF Agents in Rheumatoid Arthritis, Psoriatic Arthritis, and Ankylosing Spondylitis: A Systematic Review and Meta-Analysis. Expert Opin. Drug Saf. 2016, 15 (Suppl. S1), 11–34. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Ziring, D.; Desai, S.; Kim, S.; Wong, M.; Korin, Y.; Braun, J.; Reed, E.; Gjertson, D.; Singh, R.R. TNFalpha blockade in human diseases: An overview of efficacy and safety. Clin immunol. 2008, 126, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Plant, D.; Wilson, A.G.; Barton, A. Genetic and Epigenetic Predictors of Responsiveness to Treatment in RA. Nat. Rev. Rheumatol. 2014, 10, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Kamal, M.; Lindblad-Toh, K.; Bekiranov, S.; Bailey, D.K.; Huebert, D.J.; McMahon, S.; Karlsson, E.K.; Kulbokas, E.J.; Gingeras, T.R.; et al. Genomic Maps and Comparative Analysis of Histone Modifications in Human and Mouse. Cell 2005, 120, 169–181. [Google Scholar] [CrossRef]
- O’Rielly, D.D.; Roslin, N.M.; Beyene, J.; Pope, A.; Rahman, P. TNF-Alpha-308 G/A Polymorphism and Responsiveness to TNF-Alpha Blockade Therapy in Moderate to Severe Rheumatoid Arthritis: A Systematic Review and Meta-Analysis. Pharm. J. 2009, 9, 161–167. [Google Scholar] [CrossRef]
- Gowers, I.R.; Walters, K.; Kiss-Toth, E.; Read, R.C.; Duff, G.W.; Wilson, A.G. Age-Related Loss of CpG Methylation in the Tumour Necrosis Factor Promoter. Cytokine 2011, 56, 792–797. [Google Scholar] [CrossRef]
- Brinkman, B.M.; Huizinga, T.W.; Kurban, S.S.; van der Velde, E.A.; Schreuder, G.M.; Hazes, J.M.; Breeddveld, F.C.; Verweij, C.L. Tumour necrosis factor alpha gene polymorphisms in rheumatoid arthritis: Association with susceptibility to, or severity of, disease? Br. J. Rheumatol. 1997, 36, 516–521. [Google Scholar] [CrossRef]
- Kawazoe, M.; Kihara, M.; Nanki, T. Antirheumatic Drugs against COVID-19 from the Perspective of Rheumatologists. Pharm. Basel Switz. 2021, 14, 1256. [Google Scholar] [CrossRef]
- Sakimoto, T.; Ohnishi, T.; Ishimori, A. Significance of Ectodomain Shedding of TNF Receptor 1 in Ocular Surface. Investig. Ophthalmol. Vis. Sci. 2014, 55, 2419–2423. [Google Scholar] [CrossRef]
- Wajant, H.; Siegmund, D. TNFR1 and TNFR2 in the Control of the Life and Death Balance of Macrophages. Front. Cell Dev. Biol. 2019, 7, 91. [Google Scholar] [CrossRef]
- Gohda, T.; Tomino, Y. Novel Biomarkers for the Progression of Diabetic Nephropathy: Soluble TNF Receptors. Curr. Diab. Rep. 2013, 13, 560–566. [Google Scholar] [CrossRef]
- Nilsson, L.; Szymanowski, A.; Swahn, E.; Jonasson, L. Soluble TNF Receptors Are Associated with Infarct Size and Ventricular Dysfunction in ST-Elevation Myocardial Infarction. PLoS ONE 2013, 8, e55477. [Google Scholar] [CrossRef]
- Parsons, P.E.; Matthay, M.A.; Ware, L.B.; Eisner, M.D.; National Heart, Lung, Blood Institute. Acute Respiratory Distress Syndrome Clinical Trials Network. Elevated Plasma Levels of Soluble TNF Receptors Are Associated with Morbidity and Mortality in Patients with Acute Lung Injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 288, L426–L431. [Google Scholar] [CrossRef]
- Zoppini, G.; Faccini, G.; Muggeo, M.; Zenari, L.; Falezza, G.; Targher, G. Elevated Plasma Levels of Soluble Receptors of TNF-Alpha and Their Association with Smoking and Microvascular Complications in Young Adults with Type 1 Diabetes. J. Clin. Endocrinol. Metab. 2001, 86, 3805–3808. [Google Scholar] [CrossRef][Green Version]
- Paczesny, S.; Krijanovski, O.I.; Braun, T.M.; Choi, S.W.; Clouthier, S.G.; Kuick, R.; Misek, D.E.; Cooke, K.R.; Kitko, C.L.; Weyand, A.; et al. A Biomarker Panel for Acute Graft-versus-Host Disease. Blood 2009, 113, 273–278. [Google Scholar] [CrossRef]
- McElvaney, O.J.; McEvoy, N.L.; McElvaney, O.F.; Carroll, T.P.; Murphy, M.P.; Dunlea, D.M.; Ní Choileáin, O.; Clarke, J.; O’Connor, E.; Hogan, G.; et al. Characterization of the Inflammatory Response to Severe COVID-19 Illness. Am. J. Respir. Crit. Care Med. 2020, 202, 812–821. [Google Scholar] [CrossRef]
- Mortaz, E.; Tabarsi, P.; Jamaati, H.; Dalil Roofchayee, N.; KakaDezfuli, N.; Hashemian, S.M.; Moniri, A.; Marjani, M.; Malekmohammd, M.; Manosuri, D.; et al. Increased Serum Levels of Soluble TNF-α Receptor Is Associated with ICU Mortality in COVID-19 Patients. Front Immunol. 2021, 12, 592727. [Google Scholar] [CrossRef]
- Bowman, E.R.; Cameron, C.M.A.; Avery, A.; Gabriel, J.; Kettelhut, A.; Hecker, M.; Ute Sontich, C.; Tamilselvan, B.; Nichols, C.N.; Richardson, B.; et al. Levels of Soluble CD14 and Tumor Necrosis Factor Receptors 1 and 2 May Be Predictive of Death in Severe Coronavirus Disease 2019 (COVID-19). J. Infect. Dis. 2021, 223, 805–810. [Google Scholar] [CrossRef]
- Aderka, D.; Engelmann, H.; Maor, Y.; Brakebusch, C.; Wallach, D. Stabilization of the Bioactivity of Tumor Necrosis Factor by Its Soluble Receptors. J. Exp. Med. 1992, 175, 323–329. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Z.; Wu, X.; Zhou, J.; Meng, D.; Zhu, P. Risk of Adverse Events After Anti-TNF Treatment for Inflammatory Rheumatological Disease. A Meta-Analysis. Front. Pharmacol. 2021, 12, 3010. [Google Scholar] [CrossRef]
- Robert, M.; Miossec, P. Reactivation of Latent Tuberculosis with TNF Inhibitors: Critical Role of the Beta 2 Chain of the IL-12 Receptor. Cell. Mol. Immunol. 2021, 18, 1644–1651. [Google Scholar] [CrossRef]
- Schmidt, E.M.; Davies, M.; Mistry, P.; Green, P.; Giddins, G.; Feldmann, M.; Stoop, A.A.; Brennan, F.M. Selective Blockade of Tumor Necrosis Factor Receptor I Inhibits Proinflammatory Cytokine and Chemokine Production in Human Rheumatoid Arthritis Synovial Membrane Cell Cultures. Arthritis Rheum. 2013, 65, 2262–2273. [Google Scholar] [CrossRef]
- Fischer, R.; Kontermann, R.E.; Pfizenmaier, K. Selective Targeting of TNF Receptors as a Novel Therapeutic Approach. Front. Cell Dev. Biol. 2020, 8, 401. [Google Scholar] [CrossRef]
- Van Hauwermeiren, F.; Vandenbroucke, R.E.; Grine, L.; Lodens, S.; Van Wonterghem, E.; De Rycke, R.; De Geest, N.; Hassan, B.; Libert, C. TNFR1-Induced Lethal Inflammation Is Mediated by Goblet and Paneth Cell Dysfunction. Mucosal Immunol. 2015, 8, 828–840. [Google Scholar] [CrossRef]
- Zhang, N.; Wang, Z.; Zhao, Y. Selective Inhibition of Tumor Necrosis Factor Receptor-1 (TNFR1) for the Treatment of Autoimmune Diseases. Cytokine Growth Factor Rev. 2020, 55, 80–85. [Google Scholar] [CrossRef]
- Steeland, S.; Puimège, L.; Vandenbroucke, R.E.; Van Hauwermeiren, F.; Haustraete, J.; Devoogdt, N.; Hulpiau, P.; Leroux-Roels, G.; Laukens, D.; Meuleman, P.; et al. Generation and Characterization of Small Single Domain Antibodies Inhibiting Human Tumor Necrosis Factor Receptor 1. J. Biol. Chem. 2015, 290, 4022–4037. [Google Scholar] [CrossRef]
- Kontermann, R.E.; Münkel, S.; Neumeyer, J.; Müller, D.; Branschädel, M.; Scheurich, P.; Pfizenmaier, K. A Humanized Tumor Necrosis Factor Receptor 1 (TNFR1)-Specific Antagonistic Antibody for Selective Inhibition of Tumor Necrosis Factor (TNF) Action. J. Immunother. Hagerstown Md. 1997 2008, 31, 225–234. [Google Scholar] [CrossRef]
- Williams, S.K.; Fairless, R.; Maier, O.; Liermann, P.C.; Pichi, K.; Fischer, R.; Eisel, U.L.M.; Kontermann, R.; Herrmann, A.; Weksler, B.; et al. Anti-TNFR1 Targeting in Humanized Mice Ameliorates Disease in a Model of Multiple Sclerosis. Sci. Rep. 2018, 8, 13628. [Google Scholar] [CrossRef]
- Richter, F.; Zettlitz, K.A.; Seifert, O.; Herrmann, A.; Scheurich, P.; Pfizenmaier, K.; Kontermann, R.E. Monovalent TNF Receptor 1-Selective Antibody with Improved Affinity and Neutralizing Activity. mAbs 2019, 11, 166–177. [Google Scholar] [CrossRef]
- Richter, F.; Seifert, O.; Herrmann, A.; Pfizenmaier, K.; Kontermann, R.E. Improved Monovalent TNF Receptor 1-Selective Inhibitor with Novel Heterodimerizing Fc. mAbs 2019, 11, 653–665. [Google Scholar] [CrossRef]
- Shibata, H.; Yoshioka, Y.; Ohkawa, A.; Minowa, K.; Mukai, Y.; Abe, Y.; Taniai, M.; Nomura, T.; Kayamuro, H.; Nabeshi, H.; et al. Creation and X-Ray Structure Analysis of the Tumor Necrosis Factor Receptor-1-Selective Mutant of a Tumor Necrosis Factor-Alpha Antagonist. J. Biol. Chem. 2008, 283, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Shibata, H.; Yoshioka, Y.; Ohkawa, A.; Abe, Y.; Nomura, T.; Mukai, Y.; Nakagawa, S.; Taniai, M.; Ohta, T.; Mayumi, T.; et al. The Therapeutic Effect of TNFR1-Selective Antagonistic Mutant TNF-Alpha in Murine Hepatitis Models. Cytokine 2008, 44, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Mukai, Y.; Shibata, H.; Nakamura, T.; Yoshioka, Y.; Abe, Y.; Nomura, T.; Taniai, M.; Ohta, T.; Ikemizu, S.; Nakagawa, S.; et al. Structure-Function Relationship of Tumor Necrosis Factor (TNF) and Its Receptor Interaction Based on 3D Structural Analysis of a Fully Active TNFR1-Selective TNF Mutant. J. Mol. Biol. 2009, 385, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Mukai, Y.; Nakamura, T.; Yoshioka, Y.; Shibata, H.; Abe, Y.; Nomura, T.; Taniai, M.; Ohta, T.; Nakagawa, S.; Tsunoda, S.; et al. Fast Binding Kinetics and Conserved 3D Structure Underlie the Antagonistic Activity of Mutant TNF: Useful Information for Designing Artificial Proteo-Antagonists. J. Biochem. 2009, 146, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Ando, D.; Kamada, H.; Taki, S.; Niiyama, M.; Mukai, Y.; Tadokoro, T.; Maenaka, K.; Nakayama, T.; Kado, Y.; et al. A Trimeric Structural Fusion of an Antagonistic Tumor Necrosis Factor-α Mutant Enhances Molecular Stability and Enables Facile Modification. J. Biol. Chem. 2017, 292, 6438–6451. [Google Scholar] [CrossRef]
- Steeland, S.; Van Ryckeghem, S.; Van Imschoot, G.; De Rycke, R.; Toussaint, W.; Vanhoutte, L.; Vanhove, C.; De Vos, F.; Vandenbroucke, R.E.; Libert, C. TNFR1 Inhibition with a Nanobody Protects against EAE Development in Mice. Sci. Rep. 2017, 7, 13646. [Google Scholar] [CrossRef]
- Cao, Y.; Li, Y.H.; Lv, D.Y.; Chen, X.F.; Chen, L.D.; Zhu, Z.Y.; Chai, Y.F.; Zhang, J.P. Identification of a Ligand for Tumor Necrosis Factor Receptor from Chinese Herbs by Combination of Surface Plasmon Resonance Biosensor and UPLC-MS. Anal. Bioanal. Chem. 2016, 408, 5359–5367. [Google Scholar] [CrossRef]
- Lo, C.H.; Vunnam, N.; Lewis, A.K.; Chiu, T.-L.; Brummel, B.E.; Schaaf, T.M.; Grant, B.D.; Bawaskar, P.; Thomas, D.D.; Sachs, J.N. An Innovative High-Throughput Screening Approach for Discovery of Small Molecules That Inhibit TNF Receptors. SLAS Discov. Adv. Life Sci. R D 2017, 22, 950–961. [Google Scholar] [CrossRef]
- Lo, C.H.; Schaaf, T.M.; Grant, B.D.; Lim, C.K.-W.; Bawaskar, P.; Aldrich, C.C.; Thomas, D.D.; Sachs, J.N. Noncompetitive Inhibitors of TNFR1 Probe Conformational Activation States. Sci. Signal. 2019, 12, eaav5637. [Google Scholar] [CrossRef]
- Huang, X.W.; Yang, J.; Dragovic, A.F.; Zhang, H.; Lawrence, T.S.; Zhang, M. Antisense Oligonucleotide Inhibition of Tumor Necrosis Factor Receptor 1 Protects the Liver from Radiation-Induced Apoptosis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 2849–2855. [Google Scholar] [CrossRef][Green Version]
- Proudfoot, A.; Bayliffe, A.; O’Kane, C.M.; Wright, T.; Serone, A.; Bareille, P.J.; Brown, V.; Hamid, U.I.; Chen, Y.; Wilson, R.; et al. Novel Anti-Tumour Necrosis Factor Receptor-1 (TNFR1) Domain Antibody Prevents Pulmonary Inflammation in Experimental Acute Lung Injury. Thorax 2018, 73, 723–730. [Google Scholar] [CrossRef]
- Tao, S.; Song, P.; Zhang, X.; Zhang, L.; Chu, C.-Q. Single-Stranded DNA Aptamers Against TNF and Their Potential Applications. Methods Mol. Biol. Clifton NJ 2020, 2108, 181–196. [Google Scholar] [CrossRef]
- Chan, F.K.; Chun, H.J.; Zheng, L.; Siegel, R.M.; Bui, K.L.; Lenardo, M.J. A Domain in TNF Receptors That Mediates Ligand-Independent Receptor Assembly and Signaling. Science 2000, 288, 2351–2354. [Google Scholar] [CrossRef]
- Richter, F.; Williams, S.K.; John, K.; Huber, C.; Vaslin, C.; Zanker, H.; Fairless, R.; Pichi, K.; Marhenke, S.; Vogel, A.; et al. The TNFR1 Antagonist Atrosimab Is Therapeutic in Mouse Models of Acute and Chronic Inflammation. Front. Immunol. 2021, 12, 705485. [Google Scholar] [CrossRef]
- Inoue, M.; Tsuji, Y.; Yoshimine, C.; Enomoto, S.; Morita, Y.; Osaki, N.; Kunishige, M.; Miki, M.; Amano, S.; Yamashita, K.; et al. Structural Optimization of a TNFR1-Selective Antagonistic TNFα Mutant to Create New-Modality TNF-Regulating Biologics. J. Biol. Chem. 2020, 295, 9379–9391. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Class | Description | Properties | Disease Model Evaluation | References |
|---|---|---|---|---|---|
| Atrosab | Humanized IgG1 | Receptor selective inhibitor | Antagonistic monoclonal antibody | Multiple sclerosis | [134,135] |
| Fv13.7-Fc | Fab | Receptor selective inhibitor with affinity maturation | Monovalent Fab of Atrosab | [136] | |
| Atrosimab | Fv-Fc1k fusion protein | Receptor selective inhibitor | Fusion protein | Arthritis | [137] |
| R1antTNF R1antTNF-T8 scR1antTNF | TNF-muteins | Receptor-selective antagonistic activity | Bind to TNFR1 without activation | Acute hepatitis | [138,139,140,141,142] |
| DMS5541 | A bispecific, single variable-domain antibody | Receptor-selective blockade | anti-TNFRI moiety plus an albumin binding moiety | Human Rheumatoid Arthritis | [129] |
| TROS | Nanobody (Nb) technology | TROS selectively binds and inhibits the TNF/TNFR1 signaling pathway | KD and IC50 values in the nanomolar range | Crohn’s disease ex vivo model; experimental autoimmune encephalomyelitis; multiple sclerosis murine model | [133,143] |
| PMG (physcion-8-O-β-D-monoglucoside) | A bioactive compound isolated from Chinese herbs | Ligand for TNF receptor from herbal medicines | KD at nanomolar range | [144] | |
| Zafirlukast Triclabendazole (does not compete with a ligand or with PLAD-PLAD assembly *) DS42 | Small molecule Small molecule Small-molecule allosteric inhibitor | Small-molecule approaches inhibit receptor interaction or alter receptor conformational dynamics without interrupting ligand binding. Noncompetitive inhibitor without reducing ligand affinity or disrupting receptor dimerization. | Asthma Allergic rhinitis Chronic Idiopathic Urticaria | [145,146] | |
| ASOs | Antisense oligonucleotides | Blocking TNFR1 gene expression | Protection from Radiation-Induced Apoptosis | [147] | |
| GSK1995057 | Fully human domain antibody (dAb) fragment | Selectively antagonizes TNF signaling through TNFR1 | Phase IIa clinical trial | Respiratory Disorders | [148] |
| Aptamers AptTNR1 | Binding to TNFR1 but not TNFR2 | KD around 100 nM | [132,149] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palacios, Y.; Chavez-Galan, L. Immunosuppressant Therapies in COVID-19: Is the TNF Axis an Alternative? Pharmaceuticals 2022, 15, 616. https://doi.org/10.3390/ph15050616
Palacios Y, Chavez-Galan L. Immunosuppressant Therapies in COVID-19: Is the TNF Axis an Alternative? Pharmaceuticals. 2022; 15(5):616. https://doi.org/10.3390/ph15050616
Chicago/Turabian StylePalacios, Yadira, and Leslie Chavez-Galan. 2022. "Immunosuppressant Therapies in COVID-19: Is the TNF Axis an Alternative?" Pharmaceuticals 15, no. 5: 616. https://doi.org/10.3390/ph15050616
APA StylePalacios, Y., & Chavez-Galan, L. (2022). Immunosuppressant Therapies in COVID-19: Is the TNF Axis an Alternative? Pharmaceuticals, 15(5), 616. https://doi.org/10.3390/ph15050616