
Machine-Learning Guided Discovery of Bioactive Inhibitors of PD1-PDL1 Interaction

Abstract
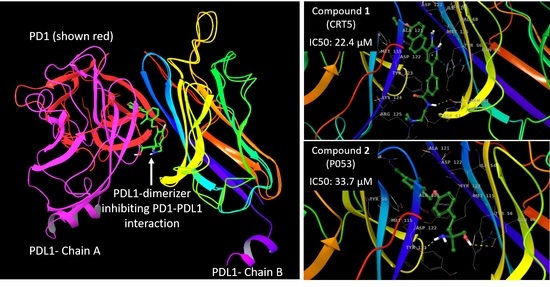
:
1. Introduction
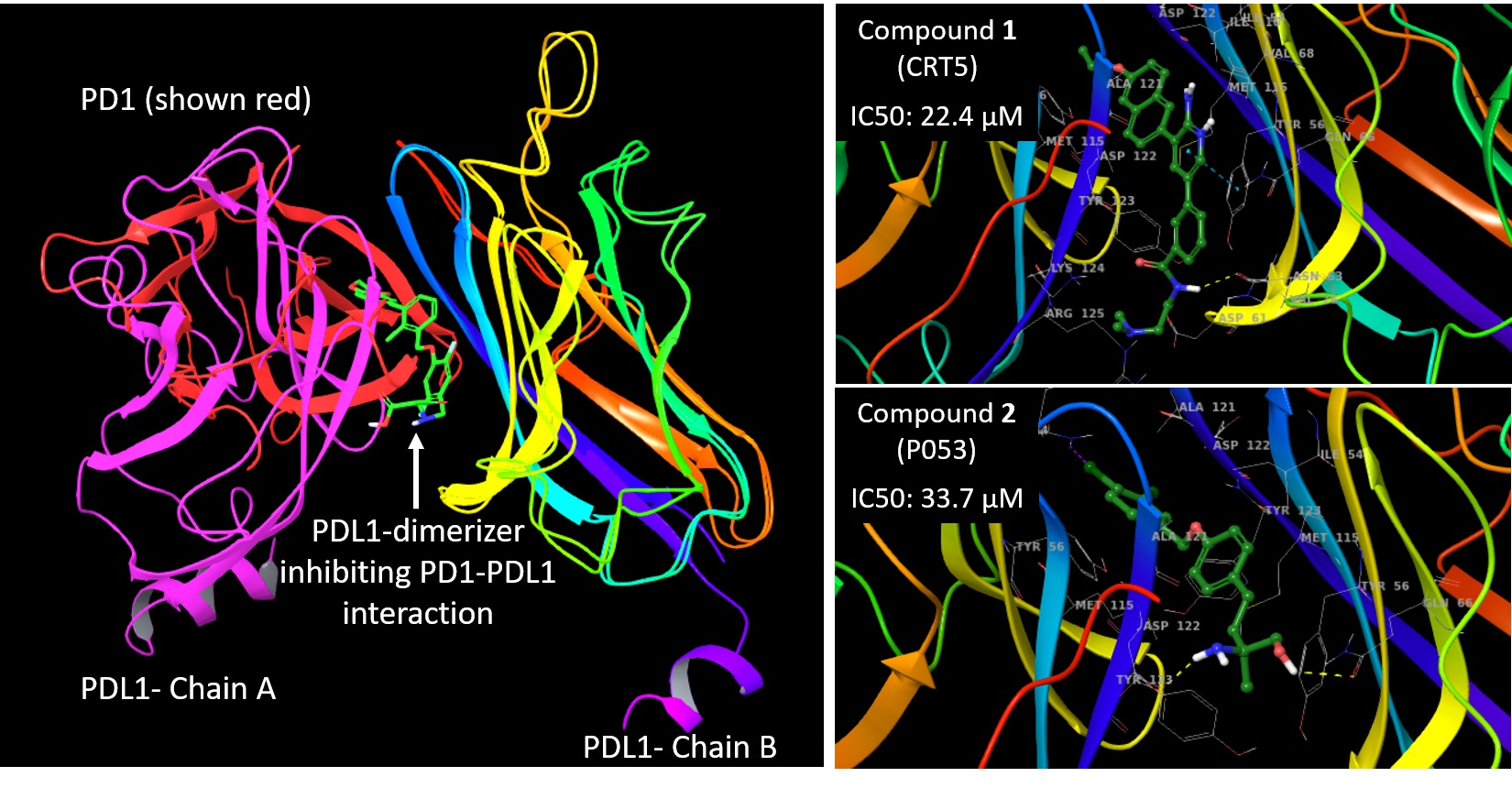
2. Results and Discussion
3. Materials and Methods
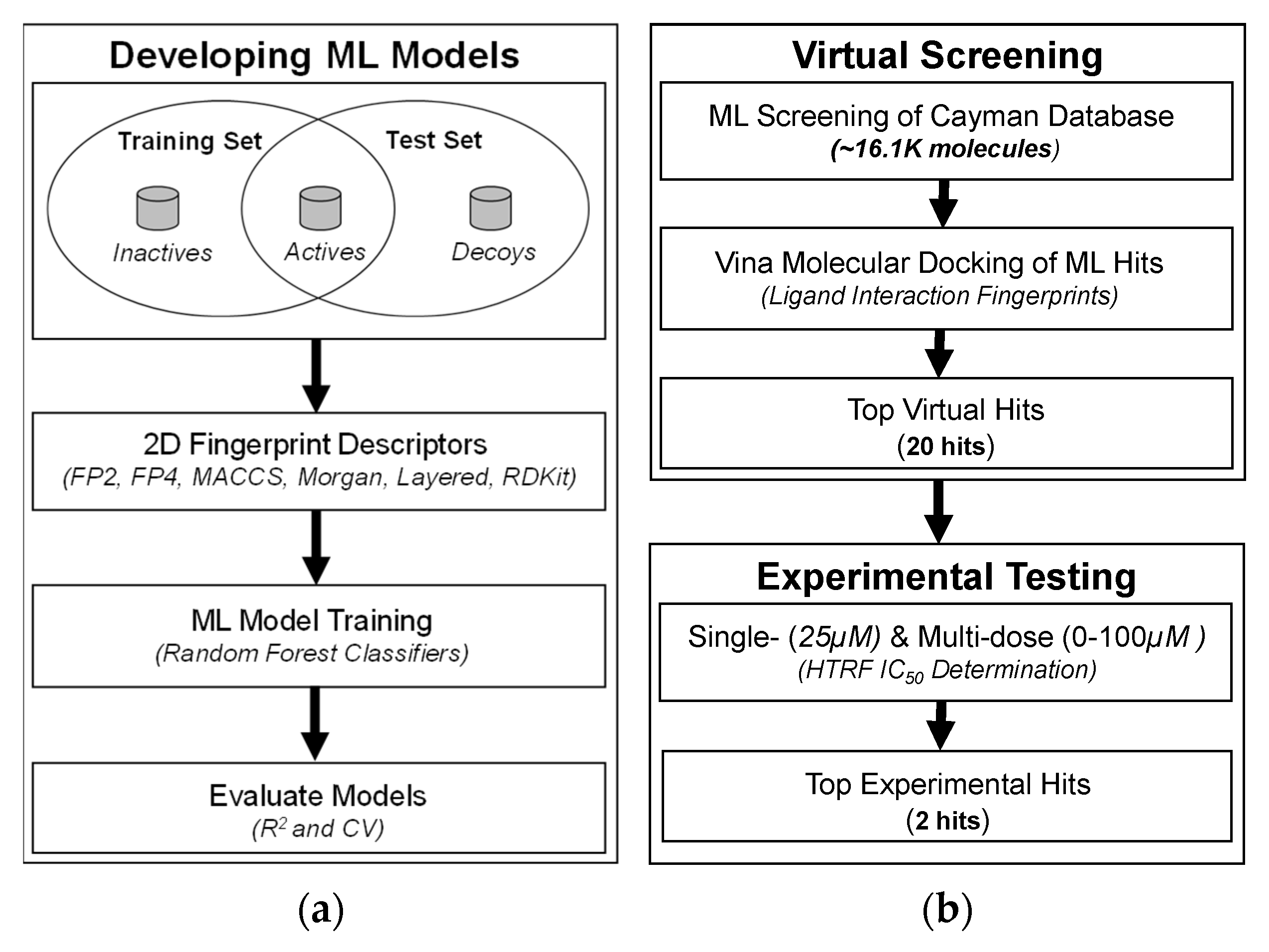
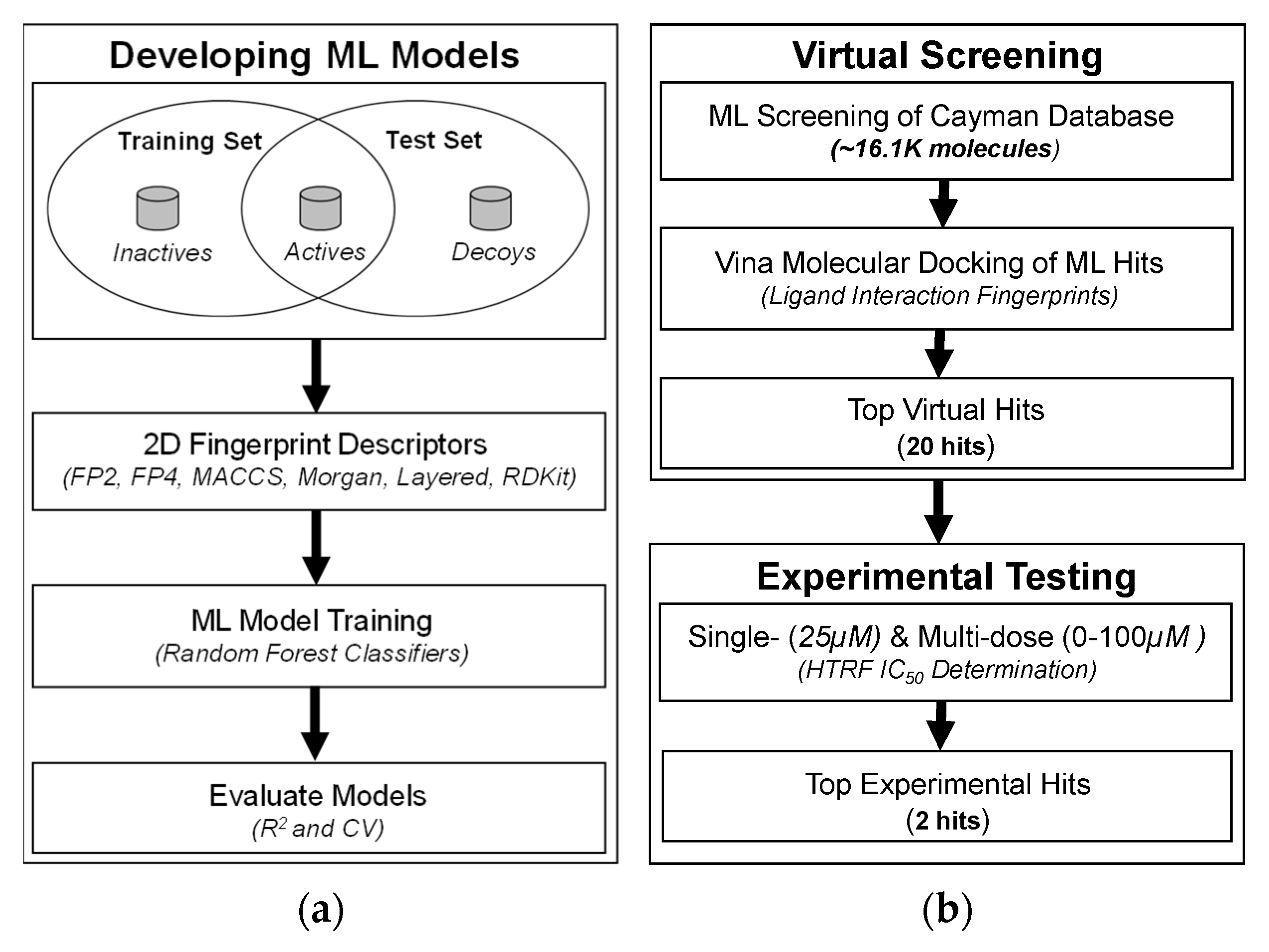
3.1. Development of 2D Fingerprint-Based Classification Models
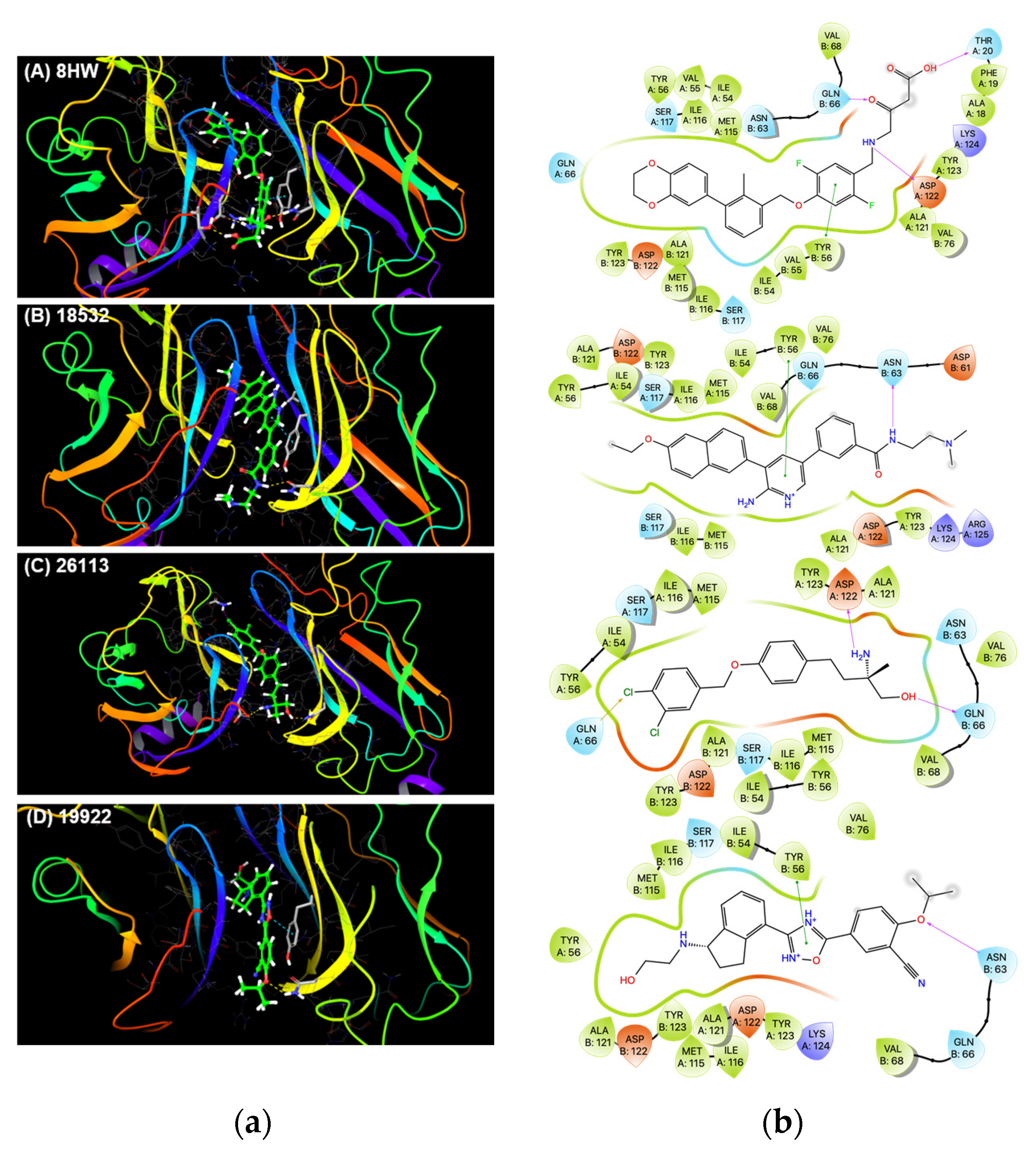
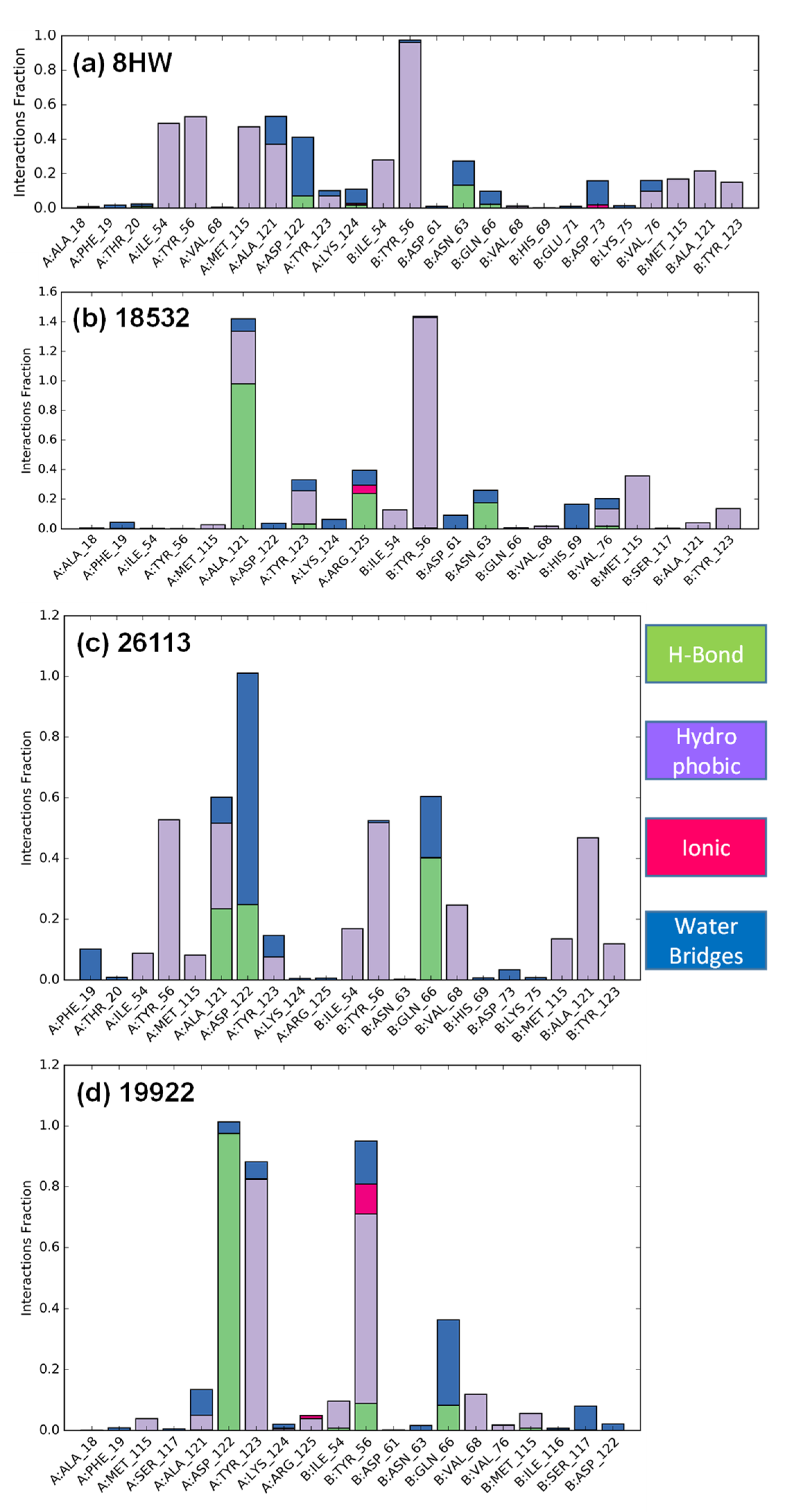
3.2. Molecular Docking and Ligand Interaction Fingerprint Analysis
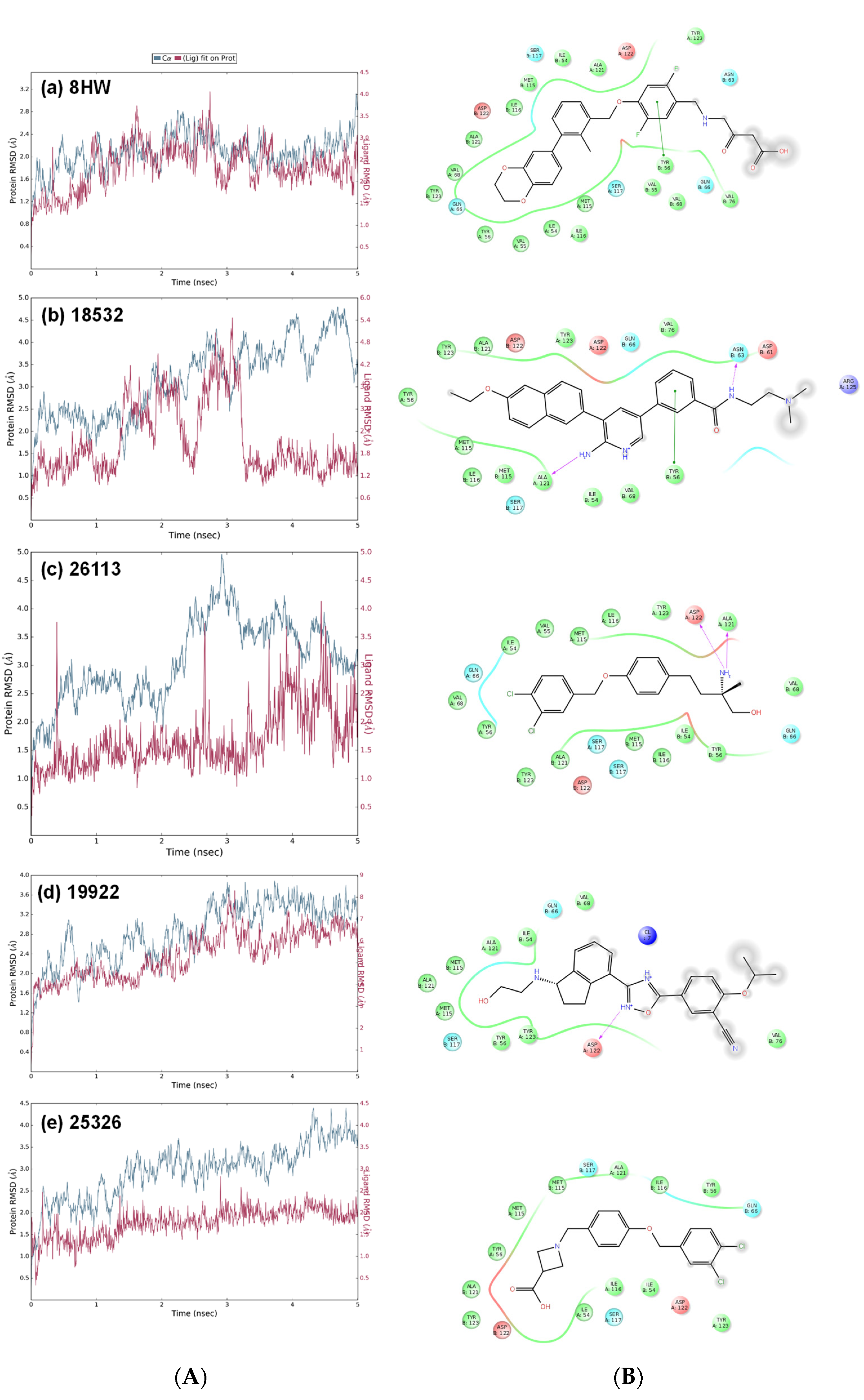
3.3. Molecular Dynamics Simulations
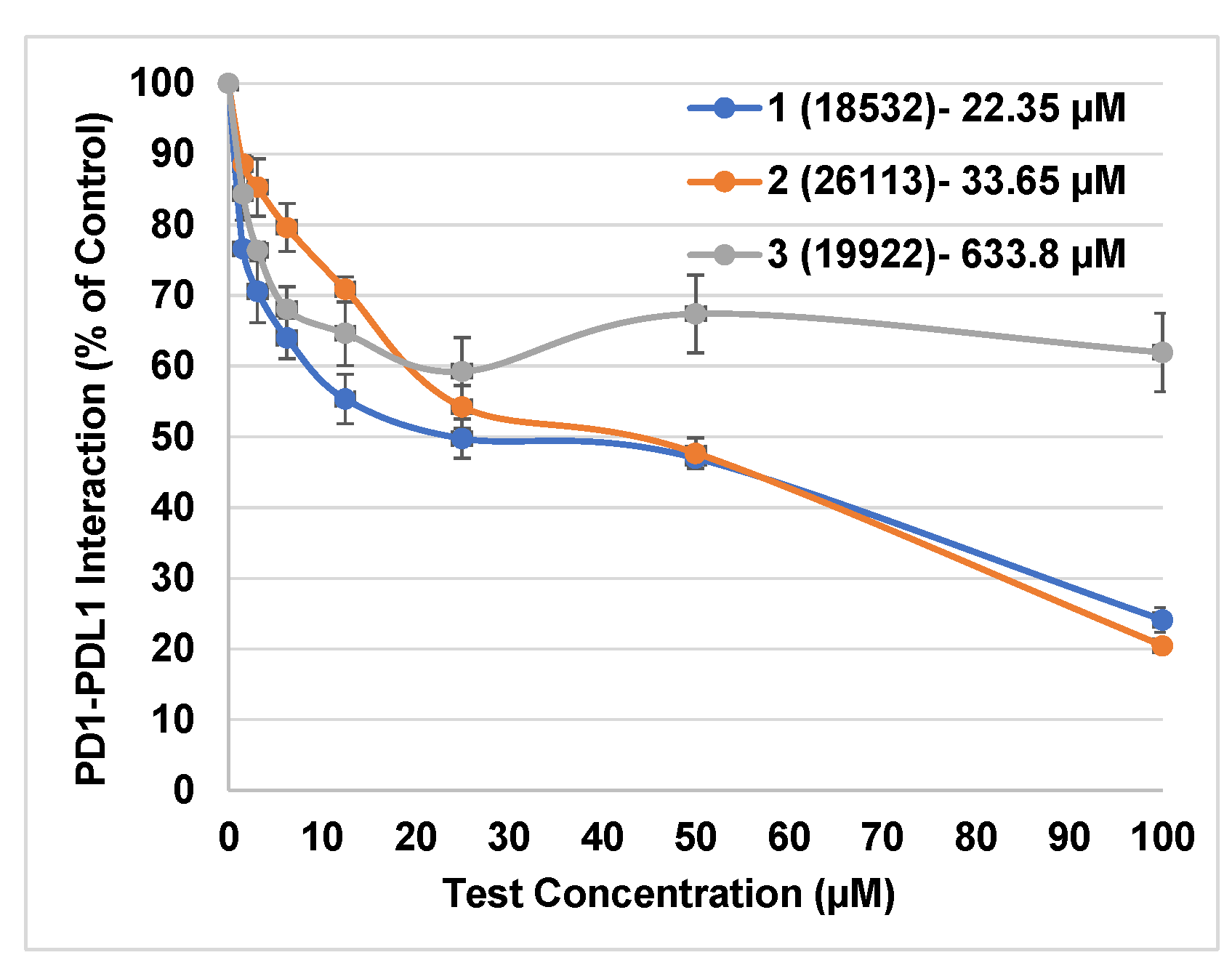
3.4. In Vitro Testing Using HTRF Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Q.C.; Petrey, D.; Deng, L.; Qiang, L.; Shi, Y.; Thu, C.A.; Bisikirska, B.; Lefebvre, C.; Accili, D.; Hunter, T.; et al. Structure-based prediction of protein-protein interactions on a genome-wide scale. Nature 2012, 490, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Stumpf, M.P.; Thorne, T.; de Silva, E.; Stewart, R.; An, H.J.; Lappe, M.; Wiuf, C. Estimating the size of the human interactome. Proc. Natl. Acad. Sci. USA 2008, 105, 6959–6964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, C.G.; Arkin, M.R. Small-molecule inhibitors of IL-2/IL-2R: Lessons learned and applied. Curr. Top. Microbiol. Immunol. 2011, 348, 25–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiel, P.; Kaiser, M.; Ottmann, C. Small-molecule stabilization of protein-protein interactions: An underestimated concept in drug discovery? Angew. Chem. Int. Ed. Engl. 2012, 51, 2012–2018. [Google Scholar] [CrossRef]
- Qin, W.; Hu, L.; Zhang, X.; Jiang, S.; Li, J.; Zhang, Z.; Wang, X. The Diverse Function of PD-1/PD-L Pathway beyond Cancer. Front. Immunol. 2019, 10, 2298. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, W.; Xu, Z.P.; Gu, W. PD-L1 Distribution and Perspective for Cancer Immunotherapy-Blockade, Knockdown, or Inhibition. Front. Immunol. 2019, 10, 2022. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Yu, J.X.; Hubbard-Lucey, V.M.; Neftelinov, S.T.; Hodge, J.P.; Lin, Y. Trial watch: The clinical trial landscape for PD1/PDL1 immune checkpoint inhibitors. Nat. Rev. Drug Discov. 2018, 17, 854–855. [Google Scholar] [CrossRef]
- Akinleye, A.; Rasool, Z. Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J. Hematol. Oncol. 2019, 12, 92. [Google Scholar] [CrossRef] [Green Version]
- Sritharan, J.; MacLeod, J.; Harris, S.; Cole, D.C.; Harris, A.; Tjepkema, M.; Peters, P.A.; Demers, P.A. Prostate cancer surveillance by occupation and industry: The Canadian Census Health and Environment Cohort (CanCHEC). Cancer Med. 2018, 7, 1468–1478. [Google Scholar] [CrossRef] [Green Version]
- Patil, S.P.; Fink, M.A.; Enley, E.S.; Fisher, J.E.; Herb, M.C.; Klingos, A.; Proulx, J.T.; Fedorky, M.T. Identification of Small-Molecule Inhibitors of PD-1/PD-L1 Protein-Protein Interaction. ChemistrySelect 2018, 3, 2185. [Google Scholar] [CrossRef]
- Patil, S.P.; Yoon, S.C.; Aradhya, A.G.; Hofer, J.; Fink, M.A.; Enley, E.S.; Fisher, J.E.; Herb, M.C.; Klingos, A.; Proulx, J.T.; et al. Macrocyclic Compounds from Ansamycin Antibiotic Class as Inhibitors of PD1-PDL1 Protein-Protein Interaction. Chem. Pharm. Bull. 2018, 66, 773–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzik, K.; Tomala, M.; Muszak, D.; Konieczny, M.; Hec, A.; Błaszkiewicz, U.; Pustuła, M.; Butera, R.; Dömling, A.; Holak, T.A. Development of the Inhibitors that Target the PD-1/PD-L1 Interaction-A Brief Look at Progress on Small Molecules, Peptides and Macrocycles. Molecules 2019, 24, 2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zak, K.M.; Grudnik, P.; Guzik, K.; Zieba, B.J.; Musielak, B.; Dömling, A.; Dubin, G.; Holak, T.A. Structural basis for small molecule targeting of the programmed death ligand 1 (PD-L1). Oncotarget 2016, 7, 30323–30335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Ciallella, H.L.; Aleksunes, L.M.; Zhu, H. Advancing computer-aided drug discovery (CADD) by big data and data-driven machine learning modeling. Drug Discov. Today 2020, 25, 1624–1638. [Google Scholar] [CrossRef]
- Wójcikowski, M.; Zielenkiewicz, P.; Siedlecki, P. Open Drug Discovery Toolkit (ODDT): A new open-source player in the drug discovery field. J. Cheminform. 2015, 7, 26. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Fattakhova, E.; Hofer, J.; DiFlumeri, J.; Cobb, M.; Dando, T.; Romisher, Z.; Wellington, J.; Oravic, M.; Radnoff, M.; Patil, S.P. Identification of the FDA-Approved Drug Pyrvinium as a Small-Molecule Inhibitor of the PD-1/PD-L1 Interaction. ChemMedChem 2021, 16, 2769–2774. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Deng, Z.; Chuaqui, C.; Singh, J. Structural interaction fingerprint (SIFt): A novel method for analyzing three-dimensional protein-ligand binding interactions. J. Med. Chem. 2004, 47, 337–344. [Google Scholar] [CrossRef]
- Almahmoud, S.; Zhong, H.A. Molecular Modeling Studies on the Binding Mode of the PD-1/PD-L1 Complex Inhibitors. Int. J. Mol. Sci. 2019, 20, 4654. [Google Scholar] [CrossRef] [Green Version]
- Chupak, L.S.; Ding, M.; Martin, S.W.; Zheng, X.; Hewawasam, P.; Connolly, T.P.; Xu, N.; Yeung, K.S.; Zhu, J.; Langley, D.R.; et al. Compounds Useful as Immunomodulators. WO Patent 2015160641 A3, 23 December 2015. [Google Scholar]
- Zak, K.M.; Kitel, R.; Przetocka, S.; Golik, P.; Guzik, K.; Musielak, B.; Dömling, A.; Dubin, G.; Holak, T.A. Structure of the Complex of Human Programmed Death 1, PD-1, and Its Ligand PD-L1. Structure 2015, 23, 2341–2348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, I.M.; Bagherzadeh, A.; Charles, M.; Raynham, T.; Ireson, C.; Boakes, A.; Kelland, L.; Zachary, I.C. Characterization of the biological effects of a novel protein kinase D inhibitor in endothelial cells. Biochem. J. 2010, 429, 565–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, N.; Lim, X.Y.; Toop, H.D.; Osborne, B.; Brandon, A.E.; Taylor, E.N.; Fiveash, C.E.; Govindaraju, H.; Teo, J.D.; McEwen, H.P.; et al. A selective inhibitor of ceramide synthase 1 reveals a novel role in fat metabolism. Nat. Commun. 2018, 9, 3165. [Google Scholar] [CrossRef] [PubMed]
- Lowe, D.M.; Corbett, P.T.; Murray-Rust, P.; Glen, R.C. Chemical Name to Structure: OPSIN, an Open Source Solution. J. Chem. Inf. Model. 2011, 51, 739–753. [Google Scholar] [CrossRef]
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Ganesan, A.; Ahmed, M.; Okoye, I.; Arutyunova, E.; Babu, D.; Turnbull, W.L.; Kundu, J.K.; Shields, J.; Agopsowicz, K.C.; Xu, L.; et al. Comprehensive in vitro characterization of PD-L1 small molecule inhibitors. Sci. Rep. 2019, 9, 12392. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
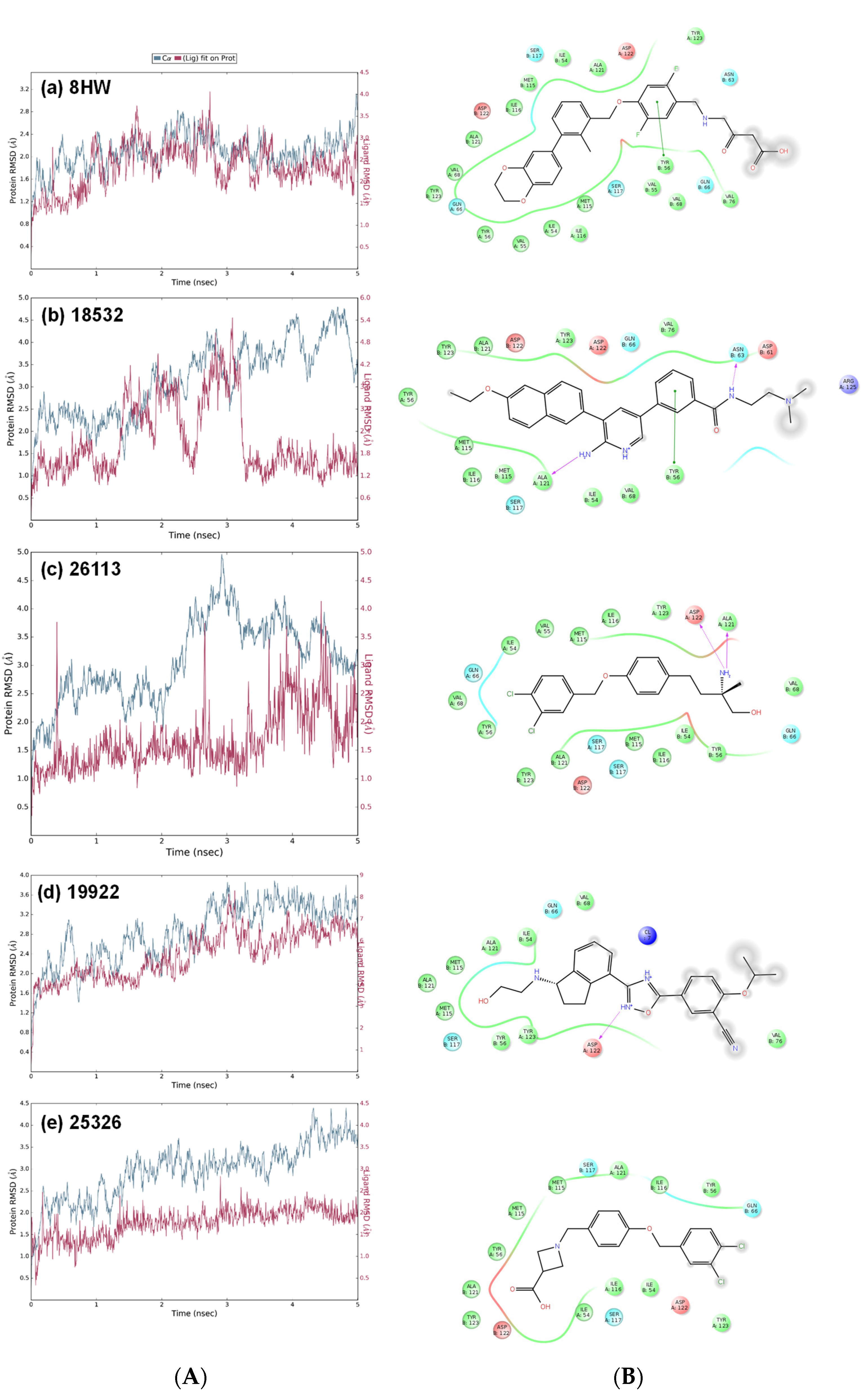{kind=link}
{kind=link}
| Fingerprint | FP2 | FP4 | MACCS | Morgan | Layered | RDKit |
|---|---|---|---|---|---|---|
| Training | 0.9629 | 0.9429 | 0.9549 | 0.9729 | 0.9649 | 0.9659 |
| Test | 0.9537 | 0.9283 | 0.9442 | 0.9664 | 0.9562 | 0.9575 |
| Compound # | Cayman ID | % PD1-PDL1 Inhibition | AutoDock Vina Score (Kcal/mol) |
|---|---|---|---|
| 1 | 18532 | 50.9 ± 1.3 | −11.0 |
| 2 | 26113 | 41.4 ± 0.9 | −9.6 |
| 3 | 19922 | 34.7 ± 2.1 | −10.6 |
| 4 | 18006 | 24.9 ± 1.8 | −9.8 |
| 5 | 24159 | 20.2 ± 1.3 | −10.5 |
| 6 | 29424 | 18.7 ± 1.1 | −10.3 |
| 7 | 19160 | 18.5 ± 0.7 | −9.4 |
| 8 | 21546 | 17.0 ± 1.0 | −9.6 |
| 9 | 70635 | 16.6 ± 1.3 | −9.8 |
| 10 | 24057 | 15.9 ± 2.1 | −10.6 |
| 11 | 32729 | 15.4 ± 1.3 | −10.8 |
| 12 | 25747 | 14.4 ± 0.9 | −10.1 |
| 13 | 21688 | 13.5 ± 1.3 | −9.7 |
| 14 | 31758 | 13.1 ± 1.3 | −10.4 |
| 15 | 21137 | 12.4 ± 1.5 | −10.9 |
| 16 | 19404 | 5.8 ± 1.8 | −9.8 |
| 17 | 19876 | −4.3 ± 1.5 | −10.4 |
| 18 | 18124 | −4.4 ± 1.2 | −10.3 |
| 19 | 25326 | −4.5 ± 2.0 | −9.9 |
| 20 | 17034 | −4.6 ± 1.4 | −10.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patil, S.P.; Fattakhova, E.; Hofer, J.; Oravic, M.; Bender, A.; Brearey, J.; Parker, D.; Radnoff, M.; Smith, Z. Machine-Learning Guided Discovery of Bioactive Inhibitors of PD1-PDL1 Interaction. Pharmaceuticals 2022, 15, 613. https://doi.org/10.3390/ph15050613
Patil SP, Fattakhova E, Hofer J, Oravic M, Bender A, Brearey J, Parker D, Radnoff M, Smith Z. Machine-Learning Guided Discovery of Bioactive Inhibitors of PD1-PDL1 Interaction. Pharmaceuticals. 2022; 15(5):613. https://doi.org/10.3390/ph15050613
Chicago/Turabian StylePatil, Sachin P., Elena Fattakhova, Jeremy Hofer, Michael Oravic, Autumn Bender, Jason Brearey, Daniel Parker, Madison Radnoff, and Zackary Smith. 2022. "Machine-Learning Guided Discovery of Bioactive Inhibitors of PD1-PDL1 Interaction" Pharmaceuticals 15, no. 5: 613. https://doi.org/10.3390/ph15050613
APA StylePatil, S. P., Fattakhova, E., Hofer, J., Oravic, M., Bender, A., Brearey, J., Parker, D., Radnoff, M., & Smith, Z. (2022). Machine-Learning Guided Discovery of Bioactive Inhibitors of PD1-PDL1 Interaction. Pharmaceuticals, 15(5), 613. https://doi.org/10.3390/ph15050613