
Current and Emerging Pharmacotherapeutic Interventions for the Treatment of Peripheral Nerve Disorders

 ,
, 
Abstract
: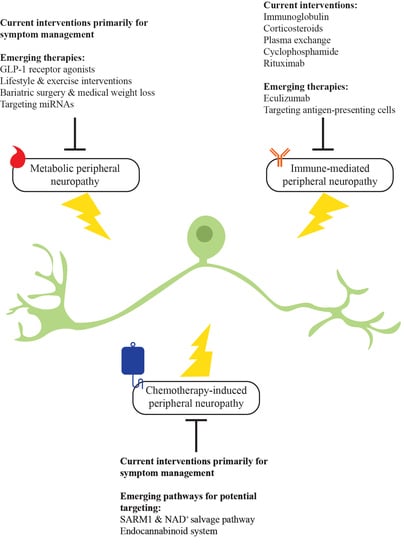
1. Introduction
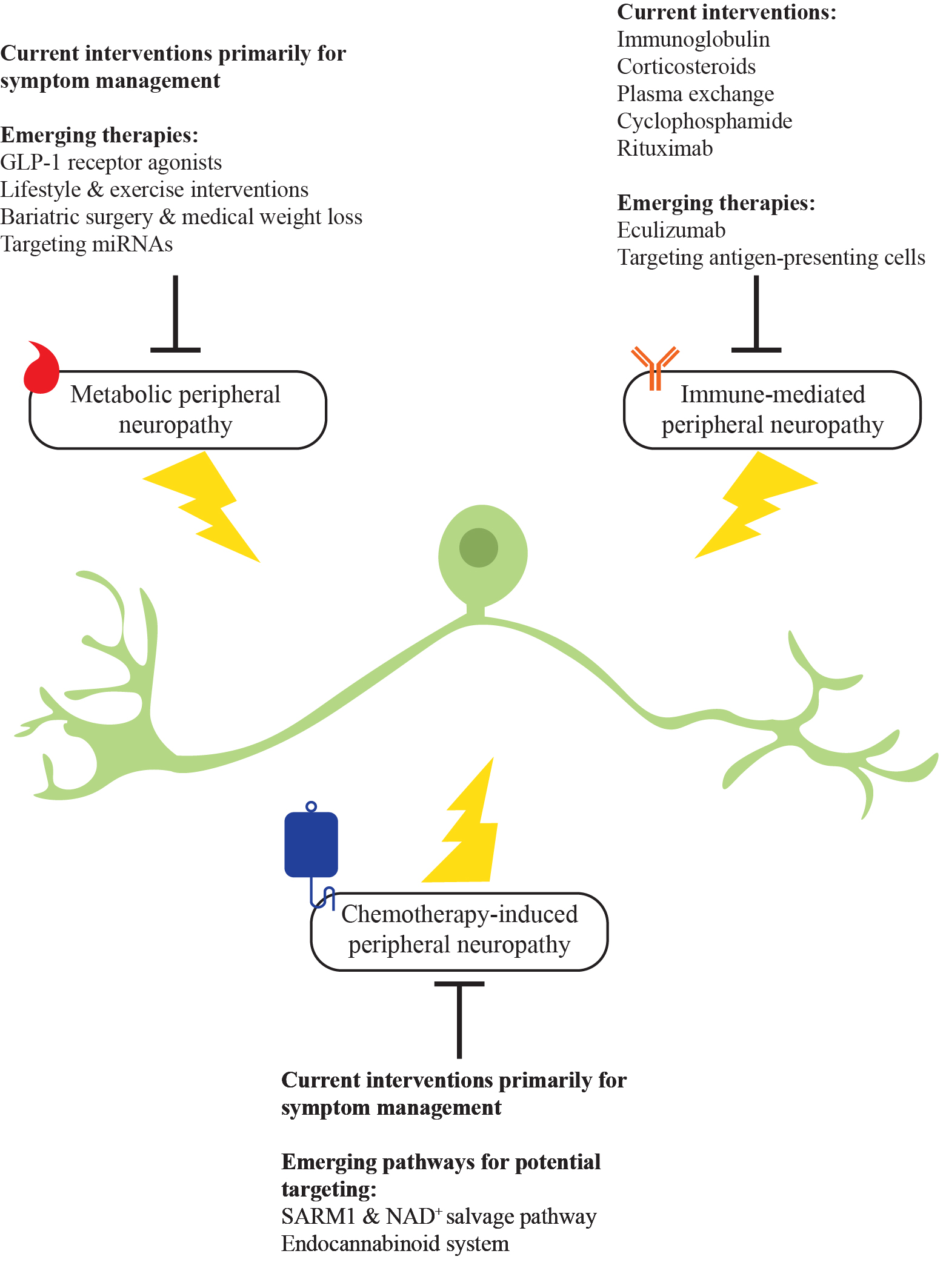
2. Metabolic Peripheral Neuropathy
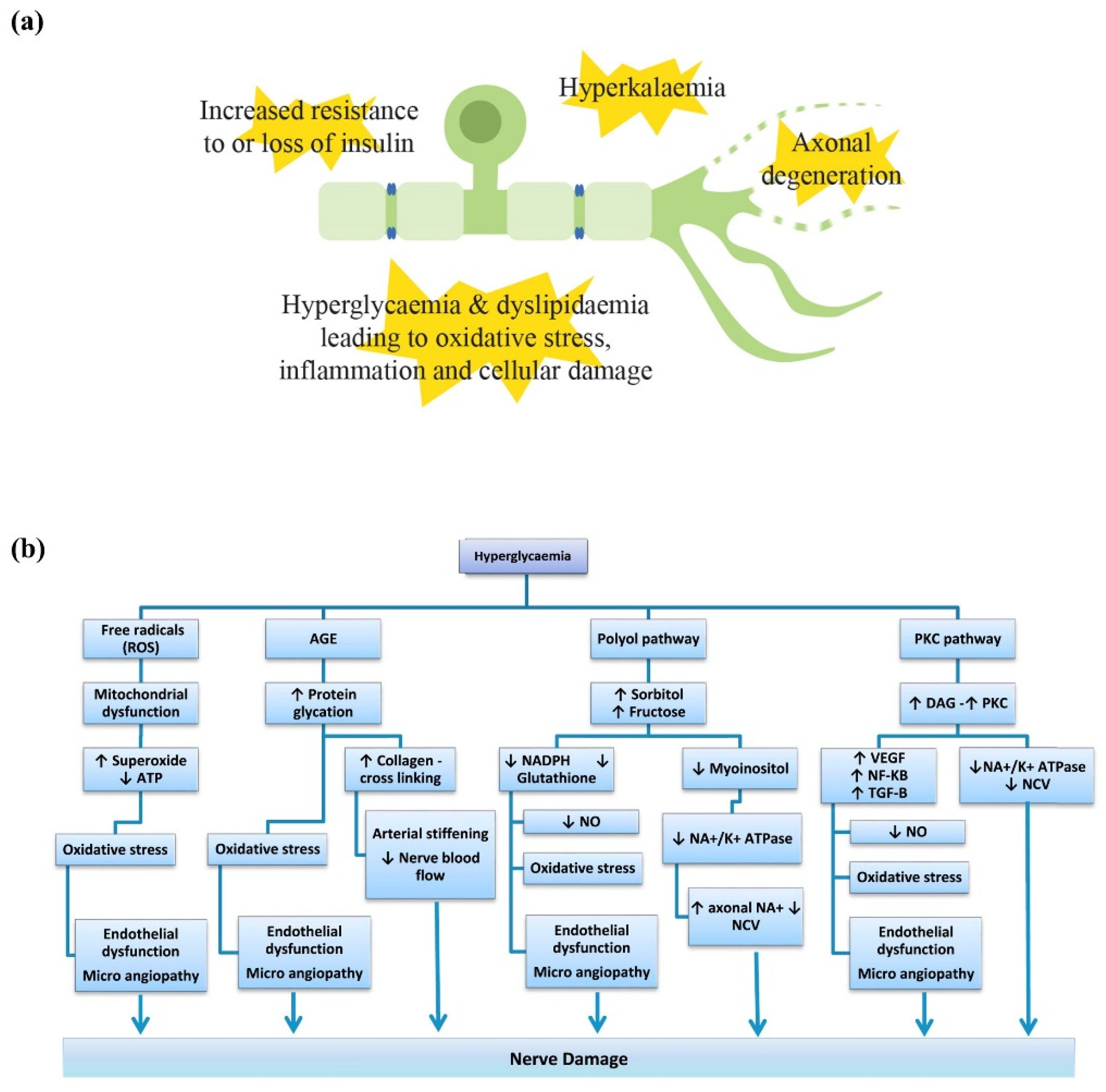
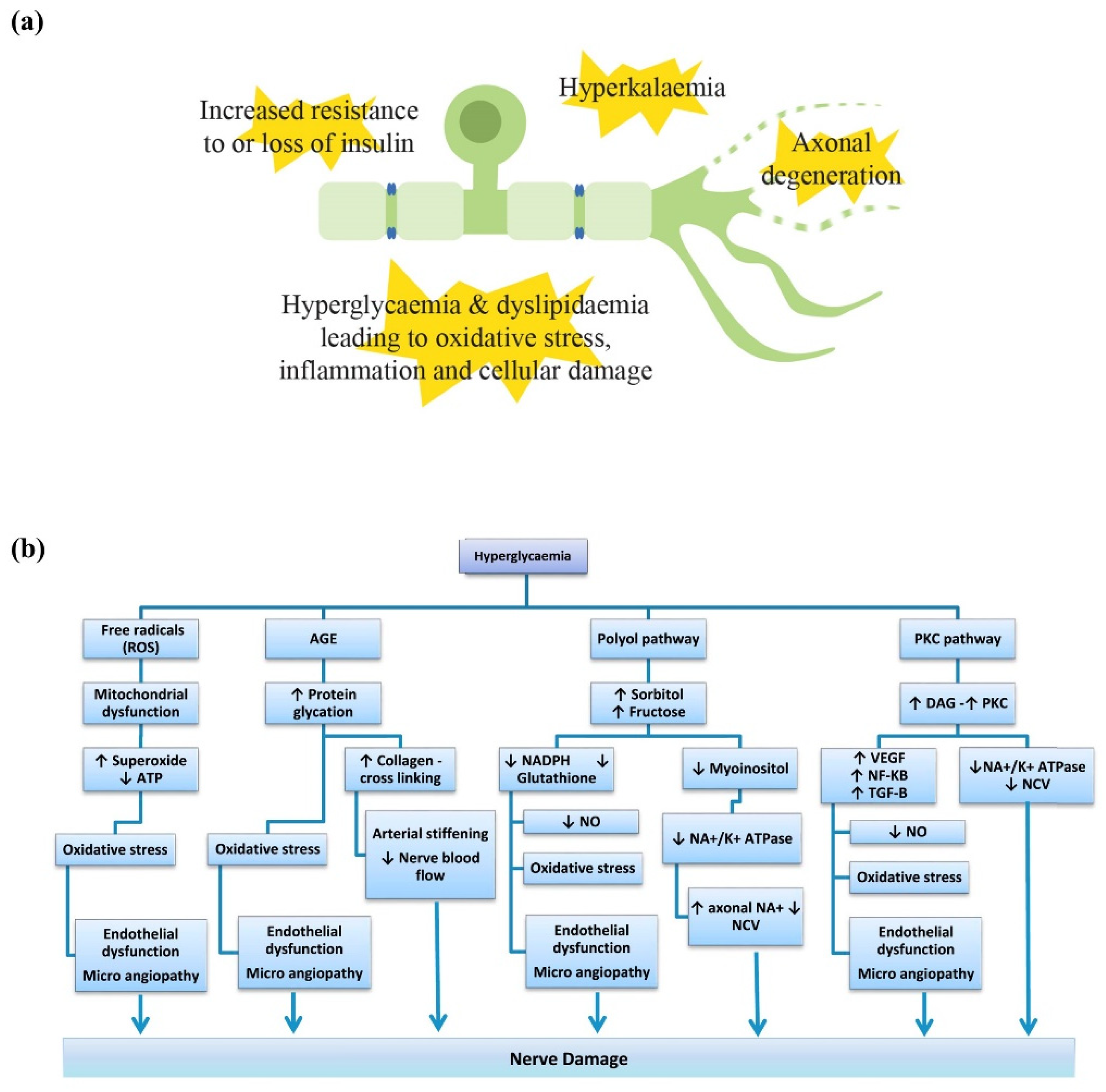
2.1. Pathophysiology Underlying Metabolic Peripheral Neuropathy
2.1.1. Hyperglycaemia
2.1.2. Dyslipidaemia
2.1.3. Insulin
2.1.4. Potassium
2.2. Current Therapies for Metabolic Peripheral Neuropathy
2.3. Emerging Pharmacotherapeutics in Metabolic Peripheral Neuropathy
2.3.1. Glucagon-like Peptide-1 (GLP-1) Receptor Agonists
2.3.2. Lifestyle and Exercise Interventions
2.3.3. Bariatric Surgery and Medical Weight Loss
2.3.4. Targeting MicroRNA (miRNA)
3. Immune-Mediated Peripheral Neuropathy
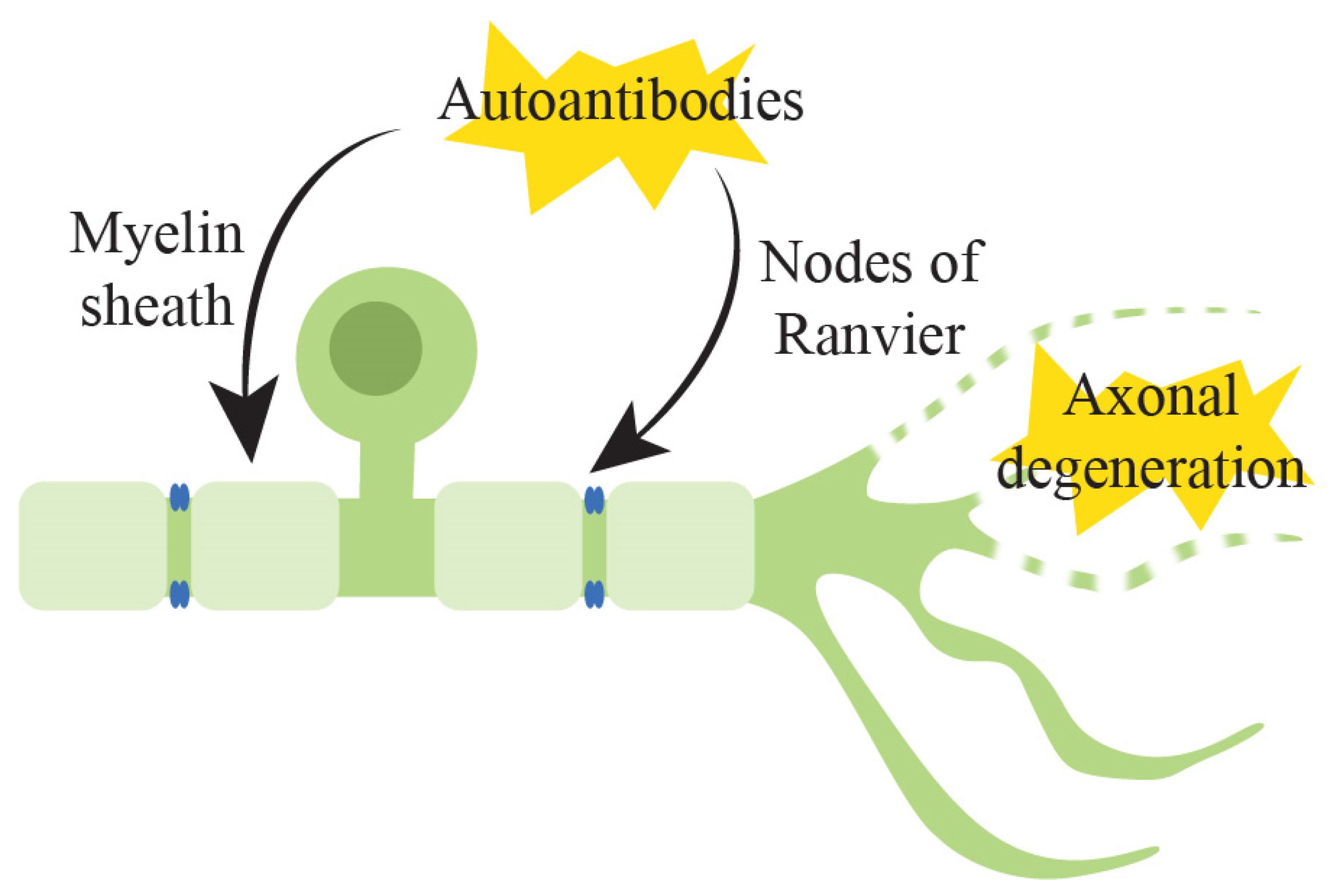
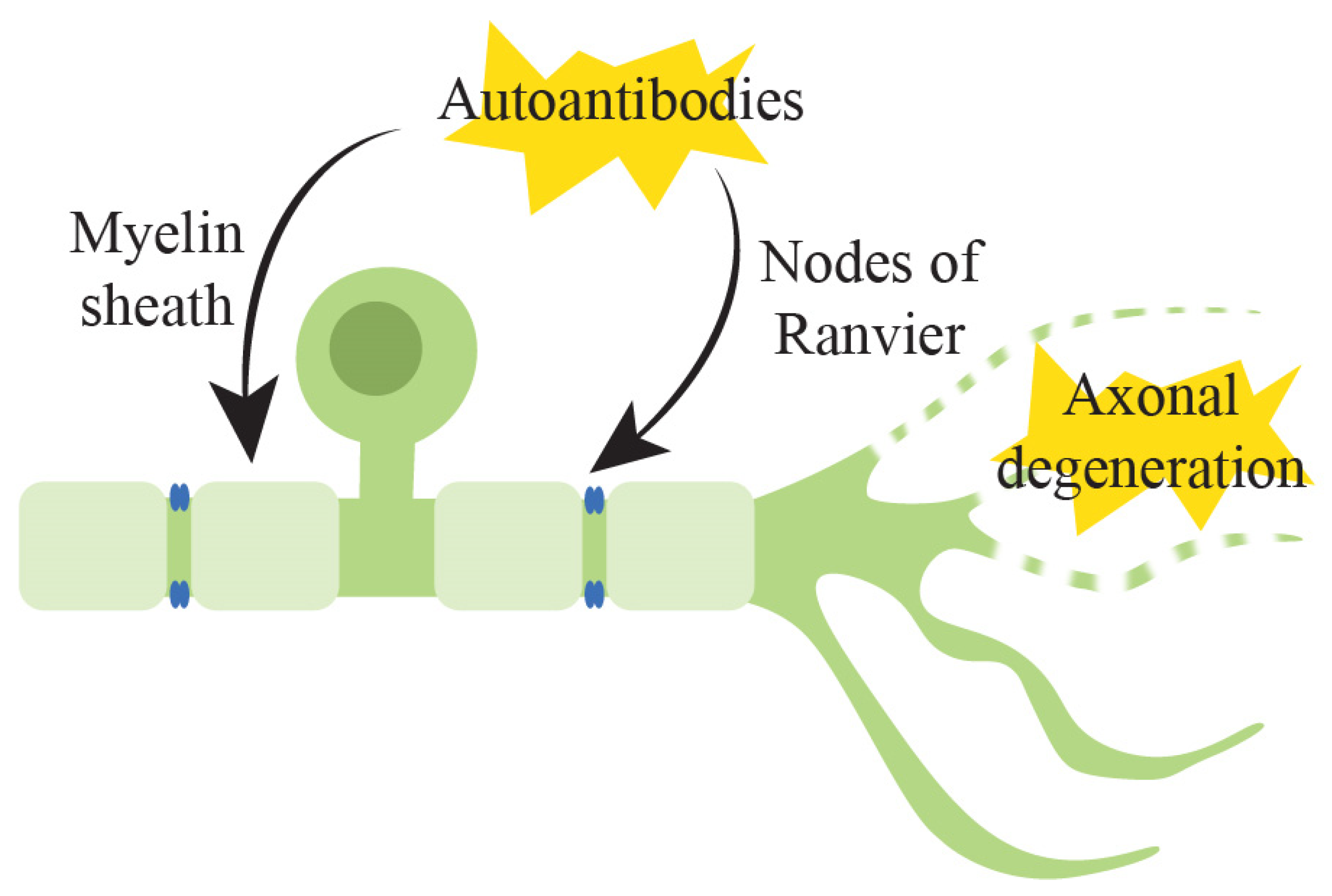
3.1. Pathophysiology of Underlying Immune-Mediated Peripheral Neuropathy
3.2. Current Therapies for Immune-Mediated Peripheral Neuropathy
3.2.1. Immunoglobulin
3.2.2. Corticosteroids
3.2.3. Plasma Exchange
3.2.4. Cyclophosphamide
3.2.5. Rituximab
3.3. Emerging Pharmacotherapeutics in Immune-Mediated Peripheral Neuropathy
3.3.1. Eculizumab
3.3.2. Targeting Antigen-Presenting Cells
4. Chemotherapy-Induced Peripheral Neuropathy
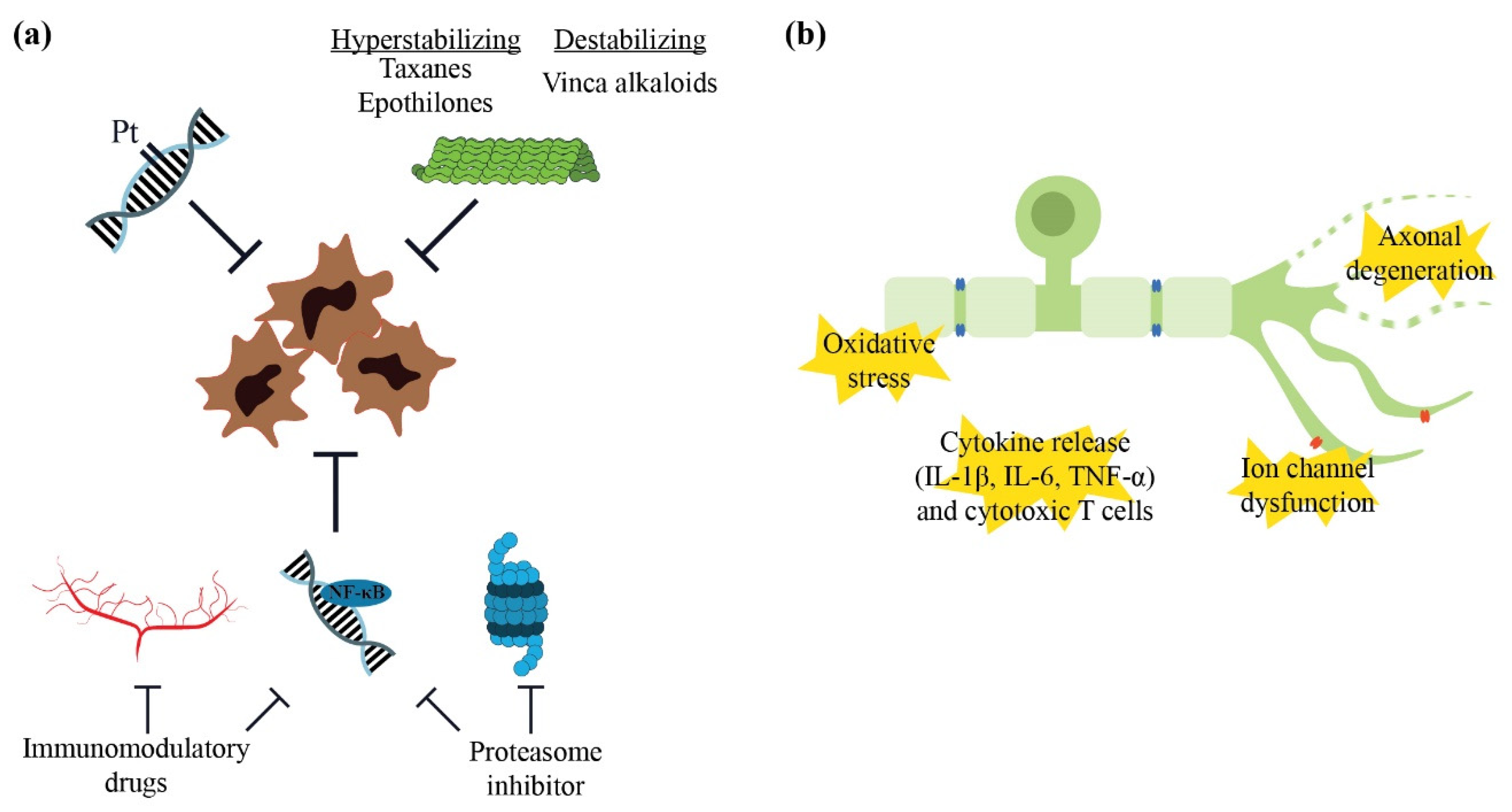
4.1. Pathophysiology Underlying Neurotoxicity
4.1.1. Neurotoxic Chemotherapy
4.1.2. Mechanisms of Neurotoxicity
4.2. Current Therapies for Chemotherapy-Induced Peripheral Neuropathy
4.3. Emerging Pharmacotherapeutics in Chemotherapy-Induce Peripheral Neuropathy
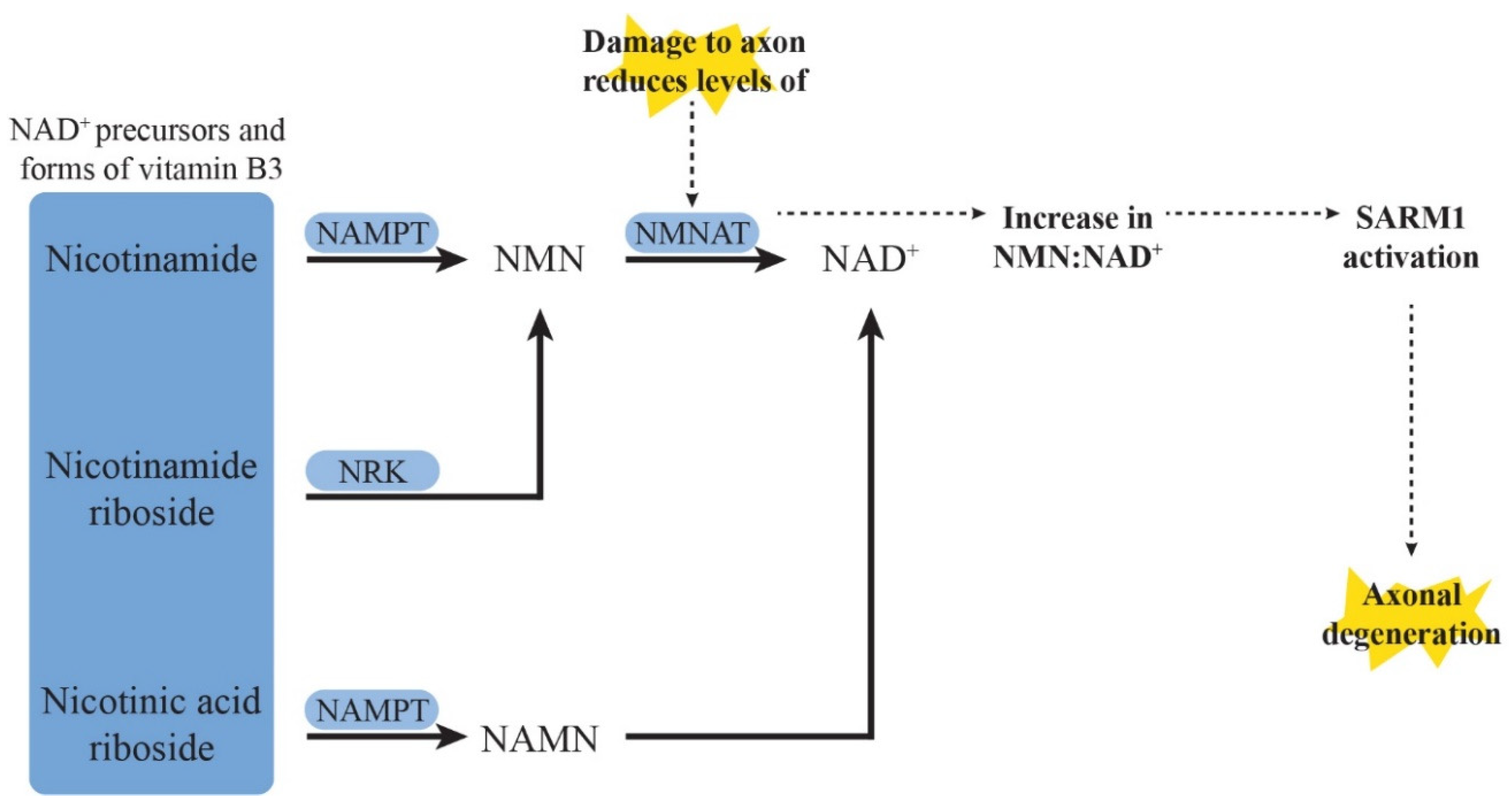
4.3.1. Sterile Alpha- and T1R-Motif-Containing 1 (SARM1) and Nicotinamide Adenine Dinucleotide (NAD+) Salvage Pathway
SARM1 Deletion and Inhibition
Targeting the NAD+ Signalling Pathway
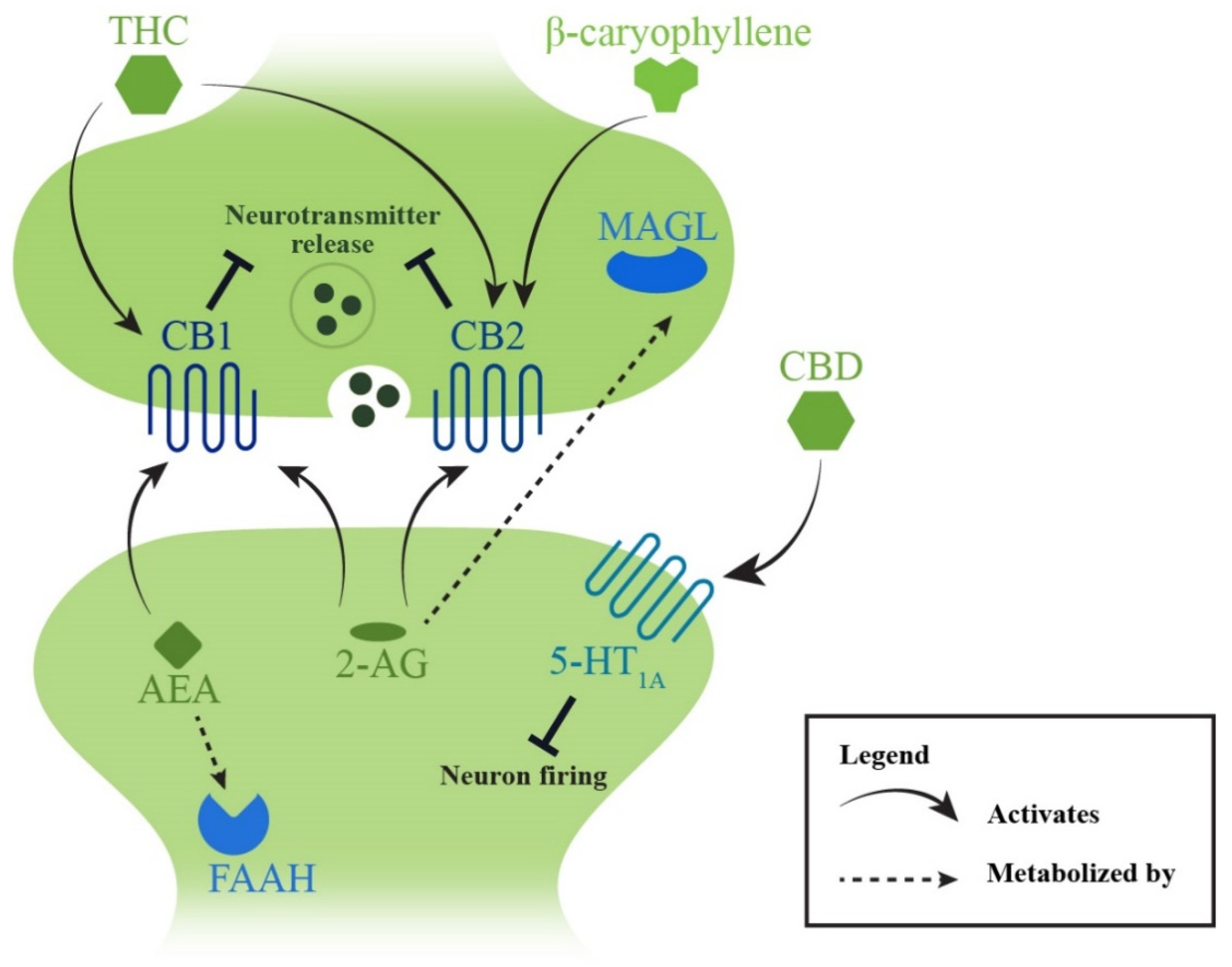
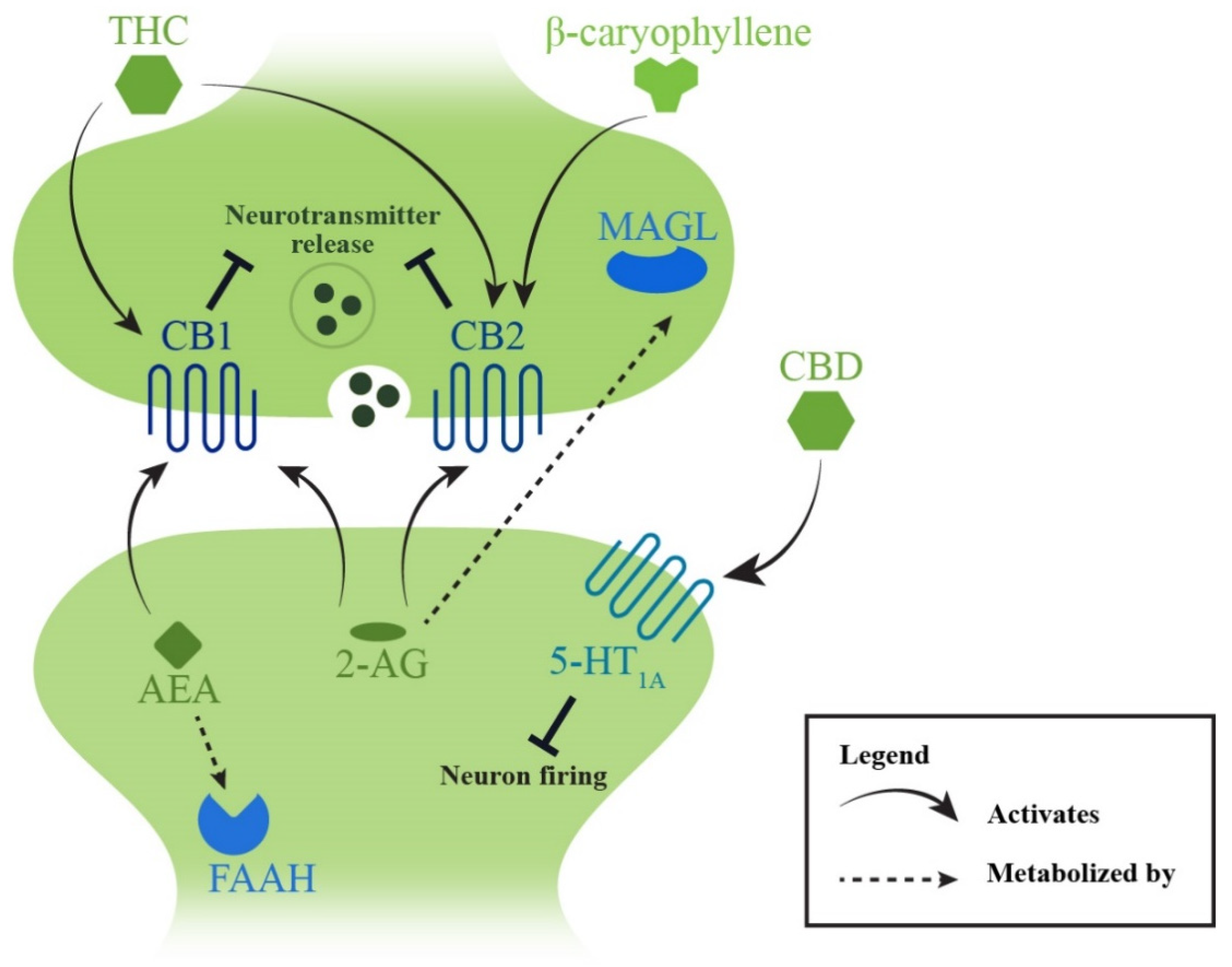
4.3.2. Endocannabinoid System
Targeting the Metabolization of Endocannabinoids
Stimulating Endocannabinoid Receptors
Phytocannabinoids and Cannabis
5. Overall Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119. [Google Scholar] [CrossRef] [PubMed]
- GBD Chronic Kidney Disease Collaboration. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef] [Green Version]
- Feldman, E.L.; Callaghan, B.C.; Pop-Busui, R.; Zochodne, D.W.; Wright, D.E.; Bennett, D.L.; Bril, V.; Russell, J.W.; Viswanathan, V. Diabetic neuropathy. Nat. Rev. Dis. Primers 2019, 5, 41. [Google Scholar] [CrossRef]
- Arnold, R.; Pianta, T.J.; Pussell, B.A.; Endre, Z.; Kiernan, M.C.; Krishnan, A.V. Potassium control in chronic kidney disease: Implications for neuromuscular function. Intern. Med. J. 2019, 49, 817–825. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Timmins, H.C.; Mizrahi, D.; Li, T.; Kiernan, M.C.; Goldstein, D.; Park, S.B. Metabolic and lifestyle risk factors for chemotherapy-induced peripheral neuropathy in taxane and platinum-treated patients: A systematic review. J. Cancer Surviv. 2021. [Google Scholar] [CrossRef] [PubMed]
- Seretny, M.; Currie, G.L.; Sena, E.S.; Ramnarine, S.; Grant, R.; MacLeod, M.R.; Colvin, L.A.; Fallon, M. Incidence, prevalence, and predictors of chemotherapy-induced peripheral neuropathy: A systematic review and meta-analysis. Pain 2014, 155, 2461–2470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molassiotis, A.; Cheng, H.L.; Lopez, V.; Au, J.S.K.; Chan, A.; Bandla, A.; Leung, K.T.; Li, Y.C.; Wong, K.H.; Suen, L.K.P.; et al. Are we mis-estimating chemotherapy-induced peripheral neuropathy? Analysis of assessment methodologies from a prospective, multinational, longitudinal cohort study of patients receiving neurotoxic chemotherapy. BMC Cancer 2019, 19, 132. [Google Scholar] [CrossRef] [Green Version]
- Teng, C.; Cohen, J.; Egger, S.; Blinman, P.L.; Vardy, J.L. Systematic review of long-term chemotherapy-induced peripheral neuropathy (CIPN) following adjuvant oxaliplatin for colorectal cancer. Support. Care Cancer 2022, 30, 33–47. [Google Scholar] [CrossRef]
- Levison, L.S.; Thomsen, R.W.; Andersen, H. Increased mortality following Guillain-Barré syndrome: A population-based cohort study. Eur. J. Neurol. 2022, 29, 1145–1154. [Google Scholar] [CrossRef]
- Broers, M.C.; Bunschoten, C.; Nieboer, D.; Lingsma, H.F.; Jacobs, B.C. Incidence and prevalence of chronic inflammatory demyelinating polyradiculoneuropathy: A systematic review and meta-analysis. Neuroepidemiology 2019, 52, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Bragazzi, N.L.; Kolahi, A.A.; Nejadghaderi, S.A.; Lochner, P.; Brigo, F.; Naldi, A.; Lanteri, P.; Garbarino, S.; Sullman, M.J.M.; Dai, H.; et al. Global, regional, and national burden of Guillain-Barré syndrome and its underlying causes from 1990 to 2019. J. Neuroinflamm. 2021, 18, 264. [Google Scholar] [CrossRef] [PubMed]
- Hicks, C.W.; Wang, D.; Matsushita, K.; Windham, B.G.; Selvin, E. Peripheral neuropathy and all-cause and cardiovascular mortality in U.S. adults: A prospective cohort study. Ann. Intern. Med. 2021, 174, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Mold, J.W.; Vesely, S.K.; Keyl, B.A.; Schenk, J.B.; Roberts, M. The prevalence, predictors, and consequences of peripheral sensory neuropathy in older patients. J. Am. Board Fam. Pract. 2004, 17, 309–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hicks, C.W.; Wang, D.; Windham, B.G.; Matsushita, K.; Selvin, E. Prevalence of peripheral neuropathy defined by monofilament insensitivity in middle-aged and older adults in two US cohorts. Sci. Rep. 2021, 11, 19159. [Google Scholar] [CrossRef] [PubMed]
- Formiga, F.; Moreno-González, R.; Corsonello, A.; Carlsson, A.; Ärnlöv, J.; Mattace-Raso, F.; Kostka, T.; Weingart, C.; Roller-Wirnsberger, R.; Tap, L.; et al. Diabetes, sarcopenia and chronic kidney disease; the Screening for CKD among Older People across Europe (SCOPE) study. BMC Geriatr. 2022, 22, 254. [Google Scholar] [CrossRef]
- Lorenzi, M.; Bonassi, S.; Lorenzi, T.; Giovannini, S.; Bernabei, R.; Onder, G. A review of telomere length in sarcopenia and frailty. Biogerontology 2018, 19, 209–221. [Google Scholar] [CrossRef]
- Giovannini, S.; Coraci, D.; Brau, F.; Galluzzo, V.; Loreti, C.; Caliandro, P.; Padua, L.; Maccauro, G.; Biscotti, L.; Bernabei, R. Neuropathic pain in the elderly. Diagnostics 2021, 11, 613. [Google Scholar] [CrossRef]
- Bao, T.; Basal, C.; Seluzicki, C.; Li, S.Q.; Seidman, A.D.; Mao, J.J. Long-term chemotherapy-induced peripheral neuropathy among breast cancer survivors: Prevalence, risk factors, and fall risk. Breast Cancer Res. Treat. 2016, 159, 327–333. [Google Scholar] [CrossRef]
- Alleman, C.J.; Westerhout, K.Y.; Hensen, M.; Chambers, C.; Stoker, M.; Long, S.; van Nooten, F.E. Humanistic and economic burden of painful diabetic peripheral neuropathy in Europe: A review of the literature. Diabetes Res. Clin. Pract. 2015, 109, 215–225. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Wilson, K.L.; Kagan, J.; Panjabi, S. Cost of peripheral neuropathy in patients receiving treatment for multiple myeloma: A US administrative claims analysis. Ther. Adv. Hematol. 2019, 10, 2040620719839025. [Google Scholar] [CrossRef] [PubMed]
- Issar, T.; Tummanapalli, S.S.; Borire, A.A.; Kwai, N.C.G.; Poynten, A.M.; Arnold, R.; Markoulli, M.; Krishnan, A.V. Impact of the metabolic syndrome on peripheral nerve structure and function in type 2 diabetes. Eur. J. Neurol. 2021, 28, 2074–2082. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, B.C.; Xia, R.; Reynolds, E.; Banerjee, M.; Rothberg, A.E.; Burant, C.F.; Villegas-Umana, E.; Pop-Busui, R.; Feldman, E.L. Association between metabolic syndrome components and polyneuropathy in an obese population. JAMA Neurol. 2016, 73, 1468–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyck, P.J.; Kratz, K.M.; Karnes, J.L.; Litchy, W.J.; Klein, R.; Pach, J.M.; Wilson, D.M.; O’Brien, P.C.; Melton, L.J., 3rd; Service, F.J. The prevalence by staged severity of various types of diabetic neuropathy, retinopathy, and nephropathy in a population-based cohort: The Rochester Diabetic Neuropathy Study. Neurology 1993, 43, 817–824. [Google Scholar] [CrossRef]
- Krishnan, A.V.; Kiernan, M.C. Neurological complications of chronic kidney disease. Nat. Rev. Neurol. 2009, 5, 542–551. [Google Scholar] [CrossRef]
- Vincent, A.M.; Callaghan, B.C.; Smith, A.L.; Feldman, E.L. Diabetic neuropathy: Cellular mechanisms as therapeutic targets. Nat. Rev. Neurol. 2011, 7, 573–583. [Google Scholar] [CrossRef]
- Greene, D.A.; Sima, A.A.; Stevens, M.J.; Feldman, E.L.; Lattimer, S.A. Complications: Neuropathy, pathogenetic considerations. Diabetes Care 1992, 15, 1902–1925. [Google Scholar] [CrossRef]
- Hempel, A.; Maasch, C.; Heintze, U.; Lindschau, C.; Dietz, R.; Luft, F.C.; Haller, H. High glucose concentrations increase endothelial cell permeability via activation of protein kinase C alpha. Circ. Res. 1997, 81, 363–371. [Google Scholar] [CrossRef]
- Markoulli, M.; Flanagan, J.; Tummanapalli, S.S.; Wu, J.; Willcox, M. The impact of diabetes on corneal nerve morphology and ocular surface integrity. Ocul. Surf. 2018, 16, 45–57. [Google Scholar] [CrossRef]
- Callaghan, B.C.; Gallagher, G.; Fridman, V.; Feldman, E.L. Diabetic neuropathy: What does the future hold? Diabetologia 2020, 63, 891–897. [Google Scholar] [CrossRef]
- Krauss, R.; Bosanac, T.; Devraj, R.; Engber, T.; Hughes, R.O. Axons matter: The promise of treating neurodegenerative disorders by targeting SARM1-mediated axonal degeneration. Trends Pharmacol. Sci. 2020, 41, 281–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callaghan, B.C.; Little, A.A.; Feldman, E.L.; Hughes, R.A. Enhanced glucose control for preventing and treating diabetic neuropathy. Cochrane Database Syst. Rev. 2012, 6, CD007543. [Google Scholar] [CrossRef] [PubMed]
- Stino, A.M.; Rumora, A.E.; Kim, B.; Feldman, E.L. Evolving concepts on the role of dyslipidemia, bioenergetics, and inflammation in the pathogenesis and treatment of diabetic peripheral neuropathy. J. Peripher. Nerv. Syst. 2020, 25, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Feldman, E.L.; Nave, K.-A.; Jensen, T.S.; Bennett, D.L.H. New horizons in diabetic neuropathy: Mechanisms, bioenergetics, and pain. Neuron 2017, 93, 1296–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eid, S.; Sas, K.M.; Abcouwer, S.F.; Feldman, E.L.; Gardner, T.W.; Pennathur, S.; Fort, P.E. New insights into the mechanisms of diabetic complications: Role of lipids and lipid metabolism. Diabetologia 2019, 62, 1539–1549. [Google Scholar] [CrossRef] [Green Version]
- Mielke, J.G.; Wang, Y.-T. Insulin, Synaptic Function, and Opportunities for Neuroprotection. Prog. Mol. Biol. Transl. Sci. 2011, 98, 133–186. [Google Scholar] [CrossRef]
- Waldbillig, R.J.; LeRoith, D. Insulin receptors in the peripheral nervous system: A structural and functional analysis. Brain Res. 1987, 409, 215–220. [Google Scholar] [CrossRef]
- Zochodne, D.W. Diabetes and the plasticity of sensory neurons. Neurosci. Lett. 2015, 596, 60–65. [Google Scholar] [CrossRef]
- Wahren, J.; Foyt, H.; Daniels, M.; Arezzo, J.C. Long-acting C-peptide and neuropathy in Type 1 Diabetes: A 12-month clinical trial. Diabetes Care 2016, 39, 596–602. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.; Feldman, E.L. Insulin resistance in the nervous system. Trends Endocrinol. Metab. 2012, 23, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Tuttle, K.R.; Bakris, G.L.; Bilous, R.W.; Chiang, J.L.; de Boer, I.H.; Goldstein-Fuchs, J.; Hirsch, I.B.; Kalantar-Zadeh, K.; Narva, A.S.; Navaneethan, S.D.; et al. Diabetic kidney disease: A report from an ADA consensus conference. Diabetes Care 2014, 37, 2864–2883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Issar, T.; Arnold, R.; Kwai, N.C.G.; Walker, S.; Yan, A.; Borire, A.A.; Poynten, A.M.; Pussell, B.A.; Endre, Z.H.; Kiernan, M.C.; et al. Relative contributions of diabetes and chronic kidney disease to neuropathy development in diabetic nephropathy patients. Clin. Neurophysiol. 2019, 130, 2088–2095. [Google Scholar] [CrossRef] [PubMed]
- Arnold, R.; Pianta, T.J.; Issar, T.; Kirby, A.; Scales, C.M.K.; Kwai, N.C.G.; Endre, Z.; Krishnan, A.V. Peripheral neuropathy: An important contributor to physical limitation and morbidity in stages 3 and 4 chronic kidney disease. Nephrol. Dial. Transplant. 2022, 37, 713–719. [Google Scholar] [CrossRef]
- Arnold, R.; Pussell, B.A.; Howells, J.; Grinius, V.; Kiernan, M.C.; Lin, C.S.-Y.; Krishnan, A.V. Evidence for a causal relationship between hyperkalaemia and axonal dysfunction in end-stage kidney disease. Clin. Neurophysiol. 2014, 125, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Borire, A.A.; Arnold, R.; Pussell, B.A.; Kwai, N.C.; Visser, L.H.; Padua, L.; Simon, N.G.; Kiernan, M.C.; Krishnan, A.V. Haemodialysis alters peripheral nerve morphology in end-stage kidney disease. Clin. Neurophysiol. 2017, 128, 281–286. [Google Scholar] [CrossRef]
- Krishnan, A.V.; Phoon, R.K.; Pussell, B.A.; Charlesworth, J.A.; Bostock, H.; Kiernan, M.C. Neuropathy, axonal Na+/K+ pump function and activity-dependent excitability changes in end-stage kidney disease. Clin. Neurophysiol. 2006, 117, 992–999. [Google Scholar] [CrossRef]
- Arnold, R.; Pianta, T.J.; Pussell, B.A.; Kirby, A.; O’Brien, K.; Sullivan, K.; Holyday, M.; Cormack, C.; Kiernan, M.C.; Krishnan, A.V. Randomized, controlled trial of the effect of dietary potassium restriction on nerve function in CKD. Clin. J. Am. Soc. Nephrol. 2017, 12, 1569–1577. [Google Scholar] [CrossRef] [Green Version]
- Bril, V.; Breiner, A.; Perkins, B.A.; Zochodne, D. Neuropathy. Can. J. Diabetes 2018, 42, S217–S221. [Google Scholar] [CrossRef] [Green Version]
- Lunn, M.P.; Hughes, R.A.; Wiffen, P.J. Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia. Cochrane Database Syst. Rev. 2014, 1, CD007115. [Google Scholar] [CrossRef]
- Soltesova Prnova, M.; Svik, K.; Bezek, S.; Kovacikova, L.; Karasu, C.; Stefek, M. 3-mercapto-5H-1,2,4-triazino[5,6-b]indole-5-acetic acid (Cemtirestat) alleviates symptoms of peripheral diabetic neuropathy in Zucker Diabetic Fatty (ZDF) rats: A role of aldose reductase. Neurochem. Res. 2019, 44, 1056–1064. [Google Scholar] [CrossRef]
- Polydefkis, M.; Arezzo, J.; Nash, M.; Bril, V.; Shaibani, A.; Gordon, R.J.; Bradshaw, K.L.; Junor, R.W. Safety and efficacy of ranirestat in patients with mild-to-moderate diabetic sensorimotor polyneuropathy. J. Peripher. Nerv. Syst. 2015, 20, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, K.; Kohara, N.; Baba, M.; Komori, T.; Naito, Y.; Imai, T.; Satoh, J.; Yamaguchi, Y.; Hamatani, T. Aldose reductase inhibitor ranirestat significantly improves nerve conduction velocity in diabetic polyneuropathy: A randomized double-blind placebo-controlled study in Japan. J. Diabetes Investig. 2019, 10, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J. Mechanisms of action and therapeutic application of glucagon-like peptide-1. Cell Metab. 2018, 27, 740–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Himeno, T.; Kamiya, H.; Naruse, K.; Harada, N.; Ozaki, N.; Seino, Y.; Shibata, T.; Kondo, M.; Kato, J.; Okawa, T.; et al. Beneficial effects of exendin-4 on experimental polyneuropathy in diabetic mice. Diabetes 2011, 60, 2397–2406. [Google Scholar] [CrossRef] [Green Version]
- Jolivalt, C.G.; Fineman, M.; Deacon, C.F.; Carr, R.D.; Calcutt, N.A. GLP-1 signals via ERK in peripheral nerve and prevents nerve dysfunction in diabetic mice. Diabetes, Obes. Metab. 2011, 13, 990–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.J.; Jin, H.Y.; Lee, K.A.; Xie, S.H.; Baek, H.S.; Park, T.S. Neuroprotective effect of the glucagon-like peptide-1 receptor agonist, synthetic exendin-4, in streptozotocin-induced diabetic rats. Br. J. Pharmacol. 2011, 164, 1410–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kan, M.; Guo, G.; Singh, B.; Singh, V.; Zochodne, D.W. Glucagon-like peptide 1, insulin, sensory neurons, and diabetic neuropathy. J. Neuropathol. Exp. Neurol. 2012, 71, 494–510. [Google Scholar] [CrossRef] [Green Version]
- Gong, N.; Xiao, Q.; Zhu, B.; Zhang, C.-Y.; Wang, Y.C.; Fan, H.; Ma, A.-N.; Wang, Y.X. Activation of spinal glucagon-like peptide-1 receptors specifically suppresses pain hypersensitivity. J. Neurosci. 2014, 34, 5322–5334. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Shi, M.; Zhang, X.; Liu, X.; Chen, J.; Zhang, R.; Wang, X.; Zhang, H. GLP-1R agonists ameliorate peripheral nerve dysfunction and inflammation via p38 MAPK/NF-κB signaling pathways in streptozotocin-induced diabetic rats. Int. J. Mol. Med. 2018, 41, 2977–2985. [Google Scholar] [CrossRef] [Green Version]
- Shekunova, E.V.; Kashkin, V.A.; Muzhikyan, A.; Makarova, M.N.; Balabanyan, V.Y.; Makarov, V.G. Therapeutic efficacy of arginine-rich exenatide on diabetic neuropathy in rats. Eur. J. Pharmacol. 2020, 866, 172835. [Google Scholar] [CrossRef]
- Moustafa, P.E.; Abdelkader, N.F.; El Awdan, S.A.; El-Shabrawy, O.A.; Zaki, H.F. Liraglutide ameliorated peripheral neuropathy in diabetic rats: Involvement of oxidative stress, inflammation and extracellular matrix remodeling. J. Neurochem. 2018, 146, 173–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muscogiuri, G.; DeFronzo, R.A.; Gastaldelli, A.; Holst, J.J. Glucagon-like peptide-1 and the central/peripheral nervous system: Crosstalk in diabetes. Trends Endocrinol. Metab. 2017, 28, 88–103. [Google Scholar] [CrossRef] [PubMed]
- Aung, M.M.; Slade, K.; Freeman, L.A.R.; Kos, K.; Whatmore, J.L.; Shore, A.C.; Gooding, K.M. Locally delivered GLP-1 analogues liraglutide and exenatide enhance microvascular perfusion in individuals with and without type 2 diabetes. Diabetologia 2019, 62, 1701–1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Issar, T.; Kwai, N.C.G.; Poynten, A.M.; Arnold, R.; Milner, K.-L.; Krishnan, A.V. Effect of exenatide on peripheral nerve excitability in type 2 diabetes. Clin. Neurophysiol. 2021, 132, 2532–2539. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, M.; Martin, C.L.; Brown, M.B.; Callaghan, B.; Albers, J.W.; Feldman, E.L.; Pop-Busui, R. Effects of exenatide on measures of diabetic neuropathy in subjects with type 2 diabetes: Results from an 18-month proof-of-concept open-label randomized study. J. Diabetes Complicat. 2015, 29, 1287–1294. [Google Scholar] [CrossRef] [Green Version]
- Zilliox, L.A.; Russell, J.W. Physical activity and dietary interventions in diabetic neuropathy: A systematic review. Clin. Auton. Res. 2019, 29, 443–455. [Google Scholar] [CrossRef]
- Colberg, S.R.; Sigal, R.J.; Yardley, J.E.; Riddell, M.C.; Dunstan, D.W.; Dempsey, P.C.; Horton, E.S.; Castorino, K.; Tate, D.F. Physical activity/exercise and diabetes: A position statement of the American Diabetes Association. Diabetes Care 2016, 39, 2065–2079. [Google Scholar] [CrossRef] [Green Version]
- Guthold, R.; Stevens, G.A.; Riley, L.M.; Bull, F.C. Worldwide trends in insufficient physical activity from 2001 to 2016: A pooled analysis of 358 population-based surveys with 1.9 million participants. Lancet Glob. Health 2018, 6, e1077–e1086. [Google Scholar] [CrossRef] [Green Version]
- Balducci, S.; Iacobellis, G.; Parisi, L.; Di Biase, N.; Calandriello, E.; Leonetti, F.; Fallucca, F. Exercise training can modify the natural history of diabetic peripheral neuropathy. J. Diabetes Complicat. 2006, 20, 216–223. [Google Scholar] [CrossRef]
- Perrin, B.M.; Southon, J.; McCaig, J.; Skinner, I.; Skinner, T.C.; Kingsley, M.I.C. The effect of structured exercise compared with education on neuropathic signs and symptoms in people at risk of neuropathic diabetic foot ulcers: A randomized clinical trial. Medicina 2021, 58, 59. [Google Scholar] [CrossRef]
- Kluding, P.M.; Pasnoor, M.; Singh, R.; Jernigan, S.; Farmer, K.; Rucker, J.; Sharma, N.K.; Wright, D.E. The effect of exercise on neuropathic symptoms, nerve function, and cutaneous innervation in people with diabetic peripheral neuropathy. J. Diabetes Complicat. 2012, 26, 424–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singleton, J.R.; Marcus, R.L.; Jackson, J.E.; Lessard, M.K.; Graham, T.E.; Smith, A.G. Exercise increases cutaneous nerve density in diabetic patients without neuropathy. Ann. Clin. Transl. Neurol. 2014, 1, 844–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.G.; Russell, J.; Feldman, E.L.; Goldstein, J.; Peltier, A.; Smith, S.; Hamwi, J.; Pollari, D.; Bixby, B.; Howard, J.; et al. Lifestyle intervention for pre-diabetic neuropathy. Diabetes Care 2006, 29, 1294–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singleton, J.R.; Marcus, R.L.; Lessard, M.K.; Jackson, J.E.; Smith, A.G. Supervised exercise improves cutaneous reinnervation capacity in metabolic syndrome patients. Ann. Neurol. 2015, 77, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Kluding, P.M.; Singleton, J.R.; Pasnoor, M.; Dimachkie, M.M.; Barohn, R.J.; Smith, A.G.; Marcus, R.L. Activity for Diabetic Polyneuropathy (ADAPT): Study design and protocol for a 2-site randomized controlled trial. Phys. Ther. 2017, 97, 20–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skoglund, G.; Nilsson, B.B.; Olsen, C.F.; Bergland, A.; Hilde, G. Facilitators and barriers for lifestyle change in people with prediabetes: A meta-synthesis of qualitative studies. BMC Public Health 2022, 22, 553. [Google Scholar] [CrossRef] [PubMed]
- Aghili, R.; Malek, M.; Tanha, K.; Mottaghi, A. The effect of bariatric surgery on peripheral polyneuropathy: A systematic review and meta-analysis. Obes. Surg. 2019, 29, 3010–3020. [Google Scholar] [CrossRef]
- O’Brien, R.; Johnson, E.; Haneuse, S.; Coleman, K.J.; O’Connor, P.J.; Fisher, D.P.; Sidney, S.; Bogart, A.; Theis, M.K.; Anau, J.; et al. Microvascular outcomes in patients with diabetes after bariatric surgery versus usual care: A matched cohort study. Ann. Intern Med. 2018, 169, 300–310. [Google Scholar] [CrossRef]
- Azmi, S.; Ferdousi, M.; Liu, Y.; Adam, S.; Iqbal, Z.; Dhage, S.; Ponirakis, G.; Siahmansur, T.; Marshall, A.; Petropoulos, I.; et al. Bariatric surgery leads to an improvement in small nerve fibre damage in subjects with obesity. Int. J. Obes. 2021, 45, 631–638. [Google Scholar] [CrossRef]
- Adam, S.; Azmi, S.; Ho, J.H.; Liu, Y.; Ferdousi, M.; Siahmansur, T.; Kalteniece, A.; Marshall, A.; Dhage, S.S.; Iqbal, Z.; et al. Improvements in diabetic neuropathy and nephropathy after bariatric surgery: A prospective cohort study. Obes. Surg. 2021, 31, 554–563. [Google Scholar] [CrossRef]
- Callaghan, B.C.; Reynolds, E.L.; Banerjee, M.; Akinci, G.; Chant, E.; Villegas-Umana, E.; Rothberg, A.E.; Burant, C.F.; Feldman, E.L. Dietary weight loss in people with severe obesity stabilizes neuropathy and improves symptomatology. Obesity 2021, 29, 2108–2118. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Xu, X.; Chen, J.; Kang, Y.; Guo, J.; Duscher, D.; Yang, X.; Guo, G.; Ren, S.; Xiong, H.; et al. The construction and analysis of incRNA-miRNA-mRNA competing endogenous RNA network of Schwann cells in diabetic peripheral neuropathy. Front. Bioeng. Biotechnol. 2020, 8, 490. [Google Scholar] [CrossRef] [PubMed]
- Bali, K.K.; Gandla, J.; Rangel, D.R.; Castaldi, L.; Mouritzen, P.; Agarwal, N.; Schmelz, M.; Heppenstall, P.; Kuner, R. A genome-wide screen reveals microRNAs in peripheral sensory neurons driving painful diabetic neuropathy. Pain 2021, 162, 1334–1351. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Wang, L.; Chopp, M.; Zhang, Y.; Szalad, A.; Zhang, Z.G. MicroRNA 146a locally mediates distal axonal growth of dorsal root ganglia neurons under high glucose and sildenafil conditions. Neuroscience 2016, 329, 43–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Chen, L.; Luo, Q.; Wu, M.; Chen, Y.; Shi, X. Involvement of microRNA-146a in diabetic peripheral neuropathy through the regulation of inflammation. Drug Des. Dev. Ther. 2018, 12, 171–177. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.S.; Fan, B.; Szalad, A.; Jia, L.; Wang, L.; Wang, X.; Pan, W.; Zhang, L.; Zhang, R.; Hu, J.; et al. MicroRNA-146a mimics reduce the peripheral neuropathy in type 2 diabetic mice. Diabetes 2017, 66, 3111–3121. [Google Scholar] [CrossRef] [Green Version]
- Fan, B.; Chopp, M.; Zhang, Z.G.; Liu, X.S. Treatment of diabetic peripheral neuropathy with engineered mesenchymal stromal cell-derived exosomes enriched with microRNA-146a provide amplified therapeutic efficacy. Exp. Neurol. 2021, 341, 113694. [Google Scholar] [CrossRef]
- Luo, Q.; Feng, Y.; Xie, Y.; Shao, Y.; Wu, M.; Deng, X.; Yuan, W.E.; Chen, Y.; Shi, X. Nanoparticle-microRNA-146a-5p polyplexes ameliorate diabetic peripheral neuropathy by modulating inflammation and apoptosis. Nanomedicine 2019, 17, 188–197. [Google Scholar] [CrossRef]
- Wang, L.; Chopp, M.; Szalad, A.; Lu, X.; Zhang, Y.; Wang, X.; Cepparulo, P.; Lu, M.; Li, C.; Zhang, Z.G. Exosomes derived from Schwann cells ameliorate peripheral neuropathy in type 2 diabetic mice. Diabetes 2020, 69, 749–759. [Google Scholar] [CrossRef]
- Fan, B.; Li, C.; Szalad, A.; Wang, L.; Pan, W.; Zhang, R.; Chopp, M.; Zhang, Z.G.; Liu, X.S. Mesenchymal stromal cell-derived exosomes ameliorate peripheral neuropathy in a mouse model of diabetes. Diabetologia 2020, 63, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Guo, Y.; Chen, Q.; Xiong, Q.; Min, S. MicroRNA-193a downregulates HMGB1 to alleviate diabetic neuropathic pain in a mouse model. Neuroimmunomodulation 2019, 26, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Gu, Y.; Shi, B. miR-590-3p alleviates diabetic peripheral neuropathic pain by targeting RAP1A and suppressing infiltration by the T cells. Acta Biochim. Pol. 2020, 67, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wang, X.; Yin, Y.; Zhu, L.; Zhang, F.; Yang, J. Investigation of the role of miR-221 in diabetic peripheral neuropathy and related molecular mechanisms. Adv. Clin. Exp. Med. 2021, 30, 623–632. [Google Scholar] [CrossRef]
- Czaplinski, A.; Steck, A.J. Immune mediated neuropathies--an update on therapeutic strategies. J. Neurol. 2004, 251, 127–137. [Google Scholar] [CrossRef]
- Vallat, J.-M.; Sommer, C.; Magy, L. Chronic inflammatory demyelinating polyradiculoneuropathy: Diagnostic and therapeutic challenges for a treatable condition. Lancet Neurol. 2010, 9, 402–412. [Google Scholar] [CrossRef]
- Shahrizaila, N.; Lehmann, H.C.; Kuwabara, S. Guillain-Barré syndrome. Lancet 2021, 397, 1214–1228. [Google Scholar] [CrossRef]
- Uncini, A.; Susuki, K.; Yuki, N. Nodo-paranodopathy: Beyond the demyelinating and axonal classification in anti-ganglioside antibody-mediated neuropathies. Clin. Neurophysiol. 2013, 124, 1928–1934. [Google Scholar] [CrossRef]
- Rees, J.H.; Soudain, S.E.; Gregson, N.A.; Hughes, R.A. Campylobacter jejuni infection and Guillain-Barré syndrome. N. Engl. J. Med. 1995, 333, 1374–1379. [Google Scholar] [CrossRef]
- Willison, H.J. The immunobiology of Guillain-Barre syndromes. J. Peripher. Nerv. Syst. 2005, 10, 94–112. [Google Scholar] [CrossRef]
- Mathey, E.K.; Park, S.B.; Hughes, R.A.; Pollard, J.D.; Armati, P.J.; Barnett, M.H.; Taylor, B.V.; Dyck, P.J.; Kiernan, M.C.; Lin, C.S. Chronic inflammatory demyelinating polyradiculoneuropathy: From pathology to phenotype. J. Neurol. Neurosurg. Psychiatry 2015, 86, 973–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanda, T. Biology of the blood–nerve barrier and its alteration in immune mediated neuropathies. J. Neurol. Neurosurg. Psychiatry 2013, 84, 208–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Meché, F.G.; Schmitz, P.I.; The Dutch Guillain–Barré Study Group. A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain–Barré syndrome. N. Engl. J. Med. 1992, 326, 1123–1129. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.A.; Donofrio, P.; Bril, V.; Dalakas, M.C.; Deng, C.; Hanna, K.; Hartung, H.-P.; Latov, N.; Merkies, I.S.; van Doorn, P.A. Intravenous immune globulin (10% caprylate-chromatography purified) for the treatment of chronic inflammatory demyelinating polyradiculoneuropathy (ICE study): A randomised placebo-controlled trial. Lancet Neurol. 2008, 7, 136–144. [Google Scholar] [CrossRef]
- Kuwabara, S.; Mori, M.; Misawa, S.; Suzuki, M.; Nishiyama, K.; Mutoh, T.; Doi, S.; Kokubun, N.; Kamijo, M.; Yoshikawa, H.; et al. Intravenous immunoglobulin for maintenance treatment of chronic inflammatory demyelinating polyneuropathy: A multicentre, open-label, 52-week phase III trial. J. Neurol. Neurosurg. Psychiatry 2017, 88, 832–838. [Google Scholar] [CrossRef]
- Cornblath, D.R.; van Doorn, P.A.; Hartung, H.-P.; Merkies, I.S.J.; Katzberg, H.D.; Hinterberger, D.; Clodi, E.; ProCID Investigators. Randomized trial of three IVIg doses for treating chronic inflammatory demyelinating polyneuropathy. Brain 2022, 145, 887–896. [Google Scholar] [CrossRef]
- Kaji, R.; Shibasaki, H.; Kimura, J. Multifocal demyelinating motor neuropathy: Cranial nerve involvement and immunoglobulin therapy. Neurology 1992, 42, 506–509. [Google Scholar] [CrossRef]
- Charles, N.; Vial, C.; Moreau, T.; Benoit, P.; Bierme, T.; Bady, B. Intravenous immunoglobulin treatment in multifocal motor neuropathy. Lancet 1992, 340, 182. [Google Scholar] [CrossRef]
- van Schaik, I.N.; van der Berg, L.; de Haan, R.; Vermeulen, M.M. Intravenous immunoglobulin for multifocal motor neuropathy. Cochrane Database Syst. Rev. 2005, 1, CD004429. [Google Scholar] [CrossRef]
- Daëron, M.; Lesourne, R. Negative signaling in Fc receptor complexes. Adv. Immunol. 2006, 89, 39–86. [Google Scholar] [CrossRef] [Green Version]
- Bayry, J.; Lacroix-Desmazes, S.; Kazatchkine, M.D.; Kaveri, S.V. Monoclonal antibody and intravenous immunoglobulin therapy for rheumatic diseases: Rationale and mechanisms of action. Nat. Clin. Pract. Rheumatol. 2007, 3, 262–272. [Google Scholar] [CrossRef] [PubMed]
- van Schaik, I.N.; Bril, V.; van Geloven, N.; Hartung, H.-P.; Lewis, R.A.; Sobue, G.; Lawo, J.-P.; Praus, M.; Mielke, O.; Durn, B.L.; et al. Subcutaneous immunoglobulin for maintenance treatment in chronic inflammatory demyelinating polyneuropathy (PATH): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2018, 17, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Kieseier, B.C.; Kiefer, R.; Gold, R.; Hemmer, B.; Willison, H.J.; Hartung, H.-P. Advances in understanding and treatment of immune-mediated disorders of the peripheral nervous system. Muscle Nerve 2004, 30, 131–156. [Google Scholar] [CrossRef] [PubMed]
- Cocito, D.; Merola, A.; Romagnolo, A.; Peci, E.; Toscano, A.; Mazzeo, A.; Gentile, L.; Russo, M.; Fazio, R.; Filosto, M.; et al. Subcutaneous immunoglobulin in CIDP and MMN: A different long-term clinical response? J. Neurol. Neurosurg. Psychiatry 2016, 87, 791–793. [Google Scholar] [CrossRef] [PubMed]
- Nobile-Orazio, E.; Cocito, D.; Jann, S.; Uncini, A.; Messina, P.; Antonini, G.; Fazio, R.; Gallia, F.; Schenone, A.; Francia, A.; et al. Frequency and time to relapse after discontinuing 6-month therapy with IVIg or pulsed methylprednisolone in CIDP. J. Neurol. Neurosurg. Psychiatry 2015, 86, 729–734. [Google Scholar] [CrossRef]
- Dyck, P.J.; O’Brien, P.C.; Oviatt, K.F.; DiNapoli, R.P.; Daube, J.R.; Bartleson, J.D.; Mokri, B.; Swift, T.; Low, P.A.; Windebank, A.J. Prednisone improves chronic inflammatory demyelinating polyradiculoneuropathy more than no treatment. Ann. Neurol. 1982, 11, 136–141. [Google Scholar] [CrossRef]
- Hughes, R.A.; Mehndiratta, M.M.; Rajabally, Y.A. Corticosteroids for chronic inflammatory demyelinating polyradiculoneuropathy. Cochrane Database Syst. Rev. 2017, 11, CD002062. [Google Scholar] [CrossRef]
- Muley, S.A.; Kelkar, P.; Parry, G.J. Treatment of chronic inflammatory demyelinating polyneuropathy with pulsed oral steroids. Arch. Neurol. 2008, 65, 1460–1464. [Google Scholar] [CrossRef] [Green Version]
- The Guillain-Barré Syndrome Study Group. Plasmapheresis and acute Guillain-Barré syndrome. Neurology 1985, 35, 1096–1104. [Google Scholar] [CrossRef]
- The French Cooperative Group on Plasma Exchange in Guillain-Barré Syndrome. Appropriate number of plasma exchanges in Guillain-Barré syndrome. Ann. Neurol. 1997, 41, 298–306. [Google Scholar] [CrossRef]
- Dyck, P.J.; Daube, J.; O’Brien, P.; Pineda, A.; Low, P.A.; Windebank, A.J.; Swanson, C. Plasma exchange in chronic inflammatory demyelinating polyradiculoneuropathy. N. Engl. J. Med. 1986, 314, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Brannagan, T.H., 3rd; Alaedini, A.; Gladstone, D.E. High-dose cyclophosphamide without stem cell rescue for refractory multifocal motor neuropathy. Muscle Nerve 2006, 34, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Feldman, E.L.; Bromberg, M.B.; Albers, J.W.; Pestronk, A. Immunosuppressive treatment in multifocal motor neuropathy. Ann. Neurol. 1991, 30, 397–401. [Google Scholar] [CrossRef]
- Krarup, C.; Stewart, J.D.; Sumner, A.J.; Pestronk, A.; Lipton, S.A. A syndrome of asymmetric limb weakness with motor conduction block. Neurology 1990, 40, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Gladstone, D.E.; Prestrud, A.A.; Brannagan, T.H., 3rd. High-dose cyclophosphamide results in long-term disease remission with restoration of a normal quality of life in patients with severe refractory chronic inflammatory demyelinating polyneuropathy. J. Peripher. Nerv. Syst. 2005, 10, 11–16. [Google Scholar] [CrossRef]
- Maurer, M.A.; Rakocevic, G.; Leung, C.S.; Quast, I.; Lukačišin, M.; Goebels, N.; Münz, C.; Wardemann, H.; Dalakas, M.; Lünemann, J.D. Rituximab induces sustained reduction of pathogenic B cells in patients with peripheral nervous system autoimmunity. J. Clin. Investig. 2012, 122, 1393–1402. [Google Scholar] [CrossRef]
- Dalakas, M.C.; Rakocevic, G.; Salajegheh, M.; Dambrosia, J.M.; Hahn, A.F.; Raju, R.; McElroy, B. Placebo-controlled trial of rituximab in IgM anti-myelin-associated glycoprotein antibody demyelinating neuropathy. Ann. Neurol. 2009, 65, 286–293. [Google Scholar] [CrossRef]
- Léger, J.-M.; Viala, K.; Nicolas, G.; Créange, A.; Vallat, J.-M.; Pouget, J.; Clavelou, P.; Vial, C.; Steck, A.; Musset, L.; et al. Placebo-controlled trial of rituximab in IgM anti-myelin-associated glycoprotein neuropathy. Neurology 2013, 80, 2217–2225. [Google Scholar] [CrossRef]
- Chaudhry, V.; Cornblath, D.R. An open-label trial of rituximab (Rituxan®) in multifocal motor neuropathy. J. Peripher. Nerv. Syst. 2010, 15, 196–201. [Google Scholar] [CrossRef]
- Stieglbauer, K.; Topakian, R.; Hinterberger, G.; Aichner, F.T. Beneficial effect of rituximab monotherapy in multifocal motor neuropathy. Neuromuscul. Disord. 2009, 19, 473–475. [Google Scholar] [CrossRef]
- Renaud, S.; Gregor, M.; Fuhr, P.; Lorenz, D.; Deuschl, G.; Gratwohl, A.; Steck, A.J. Rituximab in the treatment of polyneuropathy associated with anti-MAG antibodies. Muscle Nerve 2003, 27, 611–615. [Google Scholar] [CrossRef]
- Querol, L.; Rojas-García, R.; Diaz-Manera, J.; Barcena, J.; Pardo, J.; Ortega-Moreno, A.; Sedano, M.J.; Seró-Ballesteros, L.; Carvajal, A.; Ortiz, N.; et al. Rituximab in treatment-resistant CIDP with antibodies against paranodal proteins. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, A.I.; Halstead, S.K.; Goodfellow, J.A.; Chavada, G.; Mallik, A.; Overell, J.; Lunn, M.P.; McConnachie, A.; van Doorn, P.; Willison, H.J. Inhibition of complement in Guillain-Barré syndrome: The ICA-GBS study. J. Peripher. Nerv. Syst. 2017, 22, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Halstead, S.K.; Zitman, F.M.; Humphreys, P.D.; Greenshields, K.; Verschuuren, J.J.; Jacobs, B.C.; Rother, R.P.; Plomp, J.J.; Willison, H.J. Eculizumab prevents anti-ganglioside antibody-mediated neuropathy in a murine model. Brain 2008, 131, 1197–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misawa, S.; Kuwabara, S.; Sato, Y.; Yamaguchi, N.; Nagashima, K.; Katayama, K.; Sekiguchi, Y.; Iwai, Y.; Amino, H.; Suichi, T.; et al. Safety and efficacy of eculizumab in Guillain-Barré syndrome: A multicentre, double-blind, randomised phase 2 trial. Lancet Neurol. 2018, 17, 519–529. [Google Scholar] [CrossRef]
- Doets, A.Y.; Hughes, R.A.; Brassington, R.; Hadden, R.D.; Pritchard, J. Pharmacological treatment other than corticosteroids, intravenous immunoglobulin and plasma exchange for Guillain-Barré syndrome. Cochrane Database Syst. Rev. 2020, 1, CD008630. [Google Scholar] [CrossRef]
- Quan, S.; Kim, H.-J.; Dukala, D.; Sheng, J.R.; Soliven, B. Impaired dendritic cell function in a spontaneous autoimmune polyneuropathy. J. Immunol. 2015, 194, 4175–4184. [Google Scholar] [CrossRef] [Green Version]
- Meyer zu Hörste, G.; Mausberg, A.K.; Müller, J.I.; Lehmann, H.C.; Löber, S.; Gmeiner, P.; Hartung, H.-P.; Stüve, O.; Korth, C.; Kieseier, B.C. Quinpramine ameliorates rat experimental autoimmune neuritis and redistributes MHC class II molecules. PLoS ONE 2011, 6, e21223. [Google Scholar] [CrossRef]
- Hartlehnert, M.; Derksen, A.; Hagenacker, T.; Kindermann, D.; Schäfers, M.; Pawlak, M.; Kieseier, B.C.; Meyer zu Horste, G. Schwann cells promote post-traumatic nerve inflammation and neuropathic pain through MHC class II. Sci. Rep. 2017, 7, 12518. [Google Scholar] [CrossRef]
- Grüter, T.; Blusch, A.; Motte, J.; Sgodzai, M.; Bachir, H.; Klimas, R.; Ambrosius, B.; Gold, R.; Ellrichmann, G.; Pitarokoili, K. Immunomodulatory and anti-oxidative effect of the direct TRPV1 receptor agonist capsaicin on Schwann cells. J. Neuroinflamm. 2020, 17, 145. [Google Scholar] [CrossRef]
- Zimmermann, M.; Li, T.; Semrad, T.J.; Wu, C.-Y.; Yu, A.; Cimino, G.; Malfatti, M.; Haack, K.; Turteltaub, K.W.; Pan, C.-X.; et al. Oxaliplatin–DNA adducts as predictive biomarkers of FOLFOX response in colorectal cancer: A potential treatment optimization strategy. Mol. Cancer Ther. 2020, 19, 1070–1079. [Google Scholar] [CrossRef] [PubMed]
- Tamburin, S.; Park, S.B.; Alberti, P.; Demichelis, C.; Schenone, A.; Argyriou, A.A. Taxane and epothilone-induced peripheral neurotoxicity: From pathogenesis to treatment. J. Peripher. Nerv. Syst. 2019, 24, S40–S51. [Google Scholar] [CrossRef]
- Islam, B.; Lustberg, M.; Staff, N.P.; Kolb, N.; Alberti, P.; Argyriou, A.A. Vinca alkaloids, thalidomide and eribulin-induced peripheral neurotoxicity: From pathogenesis to treatment. J. Peripher. Nerv. Syst. 2019, 24, S63–S73. [Google Scholar] [CrossRef] [PubMed]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Alé, A.; Bruna, J.; Calls, A.; Karamita, M.; Haralambous, S.; Probert, L.; Navarro, X.; Udina, E. Inhibition of the neuronal NFκB pathway attenuates bortezomib-induced neuropathy in a mouse model. NeuroToxicology 2016, 55, 58–64. [Google Scholar] [CrossRef]
- Leo, M.; Schmitt, L.-I.; Küsterarent, P.; Kutritz, A.; Rassaf, T.; Kleinschnitz, C.; Hendgen-Cotta, U.B.; Hagenacker, T. Platinum-based drugs cause mitochondrial dysfunction in cultured dorsal root ganglion neurons. Int. J. Mol. Sci. 2020, 21, 8636. [Google Scholar] [CrossRef]
- Jannuzzi, A.T.; Arslan, S.; Yilmaz, A.M.; Sari, G.; Beklen, H.; Méndez, L.; Fedorova, M.; Arga, K.Y.; Karademir Yilmaz, B.; Alpertunga, B. Higher proteotoxic stress rather than mitochondrial damage is involved in higher neurotoxicity of bortezomib compared to carfilzomib. Redox Biol. 2020, 32, 101502. [Google Scholar] [CrossRef]
- Shim, H.S.; Bae, C.; Wang, J.; Lee, K.-H.; Hankerd, K.M.; Kim, H.K.; Chung, J.M.; La, J.-H. Peripheral and central oxidative stress in chemotherapy-induced neuropathic pain. Mol. Pain 2019, 15, 1744806919840098. [Google Scholar] [CrossRef] [Green Version]
- Imai, S.; Koyanagi, M.; Azimi, Z.; Nakazato, Y.; Matsumoto, M.; Ogihara, T.; Yonezawa, A.; Omura, T.; Nakagawa, S.; Wakatsuki, S.; et al. Taxanes and platinum derivatives impair Schwann cells via distinct mechanisms. Sci. Rep. 2017, 7, 5947. [Google Scholar] [CrossRef] [Green Version]
- Sun, F.; Cui, L.; Li, T.; Chen, S.; Song, J.; Li, D. Oxaliplatin induces immunogenic cells death and enhances therapeutic efficacy of checkpoint inhibitor in a model of murine lung carcinoma. J. Recept. Signal Transduct. 2019, 39, 208–214. [Google Scholar] [CrossRef]
- Wu, P.; Chen, Y. Evodiamine ameliorates paclitaxel-induced neuropathic pain by inhibiting inflammation and maintaining mitochondrial anti-oxidant functions. Hum. Cell 2019, 32, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Hwang, S.-H.; Kim, H.-K.; Abdi, S.; Kim, H.K. Losartan, an angiotensin II type 1 receptor antagonist, alleviates mechanical hyperalgesia in a rat model of chemotherapy-induced neuropathic pain by inhibiting inflammatory cytokines in the dorsal root ganglia. Mol. Neurobiol. 2019, 56, 7408–7419. [Google Scholar] [CrossRef] [PubMed]
- Qin, B.; Luo, N.; Li, Y.; Gong, D.; Zheng, J.; Tan, X.; Zheng, W. Protective effect of gastrodin on peripheral neuropathy induced by anti-tumor treatment with vincristine in rat models. Drug Chem. Toxicol. 2021, 44, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.C.B.; Goldstein, D.; Tavakoli, A.; Trinh, T.; Klisser, J.; Lewis, C.R.; Friedlander, M.; Naduvilath, T.J.; Au, K.; Park, S.B.; et al. Corneal dendritic cells and the subbasal nerve plexus following neurotoxic treatment with oxaliplatin or paclitaxel. Sci. Rep. 2021, 11, 22884. [Google Scholar] [CrossRef] [PubMed]
- Trinh, T.; Park, S.B.; Murray, J.; Pickering, H.; Lin, C.S.; Martin, A.; Friedlander, M.; Kiernan, M.C.; Goldstein, D.; Krishnan, A.V. Neu-horizons: Neuroprotection and therapeutic use of riluzole for the prevention of oxaliplatin-induced neuropathy—A randomised controlled trial. Support. Care Cancer 2021, 29, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Alberti, P.; Canta, A.; Chiorazzi, A.; Fumagalli, G.; Meregalli, C.; Monza, L.; Pozzi, E.; Ballarini, E.; Rodriguez-Menendez, V.; Oggioni, N.; et al. Topiramate prevents oxaliplatin-related axonal hyperexcitability and oxaliplatin induced peripheral neurotoxicity. Neuropharmacology 2020, 164, 107905. [Google Scholar] [CrossRef] [PubMed]
- Makker, P.G.S.; White, D.; Lees, J.G.; Parmar, J.; Goldstein, D.; Park, S.B.; Howells, J.; Moalem-Taylor, G. Acute changes in nerve excitability following oxaliplatin treatment in mice. J. Neurophysiol. 2020, 124, 232–244. [Google Scholar] [CrossRef]
- Li, Y.; North, R.Y.; Rhines, L.D.; Tatsui, C.E.; Rao, G.; Edwards, D.D.; Cassidy, R.M.; Harrison, D.S.; Johansson, C.A.; Zhang, H.; et al. DRG Voltage-gated sodium channel 1.7 is upregulated in paclitaxel-induced neuropathy in rats and in humans with neuropathic pain. J. Neurosci. 2017, 38, 1124–1136. [Google Scholar] [CrossRef]
- Chen, L.; Huang, J.; Benson, C.; Lankford, K.L.; Zhao, P.; Carrara, J.; Tan, A.M.; Kocsis, J.D.; Waxman, S.G.; Dib-Hajj, S.D. Sodium channel Nav1.6 in sensory neurons contributes to vincristine-induced allodynia. Brain 2020, 143, 2421–2436. [Google Scholar] [CrossRef]
- Chukyo, A.; Chiba, T.; Kambe, T.; Yamamoto, K.; Kawakami, K.; Taguchi, K.; Abe, K. Oxaliplatin-induced changes in expression of transient receptor potential channels in the dorsal root ganglion as a neuropathic mechanism for cold hypersensitivity. Neuropeptides 2018, 67, 95–101. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, F.; Zhang, S.; Mao, M.; Feng, S.; Wang, X. Participation of transient receptor potential vanilloid 1 in the analgesic effect of duloxetine for paclitaxel induced peripheral neuropathic pain. Neurosci. Lett. 2022, 773, 136512. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Oka, Y.; Sashida, H.; Kanbe, T.; Abe, K.; Utsunomiya, I.; Taguchi, K. Vincristine-induced peripheral neuropathic pain and expression of transient receptor potential vanilloid 1 in rat. J. Pharmacol. Sci. 2017, 133, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Loprinzi, C.L.; Lacchetti, C.; Bleeker, J.; Cavaletti, G.; Chauhan, C.; Hertz, D.L.; Kelley, M.R.; Lavino, A.; Lustberg, M.B.; Paice, J.A.; et al. Prevention and management of chemotherapy-induced peripheral neuropathy in survivors of adult cancers: ASCO guideline update. J. Clin. Oncol. 2020, 38, 3325–3348. [Google Scholar] [CrossRef] [PubMed]
- Jordan, B.; Margulies, A.; Cardoso, F.; Cavaletti, G.; Haugnes, H.S.; Jahn, P.; Le Rhun, E.; Preusser, M.; Scotté, F.; Taphoorn, M.J.B.; et al. Systemic anticancer therapy-induced peripheral and central neurotoxicity: ESMO-EONS-EANO clinical practice guidelines for diagnosis, prevention, treatment and follow-up. Ann. Oncol. 2020, 31, 1306–1319. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Zhang, Q.; Yang, C.; Xiao, L.; Xue, Z.; Zhu, J. Duloxetine, a balanced serotonin-norepinephrine reuptake inhibitor, improves painful chemotherapy-induced peripheral neuropathy by inhibiting activation of p38 MAPK and NF-κB. Front. Pharmacol. 2019, 10, 365. [Google Scholar] [CrossRef]
- Salehifar, E.; Janbabaei, G.; Hendouei, N.; Alipour, A.; Tabrizi, N.; Avan, R. Comparison of the efficacy and safety of pregabalin and duloxetine in taxane-induced sensory neuropathy: A randomized controlled trial. Clin. Drug Investig. 2020, 40, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Velasco, R.; Besora, S.; Argyriou, A.A.; Santos, C.; Sala, R.; Izquierdo, C.; Simó, M.; Gil-Gil, M.; Pardo, B.; Jiménez, L.; et al. Duloxetine against symptomatic chemotherapy-induced peripheral neurotoxicity in cancer survivors: A real world, open-label experience. Anti-Cancer Drugs 2021, 32, 88–94. [Google Scholar] [CrossRef]
- Kim, B.-S.; Jin, J.-Y.; Kwon, J.H.; Woo, I.S.; Ko, Y.H.; Park, S.-Y.; Park, H.-J.; Kang, J.H. Efficacy and safety of oxycodone/naloxone as add-on therapy to gabapentin or pregabalin for the management of chemotherapy-induced peripheral neuropathy in Korea. Asia-Pac. J. Clin. Oncol. 2018, 14, e448–e454. [Google Scholar] [CrossRef]
- Naruge, D.; Nagashima, F.; Kawai, K.; Okano, N.; Kobayashi, T.; Furuse, J. Tramadol/acetaminophen combination tablets in cancer patients with chemotherapy-induced peripheral neuropathy: A single-arm phase II study. Palliat. Med. Rep. 2020, 1, 25–31. [Google Scholar] [CrossRef]
- Song, S.Y.; Ko, Y.B.; Kim, H.; Lee, G.W.; Yang, J.B.; Chang, H.K.; Kwak, S.M.; Jung, J.; Lee, S.; Lee, S.Y.; et al. Effect of serotonin-norepinephrine reuptake inhibitors for patients with chemotherapy-induced painful peripheral neuropathy: A meta-analysis. Medicine 2020, 99, e18653. [Google Scholar] [CrossRef]
- de Andrade, D.C.; Jacobsen Teixeira, M.; Galhardoni, R.; Ferreira, K.S.L.; Braz Mileno, P.; Scisci, N.; Zandonai, A.; Teixeira, W.G.J.; Saragiotto, D.F.; Silva, V.; et al. Pregabalin for the prevention of oxaliplatin-induced painful neuropathy: A randomized, double-blind trial. Oncologist 2017, 22, 1154-e105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hincker, A.; Frey, K.; Rao, L.; Wagner-Johnston, N.; Ben Abdallah, A.; Tan, B.; Amin, M.; Wildes, T.; Shah, R.; Karlsson, P.; et al. Somatosensory predictors of response to pregabalin in painful chemotherapy-induced peripheral neuropathy: A randomized, placebo-controlled, crossover study. Pain 2019, 160, 1835–1846. [Google Scholar] [CrossRef] [PubMed]
- El-Fatatry, B.M.; Ibrahim, O.M.; Hussien, F.Z.; Mostafa, T.M. Role of metformin in oxaliplatin-induced peripheral neuropathy in patients with stage III colorectal cancer: Randomized, controlled study. Int. J. Color. Dis. 2018, 33, 1675–1683. [Google Scholar] [CrossRef] [PubMed]
- Balko, R.; Hurley, R.; Jatoi, A. Poly (ADP-ribose) polymerase inhibition for chemotherapy-induced peripheral neuropathy: A meta-analysis of placebo-controlled trials. J. Palliat. Med. 2019, 22, 977–980. [Google Scholar] [CrossRef]
- Kuriyama, A.; Endo, K. Goshajinkigan for prevention of chemotherapy-induced peripheral neuropathy: A systematic review and meta-analysis. Support. Care Cancer 2018, 26, 1051–1059. [Google Scholar] [CrossRef]
- Miao, H.; Li, R.; Chen, D.; Hu, J.; Chen, Y.; Xu, C.; Wen, Z. Protective effects of vitamin E on chemotherapy-induced peripheral neuropathy: A meta-analysis of randomized controlled trials. Ann. Nutr. Metab. 2021, 77, 127–137. [Google Scholar] [CrossRef]
- Wu, S.; Bai, X.; Guo, C.; Huang, Z.; Ouyang, H.; Huang, J.; Zeng, W. Ganglioside-monosialic acid (GM1) for prevention of chemotherapy-induced peripheral neuropathy: A meta-analysis with trial sequential analysis. BMC Cancer 2021, 21, 1173. [Google Scholar] [CrossRef]
- Jordan, B.; Jahn, F.; Beckmann, J.; Unverzagt, S.; Müller-Tidow, C.; Jordan, K. Calcium and magnesium infusions for the prevention of oxaliplatin-induced peripheral neurotoxicity: A systematic review. Oncology 2016, 90, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Hershman, D.L.; Unger, J.M.; Crew, K.D.; Till, C.; Greenlee, H.; Minasian, L.M.; Moinpour, C.M.; Lew, D.L.; Fehrenbacher, L.; Wade, J.L., 3rd; et al. Two-year trends of taxane-induced neuropathy in women enrolled in a randomized trial of acetyl-L-carnitine (SWOG S0715). J. Natl. Cancer Inst. 2018, 110, 669–676. [Google Scholar] [CrossRef]
- Chen, J.; Kang, D.; Xu, J.; Lake, M.; Hogan, J.O.; Sun, C.; Walter, K.; Yao, B.; Kim, D. Species differences and molecular determinant of TRPA1 cold sensitivity. Nat. Commun. 2013, 4, 2501. [Google Scholar] [CrossRef]
- Gadgil, S.; Ergün, M.; van den Heuvel, S.A.; van der Wal, S.E.; Scheffer, G.J.; Hooijmans, C.R. A systematic summary and comparison of animal models for chemotherapy induced (peripheral) neuropathy (CIPN). PLoS ONE 2019, 14, e0221787. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S. Chemotherapy-induced peripheral neuropathy and rehabilitation: A review. Semin. Oncol. 2021, 48, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Cavaletti, G.; Marmiroli, P. Pharmacotherapy options for managing chemotherapy-induced peripheral neurotoxicity. Expert Opin. Pharmacother. 2018, 19, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.-Y.; Mi, W.-L.; Wu, G.-C.; Wang, Y.-Q.; Mao-Ying, Q.-L. Prevention and treatment for chemotherapy-induced peripheral neuropathy: Therapies based on CIPN mechanisms. Curr. Neuropharmacol. 2019, 17, 184–196. [Google Scholar] [CrossRef]
- Leen, A.J.; Yap, D.W.T.; Teo, C.B.; Tan, B.K.J.; Molassiotis, A.; Ishiguro, H.; Fan, S.W.X.; Sundar, R.; Soon, Y.Y.; Bandla, A. A Systematic review and meta-analysis of the effectiveness of neuroprotectants for paclitaxel-induced peripheral neuropathy. Front. Oncol. 2021, 11, 763229. [Google Scholar] [CrossRef]
- Szklener, K.; Szklener, S.; Michalski, A.; Żak, K.; Kuryło, W.; Rejdak, K.; Mańdziuk, S. Dietary supplements in chemotherapy-induced peripheral neuropathy: A new hope? Nutrients 2022, 14, 625. [Google Scholar] [CrossRef]
- Hopkins, E.L.; Gu, W.; Kobe, B.; Coleman, M.P. A novel NAD signaling mechanism in axon degeneration and its relationship to innate immunity. Front. Mol. Biosci. 2021, 8, 703532. [Google Scholar] [CrossRef]
- Coleman, M.P.; Höke, A. Programmed axon degeneration: From mouse to mechanism to medicine. Nat. Rev. Neurosci. 2020, 21, 183–196. [Google Scholar] [CrossRef]
- Tian, W.; Czopka, T.; López-Schier, H. Systemic loss of Sarm1 protects Schwann cells from chemotoxicity by delaying axon degeneration. Commun. Biol. 2020, 3, 49. [Google Scholar] [CrossRef] [Green Version]
- Gilley, J.; Mayer, P.R.; Yu, G.; Coleman, M.P. Low levels of NMNAT2 compromise axon development and survival. Hum. Mol. Genet. 2019, 28, 448–458. [Google Scholar] [CrossRef]
- Li, Y.; Pazyra-Murphy, M.F.; Avizonis, D.; de Sá Tavares Russo, M.; Tang, S.; Chen, C.-Y.; Hsueh, Y.-P.; Bergholz, J.S.; Jiang, T.; Zhao, J.J.; et al. Sarm1 activation produces cADPR to increase intra-axonal Ca++ and promote axon degeneration in PIPN. J. Cell Biol. 2022, 221, e202106080. [Google Scholar] [CrossRef] [PubMed]
- Gould, S.A.; White, M.; Wilbrey, A.L.; Pór, E.; Coleman, M.P.; Adalbert, R. Protection against oxaliplatin-induced mechanical and thermal hypersensitivity in Sarm1−/− mice. Exp. Neurol. 2021, 338, 113607. [Google Scholar] [CrossRef] [PubMed]
- Bosanac, T.; Hughes, R.O.; Engber, T.; Devraj, R.; Brearley, A.; Danker, K.; Young, K.; Kopatz, J.; Hermann, M.; Berthemy, A.; et al. Pharmacological SARM1 inhibition protects axon structure and function in paclitaxel-induced peripheral neuropathy. Brain 2021, 144, 3226–3238. [Google Scholar] [CrossRef] [PubMed]
- Turkiew, E.; Falconer, D.; Reed, N.; Höke, A. Deletion of Sarm1 gene is neuroprotective in two models of peripheral neuropathy. J. Peripher. Nerv. Syst. 2017, 22, 162–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-H.; Sasaki, Y.; DiAntonio, A.; Milbrandt, J. SARM1 is required in human derived sensory neurons for injury-induced and neurotoxic axon degeneration. Exp. Neurol. 2021, 339, 113636. [Google Scholar] [CrossRef]
- Cheng, Y.; Liu, J.; Luan, Y.; Liu, Z.; Lai, H.; Zhong, W.; Yang, Y.; Yu, H.; Feng, N.; Wang, H.; et al. Sarm1 gene deficiency attenuates diabetic peripheral neuropathy in mice. Diabetes 2019, 68, 2120–2130. [Google Scholar] [CrossRef] [Green Version]
- Lococo, P.M.; Risinger, A.L.; Smith, H.R.; Chavera, T.S.; Berg, K.A.; Clarke, W.P. Pharmacological augmentation of nicotinamide phosphoribosyltransferase (NAMPT) protects against paclitaxel-induced peripheral neuropathy. eLife 2017, 6, e29626. [Google Scholar] [CrossRef]
- Brazill, J.M.; Cruz, B.; Zhu, Y.; Zhai, R.G. Nmnat mitigates sensory dysfunction in a Drosophila model of paclitaxel-induced peripheral neuropathy. Dis. Model. Mech. 2018, 11, dmm032938. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.-W.; Smith, C.B.; Schmidt, M.S.; Cambronne, X.A.; Cohen, M.S.; Migaud, M.E.; Brenner, C.; Goodman, R.H. Pharmacological bypass of NAD+ salvage pathway protects neurons from chemotherapy-induced degeneration. Proc. Natl. Acad. Sci. USA 2018, 115, 10654–10659. [Google Scholar] [CrossRef] [Green Version]
- Hamity, M.V.; Kolker, S.J.; Hegarty, D.M.; Blum, C.; Langmack, L.; Aicher, S.A.; Hammond, D.L. Nicotinamide riboside alleviates corneal and somatic hypersensitivity induced by paclitaxel in male rats. Investig. Opthalmol. Vis. Sci. 2022, 63, 38. [Google Scholar] [CrossRef]
- Zou, S.; Kumar, U. Cannabinoid receptors and the endocannabinoid system: Signaling and function in the central nervous system. Int. J. Mol. Sci. 2018, 19, 833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zogopoulos, P.; Vasileiou, I.; Patsouris, E.; Theocharis, S.E. The role of endocannabinoids in pain modulation. Fundam. Clin. Pharmacol. 2013, 27, 64–80. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Okine, B.N.; Finn, D.P.; Masocha, W. Peripheral deficiency and antiallodynic effects of 2-arachidonoyl glycerol in a mouse model of paclitaxel-induced neuropathic pain. Biomed. Pharmacother. 2020, 129, 110456. [Google Scholar] [CrossRef]
- Khasabova, I.A.; Khasabov, S.; Paz, J.; Harding-Rose, C.; Simone, D.A.; Seybold, V.S. Cannabinoid type-1 receptor reduces pain and neurotoxicity produced by chemotherapy. J. Neurosci. 2012, 32, 7091–7101. [Google Scholar] [CrossRef] [PubMed]
- Curry, Z.A.; Wilkerson, J.L.; Bagdas, D.; Kyte, S.L.; Patel, N.; Donvito, G.; Mustafa, M.A.; Poklis, J.L.; Niphakis, M.J.; Hsu, K.-L.; et al. Monoacylglycerol lipase inhibitors reverse paclitaxel-induced nociceptive behavior and proinflammatory markers in a mouse model of chemotherapy-induced neuropathy. J. Pharmacol. Exp. Ther. 2018, 366, 169–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slivicki, R.A.; Saberi, S.A.; Iyer, V.; Vemuri, V.K.; Makriyannis, A.; Hohmann, A.G. Brain-permeant and -impermeant inhibitors of fatty acid amide hydrolase synergize with the opioid analgesic morphine to suppress chemotherapy-induced neuropathic nociception without enhancing effects of morphine on gastrointestinal transit. J. Pharmacol. Exp. Ther. 2018, 367, 551–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhuniya, D.; Kharul, R.K.; Hajare, A.; Shaikh, N.; Bhosale, S.; Balwe, S.; Begum, F.; De, S.; Athavankar, S.; Joshi, D.; et al. Discovery and evaluation of novel FAAH inhibitors in neuropathic pain model. Bioorg. Med. Chem. Lett. 2019, 29, 238–243. [Google Scholar] [CrossRef]
- Sierra, S.; Gupta, A.; Gomes, I.; Fowkes, M.; Ram, A.; Bobeck, E.N.; Devi, L.A. Targeting cannabinoid 1 and delta opioid receptor heteromers alleviates chemotherapy-induced neuropathic pain. ACS Pharmacol. Transl. Sci. 2019, 2, 219–229. [Google Scholar] [CrossRef]
- Segat, G.C.; Manjavachi, M.N.; Matias, D.O.; Passos, G.F.; Freitas, C.S.; Costa, R.; Calixto, J.B. Antiallodynic effect of β-caryophyllene on paclitaxel-induced peripheral neuropathy in mice. Neuropharmacology 2017, 125, 207–219. [Google Scholar] [CrossRef]
- Iyer, V.; Slivicki, R.A.; Thomaz, A.C.; Crystal, J.D.; Mackie, K.; Hohmann, A.G. The cannabinoid CB2 receptor agonist LY2828360 synergizes with morphine to suppress neuropathic nociception and attenuates morphine reward and physical dependence. Eur. J. Pharmacol. 2020, 886, 173544. [Google Scholar] [CrossRef]
- Li, A.-L.; Lin, X.; Dhopeshwarkar, A.S.; Thomaz, A.C.; Carey, L.M.; Liu, Y.; Nikas, S.P.; Makriyannis, A.; Mackie, K.; Hohmann, A.G. Cannabinoid CB2 agonist AM1710 differentially suppresses distinct pathological pain states and attenuates morphine tolerance and withdrawal. Mol. Pharmacol. 2019, 95, 155–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Hocevar, M.; Bie, B.; Foss, J.F.; Naguib, M. Cannabinoid type 2 receptor system modulates paclitaxel-induced microglial dysregulation and central sensitization in rats. J. Pain 2019, 20, 501–514. [Google Scholar] [CrossRef] [PubMed]
- King, K.M.; Myers, A.M.; Soroka-Monzo, A.J.; Tuma, R.F.; Tallarida, R.J.; Walker, E.A.; Ward, S.J. Single and combined effects of Δ9-tetrahydrocannabinol and cannabidiol in a mouse model of chemotherapy-induced neuropathic pain. Br. J. Pharmacol. 2017, 174, 2832–2841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, S.J.; McAllister, S.D.; Kawamura, R.; Murase, R.; Neelakantan, H.; Walker, E.A. Cannabidiol inhibits paclitaxel-induced neuropathic pain through 5-HT(1A) receptors without diminishing nervous system function or chemotherapy efficacy. Br. J. Pharmacol. 2014, 171, 636–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenneman, D.E.; Kinney, W.A.; Ward, S.J. Knockdown siRNA Targeting the mitochondrial sodium-calcium exchanger-1 inhibits the protective effects of two cannabinoids against acute paclitaxel toxicity. J. Mol. Neurosci. 2019, 68, 603–619. [Google Scholar] [CrossRef]
- Lynch, M.E.; Cesar-Rittenberg, P.; Hohmann, A.G. A double-blind, placebo-controlled, crossover pilot trial with extension using an oral mucosal cannabinoid extract for treatment of chemotherapy-induced neuropathic pain. J. Pain Symptom Manag. 2014, 47, 166–173. [Google Scholar] [CrossRef]
- Waissengrin, B.; Mirelman, D.; Pelles, S.; Bukstein, F.; Blumenthal, D.T.; Wolf, I.; Geva, R. Effect of cannabis on oxaliplatin-induced peripheral neuropathy among oncology patients: A retrospective analysis. Ther. Adv. Med. Oncol. 2021, 13, 1758835921990203. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, C.; Qin, Y.; Cepparulo, P.; Millman, M.; Chopp, M.; Kemper, A.; Szalad, A.; Lu, X.; Wang, L.; et al. Small extracellular vesicles ameliorate peripheral neuropathy and enhance chemotherapy of oxaliplatin on ovarian cancer. J. Extracell. Vesicles 2021, 10, e12073. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Disease | Route/Dose | Half-Life | Adverse Effect |
|---|---|---|---|---|
| Immunoglobulin | GBS CIDP MMN | Intravenous, 0.4 g/kg daily for 5 days or subcutaneous | 25 days | hypersensitivity reactions, thromboembolism, aseptic meningitis and haemolytic anaemia |
| Plasma exchange | GBS CIDP MMN | Intravenous 4–5 exchanges on alternate days | 1–2 months | Citrate-related (hypocalcaemia or metabolic alkalosis) or cardiovascular instability |
| Corticosteroids | CIDP | Intravenous 1 g daily for 3–5 days Oral 60 mg daily, tapered over 3 months | 2–4 h | Cataracts, hyperglycaemia, weight gain, fluid retention peptic ulcer disease, osteoporosis, avascular necrosis of the femoral head and opportunistic infection |
| Cyclophosphamide | CIDP MMN | Intravenous 40–50 mg/kg | 6–8 h | Infertility, teratogenicity, malignancy and haemorrhagic cystitis |
| Rituximab | CIDP MMN | Intravenous 1 g each 2 weeks apart | 20.8 days | Neutropenia, leucopenia, opportunistic infection, nausea, rash, fever |
| Azathioprine | CIDP | Oral, 1–2 mg/kg/day | 5 h | Lymphoproliferative disorders, skin cancers, hepatotoxicity, GI discomfort |
| Methotrexate | CIDP | Oral, 7.5–20 mg weekly | 6–7 h | Teratogenic, pulmonary fibrosis, hepatotoxicity, GI discomfort |
| Mycophenolate Mofetil | CIDP | Oral, 1–3 g/day | 18 h | Bone marrow suppression, hepatotoxicity, GI upset, infertility |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiang, J.C.B.; Arnold, R.; Dhanapalaratnam, R.; Markoulli, M.; Krishnan, A.V. Current and Emerging Pharmacotherapeutic Interventions for the Treatment of Peripheral Nerve Disorders. Pharmaceuticals 2022, 15, 607. https://doi.org/10.3390/ph15050607
Chiang JCB, Arnold R, Dhanapalaratnam R, Markoulli M, Krishnan AV. Current and Emerging Pharmacotherapeutic Interventions for the Treatment of Peripheral Nerve Disorders. Pharmaceuticals. 2022; 15(5):607. https://doi.org/10.3390/ph15050607
Chicago/Turabian StyleChiang, Jeremy Chung Bo, Ria Arnold, Roshan Dhanapalaratnam, Maria Markoulli, and Arun V. Krishnan. 2022. "Current and Emerging Pharmacotherapeutic Interventions for the Treatment of Peripheral Nerve Disorders" Pharmaceuticals 15, no. 5: 607. https://doi.org/10.3390/ph15050607
APA StyleChiang, J. C. B., Arnold, R., Dhanapalaratnam, R., Markoulli, M., & Krishnan, A. V. (2022). Current and Emerging Pharmacotherapeutic Interventions for the Treatment of Peripheral Nerve Disorders. Pharmaceuticals, 15(5), 607. https://doi.org/10.3390/ph15050607