
Novel Au Carbene Complexes as Promising Multi-Target Agents in Breast Cancer Treatment

 ,
, 

 ,
,  , ,
, ,  ,
,  and
and 
Abstract
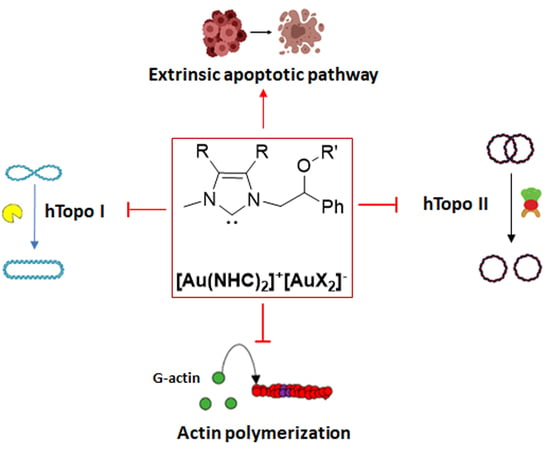
1. Introduction
2. Results
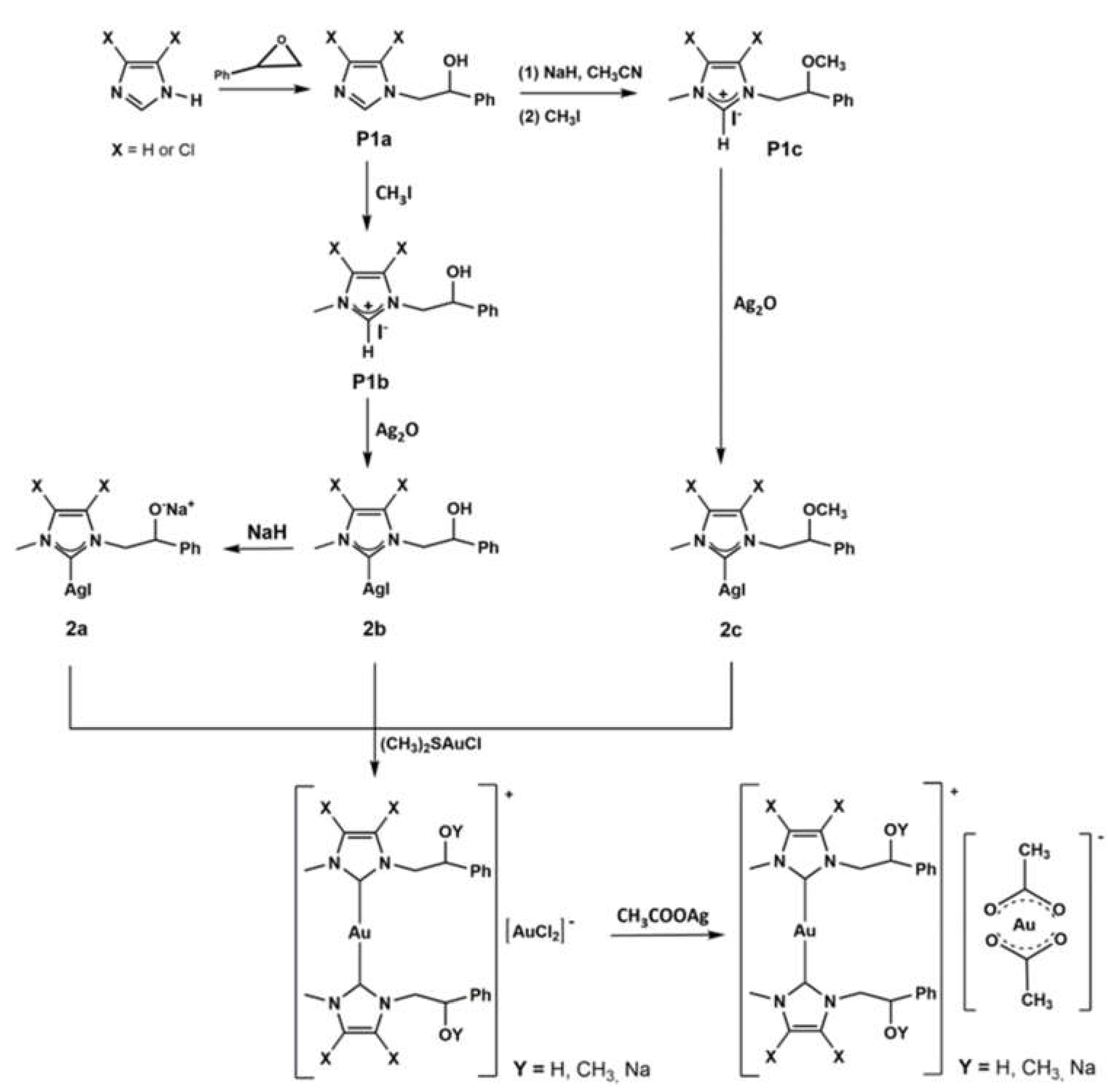
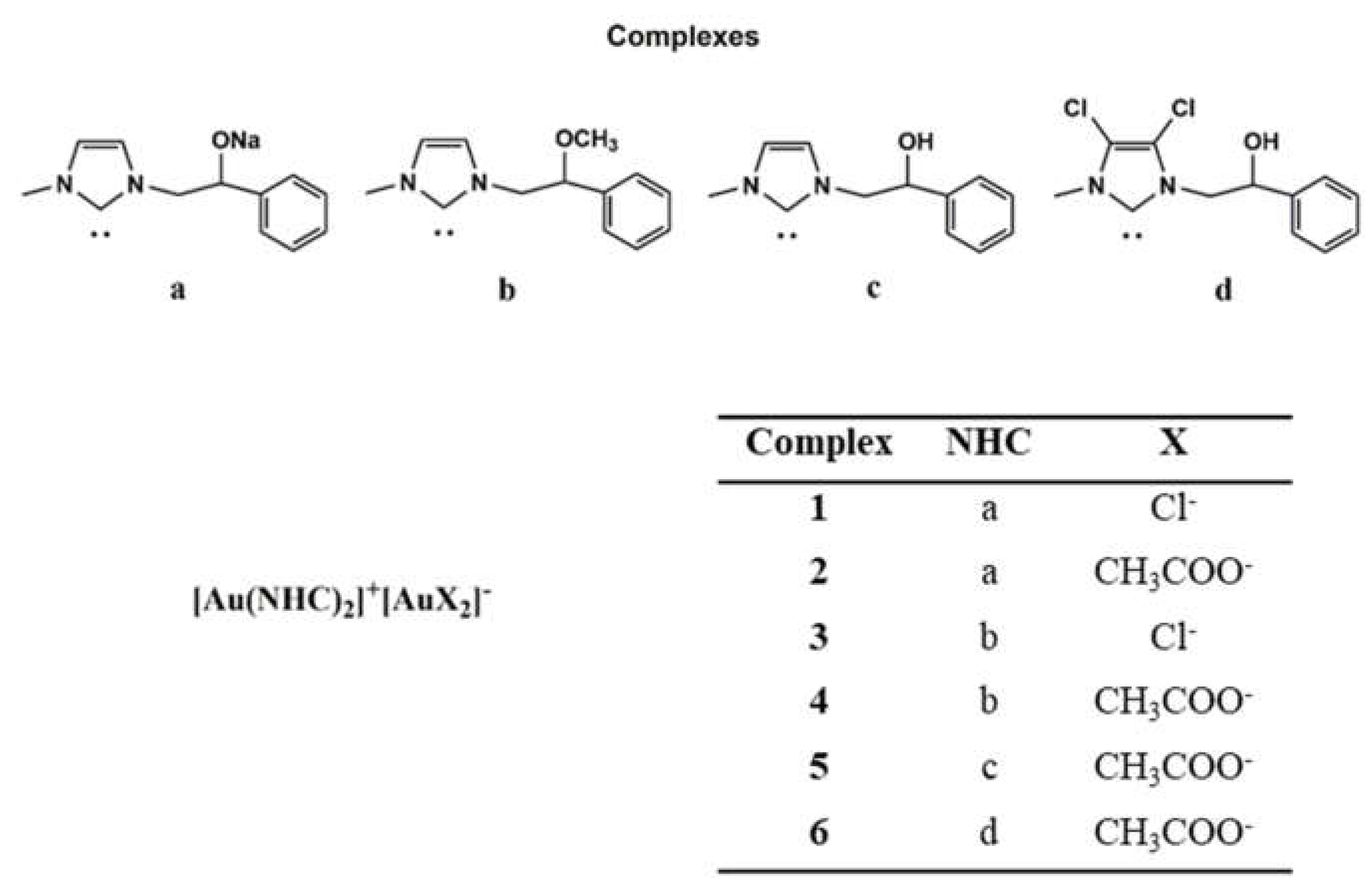
2.1. Chemistry
2.2. Biology
2.2.1. Anticancer Activity
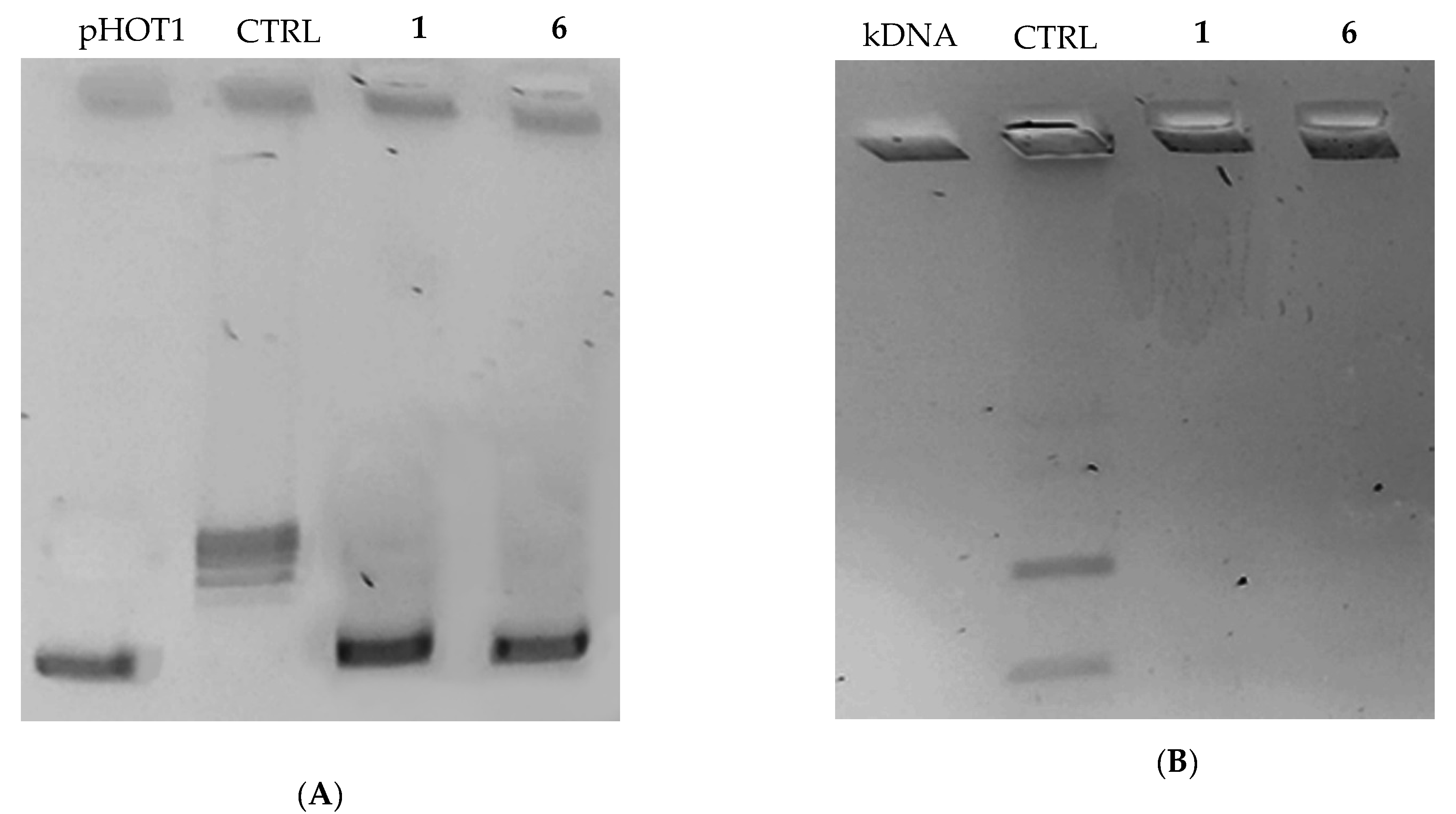
2.2.2. Compounds 1 and 6 Inhibit Both the Human Topoisomerases I and II
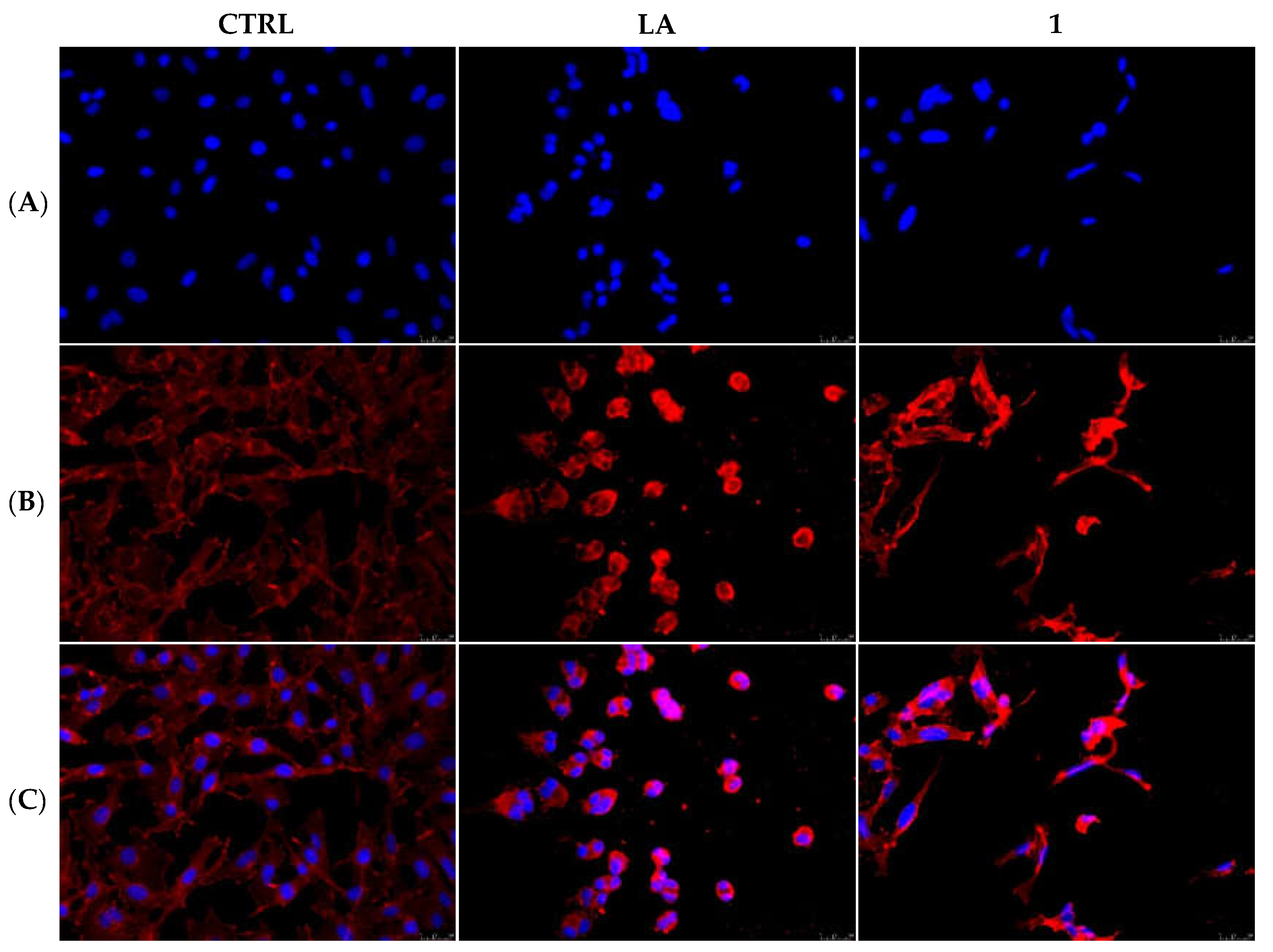
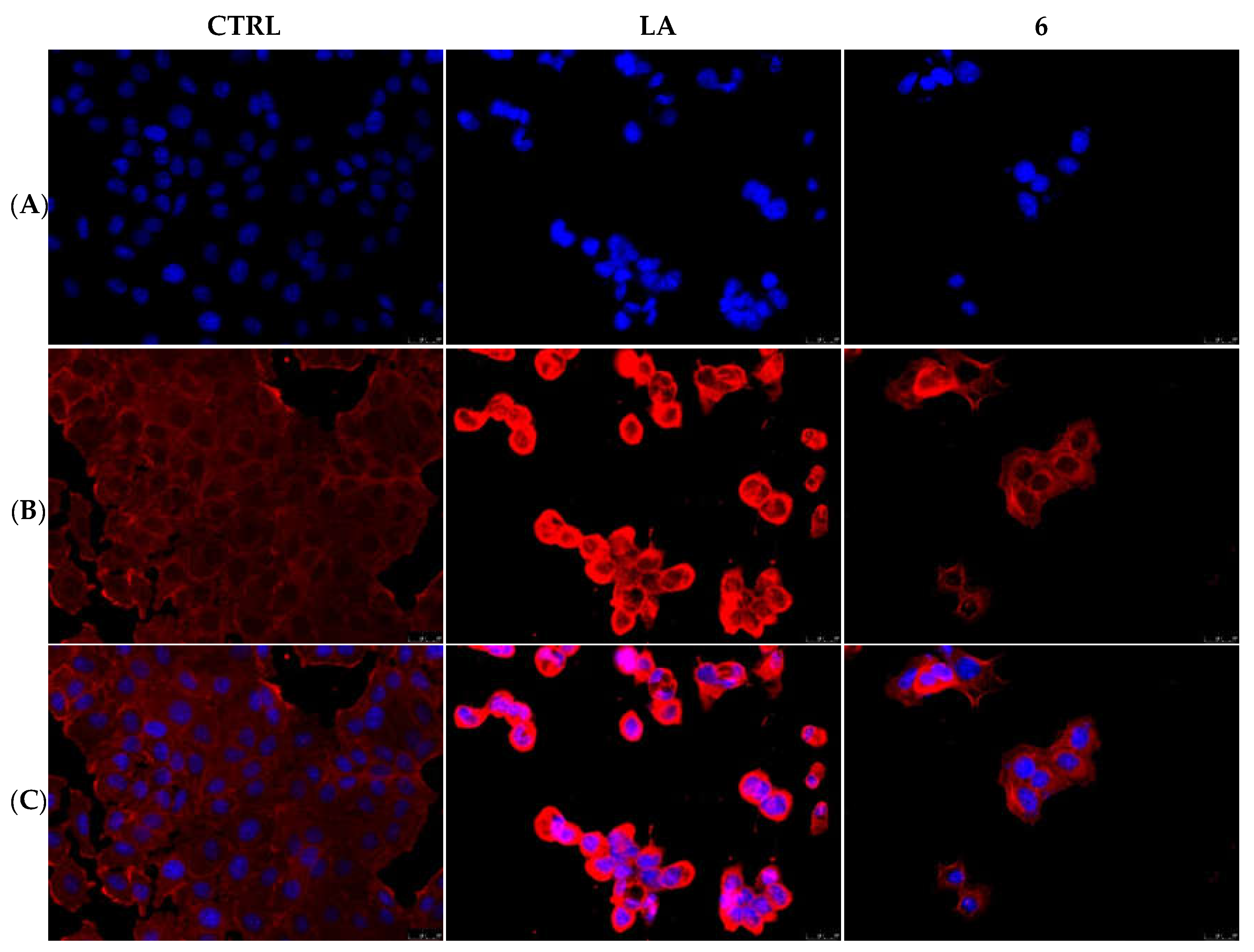
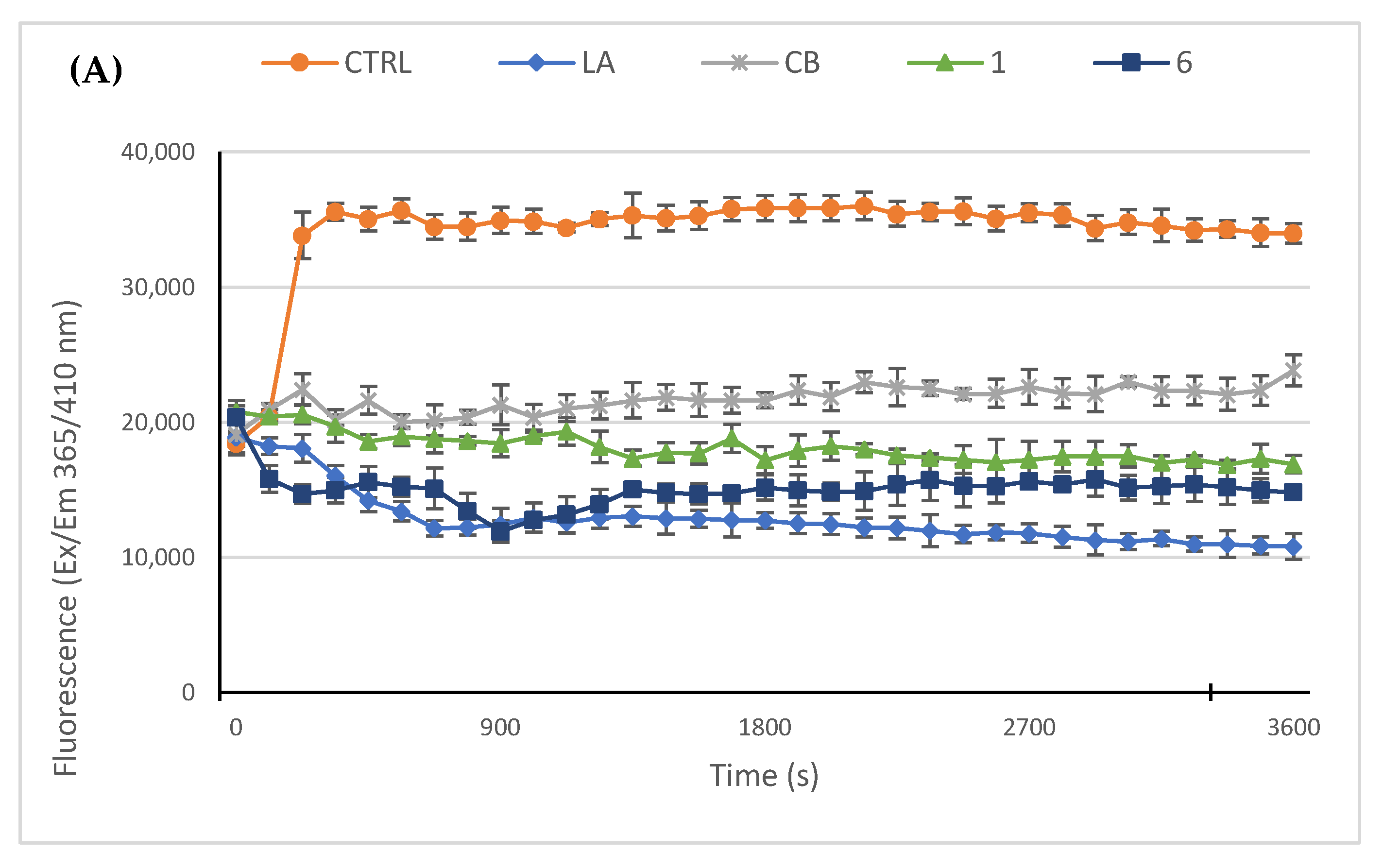
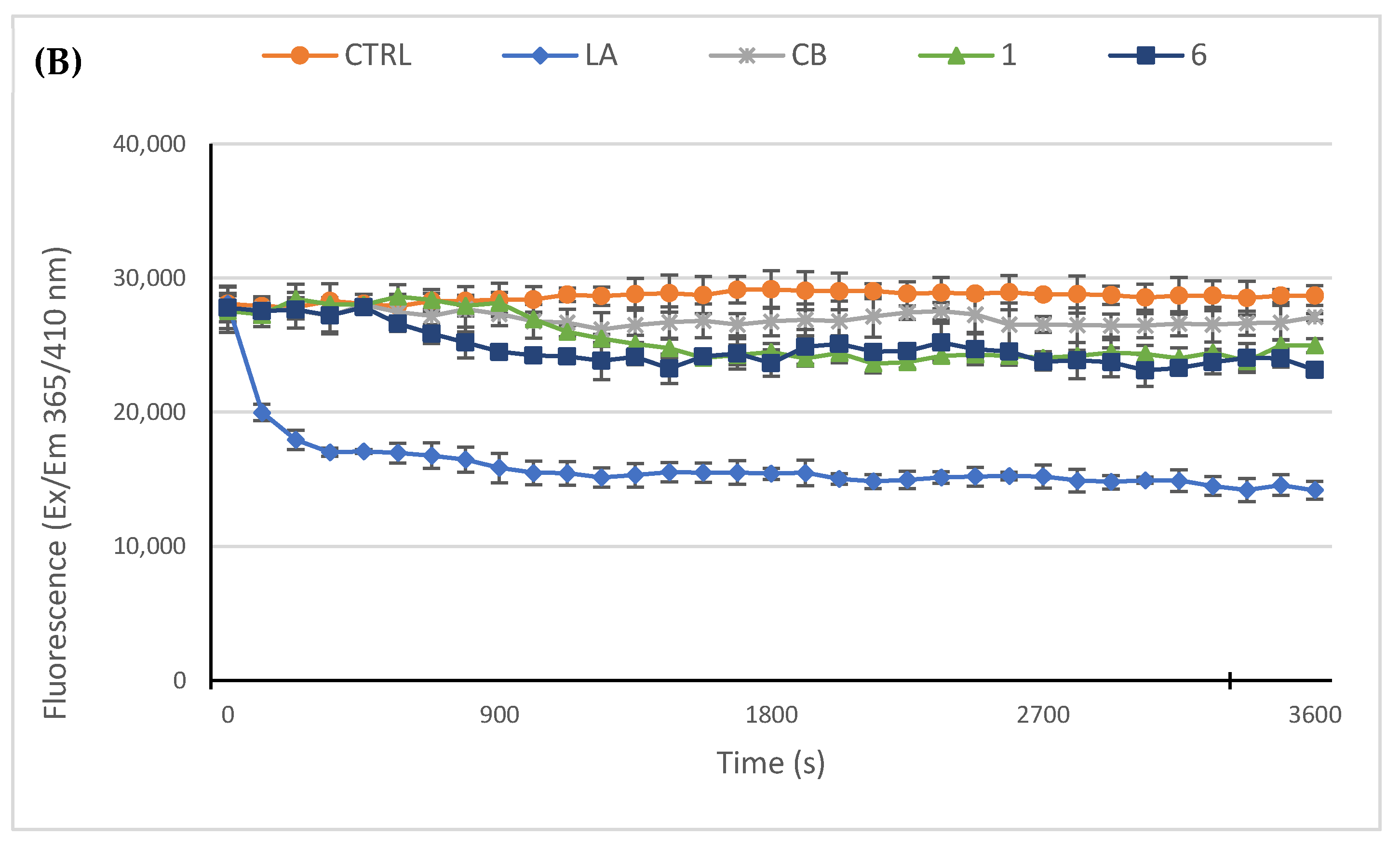
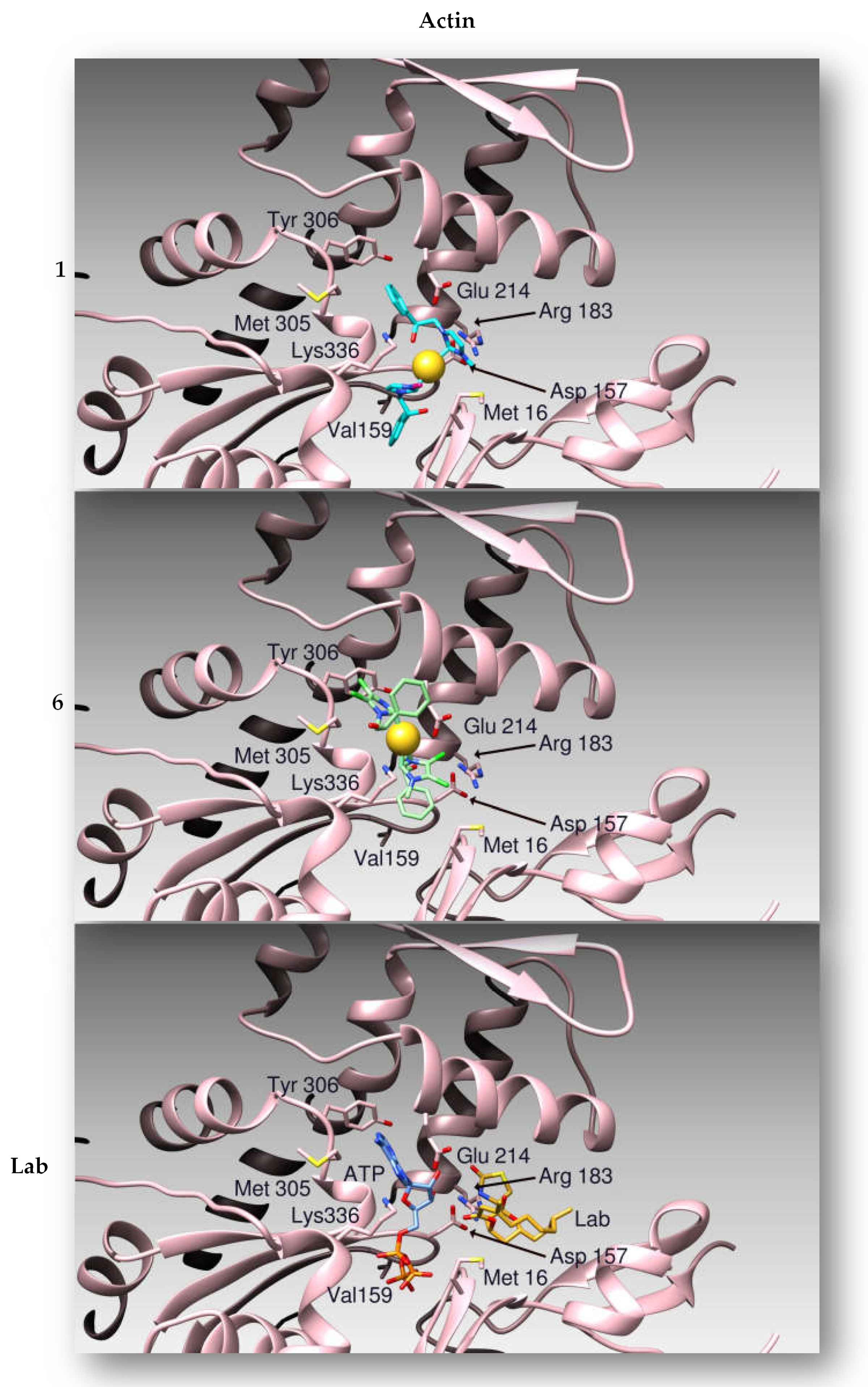
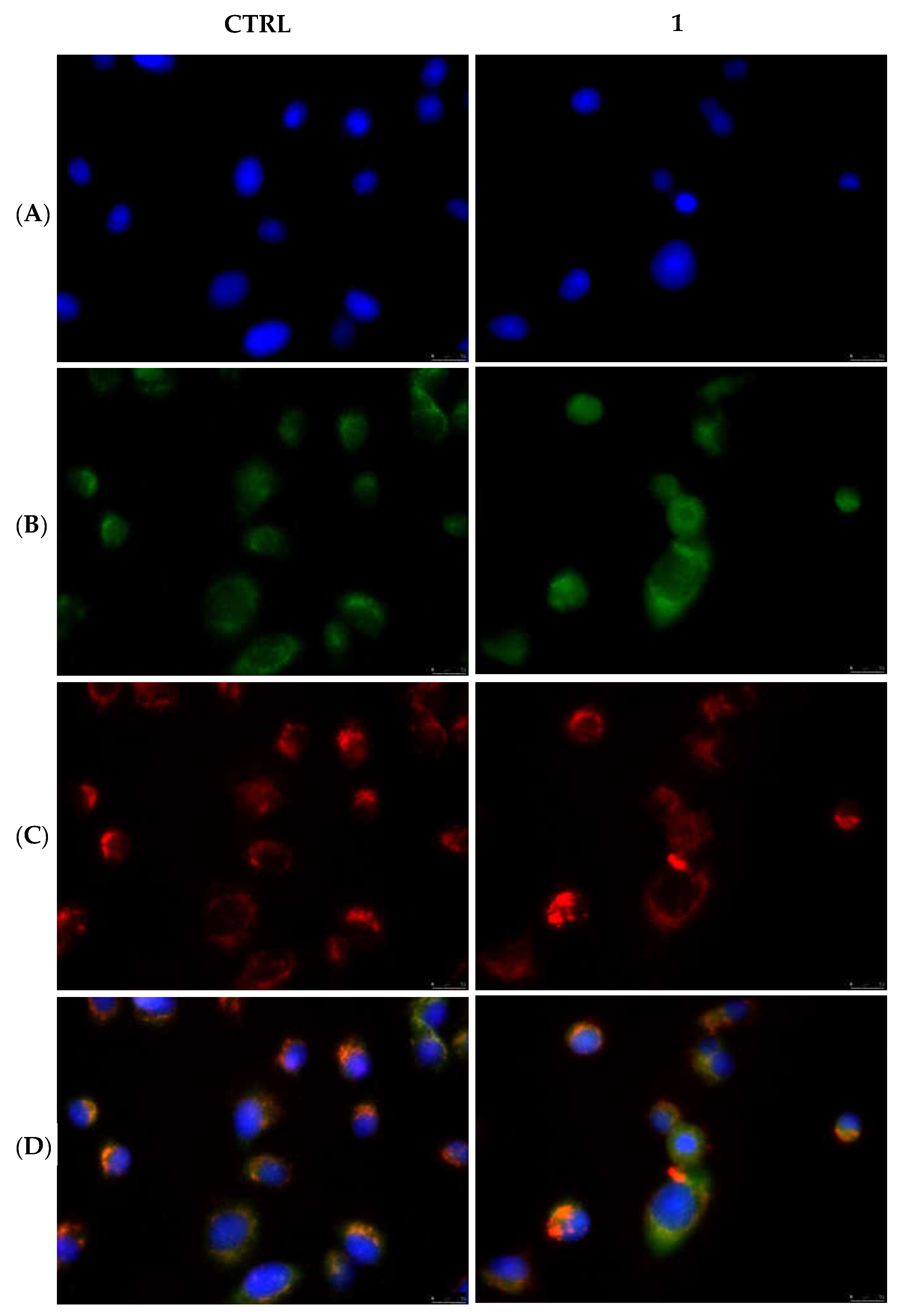
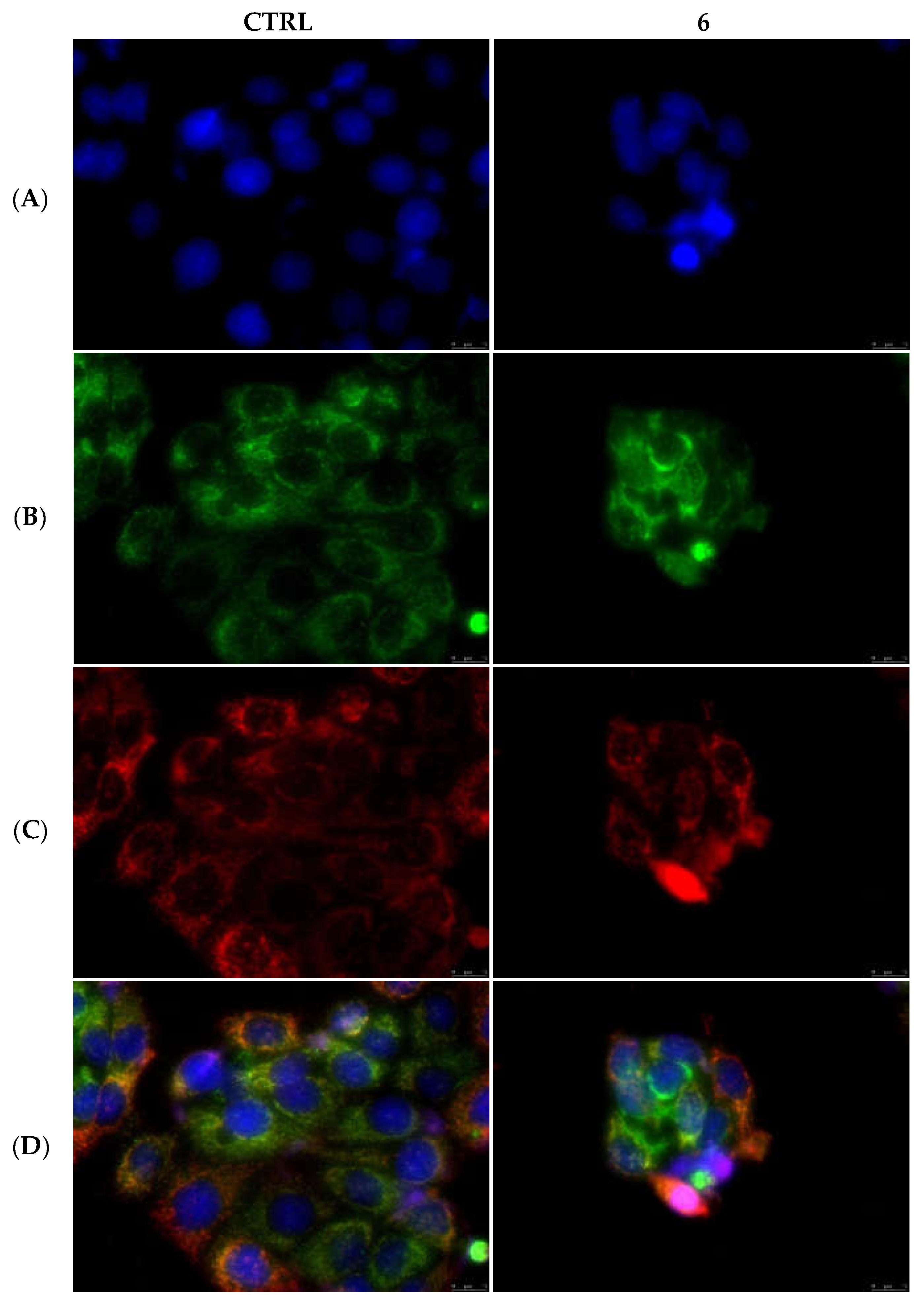
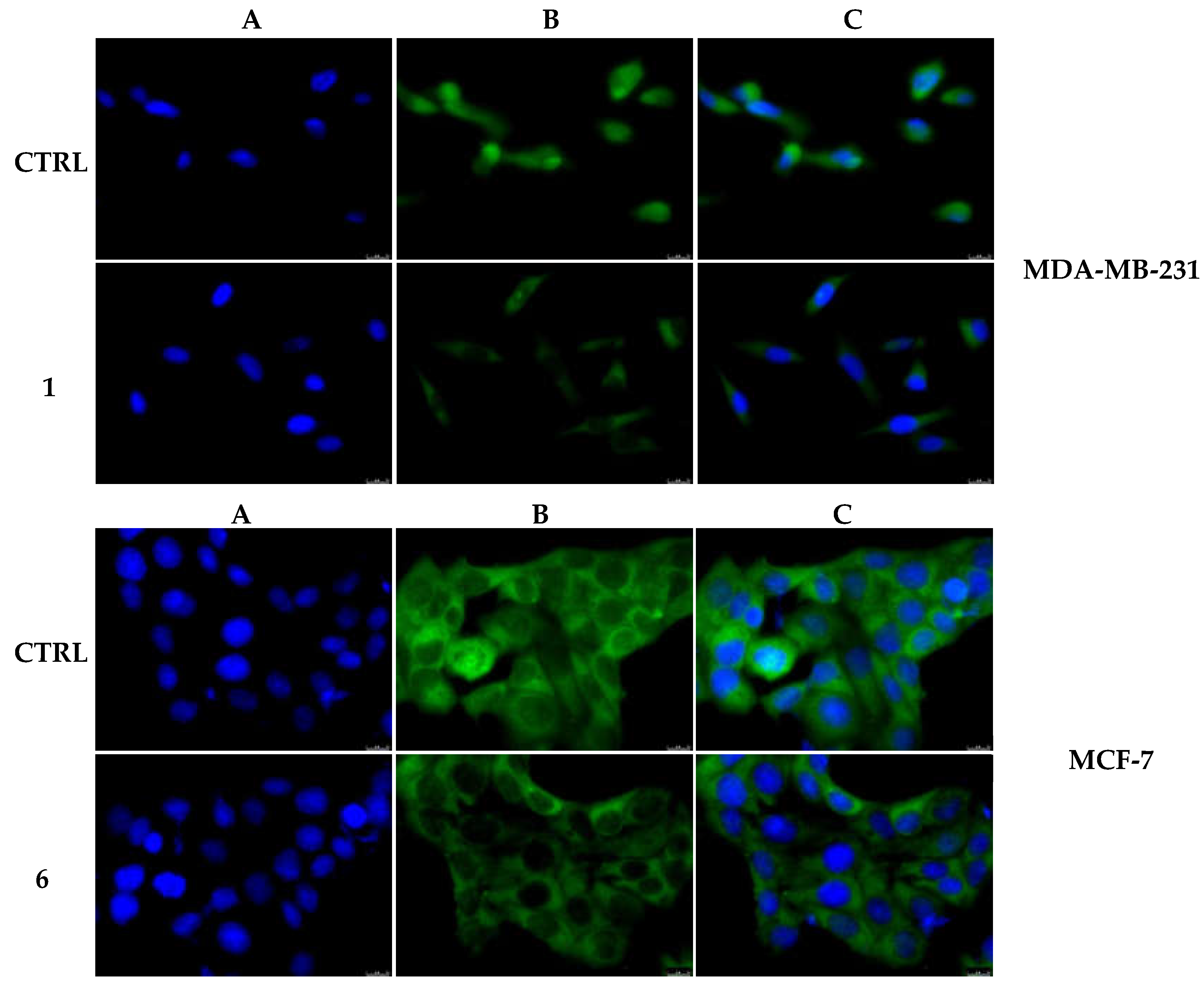
2.2.3. Compounds 1 and 6 Inhibit the Actin Polymerization
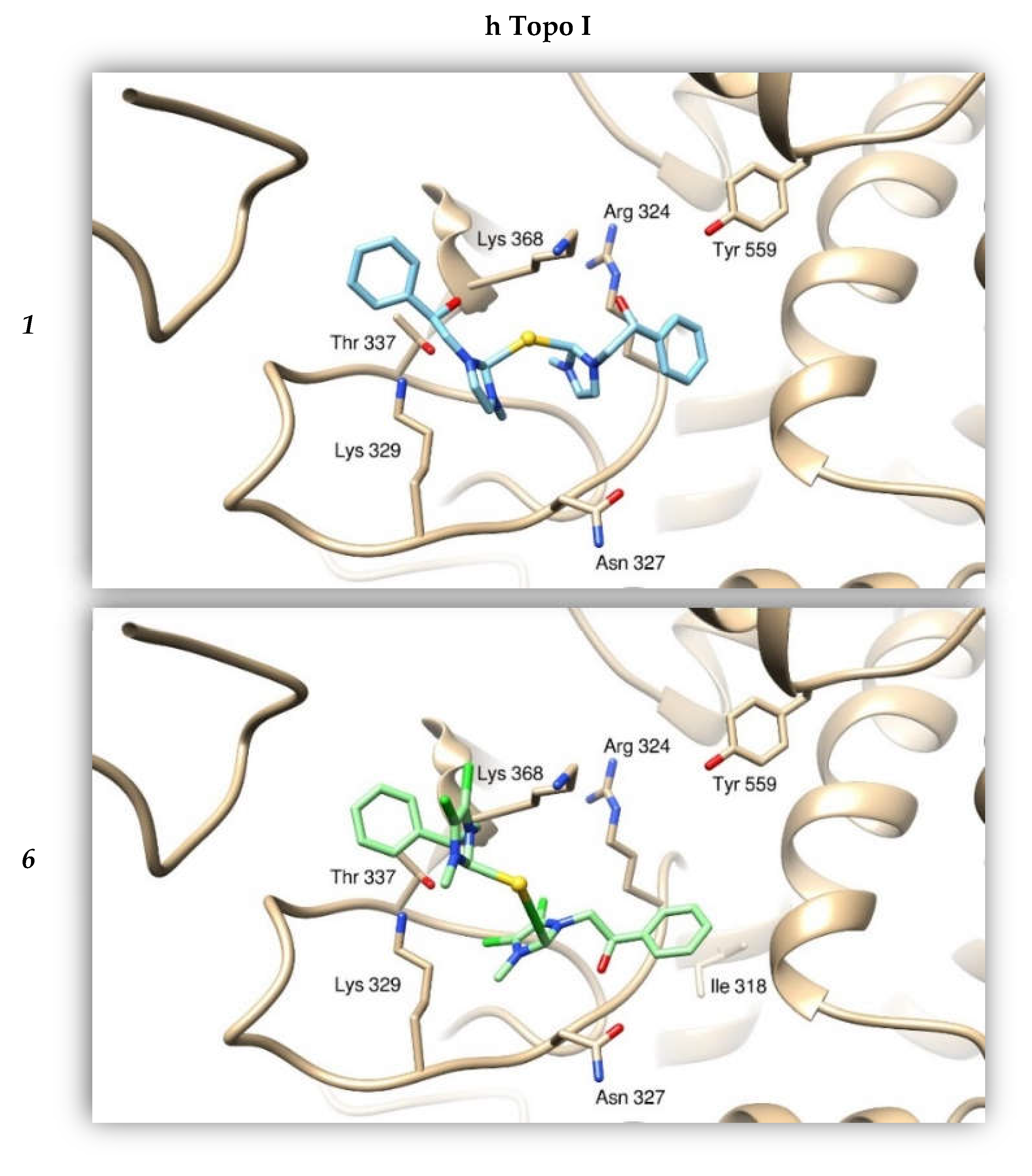
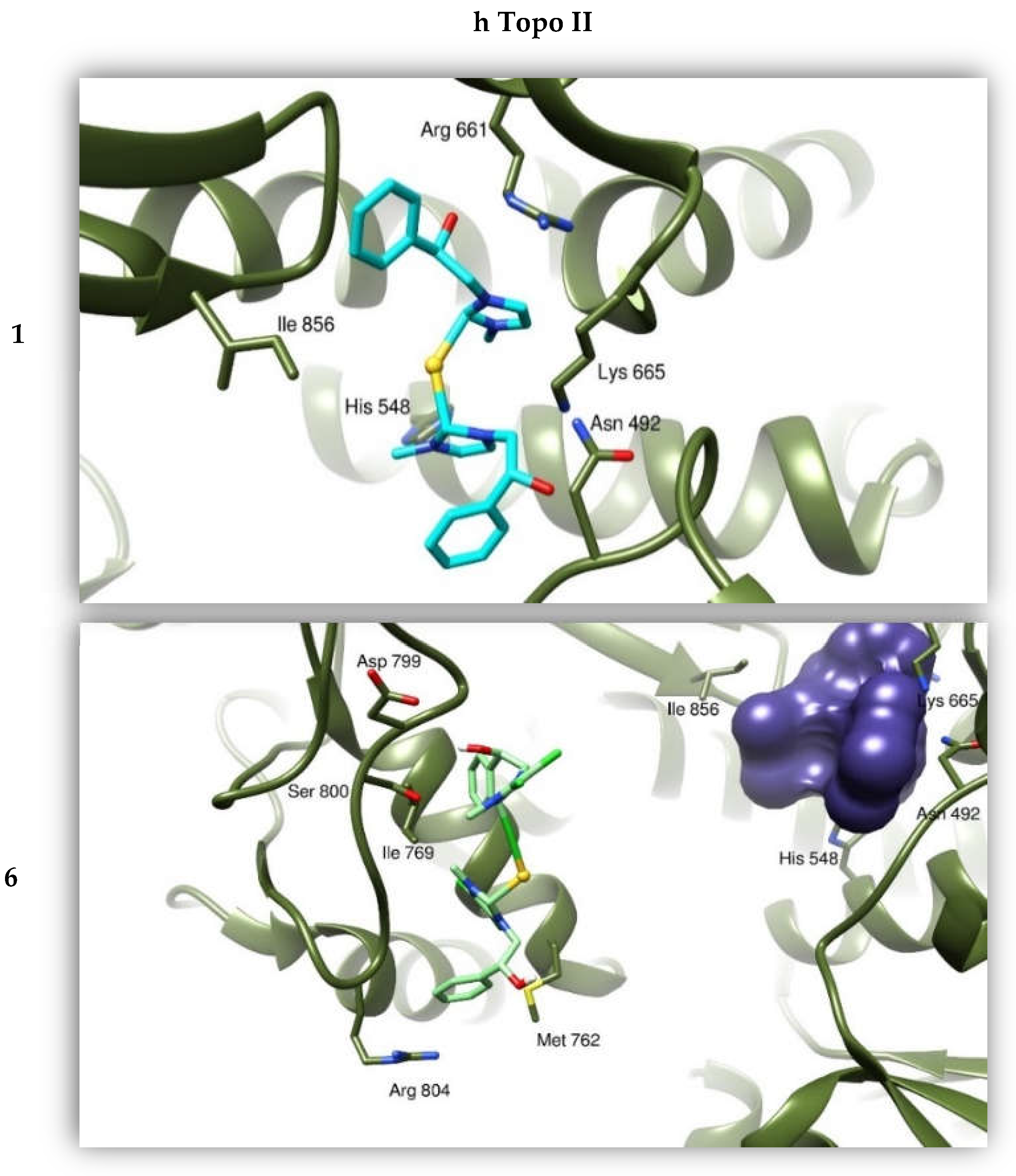
2.2.4. Docking Studies
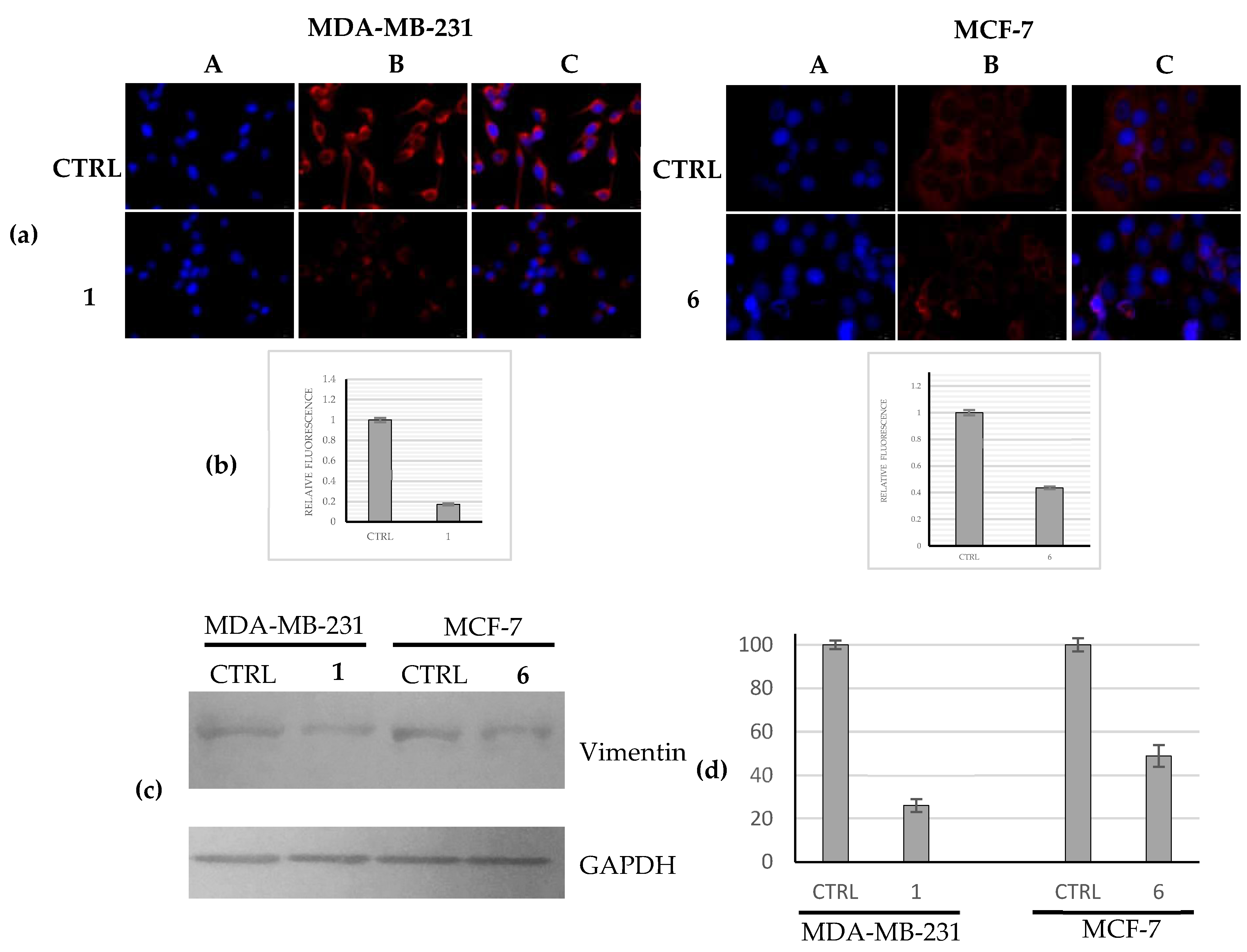
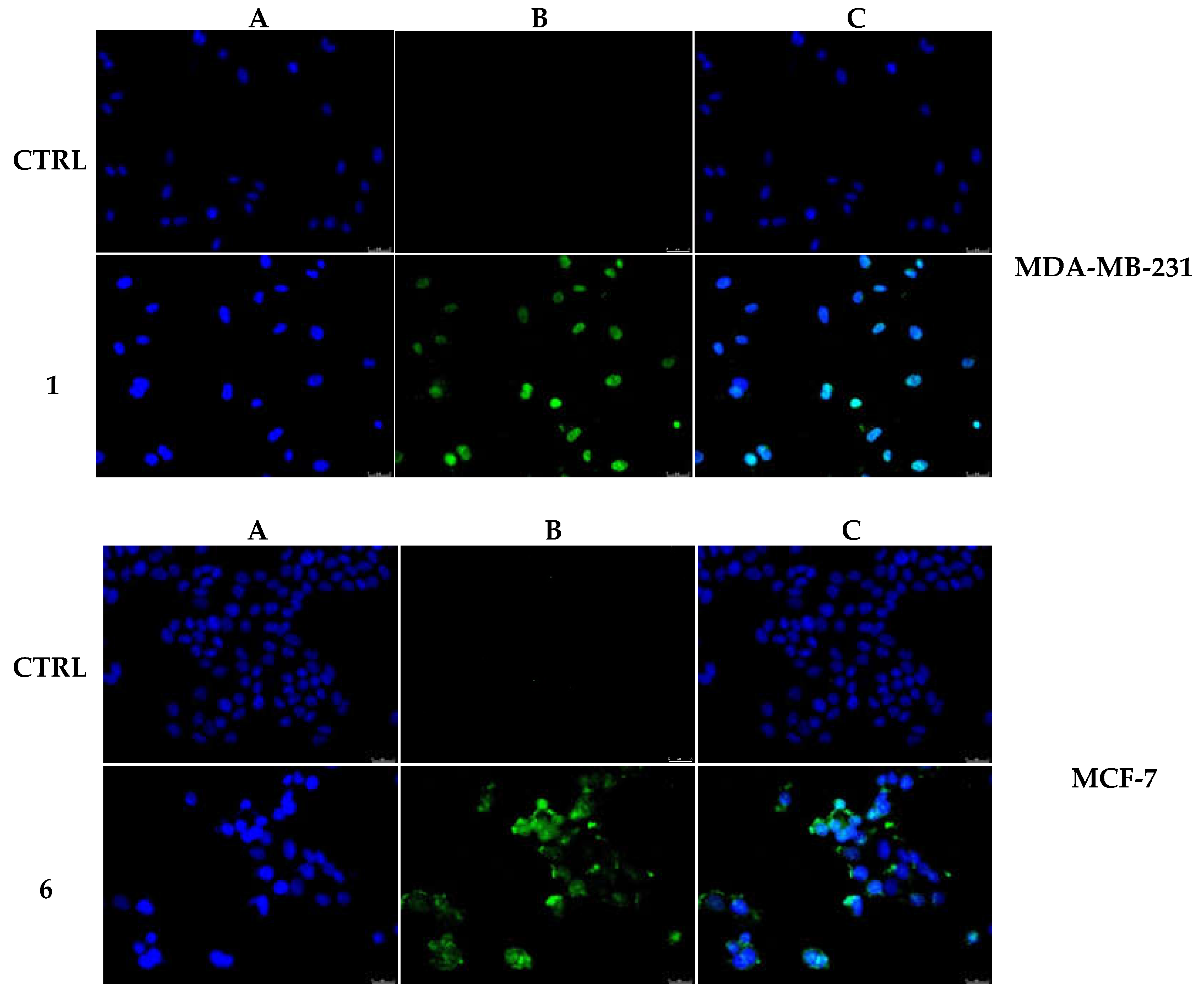
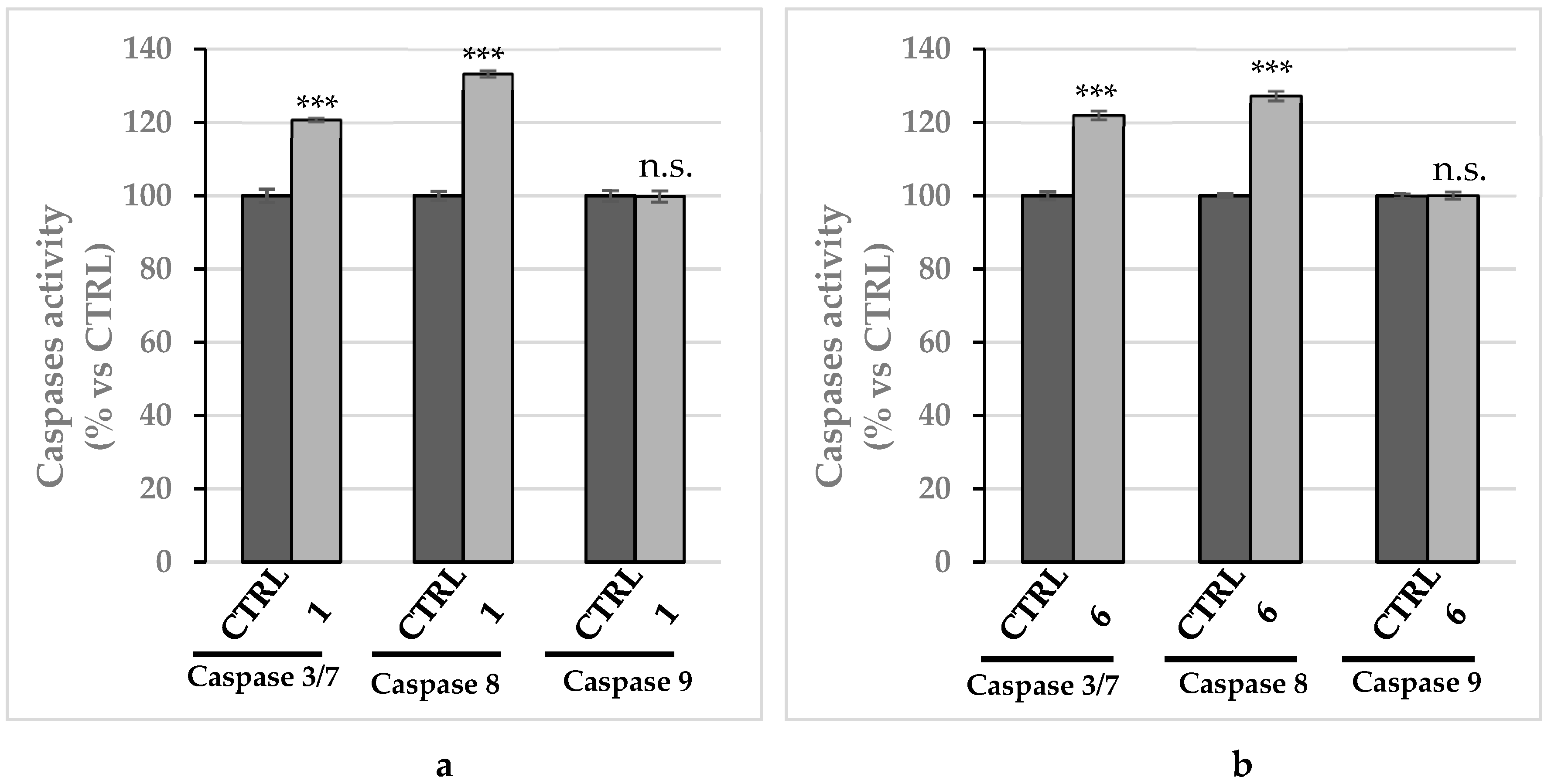
2.2.5. Compounds 1 and 6 Were Able to Trigger Apoptosis in Breast Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Chemistry
General Procedure for the Synthesis of Gold Acetate Complexes
4.2. LogP Value Determination
4.3. Biology
4.3.1. Cell Cultures
4.3.2. MTT Assay
4.3.3. Human Topoisomerase I (hTopo I) Relaxation Assay
4.3.4. Human Topoisomerase II (hTopo II) Decatenation Assay
4.3.5. Immunofluorescence Analysis
4.3.6. Actin Polymerization/Depolymerization Assay
4.3.7. Protein Lysate and Immunoblot Analysis
4.3.8. Docking Studies
4.3.9. TUNEL Assay
4.3.10. Caspases Assay
4.3.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Patil, S.A.; Patil, S.A.; Patil, R.; Keri, R.S.; Budagumpi, S.; Balakrishna, G.R.; Tacke, M. N-heterocyclic carbene metal complexes as bio-organometallic antimicrobial and anticancer drugs. Future Med. Chem. 2015, 7, 1305–1333. [Google Scholar] [CrossRef]
- Smith, C.A.; Narouz, M.R.; Lummis, P.A.; Singh, I.; Nazemi, A.; Li, C.H.; Crudden, C.M. N-Heterocyclic Carbenes in Materials Chemistry. Chem. Rev. 2019, 119, 4986–5056. [Google Scholar] [CrossRef] [PubMed]
- Roymahapatra, G.; Mandal, S.M.; Porto, W.F.; Samanta, T.; Giri, S.; Dinda, J.; Franco, O.L.; Chattaraj, P.K. Pyrazine functionalized Ag(I) and Au(I)-NHC complexes are potential antibacterial agents. Curr. Med. Chem. 2012, 19, 4184–4193. [Google Scholar] [CrossRef] [PubMed]
- Haque, R.A.; Choo, S.Y.; Budagumpi, S.; Iqbal, M.A.; Al-Ashraf Abdullah, A. Silver(I) complexes of mono- and bidentate N-heterocyclic carbene ligands: Synthesis, crystal structures, and in vitro antibacterial and anticancer studies. Eur. J. Med. Chem. 2015, 90, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Iacopetta, D.; Rosano, C.; Sirignano, M.; Mariconda, A.; Ceramella, J.; Ponassi, M.; Saturnino, C.; Sinicropi, M.S.; Longo, P. Is the Way to Fight Cancer Paved with Gold? Metal-Based Carbene Complexes with Multiple and Fascinating Biological Features. Pharmaceuticals 2020, 13, 91. [Google Scholar] [CrossRef]
- Ceramella, J.; Mariconda, A.; Iacopetta, D.; Saturnino, C.; Barbarossa, A.; Caruso, A.; Rosano, C.; Sinicropi, M.S.; Longo, P. From coins to cancer therapy: Gold, silver and copper complexes targeting human topoisomerases. Bioorg. Med. Chem. Lett. 2020, 30, 126905. [Google Scholar] [CrossRef] [PubMed]
- Iacopetta, D.; Mariconda, A.; Saturnino, C.; Caruso, A.; Palma, G.; Ceramella, J.; Muia, N.; Perri, M.; Sinicropi, M.S.; Caroleo, M.C.; et al. Novel Gold and Silver Carbene Complexes Exert Antitumor Effects Triggering the Reactive Oxygen Species Dependent Intrinsic Apoptotic Pathway. ChemMedChem 2017, 12, 2054–2065. [Google Scholar] [CrossRef]
- Cheng, X.; Holenya, P.; Can, S.; Alborzinia, H.; Rubbiani, R.; Ott, I.; Wolfl, S. A TrxR inhibiting gold(I) NHC complex induces apoptosis through ASK1-p38-MAPK signaling in pancreatic cancer cells. Mol. Cancer 2014, 13, 221. [Google Scholar] [CrossRef]
- Krishnamurthy, D.; Karver, M.R.; Fiorillo, E.; Orru, V.; Stanford, S.M.; Bottini, N.; Barrios, A.M. Gold(I)-mediated inhibition of protein tyrosine phosphatases: A detailed in vitro and cellular study. J. Med. Chem. 2008, 51, 4790–4795. [Google Scholar] [CrossRef]
- Karaaslan, M.G.; Aktas, A.; Gurses, C.; Gok, Y.; Ates, B. Chemistry, structure, and biological roles of Au-NHC complexes as TrxR inhibitors. Bioorg. Chem. 2020, 95, 103552. [Google Scholar] [CrossRef]
- Yu, B.; Liu, Y.; Peng, X.; Hua, S.; Zhou, G.; Yan, K.; Liu, Y. Synthesis, characterization, and antitumor properties of Au(i)-thiourea complexes. Met. Integr. Biometal Sci. 2020, 12, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Guarra, F.; Busto, N.; Guerri, A.; Marchetti, L.; Marzo, T.; Garcia, B.; Biver, T.; Gabbiani, C. Cytotoxic Ag(I) and Au(I) NHC-carbenes bind DNA and show TrxR inhibition. J. Inorg. Biochem. 2020, 205, 110998. [Google Scholar] [CrossRef] [PubMed]
- Saturnino, C.; Barone, I.; Iacopetta, D.; Mariconda, A.; Sinicropi, M.S.; Rosano, C.; Campana, A.; Catalano, S.; Longo, P.; Ando, S. N-heterocyclic carbene complexes of silver and gold as novel tools against breast cancer progression. Futur. Med. Chem. 2016, 8, 2213–2229. [Google Scholar] [CrossRef]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. Jama 2019, 321, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zhang, H.; Song, X.; Yang, Q. Metastatic heterogeneity of breast cancer: Molecular mechanism and potential therapeutic targets. Semin. Cancer Biol. 2020, 60, 14–27. [Google Scholar] [CrossRef]
- Yin, L.; Duan, J.J.; Bian, X.W.; Yu, S.C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef]
- Barzaman, K.; Karami, J.; Zarei, Z.; Hosseinzadeh, A.; Kazemi, M.H.; Moradi-Kalbolandi, S.; Safari, E.; Farahmand, L. Breast cancer: Biology, biomarkers, and treatments. Int. Immunopharmacol. 2020, 84, 106535. [Google Scholar] [CrossRef]
- Wong, R.S. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef]
- Costabile, C.; Mariconda, A.; Sirignano, M.; Crispini, A.; Scarpelli, F.; Longo, P. A green approach for A3-coupling reactions: An experimental and theoretical study on NHC silver and gold catalysts. New J. Chem. 2021, 45, 18509–18517. [Google Scholar] [CrossRef]
- Mariconda, A.; Sirignano, M.; Costabile, C.; Longo, P. New NHC-Silver and Gold complexes active in A3-coupling (aldehyde-alkyne-amine) reaction. Mol. Catal. 2020, 480, 110570. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Runte, O.; Artus, G. Synthesis and structure of an ionic Beryllium-carbene complex. J. Organomet. Chem. 1995, 501, C1–C4. [Google Scholar] [CrossRef]
- Mariconda, A.; Grisi, F.; Costabile, C.; Falcone, S.; Bertolasi, V.; Longo, P. Synthesis, characterization and catalytic behavior of a palladium complex bearing a hydroxy-functionalized N-heterocyclic carbene ligand. New J. Chem. 2014, 32, 762–769. [Google Scholar] [CrossRef]
- Espinosa, A.V.; Costa, D.S.; Tunes, L.G.; Monte-Neto, R.L.D.; Grazul, R.M.; de Almeida, M.V.; Silva, H. Anticancer and antileishmanial in vitro activity of gold(I) complexes with 1,3,4-oxadiazole-2(3H)-thione ligands derived from delta-D-gluconolactone. Chem. Biol. Drug Des. 2021, 97, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Gallati, C.M.; Goetzfried, S.K.; Ausserer, M.; Sagasser, J.; Plangger, M.; Wurst, K.; Hermann, M.; Baecker, D.; Kircher, B.; Gust, R. Synthesis, characterization and biological activity of bromido[3-ethyl-4-aryl-5-(2-methoxypyridin-5-yl)-1-propyl-1,3-dihydro-2H-imidazol -2-ylidene]gold(i) complexes. Dalton Trans. 2020, 49, 5471–5481. [Google Scholar] [CrossRef]
- Galassi, R.; Luciani, L.; Gambini, V.; Vincenzetti, S.; Lupidi, G.; Amici, A.; Marchini, C.; Wang, J.; Pucciarelli, S. Multi-Targeted Anticancer Activity of Imidazolate Phosphane Gold(I) Compounds by Inhibition of DHFR and TrxR in Breast Cancer Cells. Front. Chem. 2020, 8, 602845. [Google Scholar] [CrossRef]
- Sousa, S.A.; Leitao, J.H.; Silva, R.A.L.; Belo, D.; Santos, I.C.; Guerreiro, J.F.; Martins, M.; Fontinha, D.; Prudencio, M.; Almeida, M.; et al. On the path to gold: Monoanionic Au bisdithiolate complexes with antimicrobial and antitumor activities. J. Inorg. Biochem. 2020, 202, 110904. [Google Scholar] [CrossRef]
- Bian, M.; Wang, X.; Sun, Y.; Liu, W. Synthesis and biological evaluation of gold(III) Schiff base complexes for the treatment of hepatocellular carcinoma through attenuating TrxR activity. Eur. J. Med. Chem. 2020, 193, 112234. [Google Scholar] [CrossRef]
- Desouza, M.; Gunning, P.W.; Stehn, J.R. The actin cytoskeleton as a sensor and mediator of apoptosis. Bioarchitecture 2012, 2, 75–87. [Google Scholar] [CrossRef]
- Iacopetta, D.; Ceramella, J.; Rosano, C.; Mariconda, A.; Pellegrino, M.; Sirignano, M.; Saturnino, C.; Catalano, A.; Aquaro, S.; Longo, P.; et al. N-Heterocyclic Carbene-Gold(I) Complexes Targeting Actin Polymerization. Appl. Sci. 2021, 11, 5626. [Google Scholar] [CrossRef]
- Berr, A.L.; Wiese, K.; dos Santos, G.; Davis, J.M.; Koch, C.M.; Anekalla, K.R.; Kidd, M.; Cheng, Y.; Hu, Y.-S.; Ridge, K.M. Vimentin is Required for Tumor Progression and Metastasis in a Mouse Model of Non-Small Cell Lung Cancer. bioRxiv 2020. bioRxiv:2020.2006.2004.130963. [Google Scholar]
- Al-Maghrabi, J. Vimentin immunoexpression is associated with higher tumor grade, metastasis, and shorter survival in colorectal cancer. Int. J. Clin. Exp. Pathol. 2020, 13, 493–500. [Google Scholar] [PubMed]
- Stec-Martyna, E.; Ponassi, M.; Miele, M.; Parodi, S.; Felli, L.; Rosano, C. Structural comparison of the interaction of tubulin with various ligands affecting microtubule dynamics. Curr. Cancer Drug. Targets 2012, 12, 658–666. [Google Scholar] [CrossRef] [PubMed]
- Rebowski, G.; Boczkowska, M.; Drazic, A.; Ree, R.; Goris, M.; Arnesen, T.; Dominguez, R. Mechanism of actin N-terminal acetylation. Sci. Adv. 2020, 6, eaay8793. [Google Scholar] [CrossRef] [PubMed]
- Sordet, O.; Goldman, A.; Pommier, Y. Topoisomerase II and tubulin inhibitors both induce the formation of apoptotic topoisomerase I cleavage complexes. Mol. Cancer Ther. 2006, 5, 3139–3144. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sordet, O.; Khan, Q.A.; Kohn, K.W.; Pommier, Y. Apoptosis induced by topoisomerase inhibitors. Curr. Med. Chem. Anticancer. Agents 2003, 3, 271–290. [Google Scholar] [CrossRef] [PubMed]
- Celeste Morley, S.; Sun, G.P.; Bierer, B.E. Inhibition of actin polymerization enhances commitment to and execution of apoptosis induced by withdrawal of trophic support. J. Cell. Biochem. 2003, 88, 1066–1076. [Google Scholar] [CrossRef]
- Odaka, C.; Sanders, M.L.; Crews, P. Jasplakinolide induces apoptosis in various transformed cell lines by a caspase-3-like protease-dependent pathway. Clin. Diagn. Lab. Immunol. 2000, 7, 947–952. [Google Scholar] [CrossRef]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef]
- Li, Y.; Liu, B.; Shi, H.; Wang, Y.; Sun, Q.; Zhang, Q. Metal complexes against breast cancer stem cells. Dalton Trans. 2021, 50, 14498–14512. [Google Scholar] [CrossRef]
- Yadav, S. Potential of Metal Complexes for the Treatment of Cancer: Current Update and Future Prospective. In Chemistry of Biologically Potent Natural Products and Synthetic Compounds; Wiley Online Library: Hoboken, NJ, USA, 2021; pp. 183–204. [Google Scholar]
- Schoch, S.; Sen, V.; Brenner, W.; Hartwig, A.; Köberle, B. In Vitro Nephrotoxicity Studies of Established and Experimental Platinum-Based Compounds. Biomedicines 2021, 9, 1033. [Google Scholar] [CrossRef]
- Xian, C.; Chen, H.; Xiong, F.; Fang, Y.; Huang, H.; Wu, J. Platinum-based chemotherapy via nanocarriers and co-delivery of multiple drugs. Biomater. Sci. 2021, 9, 6023–6036. [Google Scholar] [CrossRef] [PubMed]
- Bruno, G.; Nicolò, F.; Lo Schiavo, S.; Sinicropi, M.S.; Tresoldi, G. Synthesis and spectroscopic properties of di-2-pyridyl sulfide (dps) compounds. Crystal structure of [Ru(dps)2Cl2]. J. Chem. Soc. Dalton Trans. 1995, 17–24. [Google Scholar] [CrossRef]
- Jakob, C.H.G.; Muñoz, A.W.; Schlagintweit, J.F.; Weiß, V.; Reich, R.M.; Sieber, S.A.; Correia, J.D.G.; Kühn, F.E. Anticancer and antibacterial properties of trinuclear Cu(I), Ag(I) and Au(I) macrocyclic NHC/urea complexes. J. Organomet. Chem. 2021, 932, 121643. [Google Scholar] [CrossRef]
- Abdulqader, A.M.; Jasim, M.N.; Abdurahman, A.M. Synthesis, characterization and molecular docking of new gold complexes as a breast anticancer. Mater. Today Proc. 2021, 45, 5635–5641. [Google Scholar] [CrossRef]
- Tolbatov, I.; Marzo, T.; Coletti, C.; La Mendola, D.; Storchi, L.; Re, N.; Marrone, A. Reactivity of antitumor coinage metal-based N-heterocyclic carbene complexes with cysteine and selenocysteine protein sites. J. Inorg. Biochem. 2021, 223, 111533. [Google Scholar] [CrossRef]
- Frei, A.; Zuegg, J.; Elliott, A.G.; Baker, M.; Braese, S.; Brown, C.; Chen, F.; Dowson, C.G.; Dujardin, G.; Jung, N.; et al. Metal complexes as a promising source for new antibiotics. Chem. Sci. 2020, 11, 2627–2639. [Google Scholar] [CrossRef]
- Berners-Price, S.J.; Filipovska, A. Gold compounds as therapeutic agents for human diseases. Met. Integr. Biometal Sci. 2011, 3, 863–873. [Google Scholar] [CrossRef]
- Hopkinson, M.N.; Richter, C.; Schedler, M.; Glorius, F. An overview of N-heterocyclic carbenes. Nature 2014, 510, 485–496. [Google Scholar] [CrossRef]
- Wenzela, M.; Casini, A. Mass spectrometry as a powerful tool to study therapeutic metallodrugs speciation mechanisms: Current frontiers and perspectives. Coord. Chem. Rev. 2017, 352, 432–460. [Google Scholar] [CrossRef]
- Ceramella, J.; Caruso, A.; Occhiuzzi, M.A.; Iacopetta, D.; Barbarossa, A.; Rizzuti, B.; Dallemagne, P.; Rault, S.; El-Kashef, H.; Saturnino, C.; et al. Benzothienoquinazolinones as new multi-target scaffolds: Dual inhibition of human Topoisomerase I and tubulin polymerization. Eur. J. Med. Chem. 2019, 181, 111583. [Google Scholar] [CrossRef]
- Puxeddu, M.; Shen, H.; Bai, R.; Coluccia, A.; Bufano, M.; Nalli, M.; Sebastiani, J.; Brancaccio, D.; Da Pozzo, E.; Tremolanti, C.; et al. Discovery of pyrrole derivatives for the treatment of glioblastoma and chronic myeloid leukemia. Eur. J. Med. Chem. 2021, 221, 113532. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Li, L.D.; Li, J.; Lu, X. Targeting of topoisomerases for prognosis and drug resistance in ovarian cancer. J. Ovarian Res. 2016, 9, 35. [Google Scholar] [CrossRef] [PubMed]
- Bjornsti, M.A.; Kaufmann, S.H. Topoisomerases and cancer chemotherapy: Recent advances and unanswered questions. F1000Res 2019, 8, 1704. [Google Scholar] [CrossRef] [PubMed]
- Delgado, J.L.; Hsieh, C.M.; Chan, N.L.; Hiasa, H. Topoisomerases as anticancer targets. Biochem. J. 2018, 475, 373–398. [Google Scholar] [CrossRef]
- Aseervatham, J. Cytoskeletal Remodeling in Cancer. Biology 2020, 9, 385. [Google Scholar] [CrossRef]
- Kadzik, R.S.; Homa, K.E.; Kovar, D.R. F-Actin Cytoskeleton Network Self-Organization Through Competition and Cooperation. Annu. Rev. Cell Dev. Biol. 2020, 36, 35–60. [Google Scholar] [CrossRef]
- Zhang, R.; Lee, D.M.; Jimah, J.R.; Gerassimov, N.; Yang, C.; Kim, S.; Luvsanjav, D.; Winkelman, J.; Mettlen, M.; Abrams, M.E.; et al. Dynamin regulates the dynamics and mechanical strength of the actin cytoskeleton as a multifilament actin-bundling protein. Nat. Cell Biol. 2020, 22, 674–688. [Google Scholar] [CrossRef]
- Banerjee, S.; Gardel, M.L.; Schwarz, U.S. The Actin Cytoskeleton as an Active Adaptive Material. Annu. Rev. Condens. Matter Phys. 2020, 11, 421–439. [Google Scholar] [CrossRef]
- Li, X.; Wang, J. Mechanical tumor microenvironment and transduction: Cytoskeleton mediates cancer cell invasion and metastasis. Int. J. Biol. Sci. 2020, 16, 2014–2028. [Google Scholar] [CrossRef]
- Morton, W.M.; Ayscough, K.R.; McLaughlin, P.J. Latrunculin alters the actin-monomer subunit interface to prevent polymerization. Nat. Cell Biol. 2000, 2, 376–378. [Google Scholar] [CrossRef]
- Liu, C.Y.; Lin, H.H.; Tang, M.J.; Wang, Y.K. Vimentin contributes to epithelial-mesenchymal transition cancer cell mechanics by mediating cytoskeletal organization and focal adhesion maturation. Oncotarget 2015, 6, 15966–15983. [Google Scholar] [CrossRef] [PubMed]
- Strouhalova, K.; Prechova, M.; Gandalovicova, A.; Brabek, J.; Gregor, M.; Rosel, D. Vimentin Intermediate Filaments as Potential Target for Cancer Treatment. Cancers 2020, 12, 184. [Google Scholar] [CrossRef]
- Jiu, Y.; Peranen, J.; Schaible, N.; Cheng, F.; Eriksson, J.E.; Krishnan, R.; Lappalainen, P. Vimentin intermediate filaments control actin stress fiber assembly through GEF-H1 and RhoA. J. Cell Sci. 2017, 130, 892–902. [Google Scholar] [CrossRef] [PubMed]
- Boice, A.; Bouchier-Hayes, L. Targeting apoptotic caspases in cancer. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118688. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [PubMed]
- Tummers, B.; Green, D.R. Caspase-8: Regulating life and death. Immunol. Rev. 2017, 277, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhou, L.; Zhao, T.; Liu, X.; Zhang, P.; Liu, Y.; Zheng, X.; Li, Q. Caspase-9: Structure, mechanisms and clinical application. Oncotarget 2017, 8, 23996–24008. [Google Scholar] [CrossRef]
- Erxleben, A. Mitochondria-Targeting Anticancer Metal Complexes. Curr. Med. Chem. 2019, 26, 694–728. [Google Scholar] [CrossRef]
- Magherini, F.; Fiaschi, T.; Valocchia, E.; Becatti, M.; Pratesi, A.; Marzo, T.; Massai, L.; Gabbiani, C.; Landini, I.; Nobili, S.; et al. Antiproliferative effects of two gold(I)-N-heterocyclic carbene complexes in A2780 human ovarian cancer cells: A comparative proteomic study. Oncotarget 2018, 9, 28042–28068. [Google Scholar] [CrossRef]
- Hoesel, B.; Schmid, J.A. The complexity of NF-kappaB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct Target 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Dolcet, X.; Llobet, D.; Pallares, J.; Matias-Guiu, X. NF-kB in development and progression of human cancer. Virchows Arch. 2005, 446, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.B.; Meng, F.R.; Fang, Y.X.; Wu, X.; Zhang, C.W.; Liu, Y.; Liu, D.; Li, G.Q.; Feng, F.B.; Qiu, H.Y. Inhibition of NF-kappaB signaling pathway induces apoptosis and suppresses proliferation and angiogenesis of human fibroblast-like synovial cells in rheumatoid arthritis. Medicine 2018, 97, e10920. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Gaynor, R.B. Role of the NF-kappaB pathway in the pathogenesis of human disease states. Curr. Mol. Med. 2001, 1, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.L.; Kamata, H.; Karin, M. IKK/NF-kappaB signaling: Balancing life and death—A new approach to cancer therapy. J. Clin. Investig. 2005, 115, 2625–2632. [Google Scholar] [CrossRef]
- Kasibhatla, S.; Brunner, T.; Genestier, L.; Echeverri, F.; Mahboubi, A.; Green, D.R. DNA Damaging Agents Induce Expression of Fas Ligand and Subsequent Apoptosis in T Lymphocytes via the Activation of NF-κB and AP-1. Mol. Cell 1998, 1, 543–551. [Google Scholar] [CrossRef]
- Hackenberg, F.; Müller-Bunz, H.; Smith, R.; Streciwilk, W.; Zhu, X.; Tacke, M. Novel ruthenium(II) and Gold(I) NHC complexes: Synthesis, characterization, and evaluation and their anticancer properties. Organometallics 2013, 32, 5551–5560. [Google Scholar] [CrossRef]
- Cirri, D.; Pillozzi, S.; Gabbiani, C.; Tricomi, J.; Bartoli, G.; Stefanini, M.; Michelucci, E.; Arcangeli, A.; Messori, L.; Marzo, T. PtI2(DACH), the iodido analogue of oxaliplatin as a candidate for colorectal cancer treatment: Chemical and biological features. Dalton Trans. 2017, 46, 311–317. [Google Scholar] [CrossRef]
- Rechoum, Y.; Rovito, D.; Iacopetta, D.; Barone, I.; Andò, S.; Weigel, N.L.; O’Malley, B.W.; Brown, P.H.; Fuqua, S.A. AR collaborates with ERα in aromatase inhibitor-resistant breast cancer. Breast Cancer Res. Treat 2014, 147, 473–485. [Google Scholar] [CrossRef]
- Redinbo, M.R.; Stewart, L.; Kuhn, P.; Champoux, J.J.; Hol, W.G. Crystal structures of human topoisomerase I in covalent and noncovalent complexes with DNA. Science 1998, 279, 1504–1513. [Google Scholar] [CrossRef]
- Wang, Y.R.; Chen, S.F.; Wu, C.C.; Liao, Y.W.; Lin, T.S.; Liu, K.T.; Chen, Y.S.; Li, T.K.; Chien, T.C.; Chan, N.L. Producing irreversible topoisomerase II-mediated DNA breaks by site-specific Pt(II)-methionine coordination chemistry. Nucleic Acids Res. 2017, 45, 10920. [Google Scholar] [CrossRef] [PubMed]
- Sanner, M.F.; Duncan, B.S.; Carrillo, C.J.; Olson, A.J. Integrating computation and visualization for biomolecular analysis: An example using python and AVS. Pac. Symp. Biocomput. 1999, 401–412. [Google Scholar] [CrossRef]
- Rosano, C.; Lappano, R.; Santolla, M.F.; Ponassi, M.; Donadini, A.; Maggiolini, M. Recent advances in the rationale design of GPER ligands. Curr. Med. Chem. 2012, 19, 6199–6206. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | Complex | Au-C (δC) ppm |
| 1 | 171.8 a | |
| 2 | 171.0 a | |
| 3 | 169.1 b | |
| 4 | 162.1 b | |
| 5 | 172.0 b | |
| 7 [13] | 161.8 b | |
| 6 | 170.2 b | |
| 8 [5] | 163.9 b |
| Complex | LogP |
|---|---|
| 1 | 0.079 |
| 2 | 0.047 |
| 3 | 0.62 |
| 4 | 0.68 |
| 5 | 0.12 |
| 6 | 0.78 |
| IC50 (µM) | |||||
|---|---|---|---|---|---|
| Compounds | MDA-MB-231 | MCF-7 | SkBr3 | MCF-10A | Hek-293 |
| Cisplatin | 28.7 ± 0.4 | 35.8 ± 0.7 | 7.4 ± 0.9 | 81.3 ± 0.6 | 16.3 ± 0.9 |
| Latrunculin A | 2.4 × 10−2 ± 0.9 | 0.14 ± 1.0 | 0.7 ± 1.2 | 4.5 × 10−2 ± 1.2 | 9.3 × 10−2 ± 0.6 |
| 1 | 15.8 ± 0.7 | 29.9 ± 1.1 | 83.0 ± 1.0 | >200 | >200 |
| 2 | 22.6 ± 1.1 | 30.6 ± 0.7 | 38.4 ± 0.4 | 39.6 ± 0.5 | >200 |
| 3 | 2.2 ± 1.1 | 3.3 ± 1.4 | 10.9 ± 0.7 | 13.3 ± 1.0 | 39.0 ± 0.9 |
| 4 | 3.0 ± 0.7 | 9.4 ± 0.6 | 2.6 ± 0.7 | 5.2 ± 0.9 | 7.6 ± 1.0 |
| 5 | 58.5 ± 1.2 | 21.9 ± 0.5 | 58.9 ± 1.2 | 27.8 ± 0.7 | >200 |
| 6 | 16.8 ± 1.2 | 1.2 ± 0.3 | 2.3 ± 0.9 | 24.4 ± 0.9 | 32.5±1.1 |
| Binding Energies (kcal/mol) | |||
|---|---|---|---|
| Compounds | h Topo I | h Topo II | Actin |
| 1 | −8.42 | −6.63 | −8.09 |
| 6 | −9.58 | −8.65 | −7.75 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ceramella, J.; Mariconda, A.; Sirignano, M.; Iacopetta, D.; Rosano, C.; Catalano, A.; Saturnino, C.; Sinicropi, M.S.; Longo, P. Novel Au Carbene Complexes as Promising Multi-Target Agents in Breast Cancer Treatment. Pharmaceuticals 2022, 15, 507. https://doi.org/10.3390/ph15050507
Ceramella J, Mariconda A, Sirignano M, Iacopetta D, Rosano C, Catalano A, Saturnino C, Sinicropi MS, Longo P. Novel Au Carbene Complexes as Promising Multi-Target Agents in Breast Cancer Treatment. Pharmaceuticals. 2022; 15(5):507. https://doi.org/10.3390/ph15050507
Chicago/Turabian StyleCeramella, Jessica, Annaluisa Mariconda, Marco Sirignano, Domenico Iacopetta, Camillo Rosano, Alessia Catalano, Carmela Saturnino, Maria Stefania Sinicropi, and Pasquale Longo. 2022. "Novel Au Carbene Complexes as Promising Multi-Target Agents in Breast Cancer Treatment" Pharmaceuticals 15, no. 5: 507. https://doi.org/10.3390/ph15050507
APA StyleCeramella, J., Mariconda, A., Sirignano, M., Iacopetta, D., Rosano, C., Catalano, A., Saturnino, C., Sinicropi, M. S., & Longo, P. (2022). Novel Au Carbene Complexes as Promising Multi-Target Agents in Breast Cancer Treatment. Pharmaceuticals, 15(5), 507. https://doi.org/10.3390/ph15050507