1. Introduction
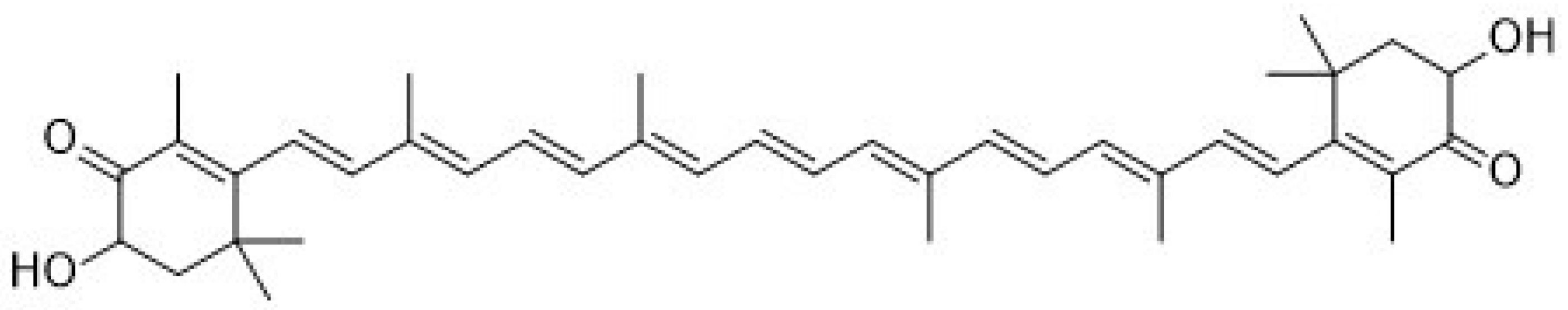
Recently, astaxanthin (3,3′-dihydroxy-β,β-carotene-4,4′dione,
Figure 1) has been widely applied as a human nutraceutical supplement for health care or an essential ingredient in the food and cosmetic industry. Astaxanthin is a lipid-soluble reddish pigment belonging to the xanthophyll group of carotenoids, produced by microalgae under pressure such as strong light, high salinity and low nutrient utilization for protection [
1]. Astaxanthin could accumulate in the tissues of marine organisms feeding on microalgae. Therefore, astaxanthin is commonly found in green microalgae
Haematococcus pluvialis; red yeast
Xanthophyllomyces dendrorhous (
Phaffia rhodozyma); and marine animals such as shrimp, krill, salmon, crab and lobster. According to numerous studies, astaxanthin showed better biological potential than other antioxidants such as lutein, lycopene, β-carotene and vitamin C [
2,
3]. Unlike other types of carotenoids, astaxanthin has two keto groups located at the 4,4′ position of the β-ionone ring that activate adjacent hydroxyl groups for capturing per-oxidants and stabilizing the trapped radicals [
4]. Through its antioxidant ability, astaxanthin can suppress cancer cell proliferation, migration or invasion; prevent cardiovascular diseases and diabetes; and promote immune system and ocular health [
2,
5,
6,
7,
8,
9]. In addition, astaxanthin has been reported to have several other health benefits, including anti-inflammatory activity, anti-skin-aging ability, protection against early brain injury and neuroprotective property [
10,
11,
12,
13,
14]. So far, relatively few studies have investigated the association between astaxanthin and bone health [
15,
16].
Astaxanthin is a partially hydrophobic carotenoid that dissolves in organic solvents such as ethanol, acetone, dimethyl sulfoxide (DMSO) and dimethylformamide (DMF) [
17]. As expected, astaxanthin has only slight solubility in water. In addition, astaxanthins in the market (extracted from algae
Haematococcus pluvialis) are mostly in esterified form, containing various fatty acids [
18]. Therefore, these hydrophobic characteristics could limit the clinical application of astaxanthin. In nature, astaxanthin exists mainly in trans-isomeric forms, which could be easily converted into cis-isomeric forms (9-cis-astaxanthin and 13-cis-astaxanthin) due to environmental factors such as heat, light and oxidation [
19]. Lin et al., reported that the main astaxanthin components in spear shrimp shells are
trans-astaxanthin, 9-cis-astaxanthin, 13-cis-astaxanthin and 16 astaxanthin esters [
20]. In order to achieve a circular economy, shrimp shell waste is a good source from which to extract astaxanthin. On the other hand, liposomes have been used in the treatment of osteoarthritis, but there is no literature on the evaluation of the effect of astaxanthin-loaded liposomes on bone health. Previous studies have indicated that the biological efficacy of the astaxanthin extracts may change with the manner of storage and extraction [
1,
21]. In the present study, astaxanthin (asta)-loaded liposomes prepared using soybean phosphatidylcholine (SPC) were expected to maintain the bioactivity of the astaxanthin extract and increase the absorption rate in the bone cells. Therefore, the aim of this study is to provide a new approach to bone health supplementation and increase the economic value of shrimp shell waste through the formulation of asta-loaded liposomes.
3. Discussion
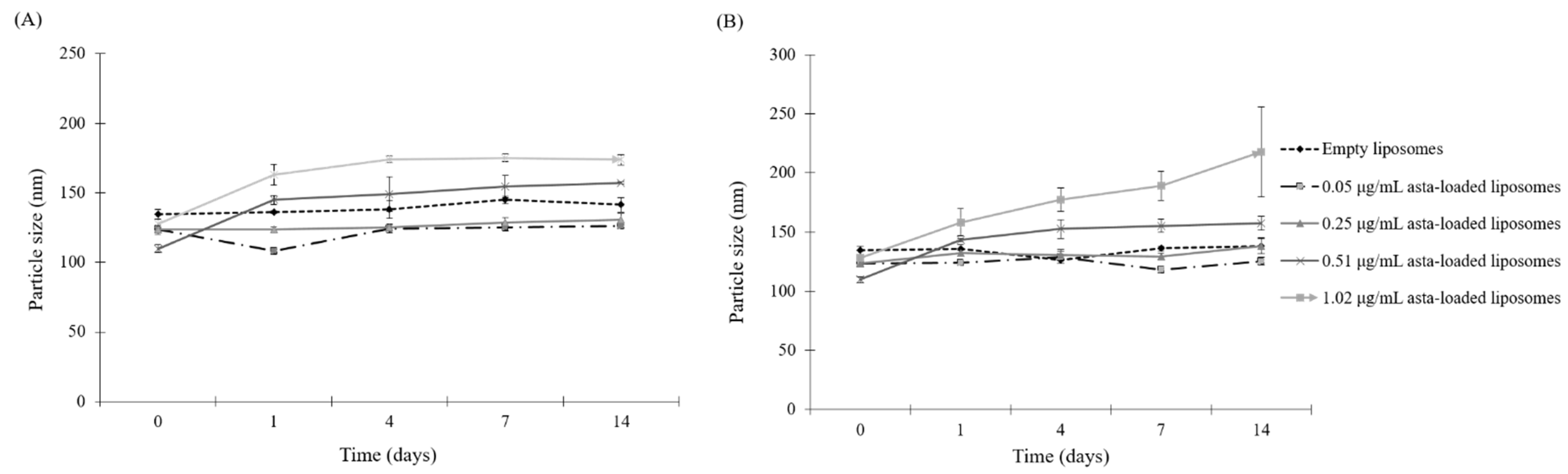
Since astaxanthin has low aqueous solubility, this limitation could be a significant barrier in the application of pharmaceutical or health products, and that can be overcome by the microencapsulation of the liposome. The therapeutic efficiency of the natural extracts such as Fraxinus angustifolia leaf and bark extracts can be improved by liposomal encapsulation techniques [
24]. In this study, SPC liposomes were formulated in order to enhance the biological activities of astaxanthin extracts. In the physical properties of asta-loaded liposomes, there were no significant differences in particle sizes, but the PDI value was raised by increasing the concentration of the astaxanthin extract. Similar to the research of David R. Khan et al., storage stability experiments demonstrated that liposomal formulations of astaxanthin extracts were more stable in the storage under refrigeration temperature, and asta-loaded liposomes with high concentrations will be less stable [
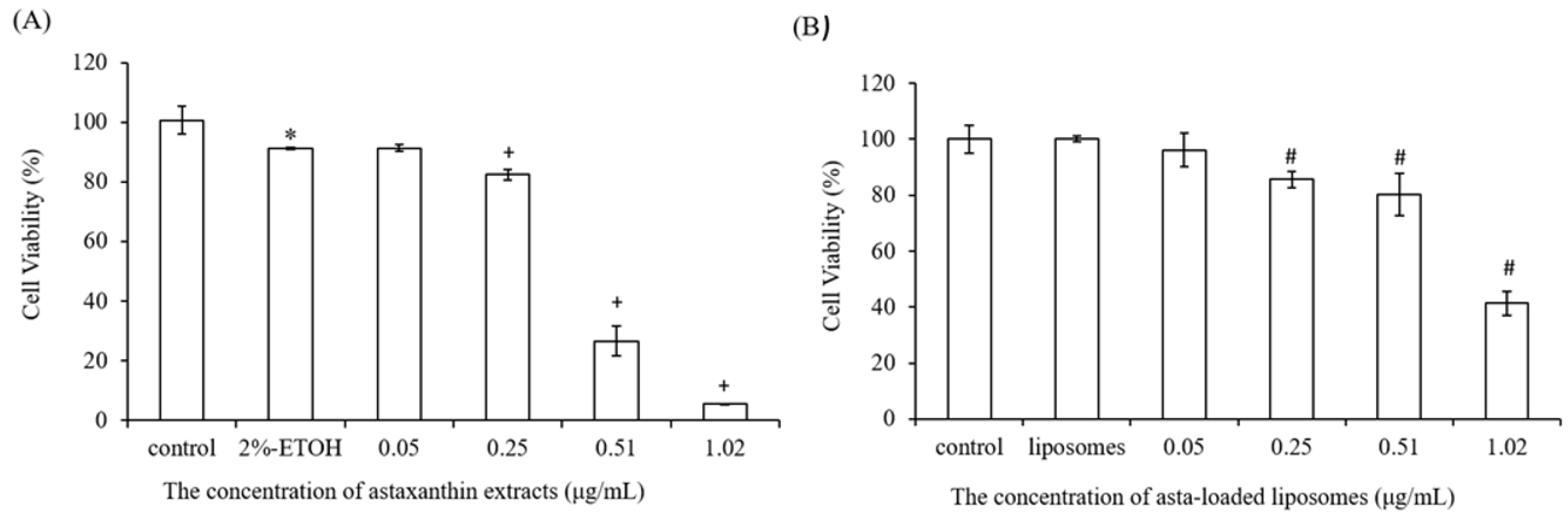
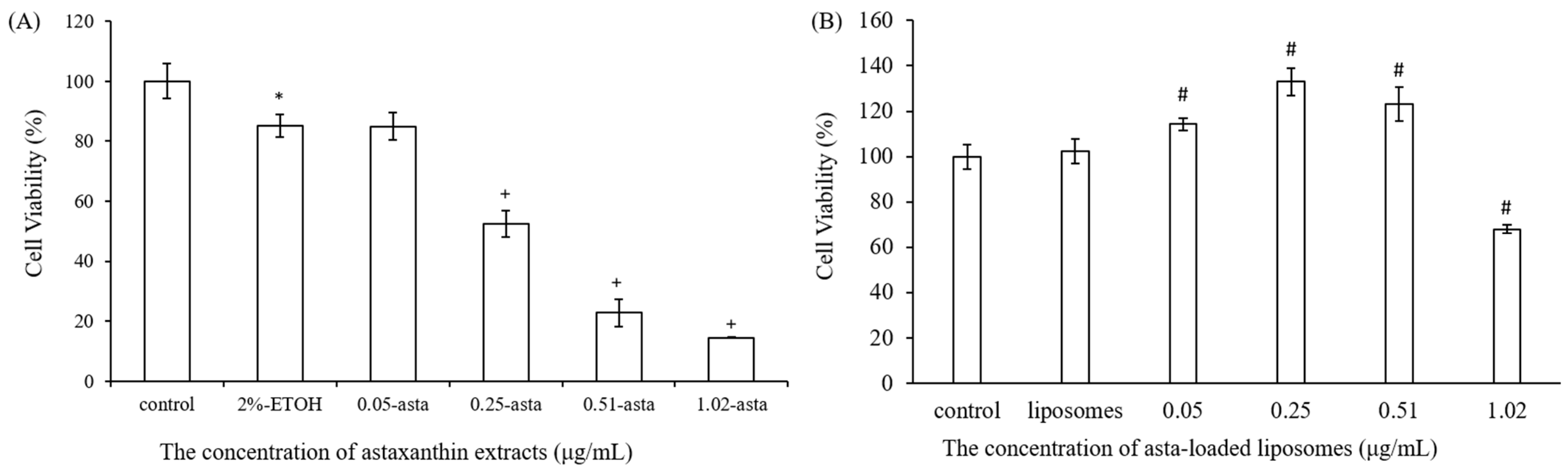
25]. We also found that liposomes reduced the entrapment efficiency of astaxanthin from 89% to 29% when the concentration of astaxanthin extract was increased from 0.05 to 1.02 μg/mL. The asta-loaded liposomes were less toxic in vitro than the free drug, and that may promote osteoblast proliferation.
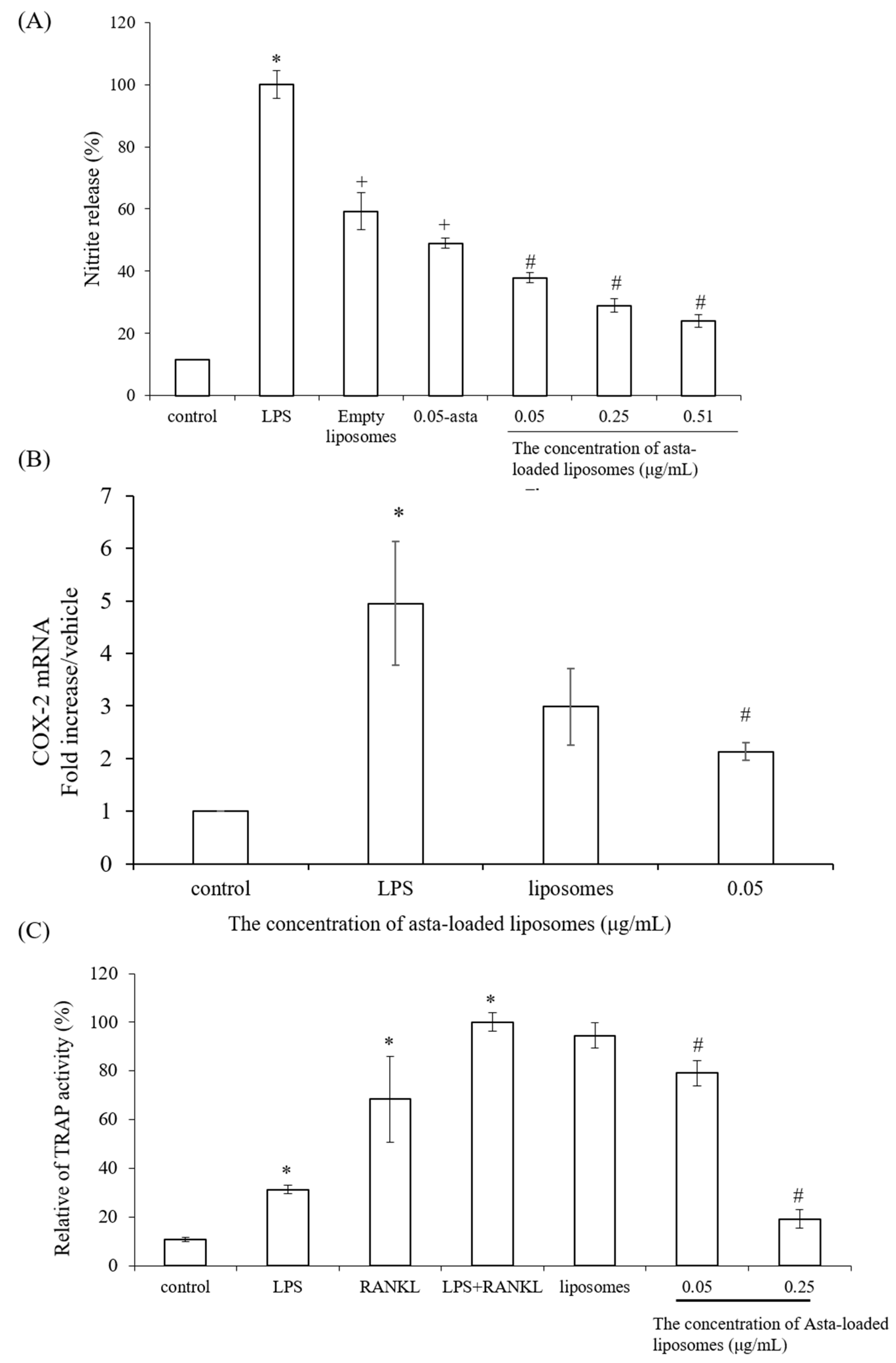
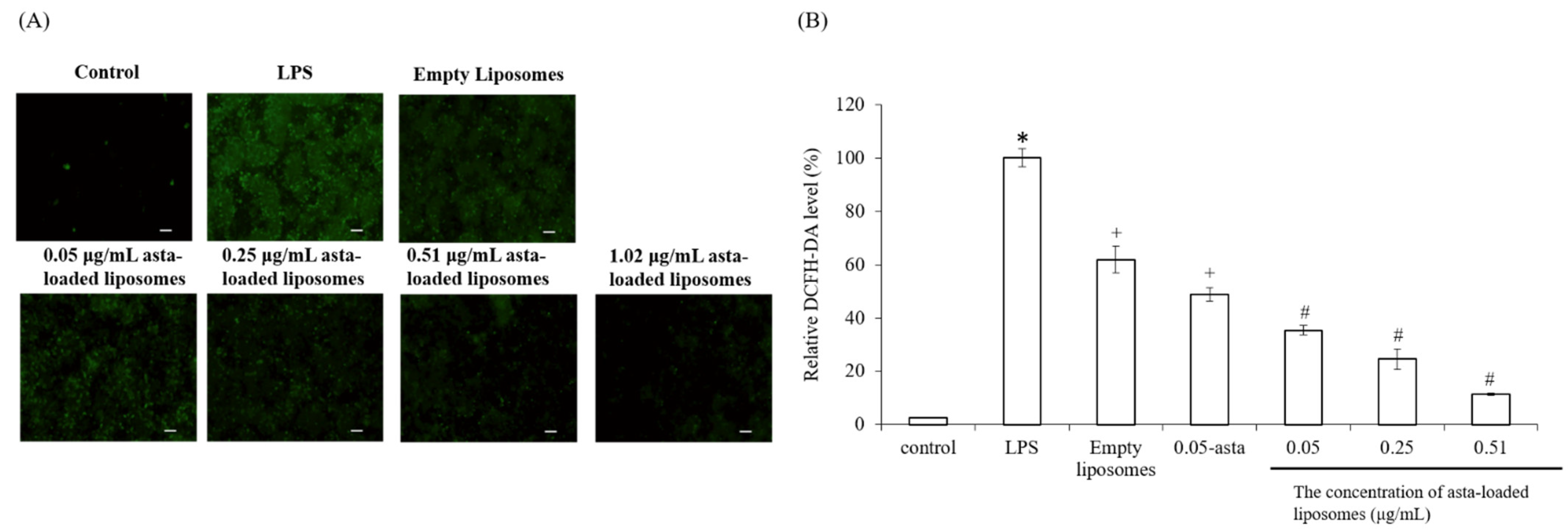
To confirm the powerful antioxidant, anti-inflammatory and anti-osteoclastogenic potential of astaxanthin, we performed a scavenging intracellular ROS assay, nitrite production assay, TRAP activity assay and RT-PCR analysis. Lee et al., have mentioned that astaxanthin can inhibit the expression of iNOS, COX-2, TNF-α and IL-1β as well as the production of NO and PGE2 in LPS-induced macrophages [
26]. Our results also demonstrated that asta-loaded liposomes can exert anti-inflammatory effects via the suppression of COX-2 and NO. Oxidative stress and inflammation are closely related pathological processes, and hence inflammatory mediators and cytokines can accelerate intracellular ROS accumulation. In our study, asta-loaded liposomes were found to protect RAW264.7 macrophages against LPS-stimulated oxidative stress by reducing intracellular ROS accumulation. Similar to our results, Liu et al., indicated that astaxanthin could significantly suppress ROS production induced by 6-hydroxydopamine (6-OHDA), which may cause Parkinson’s disease [
27]. Therefore, we might confirm that asta-loaded liposomes can reduce oxidative damage caused by excessive ROS or inflammation.
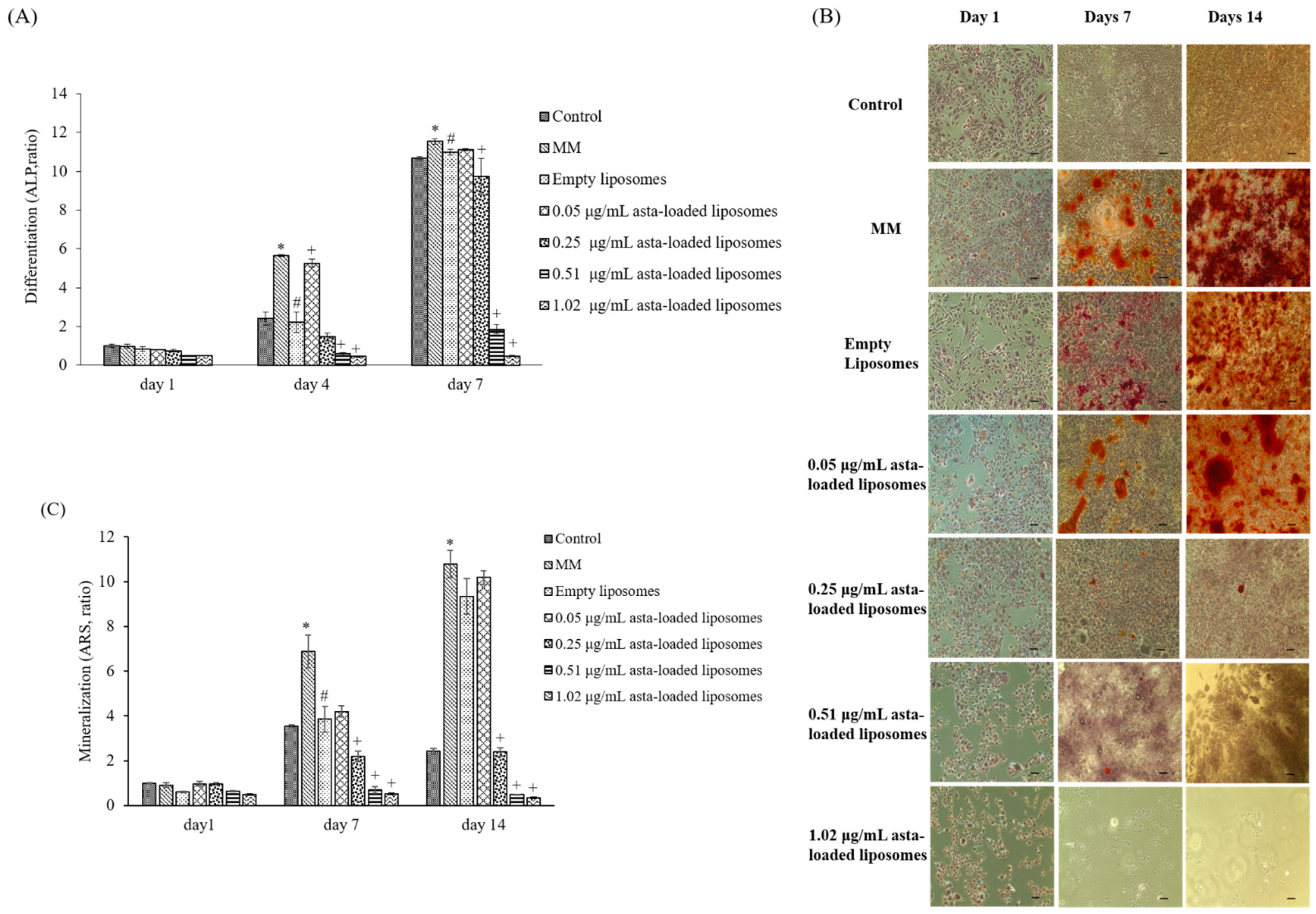
In previous studies reported by our lab, mineralization medium (MM, 5 mM β-glycerophosphate and 50 μg/mL ascorbic acid) was able to induce osteoblast differentiation and mineralization in 7F2 osteoblast-like cells [
28]. In comparison with cells treated with MM only, 0.05 μg/mL asta-loaded liposomes showed a similar pattern in osteoblast differentiation and mineralization. Similar to our finding, Zhang and Peng (2019) demonstrated that 20 ng/mL astaxanthin-encapsulated polymeric micelles could enhance osteogenic differentiation of human mesenchymal stem cells [
29]. Moreover, Hwang et al., indicated that astaxanthin may inhibit osteoclast formation through the expression of the nuclear factor of activated T cells (NFAT-c1), dendritic cell-specific transmembrane protein (DC-STAMP), TRAP and cathepsin K [
16]. Consistent with previous results, asta-loaded liposomes could suppress TRAP activities in LPS- and RANKL-induced RAW264.7 macrophages through anti-inflammatory and antioxidant pathways. In addition, El-Baz et al., demonstrated that
Heamatococcus pluvialis microalgae, containing astaxanthin, could ameliorate bone loss through the downregulation of serum OPG and upregulation of serum RANKL [
15]. Therefore, asta-loaded liposomes with low doses could inhibit osteoclastogenesis through the inhibition of TRAP activities in concurrence with no inhibitory effect on osteoblast proliferation and differentiation.
Astaxanthin is a valuable functional ingredient that has the ability to resist ROS accumulation and suppress oxidative stress associated with inflammation. Liposomal formulation of astaxanthin extract can reduce the cytotoxicity of free drugs, and asta-loaded liposomes with a low dose (0.05 μg/mL) can retain osteoblastic activity. Thus, we suggested that asta-loaded liposomes could be applied as an antioxidant and anti-inflammatory ingredient as well as a keeper of osteoblastic mineralization in nutritional health products.
4. Materials and Methods
4.1. Materials
Astaxanthin extract from dried shrimp heads was purchased from Acorty Biotechnology Co., Ltd., Chia Yi City, Taiwan. 2,2-Diphenyl-1-picrylhydrazyl (DPPH), alizarin red S (ARS) and thiazolyl blue tetrazolium bromide (MTT) were purchased from Sigma-Aldrich, Saint Louis, MO, USA. Alkaline Phosphatase Assay kit (colorimetric) was acquired from Abcam (Cambridge, UK). ABTS was purchased from Calbiochem (San Diego, CA, USA). All of the cell culture materials were acquired from Gibco (Grand Island, NT, USA), and all solvents used were of analytical grade (J.T. Baker, Phillipsburg, NJ, USA).
4.2. Cell Culture
Mouse osteoblast-like cells (7F2) and mouse macrophage cells (Raw264.7) were obtained from Bioresource Collection and Research Center (BCRC), Taiwan. 7F2 and Raw264.7 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing 10% fetal bovine serum (FBS), 1% 100 units/mL penicillin and 100 μg/mL of streptomycin and 1% 200 mM L-glutamine. Cells were maintained at 37 °C with 5% CO2 in a humidified incubator and subcultured at an initial density of 5 × 105/mL every 2–3 days.
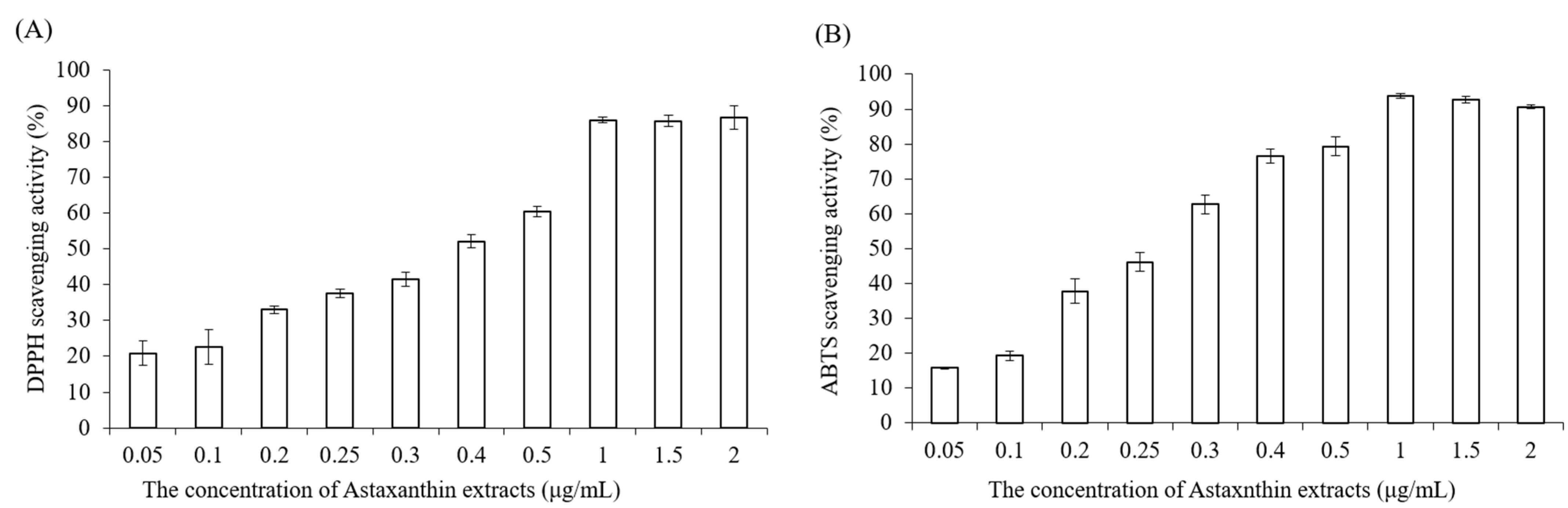
4.3. DPPH Scavenging Antioxidant Activity
Astaxanthin extract was diluted with ethanol to make various concentrations (0.05, 0.1, 0.2, 0.25, 0.3, 0.4, 0.5, 1, 1.5, 2 μg/mL). Briefly, 0.25 mM 2,2-diphenyl-1-picrylhydrazyl (DPPH) was prepared with methanol before the measurement, the blank solution was sample solvent (ethanol, 100 μL), and 100 μL DPPH reagent was mixed with 100 μL ethanol to serve as a control. Next, 100 μL astaxanthin samples and 100 μL DPPH reagent were added to 96-well plates, shaken gently and incubated for 20 min in the dark at room temperature. Finally, the absorbance of mixture solutions was determined at 517 nm by using an ELISA reader (Tecan, Infinite M200). The measurements were performed in triplicate. The radical-scavenging activity was expressed as
percentage of inhibition (AS %) and calculated by the following equation:
4.4. ABTS Radical Scavenging Activity
The 2,2-azinobis (3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) assay determines the scavenging ability of antioxidant activity by reaction with a strong antioxidant agent (potassium permanganate or potassium persulfate) in the presence of ABTS salt [
30]. The ABTS stock solution was prepared by mixing 7 mM ABTS aqueous solution with 2.45 mM aqueous solution of potassium peroxodisulfate in equal quantities and allowed to react at room temperature in the dark for 12–16 h. To make sure the working solution was able to be used in the further experiment, the stock solution was diluted with distilled water, and the absorbance was measured at 734 nm by using an ELISA reader (Tecan, Infinite M200). The absorbance value must be in the range of 0.7 ± 0.02. Astaxanthin extract was diluted with ethanol to prepare various concentrations (0.05, 0.1, 0.2, 0.25, 0.3, 0.4, 0.5, 1, 1.5, 2 μg/mL). Then, 40 μL astaxanthin samples and 160 μL ABTS working solution were added to 96-well plates carefully, and the mixtures were shaken tenderly and incubated in the dark at 37 °C for 10 min. Finally, the absorbance of the mixture solutions was determined at 734 nm by using an ELISA reader (Tecan, Infinite M200).
4.5. Liposomal Formulation
The preparation of astaxanthin-loaded liposomes was performed based on the modified thin-film hydration method [
31]. Ong et al., demonstrated that liposomes prepared using chloroform possessed better encapsulation efficiency [
23]. Therefore, chloroform was chosen as the solvent for the preparation of astaxanthin-loaded liposomes. First, 100 mg of phospholipids was dissolved in 8 mL of chloroform. Next, different amounts of astaxanthin were dissolved in 2 mL of ethanol. Later, both phospholipid and astaxanthin solutions were mixed together in a round-bottom flask. The organic solvent of the astaxanthin/phospholipid mixtures was evaporated by a rotary evaporator (Eyela, N-1000, Tokyo, Japan) at 45 °C and then vacuum-dried to form a lipid film. Next, 2 mL phosphate-buffered saline was used to rehydrate the dry lipid film to form liposomes. Finally, the liposomes were downsized by sequence passing through polycarbonate membranes with pore sizes of 400 nm and 200 nm using an extruder (Avanti Mini-Extruder, Alabaster, AL, USA) for uniform and reduced particle size. Empty liposomes were prepared by the same procedure but only with drug-free ethanol.
4.6. Particle Characterization
The particle size of liposomes was measured using a dynamic light scattering instrument (LB-550, Horiba Ltd., Kyoto, Japan). Liposomal dispersions were diluted with double-distilled water at the ratio of 5:1 in cuvettes to guarantee the light scattering intensity in the instrument’s sensitivity range. The particle size and polydispersity index (PDI) of liposomes were measured immediately after liposomal extrusion. The liposomes were incubated with culture medium at the ratio of 1:10 at different temperatures (4 °C and 37 °C) for 1, 4, 7 and 14 days. All measurements were taken in triplicate. The polydispersity index (PDI) was calculated by the following equation according to the average value of the particle size:
4.7. Entrapment Efficiency
Once the liposomes were made, high-speed centrifugation was used to analyze the amount of astaxanthin loaded in liposomes. Astaxanthin-loaded liposomes were spun at 80,000 rpm for about 30 min using a Beckman ultra-high centrifuge. Then, the supernatants which contained unentrapped astaxanthin were carefully withdrawn. Next, the pellets were dissolved with the same volume of ethanol, and absorbance was measured at 480 nm, which is one of the major absorbance peaks of astaxanthin, by an ELISA reader (Tecan, Infinite M200). The entrapment efficiency of astaxanthin in liposomes was calculated by the standard curve. The entrapment efficiency (EE) was estimated using the following equation:
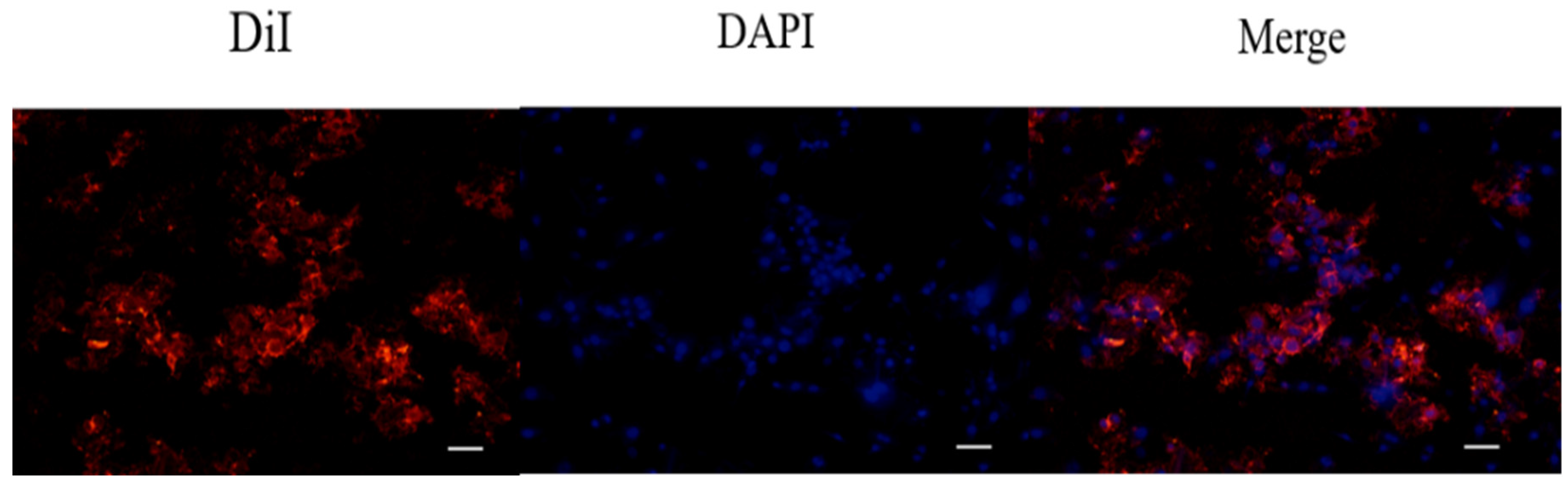
4.8. Determination of Cell Uptake of DiI-Labeled Liposomes in 7F2 Osteoblasts by Fluorescence Staining
After liposomal formulation, a lipophilic solution of DiI (1,1′-dioctadecyl-3,3,3′3′-tetramethylindocarbocyanine perchlorate, St Louis, MO, USA) was used for the fluorescent staining in order to investigate cell uptake of astaxanthin-loaded nanoparticle liposomes. First, 1 μL of DiI stock solution (10 mg of DiI powder dissolved in 1 mL of ethanol) was added to the phospholipid solution to form DiI-loaded liposomes. Briefly, 7F2 osteoblast-like cells were seeded in a 3.5 cm dish at a density of 5 × 104 cells/dish. After 24 h, the cell culture medium was replaced with a basal medium containing DiI-loaded liposomes, and cells were incubated for 4 h. Later, 4% formaldehyde was used to fix the cells for 30 min, and the cells were then rinsed with PBS and stained with DAPI dye (2-(4-amidinophenyl)-1H-indole-6-carboxamidine, 10 μg/mL) for 10 min. After washing with PBS twice, the cells were soaked with 1 mL PBS and photographed using a microscope (Nikon TI-E) and a CCD camera system (SPOT RT3). The photographs were quantified by ImageJ software.
4.9. Cell Viability and Proliferation Assay
A stock thiazolyl blue tetrazolium bromide (MTT, Sigma-Aldrich, USA) solution (5.0 mg/mL in phosphate-buffered saline (PBS) was prepared immediately prior to use and filtered through a 0.22 m Millipore filter (Burlington, MA, USA). The MTT solution was stored in 1.5 mL centrifuge tubes in the dark at −20 °C. Briefly, 7F2 osteoblast-like cells and Raw264.7 mouse macrophage cells were seeded in 96-well plates at a density of 1 × 104 cells/well and 5 × 104 cells/well, respectively. After seeding, cells were treated with DMEM medium containing astaxanthin and asta-loaded liposomes at various concentrations for 24 h at 37 °C with 5% (v/v) CO2. Next, the cell supernatants were withdrawn and 100 μL of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) solution was added to each well for 4 h of incubation. Once the MTT reagent was removed, the formazan product was dissolved in 200 μL dimethyl sulfoxide (DMSO), and the absorbance was measured at 570 nm with an ELISA reader (Tecan, Infinite M200). The measurements were performed in quadruplicate, and cell viability was expressed as a percentage of formazan absorbance compared to the control.
4.10. Determination of Anti-Inflammatory Activity by Nitrite Assay
The nitrite production in Raw264.7 macrophage cells was measured by Griess reagent. Briefly, mouse macrophage Raw264.7 cells were seeded in 24-well plates at a density of 4 × 105 cells/well and cultured at 37 °C with 5% CO2 (v/v) overnight. Next, media were withdrawn and cells were treated with 1 mL of culture medium containing various concentrations of astaxanthin extract in the presence of lipopolysaccharide (LPS, 0.5 μg/mL) for 24 h of incubation. Cells treated with culture medium and 0.5 μg/mL of LPS were indicated as positive control. After removing supernatants, 400 μL of no-phenol red medium was added for 6 h of incubation. NO release from LPS-induced macrophages was analyzed by determining nitrite concentration. Later, 100 μL aliquots of nitrite-containing supernatants were mixed with the same volume of Griess reagent in 96-well plates and gently shaken in the dark at room temperature for 15 min. To quantify the nitrite concentration, the absorbance of the mixture solutions was evaluated using an ELISA reader (Tecan, Infinite M200) at a wavelength of 550 nm. The data were shown as the mean percentage of absorbance in comparison with the LPS-treated group.
4.11. Osteoclast Differentiation Assay
TRAP activity was determined by para-nitrophenylphosphate (pNPP) according to the microplate assay method of Park et al., with the following modifications [
28]: Cells were fixed with 10%
w/
v formaldehyde for 1 min and then treated with an equal mixture of acetone and formalin for another minute. After desiccation, cells were incubated with 100 μL phosphate substrate solution (3.7 mM pNPP and 10 mM sodium tartrate in 50 mM citrate butter, pH 4.6) at 37 °C for 10 min. Then, the enzyme reaction was stopped by the addition of 100 μL 0.1 N sodium hydroxide solution, and the absorbance of the resulting yellow color product was measured by an ELISA reader at a wavelength of 405 nm.
4.12. Detection of Intracellular Reactive Oxygen Species (ROS) by DCF-DA Staining
Cellular ROS levels can be evaluated in live cells by a technique that converts 2′,7′-dichlorofluorescin diacetate (DCF-DA) to a high fluorescence dye (green), 2′,7′-dichlorofluorescein (DCF), upon oxidation. 2′,7′-Dichlorofluorescin diacetate (DCF-DA, Cayman Chemical, Ann Arbor, MI, USA) stock solution was dissolved in ethanol at a concentration of 5 mg/mL and further diluted with PBS saline to 25 μM prior to use. Briefly, Raw264.7 mouse macrophages were seeded in 24-well plates at a density of 1.5 × 105 cells/well. After 24 h, cells were treated with LPS and various concentrations of asta-loaded liposomes (0.05, 0.25, 0.51 μg/mL). Cells were then washed twice with PBS, and fixed with 4% paraformaldehyde for 30 min in dark. Thereafter, cells were rinsed again with PBS, and 25 μM of DCF-DA solution was added to each well for 1 h of staining. Finally, the cells were soaked with PBS and photographed using a microscope (Nikon TI-E) and a CCD camera system (SPOT RT3). The related fluorescence DCF-DA intensity was quantified by ImageJ. Data were shown as the mean percentage in comparison to LPS-induced cells.
4.13. Cellular Alkaline Phosphatase (ALP) Assay
The ALP activity was assessed using a colorimetric alkaline phosphatase assay kit. Firstly, the ALP detection reagent was prepared by mixing Alkaline Phosphatase Blue Microwell Substrate Component A&B (SIGMA-ALDRICH) at the ratio of 1:1 at room temperature. Briefly, 7F2 mouse osteoblast-like cells were seeded in 24-well plates at a density of 1 × 104 cells/well. In this experiment, 7F2 osteoblasts were cultivated with mineralization medium (DMEM medium containing 10% FBS, 1% penicillin–streptomycin, 1% L-glutamine, 5 mM β-glycerophosphate and 50 μg/mL ascorbic acid). Then, the cells were treated with the mineralization medium containing various concentrations of asta-loaded liposomes and incubated for 1, 4 and 7 days at 37 °C with 5% CO2 (v/v) atmosphere. Subsequently, the media were withdrawn and cells were rinsed with PBS carefully. Next, 300 μL of 1% Triton-X100 lysis buffer was added and incubated at 37 °C with 5% CO2 (v/v) atmosphere for 10 min to lyse the cells. The cell culture supernatants were moved into the 1.5 mL microtubes and centrifuged at 10,000 rpm for 5 min. Later, 100 μL aliquots of the supernatants were transferred to the 96-well plates, and the same volume of the ALP detection reagent was added to each well to react for 15 min in the dark. The absorbance was measured by an ELISA reader (Tecan, Infinite M200) at a wavelength of 560 nm. The measurements were taken in triplicate.
4.14. Mineralization of the Extracellular Matrix
Calcium content measurement and alizarin red S (ARS) staining were performed to assess the mineralization process of 7F2 osteoblasts on days 1, 7 and 14 after cells were treated with asta-loaded liposomes. Briefly, 7F2 osteoblast cells were seeded in a 24-well culture plate at a density of 104 cells/well. In this experiment, 7F2 osteoblasts were cultivated with mineralization medium (DMEM medium containing 10% FBS, 1% penicillin–streptomycin, 1% L-glutamine, 5 mM β-glycerophosphate and 50 μg/mL ascorbic acid) containing various concentrations of asta-loaded liposomes. The cells were washed twice with PBS and fixed with 75% ethanol for 20 min at 37 °C. Then, the cells were stained with 200 μL of 1% alizarin red S solution for an hour and rinsed with PBS until the supernatants became colorless. The calcified depositions, which appeared in maroon red, were imaged using a microscope (Nicon TI-E) and CCD camera system (SPOT RT3). To quantify the calcium production, 400 μL of 10% w/v cetylpyridinium chloride (CPC) was added to each well and gently shaken for 10 min to dissolve the calcified depositions. Eventually, the absorbance was measured by an ELISA reader (Tecan, Infinite M200) at a wavelength of 560 nm. The experiments were performed in triplicate.
4.15. Quantitative Real-Time PCR
Cells were seeded at a density of 2 × 105 cells in 6 cm dishes and incubated for 24 h. Inflammatory responses in RAW264.7 cells were stimulated with LPS (0.5 μg/mL), and the cells were treated with 0.05 μg/mL asta-loaded liposomes for 24 h.
After treatment, total RNA was extracted using Trizol reagent (RiboZol, AMRESCO, Solon, OH, USA) following the protocol of the manufacturer’s instructions. For reverse transcription, 1 μg of the total RNA was converted to first-strand cDNA using a reverse transcription kit (Promega, Madison, WI, USA). The resulting cDNA (equivalent to 20 ng) was used in a StepOnePlus Real-Time PCR System using FastStart DNA Master-PLUS SYBR Green I (Applied Biosystems, Foster City, CA, USA). The designed primers are shown in
Table 1, and all primers used nucleotide sequences present in the PrimerBank database. Each sample was corrected using the mean cycle threshold (CT) value for GAPDH. Relative gene expression was analyzed using the ΔC
T method and expressed as fold change (2
−ΔCCT) T relative to the expression values in nonstimulated cells.
4.16. Statistical Analysis
Each experiment was performed in triplicate and repeated at least three times with similar results. The values are shown as means ± standard deviations. Data from all experiments were analyzed by Dunn’s post-test using SPSS Version 12 (IBN, New York, NY, USA) and Sigma Plot (San Jose, CA, USA). A p value less than 0.05 was considered statistically significant.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}