
Beta and Gamma Amino Acid-Substituted Benzenesulfonamides as Inhibitors of Human Carbonic Anhydrases

 , , , ,
, , , ,  and
and 
Abstract
:
1. Introduction
2. Results and Discussion
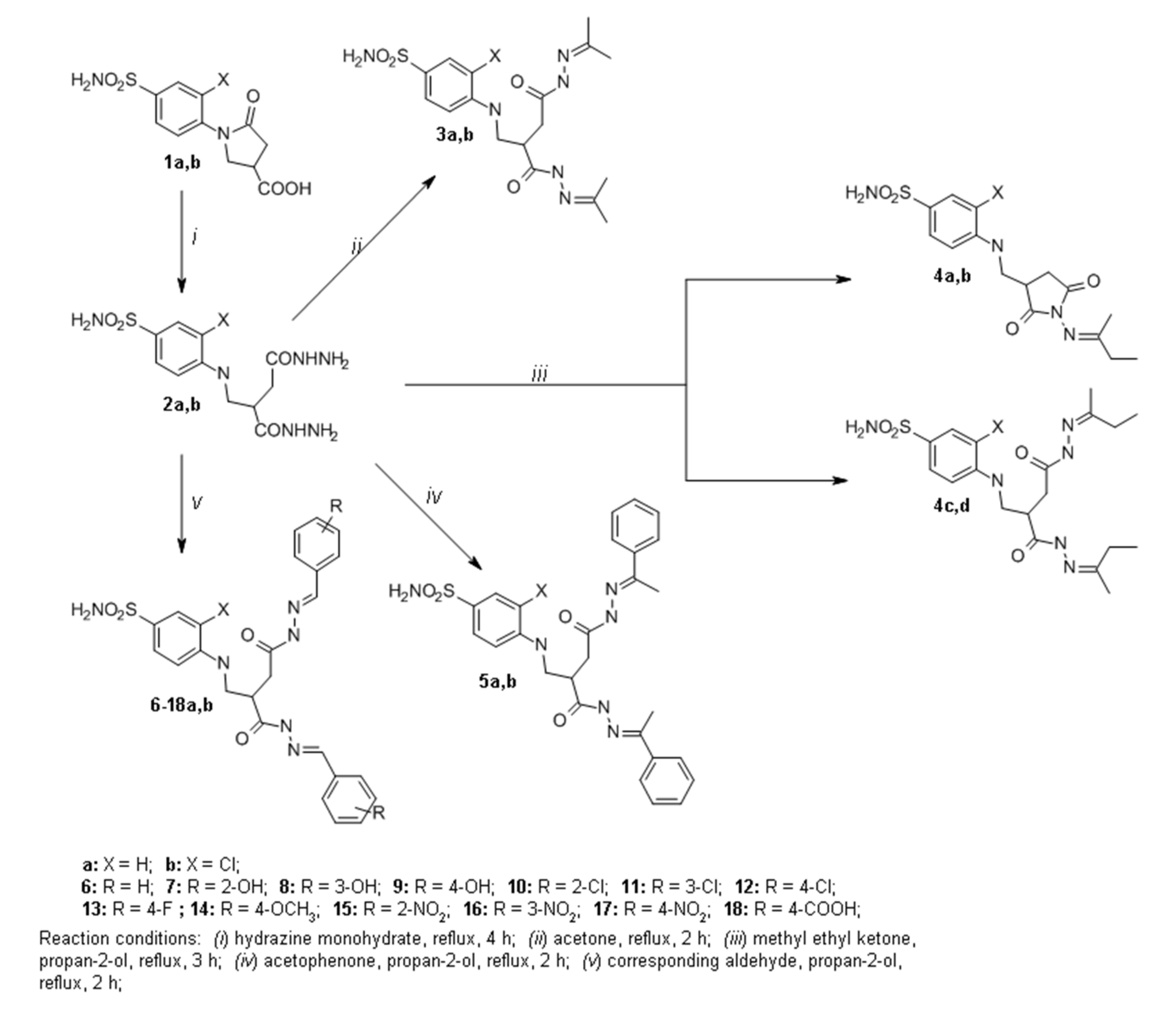
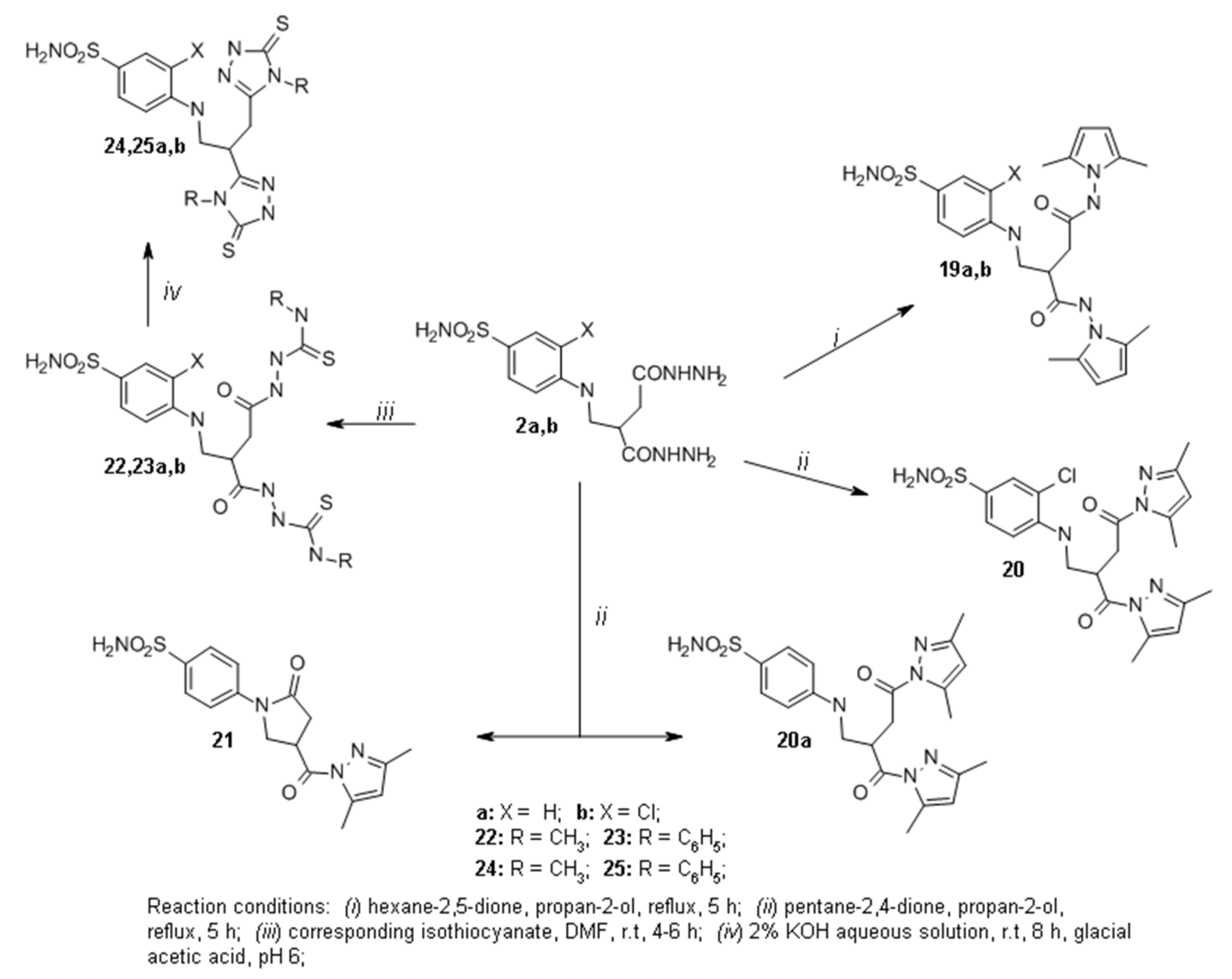
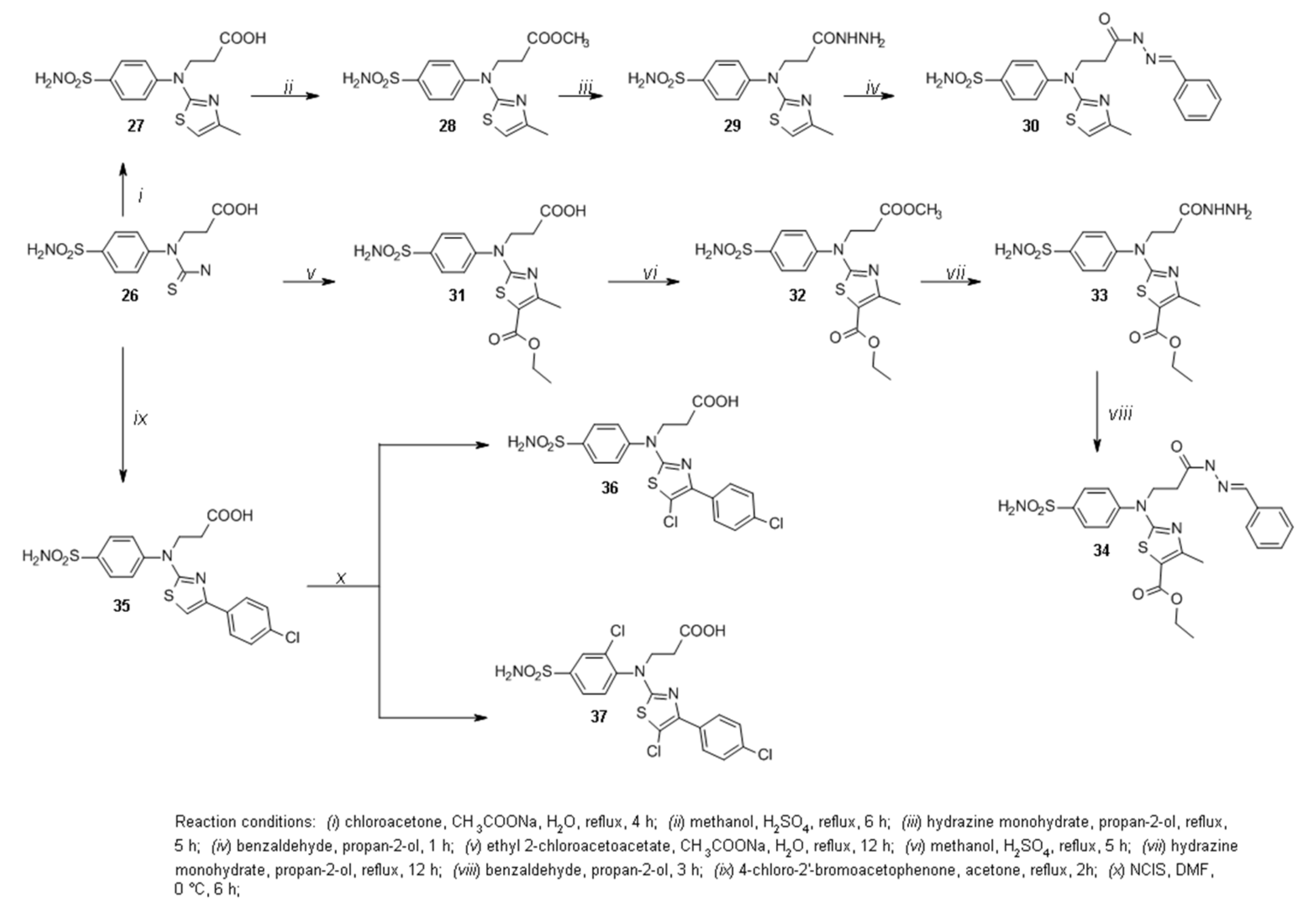
2.1. Organic Synthesis
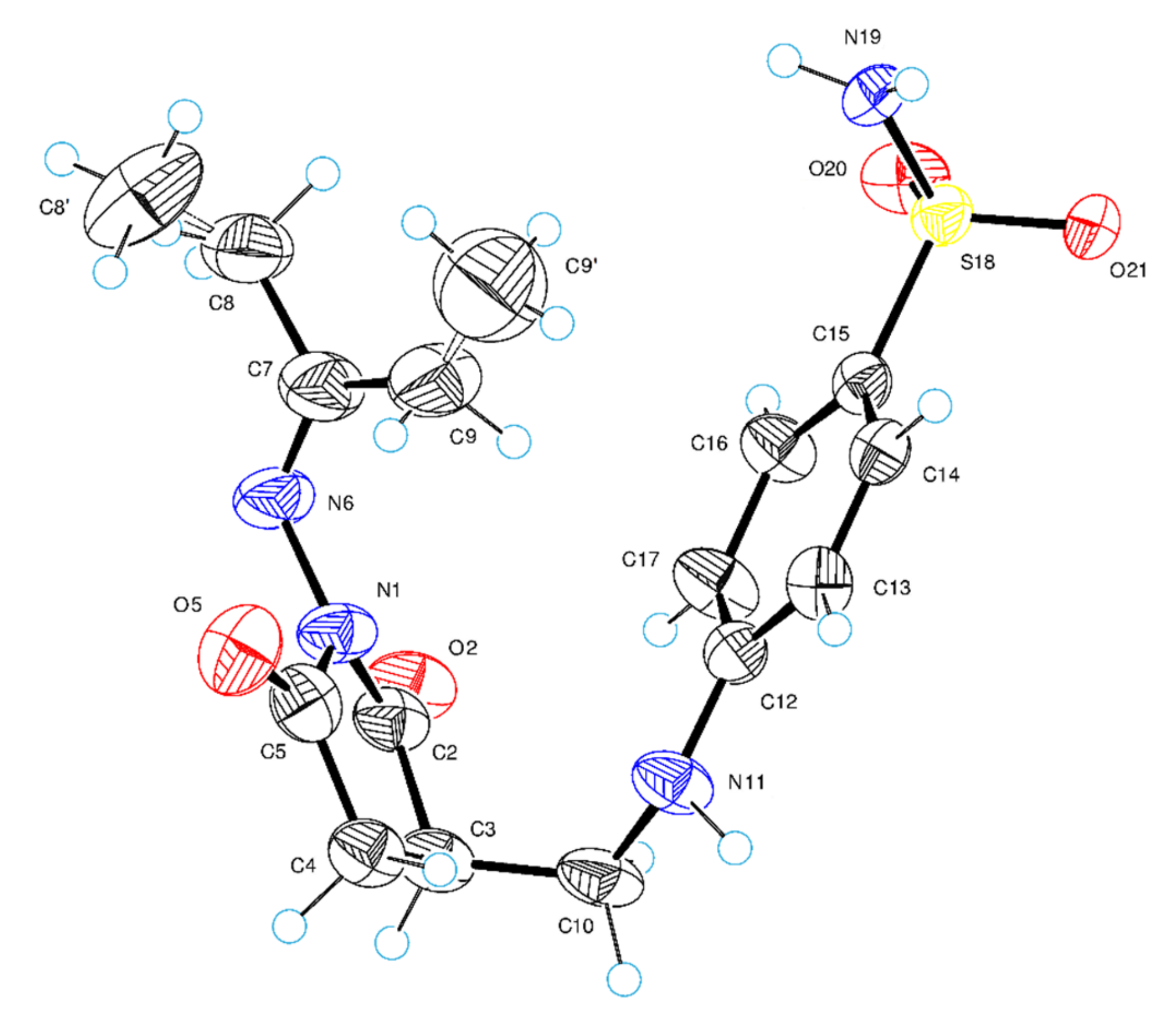
2.2. Crystal Structure of Compound 4a
2.3. Compound Binding to Carbonic Anhydrases (CA)
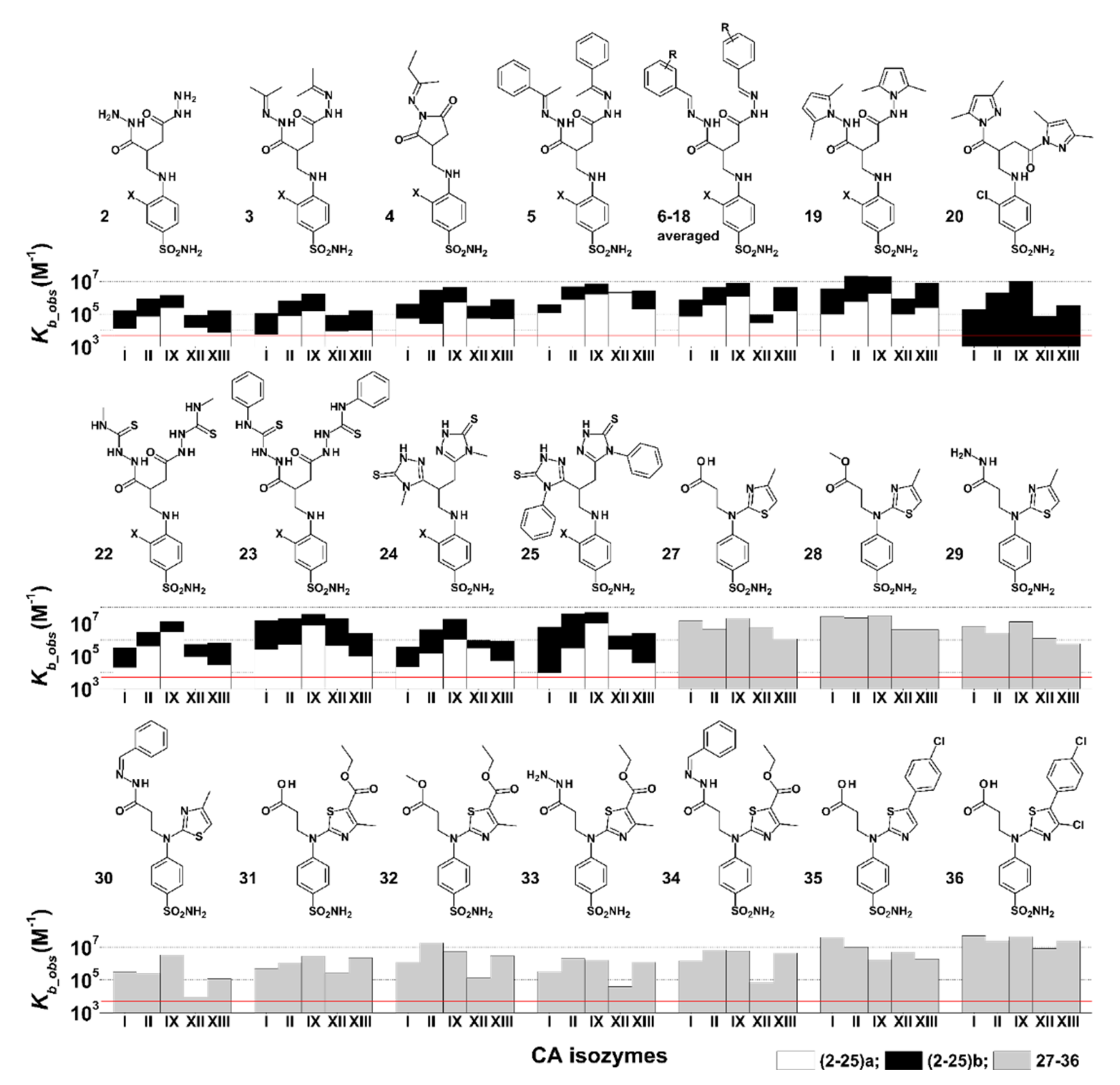
2.3.1. para-N β,γ-Amino Acid-Substituted Benzenesulfonamide Derivatives (2–25a,b) Binding to CA
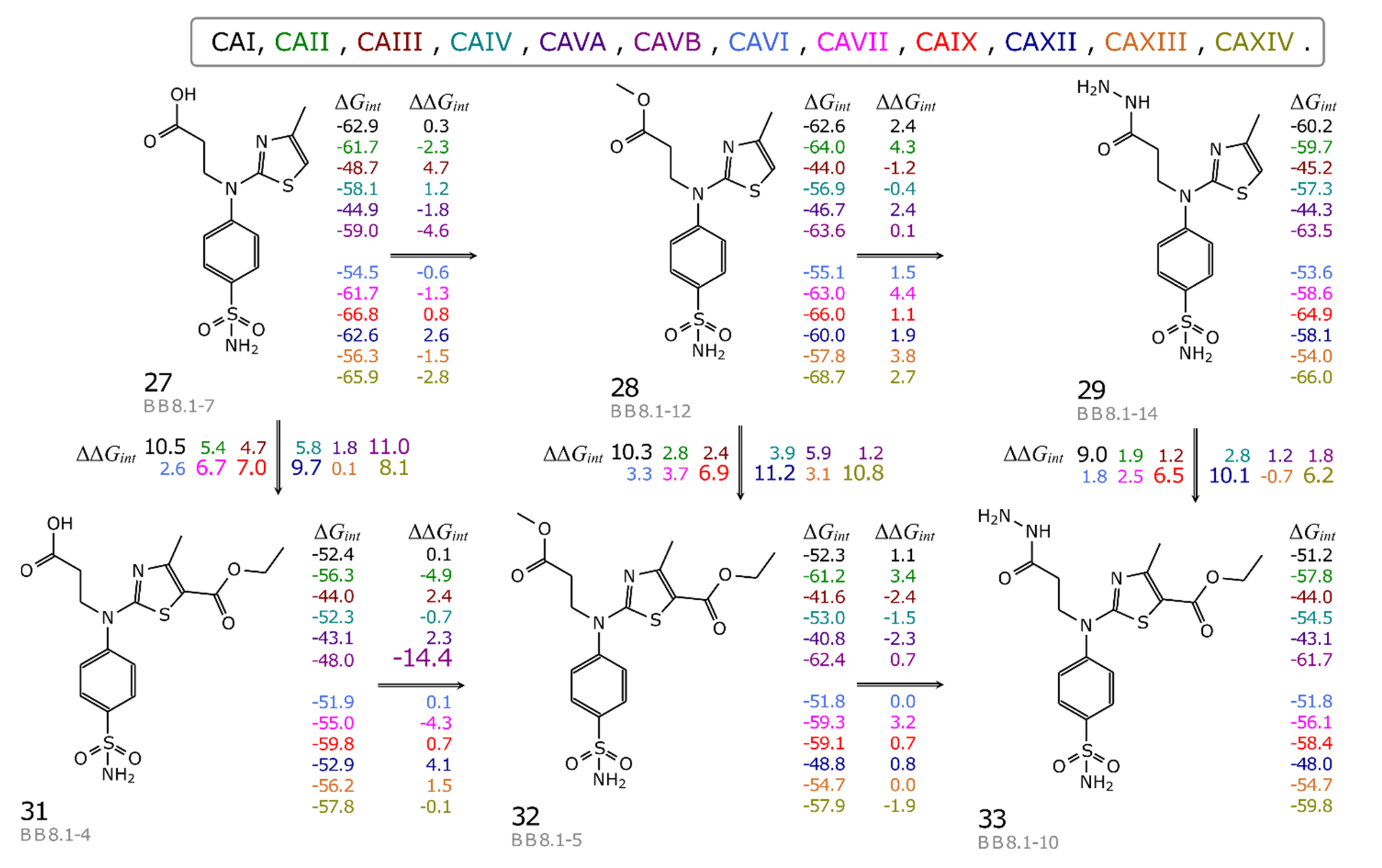
2.3.2. para-N β-Amino Acid and Thiazol Bearing Benzenesulfonamide Derivatives (27–36) Binding to CA
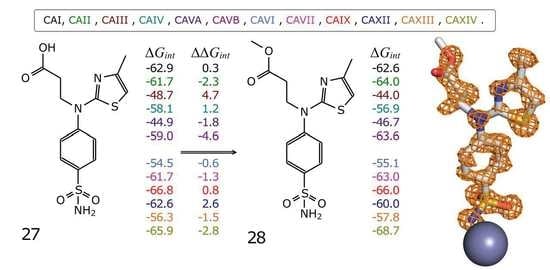
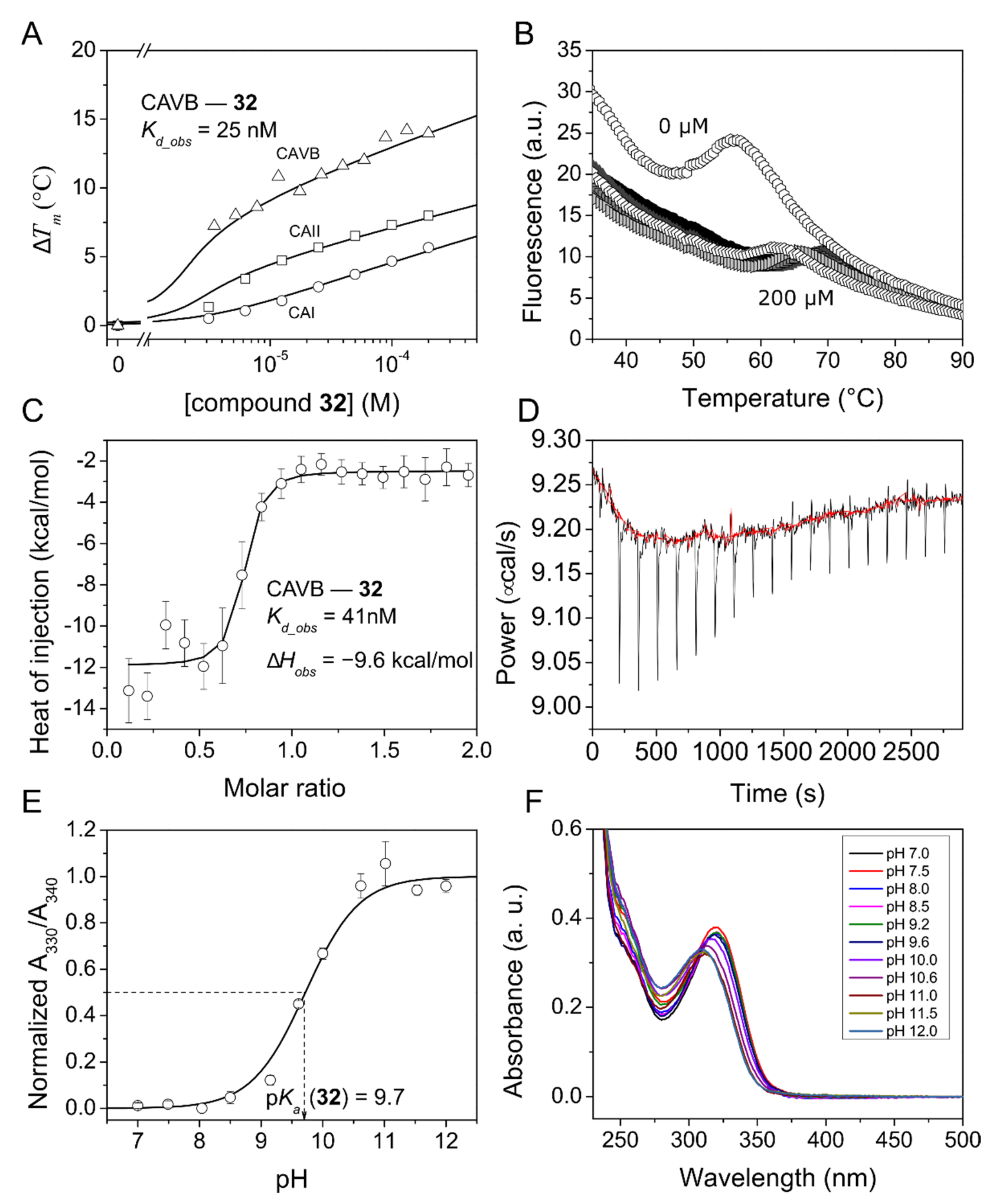
2.3.3. Intrinsic Thermodynamics of Compounds 27–36 Binding to CAs
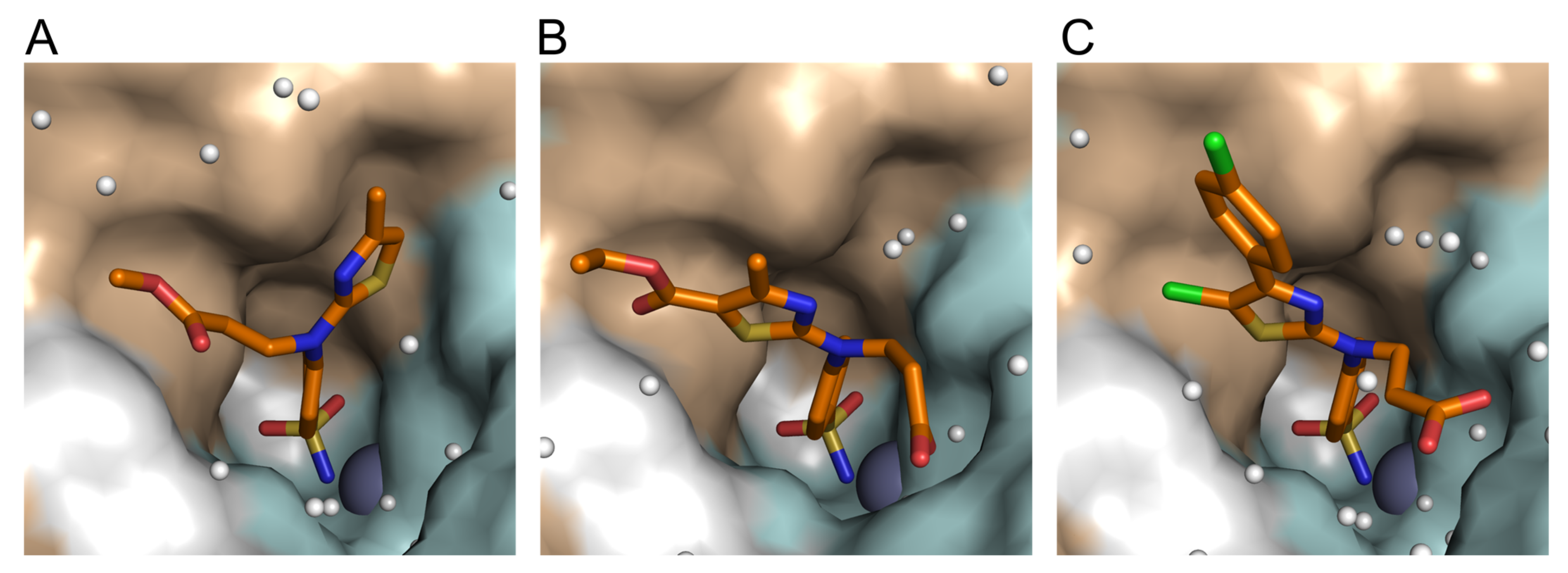
2.3.4. X-ray Crystallography of Human CAII Complexes with Inhibitors
3. Materials and Methods
3.1. Organic Synthesis
3.1.1. General Procedure for the Synthesis of Hydrazides 2a,b
4-((2-(Hydrazinecarbonyl)-4-hydrazineyl-4-oxobutyl)amino)benzenesulfonamide (2a)
3-Chloro-4-((2-(hydrazinecarbonyl)-4-hydrazineyl-4-oxobutyl)amino)benzenesulfonamide (2b)
3.1.2. General Procedure for the Synthesis of Hydrazones 3a,b
4-((4-Oxo-2-(2-(propan-2-ylidene)hydrazine-1-carbonyl)-4-(2-(propan-2-ylidene)hydrazineyl)butyl)amino)benzenesulfonamide (3a)
3-Chloro-4-((4-oxo-2-(2-(propan-2-ylidene)hydrazine-1-carbonyl)-4-(2-(propan-2-ylidene)hydrazineyl)butyl)amino)benzenesulfonamide (3b)
3.1.3. General Procedure for the Synthesis of Hydrazones 4a,b
4-(((1-(Butan-2-ylideneamino)-2,5-dioxopyrrolidin-3-yl)methyl)amino)benzenesulfonamide (4a)
4-(((1-(Butan-2-ylideneamino)-2,5-dioxopyrrolidin-3-yl)methyl)amino)-3-chlorobenzenesulfonamide (4b)
3.1.4. General Procedure for the Synthesis of Hydrazones 5a,b
4-((4-Oxo-2-(2-(1-phenylethylidene)hydrazine-1-carbonyl)-4-(2-(1-phenylethylidene)hydrazineyl)butyl)amino)benzenesulfonamide (5a)
3-Chloro-4-((4-oxo-2-(2-(1-phenylethylidene)hydrazine-1-carbonyl)-4-(2-(1-phenylethylidene)hydrazineyl)butyl)amino)benzenesulfonamide (5b)
3.1.5. General Procedure for the Synthesis of Hydrazones 6a,b–18a,b
4-((2-(2-(Benzylidene)hydrazine-1-carbonyl)-4-(2-(benzylidene)hydrazineyl)-4-oxobutyl)amino)benzenesulfonamide (6a)
4-((2-(2-(Benzylidene)hydrazine-1-carbonyl)-4-(2-(benzylidene)hydrazineyl)-4-oxobutyl)amino)-3-chlorobenzenesulfonamide (6b)
4-((2-(2-(2-Hydroxybenzylidene)hydrazine-1-carbonyl)-4-(2-(2-hydroxybenzylidene)hydrazineyl)-4-oxobutyl)amino)benzenesulfonamide (7a)
3-Chloro-4-((2-(2-(2-hydroxybenzylidene)hydrazine-1-carbonyl)-4-(2-(2-hydroxybenzylidene)hydrazineyl)-4-oxobutyl)amino)benzenesulfonamide (7b)
4-((2-(2-(3-Hydroxybenzylidene)hydrazine-1-carbonyl)-4-(2-(3-hydroxybenzylidene)hydrazineyl)-4-oxobutyl)amino)benzenesulfonamide (8a)
3-Chloro-4-((2-(2-(3-hydroxybenzylidene)hydrazine-1-carbonyl)-4-(2-(3-hydroxybenzylidene)hydrazineyl)-4-oxobutyl)amino)benzenesulfonamide (8b)
4-((2-(2-(4-Hydroxybenzylidene)hydrazine-1-carbonyl)-4-(2-(4-hydroxybenzylidene)hydrazineyl)-4-oxobutyl)amino)benzenesulfonamide (9a)
3-Chloro-4-((2-(2-(4-hydroxybenzylidene)hydrazine-1-carbonyl)-4-(2-(4-hydroxybenzylidene)hydrazineyl)-4-oxobutyl)amino)benzenesulfonamide (9b)
4-((2-(2-(2-Chlorobenzylidene)hydrazine-1-carbonyl)-4-(2-(2-chlorobenzylidene)hydrazineyl)-4-oxobutyl)amino)benzenesulfonamide (10a)
3-Chloro-4-((2-(2-(2-chlorobenzylidene)hydrazine-1-carbonyl)-4-(2-(2-chlorobenzylidene)hydrazineyl)-4-oxobutyl)amino)benzenesulfonamide (10b)
4-((2-(2-(3-Chlorobenzylidene)hydrazine-1-carbonyl)-4-(2-(3-chlorobenzylidene)hydrazineyl)-4-oxobutyl)amino)benzenesulfonamide (11a)
3-Chloro-4-((2-(2-(3-chlorobenzylidene)hydrazine-1-carbonyl)-4-(2-(3-chlorobenzylidene)hydrazineyl)-4-oxobutyl)amino)benzenesulfonamide (11b)
4-((2-(2-(4-Chlorobenzylidene)hydrazine-1-carbonyl)-4-(2-(4-chlorobenzylidene)hydrazineyl)-4-oxobutyl)amino)benzenesulfonamide (12a)
3-Chloro-4-((2-(2-(4-chlorobenzylidene)hydrazine-1-carbonyl)-4-(2-(4-chlorobenzylidene)hydrazineyl)-4-oxobutyl)amino)benzenesulfonamide (12b)
4-((2-(2-(4-Fluorobenzylidene)hydrazine-1-carbonyl)-4-(2-(4-fluorobenzylidene)hydrazineyl)-4-oxobutyl)amino)benzenesulfonamide (13a)
3-Chloro-4-((2-(2-(4-fluorobenzylidene)hydrazine-1-carbonyl)-4-(2-(4-fluorobenzylidene)hydrazineyl)-4-oxobutyl)amino)benzenesulfonamide (13b)
4-((2-(2-(4-Methoxybenzylidene)hydrazine-1-carbonyl)-4-(2-(4-methoxybenzylidene)hydrazineyl)-4-oxobutyl)amino)benzenesulfonamide (14a)
3-Chloro-4-((2-(2-(4-methoxybenzylidene)hydrazine-1-carbonyl)-4-(2-(4-methoxybenzylidene)hydrazineyl)-4-oxobutyl)amino)benzenesulfonamide (14b)
4-((2-(2-(2-Nitrobenzylidene)hydrazine-1-carbonyl)-4-(2-(2-nitrobenzylidene)hydrazineyl)-4-oxobutyl)amino)benzenesulfonamide (15a)
3-Chloro-4-((2-(2-(2-nitrobenzylidene)hydrazine-1-carbonyl)-4-(2-(2-nitrobenzylidene)hydrazineyl)-4-oxobutyl)amino)benzenesulfonamide (15b)
4-((2-(2-(3-Nitrobenzylidene)hydrazine-1-carbonyl)-4-(2-(3-nitrobenzylidene)hydrazineyl)-4-oxobutyl)amino)benzenesulfonamide (16a)
3-Chloro-4-((2-(2-(3-nitrobenzylidene)hydrazine-1-carbonyl)-4-(2-(3-nitrobenzylidene)hydrazineyl)-4-oxobutyl)amino)benzenesulfonamide (16b)
4-((2-(2-(4-Nitrobenzylidene)hydrazine-1-carbonyl)-4-(2-(4-nitrobenzylidene)hydrazineyl)-4-oxobutyl)amino)benzenesulfonamide (17a)
3-Chloro-4-((2-(2-(4-nitrobenzylidene)hydrazine-1-carbonyl)-4-(2-(4-nitrobenzylidene)hydrazineyl)-4-oxobutyl)amino)benzenesulfonamide (17b)
4,4′-(((2-(((4-Sulfamoylphenyl)amino)methyl)succinyl)bis(hydrazin-2-yl-1-ylidene))bis(methaneylylidene))dibenzoic acid (18a)
4,4′-(((2-(((2-Chloro-4-sulfamoylphenyl)amino)methyl)succinyl)bis(hydrazin-2-yl-1-ylidene))bis(methaneylylidene))dibenzoic acid (18b)
3.1.6. General Procedure for the Synthesis of Pyrroles 19a,b
N1,N4-bis(2,5-Dimethyl-1H-pyrrol-1-yl)-2-(((4-sulfamoylphenyl)amino)methyl)succinamide (19a)
2-(((2-Chloro-4-sulfamoylphenyl)amino)methyl)-N1,N4-bis(2,5-dimethyl-1H-pyrrol-1-yl)succinamide (19b)
3.1.7. 3-Chloro-4-((4-(3,5-dimethyl-1H-pyrazol-1-yl)-2-(3,5-dimethyl-1H-pyrazole-1-carbonyl)-4-oxobutyl)amino)benzenesulfonamide (20)
3.1.8. General Procedure for the Synthesis of Compounds 22a,b and 23a,b
2,2′-(2-(((4-Sulfamoylphenyl)amino)methyl)succinyl)bis(N-methylhydrazine-1-carbothioamide) (22a)
2,2′-(2-(((2-Chloro-4-sulfamoylphenyl)amino)methyl)succinyl)bis(N-methylhydrazine-1-carbothioamide (22b)
2,2′-(2-(((4-Sulfamoylphenyl)amino)methyl)succinyl)bis(N-phenylhydrazine-1-carbothioamide) (23a)
2,2′-(2-(((2-Chloro-4-sulfamoylphenyl)amino)methyl)succinyl)bis(N-phenylhydrazine-1-carbothioamide) (23b)
3.1.9. General Procedure for the Synthesis of Compounds 24a,b and 25a,b
4-((2,3-bis(4-Methyl-5-thioxo-4,5-dihydro-1H-1,2,4-triazol-3-yl)propyl)amino)benzenesulfonamide (24a)
4-((2,3-bis(4-Methyl-5-thioxo-4,5-dihydro-1H-1,2,4-triazol-3-yl)propyl)amino)-3-chlorobenzenesulfonamide (24b)
4-((2,3-bis(4-Phenyl-5-thioxo-4,5-dihydro-1H-1,2,4-triazol-3-yl)propyl)amino)benzenesulfonamide (25a)
4-((2,3-bis(4-Phenyl-5-thioxo-4,5-dihydro-1H-1,2,4-triazol-3-yl)propyl)amino)-3-chlorobenzenesulfonamide (25b)
3.1.10. 3-((4-Methylthiazol-2-yl)(4-sulfamoylphenyl)amino)propanoic acid (27)
3.1.11. Methyl 3-((4-Methylthiazol-2-yl)(4-sulfamoylphenyl)amino)propanoate (28)
3.1.12. 4-((3-Hydrazineyl-3-oxopropyl)(4-methylthiazol-2-yl)amino)benzenesulfonamide (29)
3.1.13. 4-((3-(2-Benzylidenehydrazineyl)-3-oxopropyl)(4-methylthiazol-2-yl)amino)benzenesulfonamide (30)
3.1.14. 3-((5-(Ethoxycarbonyl)-4-methylthiazol-2-yl)(4-sulfamoylphenyl)amino)propanoic acid (31)
3.1.15. Ethyl 2-((3-Methoxy-3-oxopropyl)(4-sulfamoylphenyl)amino)-4-methylthiazole-5-carboxylate (32)
3.1.16. Ethyl 2-((3-(Hydrazineyloxy)-3-oxopropyl)(4-sulfamoylphenyl)amino)-4-methylthiazole-5-carboxylate (33)
3.1.17. Ethyl 2-((3-(2-Benzylidenehydrazineyl)-3-oxopropyl)(4-sulfamoylphenyl)amino)-4-methylthiazole-5-carboxylate (34)
3.1.18. 3-((5-Chloro-4-(4-chlorophenyl)thiazol-2-yl)(4-sulfamoylphenyl)amino)propanoic acid (36)
3.2. Determination of Compounds Binding to Human CA Isozymes
3.2.1. Protein Preparation
3.2.2. Fluorescent Thermal Shift Assay (FTSA)
3.2.3. Isothermal Titration Calorimetry (ITC)
3.2.4. Determination of Compound Sulfonamide Group pKa
3.2.5. Calculation of the Intrinsic Thermodynamic Parameters
3.2.6. X-ray Crystallography: Crystallization, Data Collection, and Structure Determination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meldrum, N.U.; Roughton, F.J.W. Carbonic anhydrase. Its preparation and properties. J. Physiol. 1933, 80, 113–142. [Google Scholar] [CrossRef] [PubMed]
- Lindskog, S.; Coleman, J.E. The catalytic mechanism of carbonic anhydrase. Proc. Natl. Acad. Sci. USA 1973, 70, 2505–2508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodgson, S.J. The Carbonic Anhydrases: Overview of Their Importance in Cellular Physiology and in Molecular Genetics. In The Carbonic Anhydrases: Cellular Physiology and Molecular Genetics; Dodgson, S.J., Tashian, R.E., Gros, G., Carter, N.D., Eds.; Springer: Boston, MA, USA, 1991; pp. 3–9. [Google Scholar]
- Parkkila, S. An overview of the distribution and function of carbonic anhydrase isozymes in mammals. In The Carbonic Anhydrases: New Horizons; Chegwidden, W.R., Carter, N.D., Edwards, Y.H., Eds.; Birkhäuser Basel: Basel, Switzerland, 2000; pp. 79–93. [Google Scholar]
- Krishnamurthy, V.M.; Kaufman, G.K.; Urbach, A.R.; Gitlin, I.; Gudiksen, K.L.; Weibel, D.B.; Whitesides, G.M. Carbonic Anhydrase as a Model for Biophysical and Physical-Organic Studies of Proteins and Protein−Ligand Binding. Chem. Rev. 2008, 108, 946–1051. [Google Scholar] [CrossRef] [Green Version]
- McKenna, R.; Frost, S.C. Overview of the carbonic anhydrase family. Sub-Cell. Biochem. 2014, 75, 3–5. [Google Scholar] [CrossRef]
- Aspatwar, A.; Tolvanen, M.E.E.; Ortutay, C.; Parkkila, S. Carbonic anhydrase related proteins: Molecular biology and evolution. Sub-Cell. Biochem. 2014, 75, 135–156. [Google Scholar]
- Frost, S.C. Physiological functions of the alpha class of carbonic anhydrases. Sub-Cell. Biochem. 2014, 75, 9–30. [Google Scholar] [CrossRef]
- Chegwidden, W.R. The Carbonic anhydrases in Health and Disease. In The Carbonic Anhydrases: Current and Emerging Therapeutic Targets; Chegwidden, W.R., Carter, N.D., Eds.; Progress in Drug Research; Springer International Publishing: Cham, Switzerland, 2021; pp. 1–12. [Google Scholar]
- Aggarwal, M.; Boone, C.D.; Kondeti, B.; McKenna, R. Structural annotation of human carbonic anhydrases. J. Enzym. Inhib. Med. Chem. 2013, 28, 267–277. [Google Scholar] [CrossRef]
- Pinard, M.A.; Mahon, B.; McKenna, R. Probing the Surface of Human Carbonic Anhydrase for Clues towards the Design of Isoform Specific Inhibitors. BioMed Res. Int. 2015, 2015, 453543. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, M.; Kondeti, B.; McKenna, R. Insights towards sulfonamide drug specificity in a-carbonic anhydrases. Bioorg. Med. Chem. 2013, 21, 1526–1533. [Google Scholar] [CrossRef] [Green Version]
- Mann, T.; Keilin, D. Sulphanilamide as a Specific Inhibitor of Carbonic Anhydrase. Nature 1940, 146, 164–165. [Google Scholar] [CrossRef]
- Poulsen, S.-A. Carbonic anhydrase inhibition as a cancer therapy: A review of patent literature, 2007–2009. Expert Opin. Ther. Pat. 2010, 20, 795–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, M.; McKenna, R. Update on carbonic anhydrase inhibitors: A patent review (2008–2011). Expert Opin. Ther. Pat. 2012, 22, 903–915. [Google Scholar] [CrossRef]
- Carta, F.; Scozzafava, A.; Supuran, C.T. Sulfonamides: A patent review (2008–2012). Expert Opin. Ther. Pat. 2012, 22, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Carta, F.; Supuran, C.T.; Scozzafava, A. Novel therapies for glaucoma: A patent review 2007–2011. Expert Opin. Ther. Pat. 2012, 22, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Carta, F.; Supuran, C.T. Diuretics with carbonic anhydrase inhibitory action: A patent and literature review (2005–2013). Expert Opin. Ther. Pat. 2013, 23, 681–691. [Google Scholar] [CrossRef] [Green Version]
- Monti, S.M.; Supuran, C.T.; De Simone, G. Anticancer carbonic anhydrase inhibitors: A patent review (2008–2013). Expert Opin. Ther. Pat. 2013, 23, 737–749. [Google Scholar] [CrossRef]
- Lomelino, C.; McKenna, R. Carbonic anhydrase inhibitors: A review on the progress of patent literature (2011–2016). Expert Opin. Ther. Pat. 2016, 26, 947–956. [Google Scholar] [CrossRef]
- Gulçin, İ.; Taslimi, P. Sulfonamide inhibitors: A patent review 2013-present. Expert Opin. Ther. Pat. 2018, 28, 541–549. [Google Scholar] [CrossRef]
- Nocentini, A.; Supuran, C.T. Carbonic anhydrase inhibitors as antitumor/antimetastatic agents: A patent review (2008–2018). Expert Opin. Ther. Pat. 2018, 28, 729–740. [Google Scholar] [CrossRef]
- Nocentini, A.; Supuran, C.T. Adrenergic agonists and antagonists as antiglaucoma agents: A literature and patent review (2013–2019). Expert Opin. Ther. Pat. 2019, 29, 805–815. [Google Scholar] [CrossRef]
- Supuran, C.T.; Altamimi, A.S.A.; Carta, F. Carbonic anhydrase inhibition and the management of glaucoma: A literature and patent review 2013–2019. Expert Opin. Ther. Pat. 2019, 29, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Guglielmi, P.; Carradori, S.; Campestre, C.; Poce, G. Novel therapies for glaucoma: A patent review (2013–2019). Expert Opin. Ther. Pat. 2019, 29, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Quan, J.; Zhao, Y.; Yang, D.; Zhao, Q.; Liu, P.; Cheng, M.; Ma, C. Design, synthesis and biological evaluation of 1-benzyl-5-oxopyrrolidine-2-carboximidamide derivatives as novel neuroprotective agents. Eur. J. Med. Chem. 2019, 182, 111654. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, L.W.; Abuel-Maaty, S.M.; Mohammed, W.A.; Galal, M.A. Synthesis and biological evaluation of new oxopyrrolidine derivatives as inhibitors of acetyl cholinesterase and β amyloid protein as anti—Alzheimer’s agents. Bioorg. Chem. 2018, 76, 210–217. [Google Scholar] [CrossRef]
- Vaškevičienė, I.; Paketurytė, V.; Pajanok, N.; Žukauskas, Š.; Sapijanskaitė, B.; Kantminienė, K.; Mickevičius, V.; Zubrienė, A.; Matulis, D. Pyrrolidinone-bearing methylated and halogenated benzenesulfonamides as inhibitors of carbonic anhydrases. Bioorg. Med. Chem. 2019, 27, 322–337. [Google Scholar] [CrossRef]
- Balandis, B.; Ivanauskaitė, G.; Smirnovienė, J.; Kantminienė, K.; Matulis, D.; Mickevičius, V.; Zubrienė, A. Synthesis and structure–affinity relationship of chlorinated pyrrolidinone-bearing benzenesulfonamides as human carbonic anhydrase inhibitors. Bioorg. Chem. 2020, 97, 103658. [Google Scholar] [CrossRef]
- Rutkauskas, K.; Zubrienė, A.; Tumosienė, I.; Kantminienė, K.; Mickevičius, V.; Matulis, D. Benzenesulfonamides bearing pyrrolidinone moiety as inhibitors of carbonic anhydrase IX: Synthesis and binding studies. Med. Chem. Res. 2017, 26, 235–246. [Google Scholar] [CrossRef]
- Vaškevičienė, I.; Paketurytė, V.; Zubrienė, A.; Kantminienė, K.; Mickevičius, V.; Matulis, D. N-Sulfamoylphenyl- and N-sulfamoylphenyl-N-thiazolyl-β-alanines and their derivatives as inhibitors of human carbonic anhydrases. Bioorg. Chem. 2017, 75, 16–29. [Google Scholar] [CrossRef]
- Peleckis, A.; Anusevičius, K.; Šiugždaitė, J.; Mickevičius, V. Nucleophilic Ring Opening Of 1,4-Disubstituted 2-Pyrrolidones with Hydrazine. Synthesis of Azoles with a High Antibacterial Activity. Chemija 2018, 29, 135–144. Available online: http://lmaleidykla.lt/ojs/index.php/chemija/article/view/3717 (accessed on 10 June 2021). [CrossRef]
- Parašotas, I.; Kantminienė, K.; Urbonavičiūtė, E.; Anusevičius, K.; Tumosienė, I.; Jonuškienė, I.; Vaickelionienė, R.; Mickevičius, V. Synthesis and Biological Evaluation of Novel Di- and Trisubstituted Thiazole Derivatives. Heterocycles 2017, 94, 1074. Available online: http://www.heterocycles.jp/newlibrary/libraries/abst/25320 (accessed on 10 June 2021). [CrossRef]
- Tumosienė, I.; Peleckis, A.; Jonuškienė, I.; Vaickelionienė, R.; Kantminienė, K.; Šiugždaitė, J.; Beresnevičius, Z.J.; Mickevičius, V. Synthesis of Novel 1,2- and 2-Substituted Benzimidazoles with High Antibacterial and Antioxidant Activity. Mon. Für Chem.-Chem. Mon. 2018, 149, 577–594. Available online: http://link.springer.com/10.1007/s00706-017-2066-x (accessed on 10 June 2021). [CrossRef]
- Gudmundsson, K.S.; Tidwell, J.; Lippa, N.; Koszalka, G.W.; van Draanen, N.; Ptak, R.G.; Drach, J.C.; Townsend, L.B. Synthesis and Antiviral Evaluation of Halogenated β-d- and -l-Erythrofuranosylbenzimidazoles. J. Med. Chem. 2000, 43, 2464–2472. Available online: https://pubs.acs.org/doi/10.1021/jm990195p (accessed on 10 June 2021). [CrossRef] [PubMed]
- Pantoliano, M.W.; Petrella, E.C.; Kwasnoski, J.D.; Lobanov, V.S.; Myslik, J.; Graf, E.; Carver, T.; Asel, E.; Springer, B.A.; Lane, P.; et al. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J. Biomol. Screen. 2001, 6, 429–440. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, P.A.; Yanchunas, J.; Newitt, J.A.; Tao, L.; Kiefer, S.E.; Ortega, M.; Kut, S.; Burford, N.; Goldfarb, V.; Duke, G.J.; et al. Assessing compound binding to the Eg5 motor domain using a thermal shift assay. Anal. Biochem. 2009, 392, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Gao, K.; Oerlemans, R.; Groves, M.R. Theory and applications of differential scanning fluorimetry in early-stage drug discovery. Biophys. Rev. 2020, 12, 85–104. [Google Scholar] [CrossRef] [Green Version]
- Scott, D.E.; Spry, C.; Abell, C. Differential Scanning Fluorimetry as Part of a Biophysical Screening Cascade. In Methods and Principles in Medicinal Chemistry; Erlanson, D.A., Jahnke, W., Eds.; Wiley-VCH Verlag GmbH & Co. KgaA: Weinheim, Germany, 2016; pp. 139–172. [Google Scholar] [CrossRef]
- Zubrienė, A.; Matulis, D. Observed Versus Intrinsic Thermodynamics of Inhibitor Binding to Carbonic Anhydrases. In Carbonic Anhydrase as Drug Target: Thermodynamics and Structure of Inhibitor Binding; Matulis, D., Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 107–123. [Google Scholar] [CrossRef]
- Leavitt, S.; Freire, E. Direct Measurement of Protein Binding Energetics by Isothermal Titration Calorimetry. Curr. Opin. Struct. Biol. 2001, 11, 560–566. [Google Scholar] [CrossRef]
- Taylor, P.W.; King, R.W.; Burgen, A.S.V. Influence of pH on the kinetics of complex formation between aromatic sulfonamides and human carbonic anhydrase. Biochemistry 1970, 9, 3894–3902. [Google Scholar] [CrossRef]
- Engberg, P.; Lindskog, S. Effects of pH and inhibitors on the absorption spectrum of cobalt(II)-substituted carbonic anhydrase III from bovine skeletal muscle. FEBS Lett. 1984, 170, 326–330. [Google Scholar] [CrossRef] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Burla, M.C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; De Caro, L.; Giacovazzo, C.; Polidori, G.; Siliqi, D.; Spagna, R. IL MILIONE: A suite of computer programs for crystal structure solution of proteins. J. Appl. Crystallogr. 2007, 40, 609–613. [Google Scholar] [CrossRef]
- Bourhis, L.J.; Dolomanov, O.V.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. The anatomy of a comprehensive constrained, restrained refinement program for the modern computing environment—Olex2 dissected. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 59–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mickevičiūtė, A.; Juozapaitienė, V.; Michailovienė, V.; Jachno, J.; Matulienė, J.; Matulis, D. Recombinant Production of 12 Catalytically Active Human CA Isoforms. In Carbonic Anhydrase as Drug Target: Thermodynamics and Structure of Inhibitor Binding; Matulis, D., Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 15–37. [Google Scholar] [CrossRef]
- Cimmperman, P.; Baranauskienė, L.; Jachimovičiūtė, S.; Jachno, J.; Torresan, J.; Michailovienė, V.; Matulienė, J.; Sereikaitė, J.; Bumelis, V.; Matulis, D. A Quantitative Model of Thermal Stabilization and Destabilization of Proteins by Ligands. Biophys. J. 2008, 95, 3222–3231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, S.; Vargas, C.; Zhao, H.; Piszczek, G.; Brautigam, C.A.; Schuck, P. High-Precision Isothermal Titration Calorimetry with Automated Peak Shape Analysis. Anal. Chem. 2012, 84, 5066–5073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brautigam, C.A.; Zhao, H.; Vargas, C.; Keller, S.; Schuck, P. Integration and global analysis of isothermal titration calorimetry data for studying macromolecular interactions. Nat. Protocols. 2016, 11, 882–894. [Google Scholar] [CrossRef]
- Zhao, H.; Piszczek, G.; Schuck, P. SEDPHAT—A platform for global ITC analysis and global multi-method analysis of molecular interactions. Methods 2015, 76, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Snyder, P.W.; Mecinović, J.; Moustakas, D.T.; Thomas, S.W.; Harder, M.; Mack, E.T.; Lockett, M.R.; Héroux, A.; Sherman, W.; Whitesides, G.M. Mechanism of the hydrophobic effect in the biomolecular recognition of arylsulfonamides by carbonic anhydrase. Proc. Natl. Acad. Sci. USA 2011, 108, 17889–17894. [Google Scholar] [CrossRef] [Green Version]
- Fisher, S.Z.; Aggarwal, M.; Kovalevsky, A.Y.; Silverman, D.N.; McKenna, R. Neutron Diffraction of Acetazolamide-Bound Human Carbonic Anhydrase II Reveals Atomic Details of Drug Binding. J. Am. Chem. Soc. 2012, 134, 14726–14729. [Google Scholar] [CrossRef] [Green Version]
- Kovalevsky, A.; Aggarwal, M.; Velazquez, H.; Cuneo, M.J.; Blakeley, M.P.; Weiss, K.L.; Smith, J.C.; Fisher, S.Z.; McKenna, R. “To Be or Not to Be” Protonated: Atomic Details of Human Carbonic Anhydrase-Clinical Drug Complexes by Neutron Crystallography and Simulation. Structure 2018, 26, 383–390.e3. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, M.; Kovalevsky, A.Y.; Velazquez, H.; Fisher, S.Z.; Smith, J.C.; McKenna, R. Neutron structure of human carbonic anhydrase II in complex with methazolamide: Mapping the solvent and hydrogen-bonding patterns of an effective clinical drug. IUCrJ. 2016, 3, 319–325. [Google Scholar] [CrossRef]
- Kabsch, W. XDS . Acta Crystallogr. Sect. D 2010, D66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Battye, T.G.G.; Kontogiannis, L.; Johnson, O.; Powell, H.R.; Leslie, A.G.W. iMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 271–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, P. Scaling and assessment of data quality. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Vagin, A.; Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of ıt Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- Lebedev, A.A.; Young, P.; Isupov, M.N.; Moroz, O.V.; Vagin, A.A.; Murshudov, G.N. JLigand: A graphical tool for the CCP4 template-restraint library. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 431–440. [Google Scholar] [CrossRef] [Green Version]
- Vagin, A.A.; Steiner, R.A.; Lebedev, A.A.; Potterton, L.; McNicholas, S.; Long, F.; Murshudov, G.N. REFMAC5 dictionary: Organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2184–2195. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand–Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cmpd. | Lab. Name | Observed Dissociation Constant (Kd_obs), nM | ||||
|---|---|---|---|---|---|---|
| CAI | CAII | CAIX | CAXII | CAXIII | ||
| 2a | 2a | 78,000 | 14,000 | 4000 | 71,000 | 130,000 |
| 2b | 2b | 6700 | 1300 | 830 | 14,000 | 6700 |
| 3a | BB2.2-7 | 170,000 | 13,000 | 6700 | 110,000 | 100,000 |
| 3b | BB2.1-7 | 10,000 | 1800 | 670 | 14,000 | 6700 |
| 4a | BB2.2-8 | 18,000 | 40,000 | 2000 | 18,000 | 20,000 |
| 4b | BB2.1-8 | 2900 | 330 | 250 | 4000 | 1300 |
| 5a | BB2.2-19 | 8300 | 1300 | 590 | 500 | 5000 |
| 5b | BB2.1-19 | 4000 | 250 | 180 | 6700 | 400 |
| 6a | 3a | 14,000 | 1800 | 560 | 25,000 | 7700 |
| 7a | BB2.2-20 | 6700 | 2000 | 500 | 20,000 | 10,000 |
| 7b | BB2.1-20 | 1000 | 170 | 67 | 5000 | 170 |
| 8a | BB2.2-21 | 10,000 | 2000 | 670 | 33,000 | 5000 |
| 8b | BB2.1-21 | 1000 | 250 | 100 | 20,000 | 200 |
| 9a | BB2.2-22 | 5900 | 1300 | 500 | 25,000 | 4300 |
| 10a | BB2.2-24 | 17,000 | 2500 | 500 | 50,000 | 1700 |
| 10b | BB2.1-24 | 1700 | 330 | 130 | 50,000 | 130 |
| 11a | BB2.2-25 | 20,000 | 3300 | 1000 | 100,000 | 5000 |
| 11b | BB2.1-25 | 2500 | 330 | 130 | 33,000 | 330 |
| 12a | 4a | 36,000 | 5000 | 1000 | 66,000 | 17,000 |
| 12b | 4b | 6700 | 1100 | 670 | 57,000 | 1500 |
| 13a | 5a | 22,000 | 2900 | 1400 | 23,000 | 20,000 |
| 13b | 5b | 2600 | 390 | 330 | 23,000 | 340 |
| 14a | 6a | 32,000 | 3400 | 1400 | 38,000 | 23,000 |
| 14b | 6b | 510 | 59 | 290 | 10,000 | 66 |
| 15a | BB2.2-17 | 13,000 | 3300 | 1000 | 50,000 | 6700 |
| 15b | BB2.1-17 | 2500 | 500 | 140 | 50,000 | 400 |
| 16a | BB2.2-18 | 20,000 | 4000 | 1400 | 50,000 | 14,000 |
| 16b | BB2.1-18 | 3300 | 500 | 130 | 130,000 | 1000 |
| 17a | 7a | 36,000 | 5400 | 3300 | 79,000 | 19,000 |
| 17b | 7b | 2600 | 570 | 1400 | 58,000 | 1400 |
| 19a | BB2.2-9 | 10,000 | 1700 | 560 | 10,000 | 4000 |
| 21 | 10 * | 250 | 66 | 220 | 1400 | ND |
| 23a | BB2.2-11 | 4000 | 2000 | 130 | 2200 | 10,000 |
| 24a | BB2.2-31 | 43,000 | 6700 | 910 | 3300 | 19,000 |
| 24b | BB2.1-31 | 2900 | 250 | 59 | 1700 | 1300 |
| 25a | BB2.2-12 | 100,000 | 3300 | 100 | 4000 | 25,000 |
| 26 | 19 * | 10,000 | 5000 | 10,000 | 33,000 | ND |
| 35 | 31 * | 25 | 100 | 560 | 200 | 530 |
| Cmpd. | Lab. Name | Observed Dissociation Constants (Kd_obs), nM | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CAI | CAII | CAIII | CAIV | CAVA | CAVB | CAVI | CAVII | CAIX | CAXII | CAXIII | CAXIV | ||
| 6b | 3b | 1400 | 240 | ≥200,000 | 4200 | 20,000 | 130 | 670 | 290 | 100 | 7900 | 180 | 140 |
| 9b | BB2.1-22 | 670 | 200 | ≥200,000 | 1000 | 2900 | 830 | 670 | 250 | 100 | 14,000 | 250 | 130 |
| 18a | BB2.2-23 | 20,000 | 5000 | ≥200,000 | 33,000 | 20,000 | 14,000 | 33,000 | 6700 | 1400 | 20,000 | 25,000 | 5000 |
| 18b | BB2.1-23 | 5000 | 1300 | ≥200,000 | 13,000 | 6700 | 6700 | 20,000 | 330 | 250 | 13,000 | 1800 | 1000 |
| 19b | BB2.1-9 | 290 | 50 | ≥200,000 | 1800 | 20,000 | 1400 | 5000 | 200 | 56 | 1300 | 130 | 25 |
| 20 | BB2.1-10 | 5000 | 500 | ≥200,000 | 2000 | 22,000 | 3300 | 20,000 | 10,000 | 100 | 13,000 | 2900 | 330 |
| 22a | BB2.2-30 | 50,000 | 2500 | ≥200,000 | 6700 | 33,000 | 25,000 | 13,000 | 10,000 | 330 | 11,000 | 33,000 | 3300 |
| 22b | BB2.1-30 | 3300 | 400 | ≥200,000 | 2000 | 6700 | ≥200,000 | 3300 | 670 | 100 | 2200 | 1700 | 130 |
| 23b | BB2.1-11 | 67 | 50 | 15,000 | 1000 | 20,000 | 1100 | 1000 | 110 | 33 | 50 | 400 | 50 |
| 25b | BB2.1-12 | 170 | 25 | 5000 | 1400 | 38,000 | 1400 | 200 | 170 | 25 | 630 | 400 | 25 |
| 27 | BB8.1-7 | 67 | 220 | 63,000 | 1400 | ≥200,000 | 560 | 18,000 | 250 | 48 | 180 | 870 | 170 |
| 28 | BB8.1-12 | 37 | 45 (59) | ≥200,000 | 1100 | 50,000 | 46 | 7100 | 77 | 33 | 250 | 250 | 29 |
| 29 | BB8.1-14 | 150 | 380 | ≥200,000 | 1500 | ≥200,000 | 77 | 20,000 | 670 | 80 | 830 | 1700 | 130 |
| 30 | BB8.1-15 | 3300 | 4000 | ≥200,000 | 27,000 | ≥200,000 | 11000 | 67,000 | 100,000 | 290 | 110,000 | 8300 | 6700 |
| 31 | BB8.1-4 | 2000 | 910 (860) | ≥200,000 | 6700 | ≥200,000 | 20,000 | 24,000 | 1700 | 360 | 3800 | 450 | 2000 |
| 32 | BB8.1-5 | 830 | 53 | ≥200,000 | 2000 | ≥200,000 | 30 (41) | 10,000 | 130 | 190 | 7700 | 330 | 770 |
| 33 | BB8.1-10 | 3100 | 500 | ≥200,000 | 2800 | ≥200,000 | 100 | 25,000 | 1100 | 630 | 26,000 | 830 | 910 |
| 34 | BB8.1-13 | 670 | 150 | ≥200,000 | 1000 | ≥200,000 | 120 | 10,000 | 250 | 170 | 14,000 | 220 | 1000 |
| 36 | BB8.3-3 | 20 | 40 (130) | 17,000 | 3300 | ≥200,000 | 400 | 6300 | 33 | 23 | 120 | 40 | 130 |
| Cmpd. | Lab. Name | Sulfonamide pKa | Intrinsic Dissociation Constants (Kd_int), nM | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CAI | CAII | CAIII | CAIV | CAVA | CAVB | CAVI | CAVII | CAIX | CAXII | CAXIII | CAXIV | |||
| 27 | BB8.1-7 | 10.4 | 0.025 | 0.039 | 6.0 | 0.16 | ≥27 | 0.11 | 0.65 | 0.038 | 0.0054 | 0.028 | 0.31 | 0.0076 |
| 28 | BB8.1-12 | 10.1 | 0.027 | 0.016 | ≥38 | 0.25 | 13 | 0.018 | 0.51 | 0.024 | 0.0075 | 0.077 | 0.18 | 0.0026 |
| 29 | BB8.1-14 | 10.3 | 0.070 | 0.084 | ≥24 | 0.21 | ≥33 | 0.019 | 0.91 | 0.13 | 0.011 | 0.16 | 0.77 | 0.0073 |
| 31 | BB8.1-4 | 10.1 | 1.5 | 0.32 | ≥38 | 1.5 | ≥53 | 7.9 | 1.7 | 0.52 | 0.081 | 1.2 | 0.32 | 0.18 |
| 32 | BB8.1-5 | 9.7 | 1.5 | 0.047 | ≥96 | 1.1 | ≥130 | 0.030 | 1.8 | 0.10 | 0.11 | 5.9 | 0.60 | 0.17 |
| 33 | BB8.1-10 | 10.1 * | 2.3 | 0.18 | ≥38 | 0.63 | ≥53 | 0.040 | 1.8 | 0.34 | 0.14 | 8.0 | 0.60 | 0.081 |
| 36 | BB8.3-3 | 10.2 | 0.012 | 0.011 | 2.6 | 0.59 | ≥42 | 0.13 | 0.36 | 0.0081 | 0.0041 | 0.029 | 0.023 | 0.0092 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balandis, B.; Šimkūnas, T.; Paketurytė-Latvė, V.; Michailovienė, V.; Mickevičiūtė, A.; Manakova, E.; Gražulis, S.; Belyakov, S.; Kairys, V.; Mickevičius, V.; et al. Beta and Gamma Amino Acid-Substituted Benzenesulfonamides as Inhibitors of Human Carbonic Anhydrases. Pharmaceuticals 2022, 15, 477. https://doi.org/10.3390/ph15040477
Balandis B, Šimkūnas T, Paketurytė-Latvė V, Michailovienė V, Mickevičiūtė A, Manakova E, Gražulis S, Belyakov S, Kairys V, Mickevičius V, et al. Beta and Gamma Amino Acid-Substituted Benzenesulfonamides as Inhibitors of Human Carbonic Anhydrases. Pharmaceuticals. 2022; 15(4):477. https://doi.org/10.3390/ph15040477
Chicago/Turabian StyleBalandis, Benas, Tomas Šimkūnas, Vaida Paketurytė-Latvė, Vilma Michailovienė, Aurelija Mickevičiūtė, Elena Manakova, Saulius Gražulis, Sergey Belyakov, Visvaldas Kairys, Vytautas Mickevičius, and et al. 2022. "Beta and Gamma Amino Acid-Substituted Benzenesulfonamides as Inhibitors of Human Carbonic Anhydrases" Pharmaceuticals 15, no. 4: 477. https://doi.org/10.3390/ph15040477
APA StyleBalandis, B., Šimkūnas, T., Paketurytė-Latvė, V., Michailovienė, V., Mickevičiūtė, A., Manakova, E., Gražulis, S., Belyakov, S., Kairys, V., Mickevičius, V., Zubrienė, A., & Matulis, D. (2022). Beta and Gamma Amino Acid-Substituted Benzenesulfonamides as Inhibitors of Human Carbonic Anhydrases. Pharmaceuticals, 15(4), 477. https://doi.org/10.3390/ph15040477