
Synthesis and Antiproliferative Activity of Novel Imipridone–Ferrocene Hybrids with Triazole and Alkyne Linkers

 , , and
, , and 
Abstract

1. Introduction
2. Results and Discussion
2.1. Synthesis of the Reference and Hybrid Imipridones
2.2. Cell Viability Screenings of the Imipridone Analogues
2.3. Structure–Activity Relationships
2.4. Dose–Response Curves of Selected Imipridone Hybrids
2.5. Colony Formation Assay
2.6. Cytotoxicity Assay with the Most Potent Hybrids on Cancer Cell Lines and Nontumorous Fibroblast Cells
2.7. Apoptosis and Necrosis Quantitation Assay
3. Materials and Methods
3.1. Synthetic Procedures
3.1.1. General Procedure for the Synthesis of N-Substituted Methylcarboxylate Piperidones (3a–e)
3.1.2. Synthesis of 2-(Methylthio)-4,5-dihydro-1H-imidazole-1-carboxylate (5)
3.1.3. General Procedure for the Synthesis of Cyclic Guanidines (6e–g)
3.1.4. General Procedure for the Synthesis of Imipridones (7ag, 7be, 7bf, 7ce, 7cf, 7df, 7ef and 7eg)
3.1.5. General Procedure for the Synthesis of Azido Imipridones (7bh, 7ch, 7hf and 7hg)
3.1.6. General Procedure for Copper(I) Catalysed 1,4-Azide-alkyne Cycloadditions
3.1.7. General Procedure for Ruthenium(II) Catalysed 1,5-Azide-alkyne Cycloadditions
3.1.8. General Procedure Used for the Sonogashira Coupling
3.1.9. General Procedure for the Removal of Trimethylsilyl-Protecting Group
3.2. Cell Culturing
3.3. MTT Cell Viability Assay
3.4. CellTiter-Glo Cell Viability Assay
3.5. Colony Formation Assay
3.6. Apoptosis and Necrosis Quantitation Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prabhu, V.V.; Morrow, S.; Rahman Kawakibi, A.; Zhou, L.; Ralff, M.; Ray, J.; Jhaveri, A.; Ferrarini, I.; Lee, Y.; Parker, C.; et al. ONC201 and Imipridones: Anti-Cancer Compounds with Clinical Efficacy. Neoplasia 2020, 22, 725–744. [Google Scholar] [CrossRef] [PubMed]
- Bonner, E.R.; Waszak, S.M.; Grotzer, M.A.; Mueller, S.; Nazarian, J. Mechanisms of Imipridones in Targeting Mitochondrial Metabolism in Cancer Cells. Neuro Oncol. 2021, 23, 542–556. [Google Scholar] [CrossRef] [PubMed]
- Lev, A.; Lulla, A.R.; Wagner, J.; Ralff, M.D.; Kiehl, J.B.; Zhou, Y.; Benes, C.H.; Prabhu, V.V.; Oster, W.; Astsaturov, I.; et al. Anti-Pancreatic Cancer Activity of ONC212 Involves the Unfolded Protein Response (UPR) and Is Reduced by IGF1-R and GRP78/BIP. Oncotarget 2017, 8, 81776–81793. [Google Scholar] [CrossRef] [PubMed]
- Ferrarini, I.; Louie, A.; Zhou, L.; El-Deiry, W.S. ONC212 Is a Novel Mitocan Acting Synergistically with Glycolysis Inhibition in Pancreatic Cancer. Mol. Cancer 2021, 20, 1572–1583. [Google Scholar] [CrossRef]
- Wagner, J.; Kline, C.L.; Ralff, M.D.; Lev, A.; Lulla, A.; Zhou, L.; Olson, G.L.; Nallaganchu, B.R.; Benes, C.H.; Allen, J.E.; et al. Preclinical Evaluation of the Imipridone Family, Analogs of Clinical Stage Anti-Cancer Small Molecule ONC201, Reveals Potent Anti-Cancer Effects of ONC212. Cell Cycle 2017, 16, 1790–1799. [Google Scholar] [CrossRef]
- Jacques, S.; van der Sloot, A.M.; Huard, C.C.; Coulombe-Huntington, J.; Tsao, S.; Tollis, S.; Bertomeu, T.; Culp, E.J.; Pallant, D.; Cook, M.A.; et al. Imipridone Anticancer Compounds Ectopically Activate the ClpP Protease and Represent a New Scaffold for Antibiotic Development. Genetics 2020, 214, 1103–1120. [Google Scholar] [CrossRef]
- Nii, T.; Prabhu, V.V.; Ruvolo, V.; Madhukar, N.; Zhao, R.; Mu, H.; Heese, L.; Nishida, Y.; Kojima, K.; Garnett, M.J.; et al. Imipridone ONC212 Activates Orphan G Protein-Coupled Receptor GPR132 and Integrated Stress Response in Acute Myeloid Leukemia. Leukemia 2019, 33, 2805–2816. [Google Scholar] [CrossRef]
- Ishizawa, J.; Zarabi, S.F.; Davis, R.E.; Halgas, O.; Nii, T.; Jitkova, Y.; Zhao, R.; St-Germain, J.; Heese, L.E.; Egan, G.; et al. Mitochondrial ClpP-Mediated Proteolysis Induces Selective Cancer Cell Lethality. Cancer Cell 2019, 35, 721–737.e9. [Google Scholar] [CrossRef]
- Graves, P.R.; Aponte-Collazo, L.J.; Fennell, E.M.J.; Graves, A.C.; Hale, A.E.; Dicheva, N.; Herring, L.E.; Gilbert, T.S.K.; East, M.P.; McDonald, I.M.; et al. Mitochondrial Protease ClpP Is a Target for the Anticancer Compounds ONC201 and Related Analogues. ACS Chem. Biol. 2019, 14, 1020–1029. [Google Scholar] [CrossRef]
- Prabhu, V.V.; Talekar, M.K.; Lulla, A.R.; Kline, C.L.B.; Zhou, L.; Hall, J.; van den Heuvel, A.P.J.; Dicker, D.T.; Babar, J.; Grupp, S.A.; et al. Single Agent and Synergistic Combinatorial Efficacy of First-in-Class Small Molecule Imipridone ONC201 in Hematological Malignancies. Cell Cycle 2018, 17, 468–478. [Google Scholar] [CrossRef]
- Tu, Y.; He, J.; Liu, H.; Lee, H.C.; Wang, H.; Ishizawa, J.; Allen, J.E.; Andreeff, M.; Orlowski, R.Z.; Davis, R.E.; et al. The Imipridone ONC201 Induces Apoptosis and Overcomes Chemotherapy Resistance by Up-Regulation of Bim in Multiple Myeloma. Neoplasia 2017, 19, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS Signalling in the Biology of Cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in Cancer: Initiators, Amplifiers or an Achilles’ Heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Bardaweel, S.K.; Gul, M.; Alzweiri, M.; Ishaqat, A.; ALSalamat, H.A.; Bashatwah, R.M. Reactive Oxygen Species: The Dual Role in Physiological and Pathological Conditions of the Human Body. Eurasian J. Med. 2018, 50, 193–201. [Google Scholar] [CrossRef]
- Zhang, L.; Li, J.; Zong, L.; Chen, X.; Chen, K.; Jiang, Z.; Nan, L.; Li, X.; Li, W.; Shan, T.; et al. Reactive Oxygen Species and Targeted Therapy for Pancreatic Cancer. Oxidative Med. Cell. Longev. 2016, 2016, 1616781. [Google Scholar] [CrossRef]
- Afanas’ev, I. Reactive Oxygen Species Signaling in Cancer: Comparison with Aging. Aging Dis. 2010, 2, 219–230. [Google Scholar]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in Cancer Therapy: The Bright Side of the Moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Simon, H.U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of Reactive Oxygen Species (ROS) in Apoptosis Induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef]
- Baecker, D.; Sesli, Ö.; Knabl, L.; Huber, S.; Orth-Höller, D.; Gust, R. Investigating the antibacterial activity of salen/salophene metal complexes: Induction of ferroptosis as part of the mode of action. Eur. J. Med. Chem. 2021, 209, 112907. [Google Scholar] [CrossRef]
- Ramirez-Vick, J.; Acevedo, C.; Melendez, E.; Singh, S. Cytotoxicity and Reactive Oxygen Species Generated by Ferrocenium and Ferrocene on MCF7 and MCF10A Cell Lines. J. Cancer Sci. Ther. 2012, 4, 271–275. [Google Scholar] [CrossRef]
- Ludwig, B.S.; Correia, J.D.G.; Kühn, F.E. Ferrocene Derivatives as Anti-Infective Agents. Coord. Chem. Rev. 2019, 396, 22–48. [Google Scholar] [CrossRef]
- Peter, S.; Aderibigbe, B.A. Ferrocene-Based Compounds with Antimalaria/Anticancer Activity. Molecules 2019, 24, 3604. [Google Scholar] [CrossRef] [PubMed]
- Pérez, W.I.; Soto, Y.; Ortíz, C.; Matta, J.; Meléndez, E. Ferrocenes as Potential Chemotherapeutic Drugs: Synthesis, Cytotoxic Activity, Reactive Oxygen Species Production and Micronucleus Assay. Bioorg. Med. Chem. 2015, 23, 471–479. [Google Scholar] [CrossRef][Green Version]
- Bárány, P.; Oláh, R.S.; Kovács, I.; Czuczi, T.; Szabó, C.L.; Takács, A.; Lajkó, E.; Láng, O.; Kőhidai, L.; Schlosser, G.; et al. Ferrocene-Containing Impiridone (ONC201) Hybrids: Synthesis, DFT Modelling, In Vitro Evaluation, and Structure–Activity Relationships. Molecules 2018, 23, 2248. [Google Scholar] [CrossRef]
- Voltan, R.; Secchiero, P.; Casciano, F.; Milani, D.; Zauli, G.; Tisato, V. Redox Signaling and Oxidative Stress: Cross Talk with TNF-Related Apoptosis Inducing Ligand Activity. Int. J. Biochem. Cell. Biol. 2016, 81 Pt B, 364–374. [Google Scholar] [CrossRef]
- Zhang, M.; Harashima, N.; Moritani, T.; Huang, W.; Harada, M. The Roles of ROS and Caspases in TRAIL-Induced Apoptosis and Necroptosis in Human Pancreatic Cancer Cells. PLoS ONE 2015, 10, e0127386. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, X.; Xue, P.; Sun, H.H.Y.; Williams, I.D.; Sharpless, K.B.; Fokin, V.V.; Jia, G. Ruthenium-Catalyzed Cycloaddition of Alkynes and Organic Azides. J. Am. Chem. Soc. 2005, 127, 15998–15999. [Google Scholar] [CrossRef]
- Csámpai, A.; Bárány, P.; Czuczi, T.; Kovács, I.; Adamis, B.; Németh, Z.; Murányi, J.; Oláhné Szabó, R.; Bősze, S.; Mező, G.; et al. Synthesis of Novel Imipridone Derivatives and Their Evaluation for Their Anticancer Activity. WO2022029459 A1, 10 February 2022. Available online: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022029459&_cid=P10-KZWSCT-56026-1 (accessed on 10 February 2022).
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of fluorine in medicinal chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef] [PubMed]
- Baecker, D.; Obermoser, V.; Kirchner, E.A.; Hupfauf, A.; Kircher, B.; Gust, R. Fluorination as tool to improve bioanalytical sensitivity and COX-2-selective antitumor activity of cobalt alkyne complexes. Dalton Trans. 2019, 48, 15856–15868. [Google Scholar] [CrossRef]
- Kucuksayan, E.; Ozben, T. Hybrid Compounds as Multitarget Directed Anticancer Agents. Curr. Top. Med. Chem. 2017, 17, 907–918. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
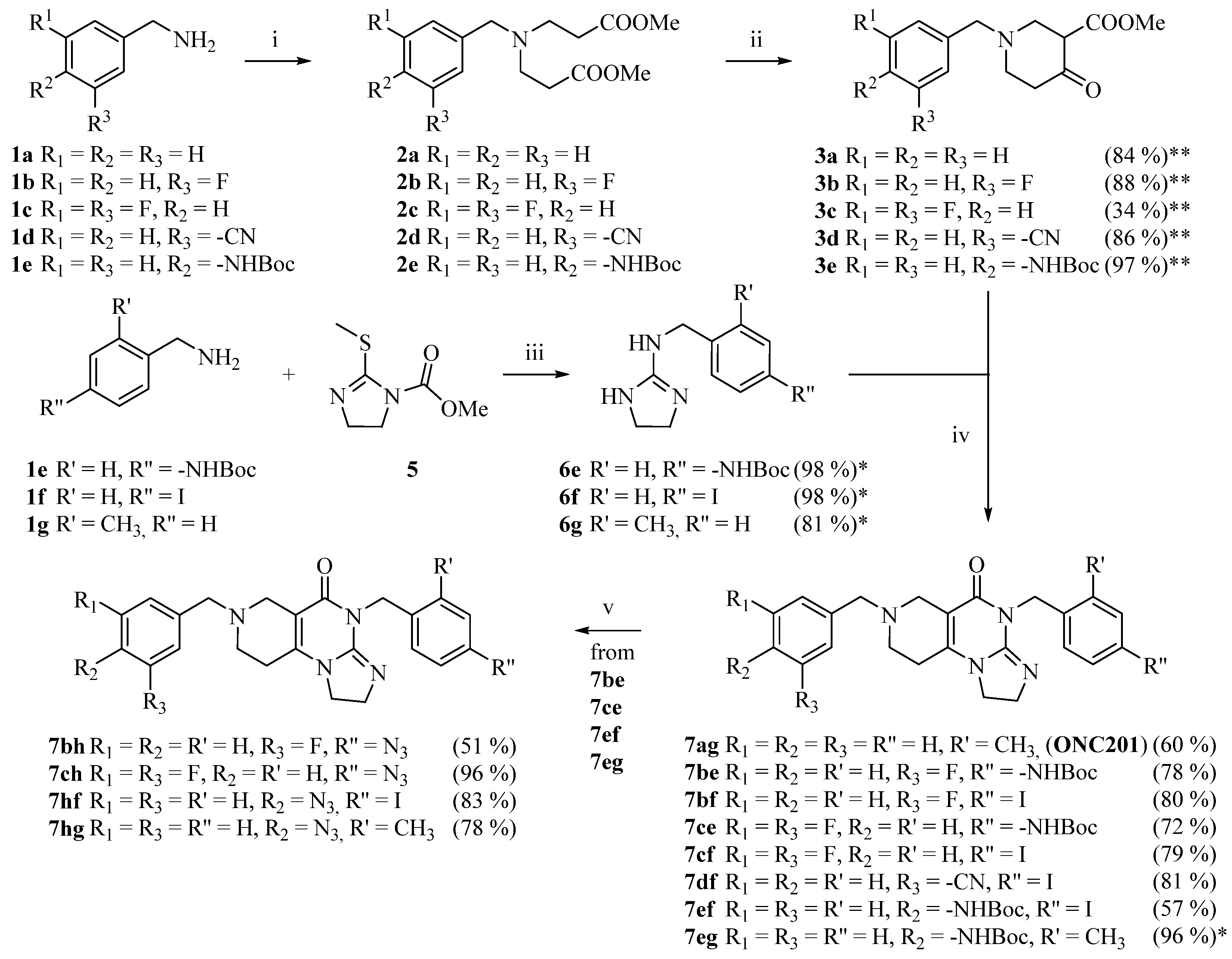{kind=link}
{kind=link}
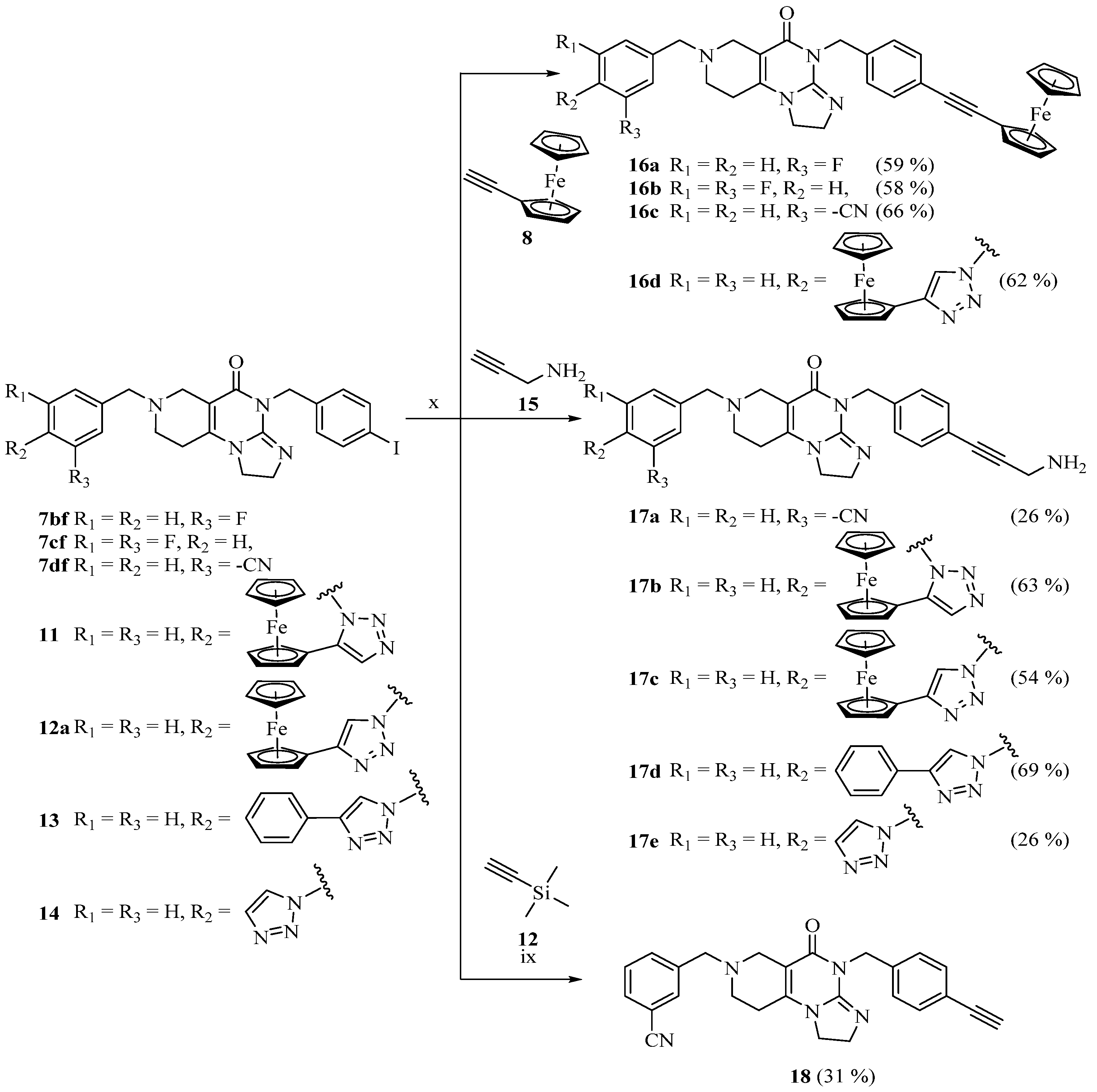{kind=link}
| Compound Identifier and Structure | Cell Viability (% of Control) ± SD (n = 4) | |||||
|---|---|---|---|---|---|---|
| µM | PANC-1 | A2058 | EBC-1 | Fadu | ||
| ONC 201 |  | 25 | 54 ± 23 | 47 ± 4 | 31 ± 3 | 16 ± 2 |
| 8.3 | 63 ± 33 | 52 ± 3 | 34 ± 4 | 21 ± 1 | ||
| 2.8 | 62 ± 25 | 59 ± 6 | 37 ± 3 | 25 ± 4 | ||
| 9a |  | 25 | 88 ± 5 | 82 ± 12 | 62 ± 15 | 69 ± 21 |
| 8.3 | 94 ± 10 | 90 ± 14 | 66 ± 8 | 74 ± 6 | ||
| 2.8 | 114 ± 25 | 88 ± 7 | 75 ± 7 | 79 ± 7 | ||
| 9b |  | 25 | 109 ± 7 | 94 ± 9 | 65 ± 13 | 72 ± 6 |
| 8.3 | 126 ± 12 | 103 ± 15 | 79 ± 13 | 71 ± 13 | ||
| 2.8 | 122 ± 20 | 104 ± 13 | 88 ± 9 | 74 ± 8 | ||
| 10a |  | 25 | 13 ± 3 | 26 ± 2 | 3 ± 1 | 9 ± 2 |
| 8.3 | 48 ± 1 | 54 ± 3 | 34 ± 4 | 19 ± 5 | ||
| 2.8 | 101 ± 9 | 114 ± 5 | 96 ± 9 | 79 ± 2 | ||
| 10b |  | 25 | 77 ± 18 | 60 ± 6 | 21 ± 6 | 29 ± 11 |
| 8.3 | 93 ± 7 | 83 ± 8 | 67 ± 7 | 66 ± 28 | ||
| 2.8 | 117 ± 12 | 120 ± 8 | 104 ± 9 | 82 ± 16 | ||
| 16a |  | 25 | 124 ± 46 | 77 ± 5 | 58 ± 3 | 59 ± 2 |
| 8.3 | 120 ± 42 | 90 ± 13 | 63 ± 14 | 75 ± 8 | ||
| 2.8 | 126 ± 49 | 87 ± 10 | 76 ± 4 | 85 ± 14 | ||
| 16b |  | 25 | 103 ± 17 | 80 ± 5 | 68 ± 12 | 72 ± 6 |
| 8.3 | 123 ± 34 | 89 ± 3 | 69 ± 13 | 84 ± 13 | ||
| 2.8 | 126 ± 31 | 94 ± 5 | 80 ± 9 | 79 ± 4 | ||
| 16c |  | 25 | 83 ± 9 | 77 ± 10 | 61 ± 18 | 58 ± 24 |
| 8.3 | 116 ± 25 | 80 ± 7 | 70 ± 13 | 79 ± 13 | ||
| 2.8 | 123 ± 35 | 91 ± 16 | 81 ± 6 | 78 ± 9 | ||
| 17a |  | 25 | 38 ± 32 | 82 ± 4 | 77 ± 4 | 36 ± 36 |
| 8.3 | 73 ± 16 | 96 ± 3 | 101 ± 7 | 52 ± 25 | ||
| 2.8 | 97 ± 13 | 92 ± 8 | 94 ± 6 | 88 ± 10 | ||
| 18 |  | 25 | 50 ± 26 | 44 ± 4 | 35 ± 2 | 14 ± 2 |
| 8.3 | 50 ± 22 | 50 ± 4 | 36 ± 5 | 17 ± 2 | ||
| 2.8 | 52 ± 26 | 50 ± 6 | 35 ± 12 | 16 ± 1 | ||
| 11 |  | 25 | 95 ± 5 | 58 ± 22 | 27 ± 19 | 51 ± 27 |
| 8.3 | 126 ± 27 | 65 ± 3 | 52 ± 12 | 65 ± 26 | ||
| 2.8 | 126 ± 13 | 84 ± 10 | 86 ± 7 | 81 ± 13 | ||
| 12a |  | 25 | 91 ± 12 | 86 ± 9 | 58 ± 15 | 55 ± 17 |
| 8.3 | 99 ± 12 | 87 ± 6 | 69 ± 17 | 64 ± 26 | ||
| 2.8 | 100 ± 9 | 91 ± 5 | 79 ± 17 | 72 ± 19 | ||
| 12b |  | 25 | 91 ± 1 | 89 ± 3 | 66 ± 4 | 89 ± 1 |
| 8.3 | 108 ± 5 | 82 ± 2 | 73 ± 1 | 93 ± 4 | ||
| 2.8 | 109 ± 9 | 79 ± 6 | 84 ± 7 | 98 ± 3 | ||
| 13 |  | 25 | 82 ± 21 | 85 ± 4 | 76 ± 4 | 75 ± 14 |
| 8.3 | 104 ± 1 | 79 ± 7 | 82 ± 6 | 81 ± 14 | ||
| 2.8 | 88 ± 23 | 73 ± 6 | 88 ± 19 | 90 ± 4 | ||
| 17b |  | 25 | 1 ± 1 | 3 ± 1 | 3 ± 3 | 3 ± 3 |
| 8.3 | 98 ± 16 | 74 ± 3 | 60 ± 31 | 73 ± 21 | ||
| 2.8 | 95 ± 4 | 83 ± 8 | 97 ± 8 | 92 ± 10 | ||
| 17c |  | 25 | 1 ± 1 | 2 ± 1 | 3 ± 2 | 2 ± 2 |
| 8.3 | 23 ± 8 | 7 ± 3 | 12 ± 10 | 22 ± 24 | ||
| 2.8 | 101 ± 8 | 79 ± 9 | 83 ± 13 | 63 ± 32 | ||
| 17d |  | 25 | 1 ± 1 | 3 ± 1 | 3 ± 1 | 3 ± 3 |
| 8.3 | 96 ± 20 | 45 ± 9 | 63 ± 14 | 51 ± 49 | ||
| 2.8 | 102 ± 19 | 85 ± 7 | 107 ± 13 | 87 ± 9 | ||
| 17e |  | 25 | 91 ± 10 | 76 ± 2 | 86 ± 12 | 58 ± 20 |
| 8.3 | 103 ± 13 | 89 ± 4 | 111 ± 17 | 89 ± 17 | ||
| 2.8 | 92 ± 11 | 93 ± 6 | 108 ± 17 | 100 ± 7 | ||
| 16d |  | 25 | 77 ± 20 | 76 ± 4 | 53 ± 11 | 53 ± 4 |
| 8.3 | 85 ± 21 | 81 ± 9 | 70 ± 17 | 71 ± 19 | ||
| 2.8 | 78 ± 10 | 91 ± 17 | 80 ± 18 | 72 ± 17 | ||
| Treated Cells | 10a | 17b | 17c | 17d | ONC201 |
|---|---|---|---|---|---|
| PANC-1 | 12.4 | 17.8 | 7.2 | 10.6 | 1.5 |
| A2058 | 7.8 | 11.8 | 4.0 | 6.1 | 1.2 |
| EBC-1 | 7.3 | 12.0 | 6.2 | 12.6 | 1.4 |
| Fadu | 5.5 | 14.6 | 6.4 | 10.3 | 1.4 |
| Treated Cells | 17c | 17d |
|---|---|---|
| PANC-1 | 4.4 | 5.0 |
| A2058 | 3.7 | 5.5 |
| EBC-1 | 3.7 | 5.5 |
| Fadu | 6.8 | 7.5 |
| Primary fibroblast | 15.8 | 11.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czuczi, T.; Murányi, J.; Bárány, P.; Móra, I.; Borbély, A.; Csala, M.; Csámpai, A. Synthesis and Antiproliferative Activity of Novel Imipridone–Ferrocene Hybrids with Triazole and Alkyne Linkers. Pharmaceuticals 2022, 15, 468. https://doi.org/10.3390/ph15040468
Czuczi T, Murányi J, Bárány P, Móra I, Borbély A, Csala M, Csámpai A. Synthesis and Antiproliferative Activity of Novel Imipridone–Ferrocene Hybrids with Triazole and Alkyne Linkers. Pharmaceuticals. 2022; 15(4):468. https://doi.org/10.3390/ph15040468
Chicago/Turabian StyleCzuczi, Tamás, József Murányi, Péter Bárány, István Móra, Adina Borbély, Miklós Csala, and Antal Csámpai. 2022. "Synthesis and Antiproliferative Activity of Novel Imipridone–Ferrocene Hybrids with Triazole and Alkyne Linkers" Pharmaceuticals 15, no. 4: 468. https://doi.org/10.3390/ph15040468
APA StyleCzuczi, T., Murányi, J., Bárány, P., Móra, I., Borbély, A., Csala, M., & Csámpai, A. (2022). Synthesis and Antiproliferative Activity of Novel Imipridone–Ferrocene Hybrids with Triazole and Alkyne Linkers. Pharmaceuticals, 15(4), 468. https://doi.org/10.3390/ph15040468