
Investigation of the Antitumor Effects of Tamoxifen and Its Ferrocene-Linked Derivatives on Pancreatic and Breast Cancer Cell Lines

 ,
,  ,
,  ,
, 
Abstract
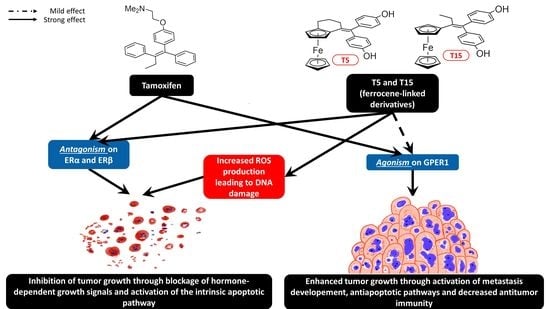
1. Introduction
2. Results
2.1. Cytotoxicity
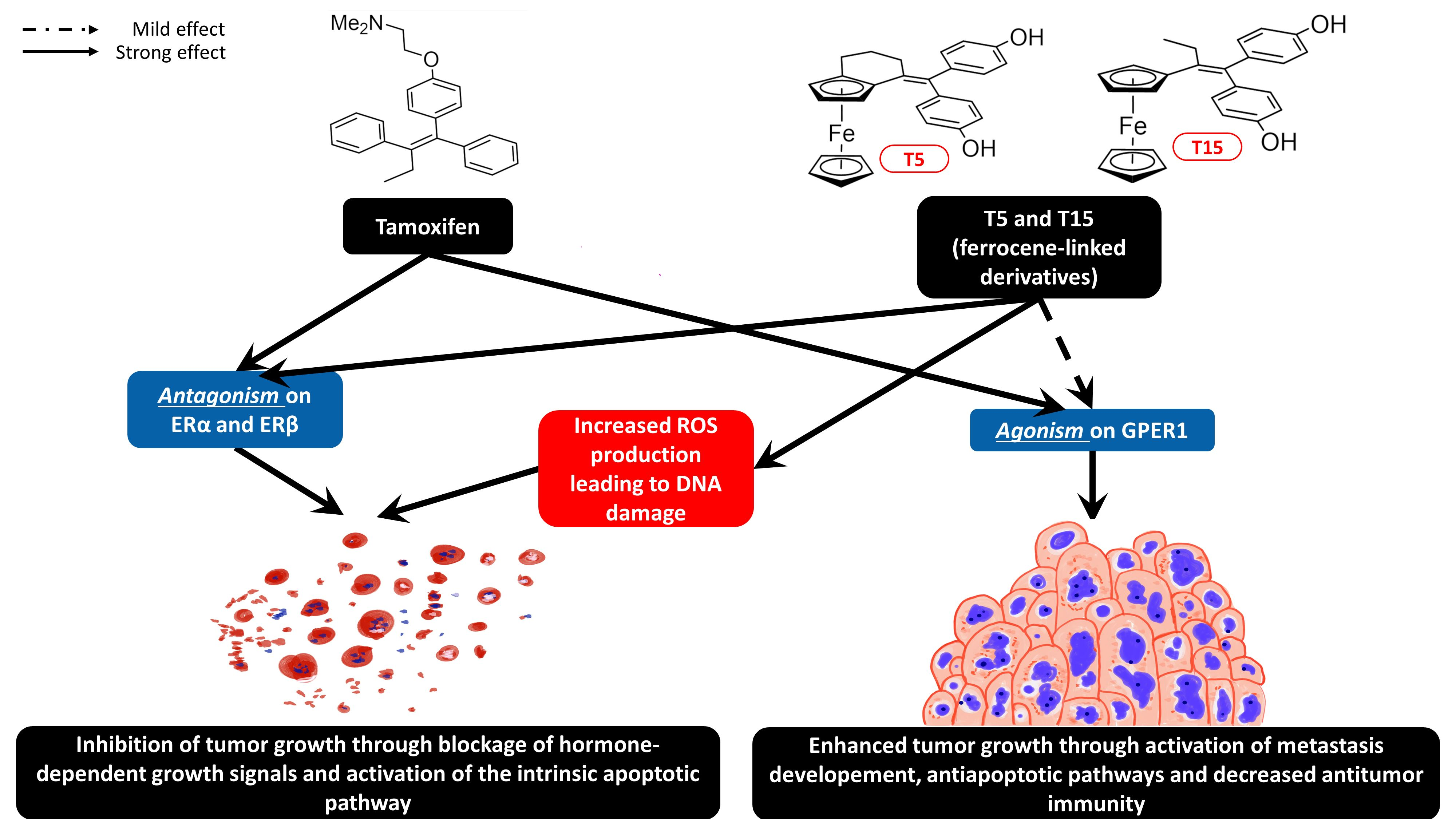
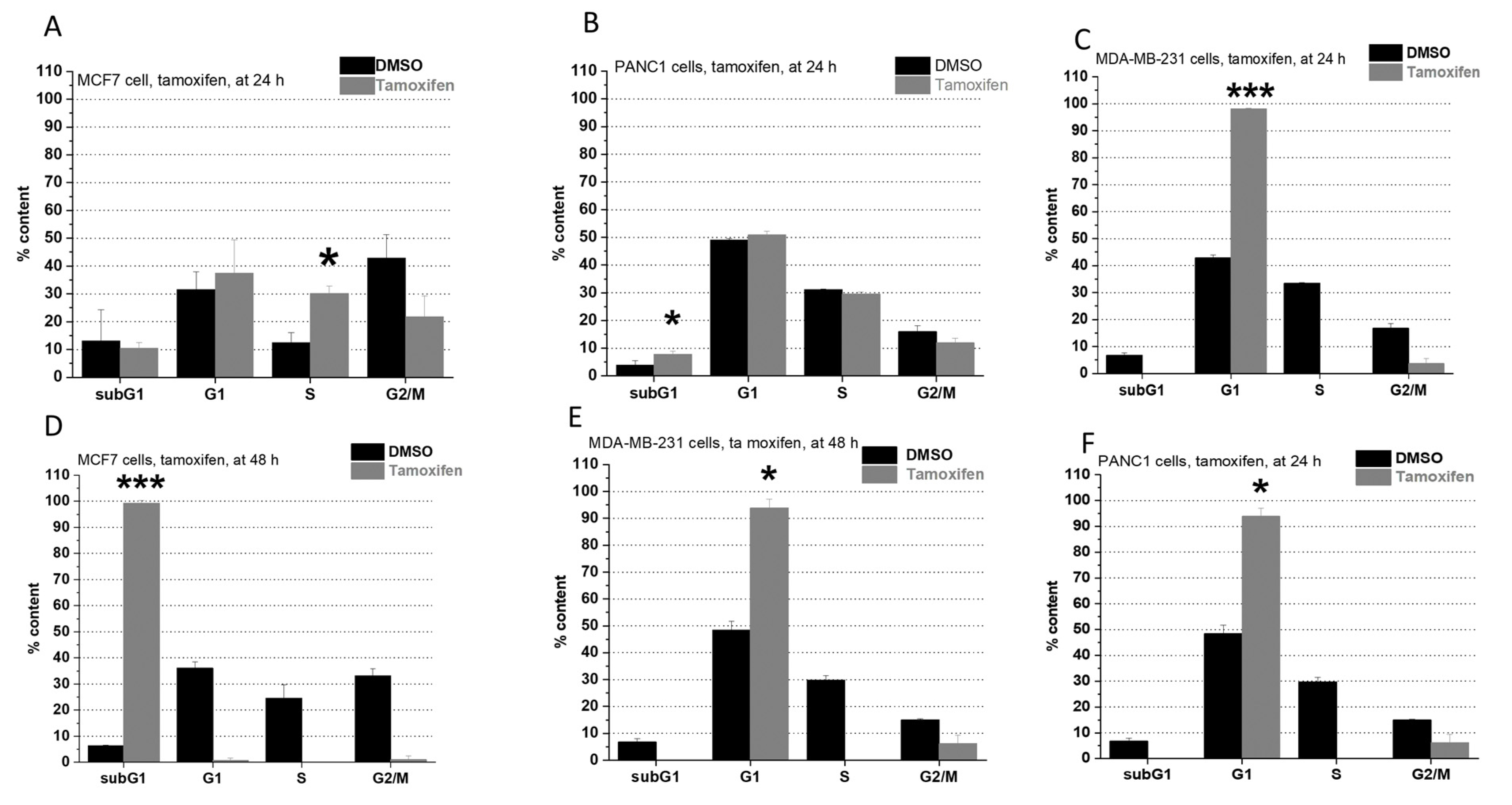
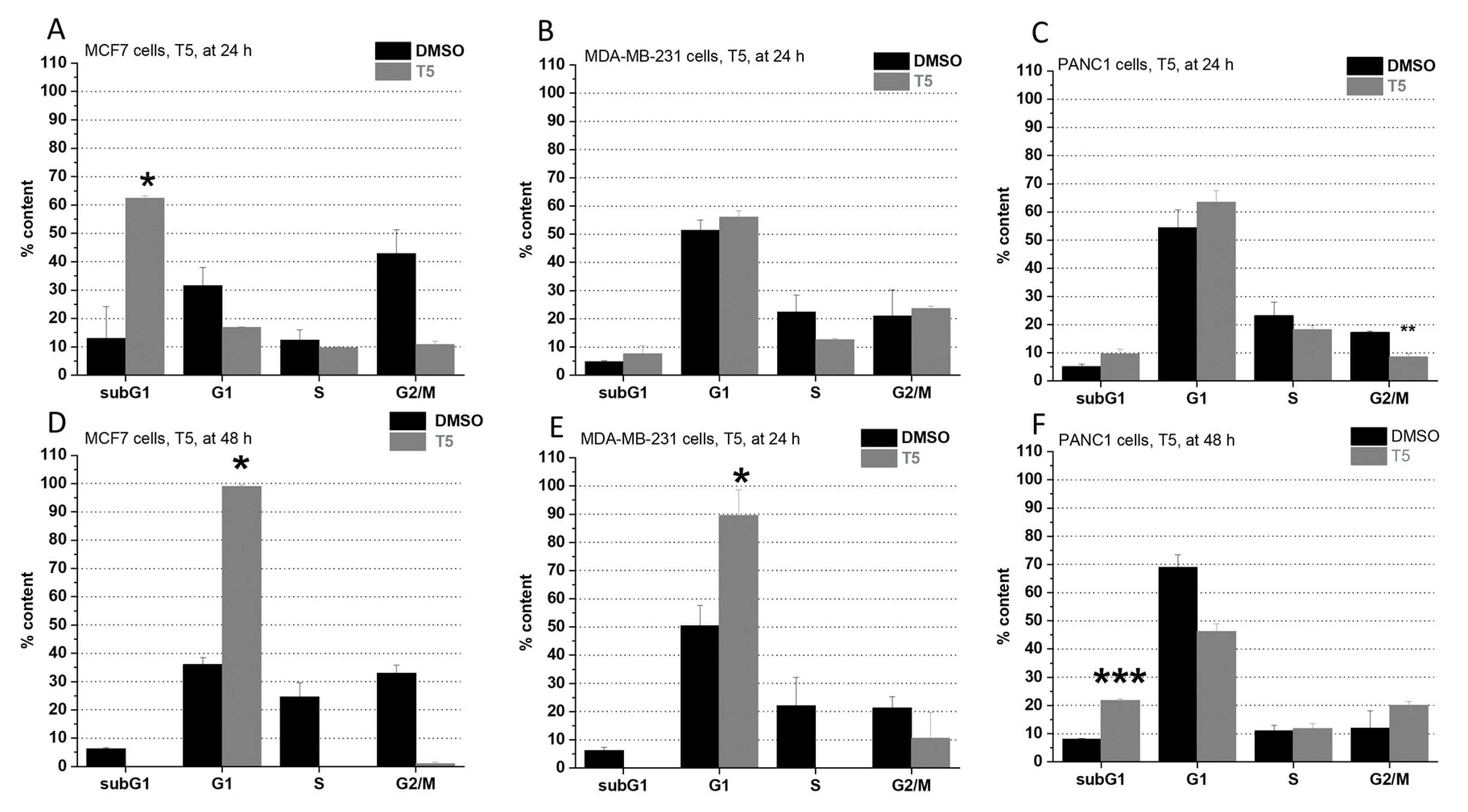
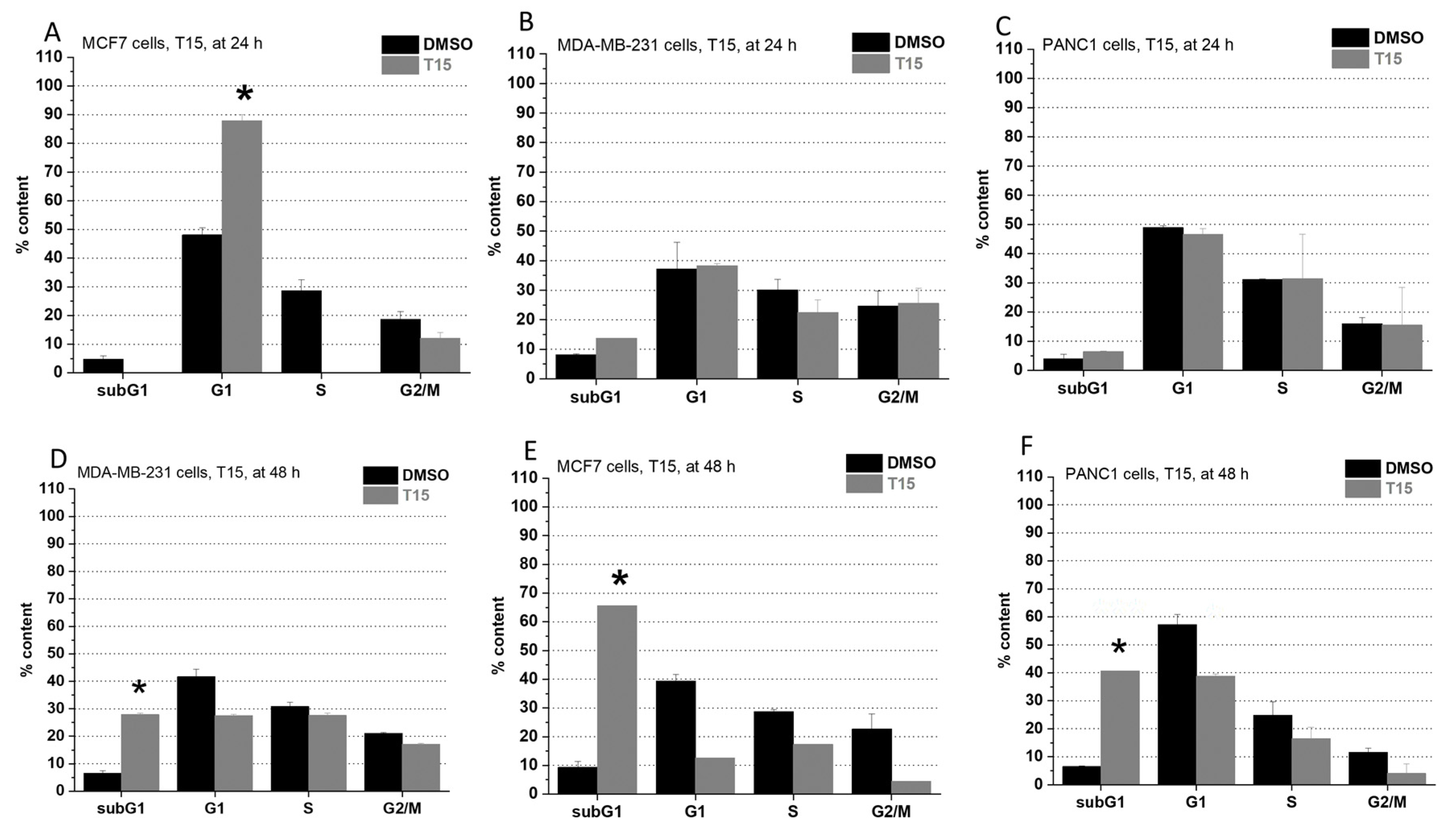
2.2. Cell Cycle Analysis
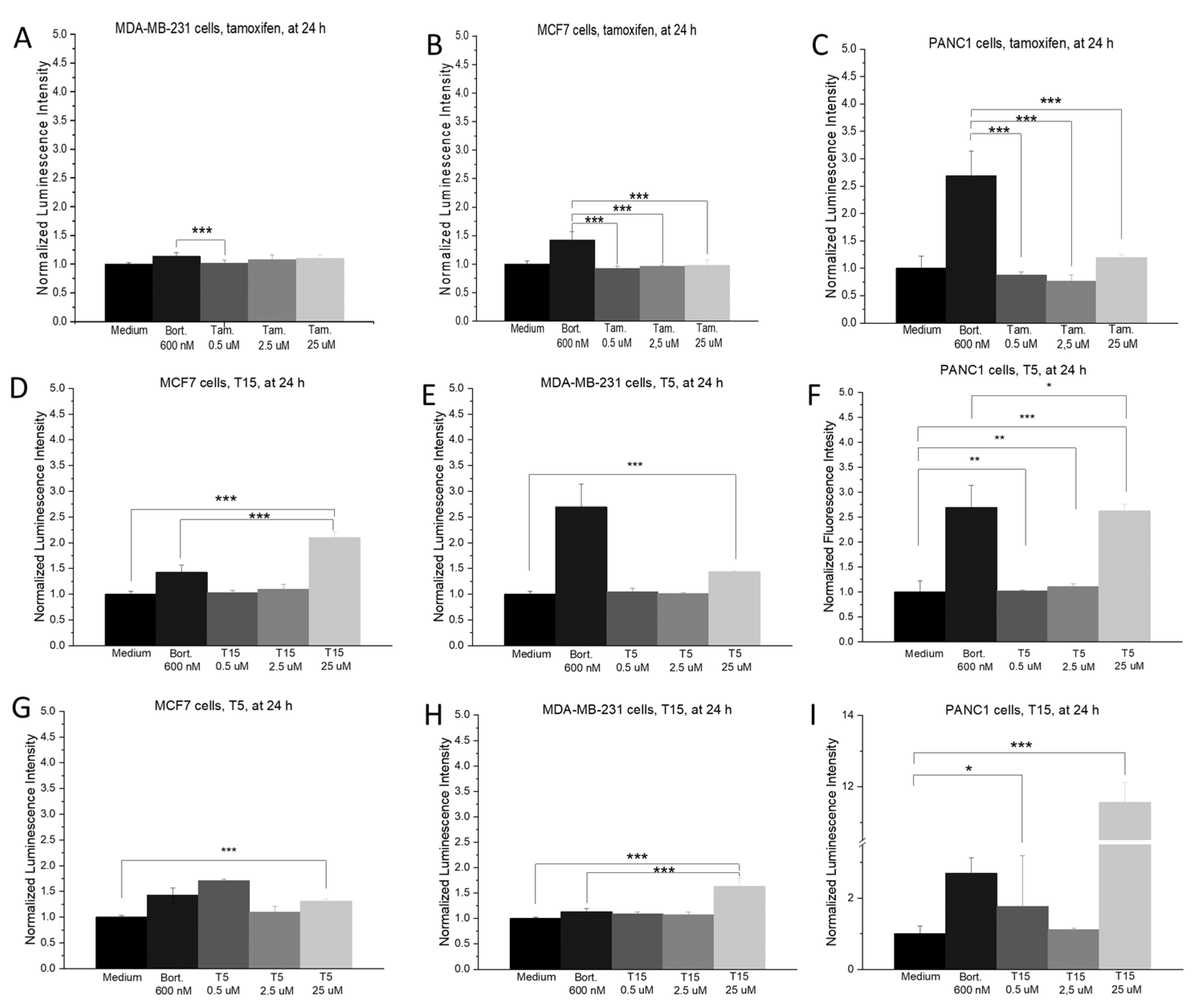
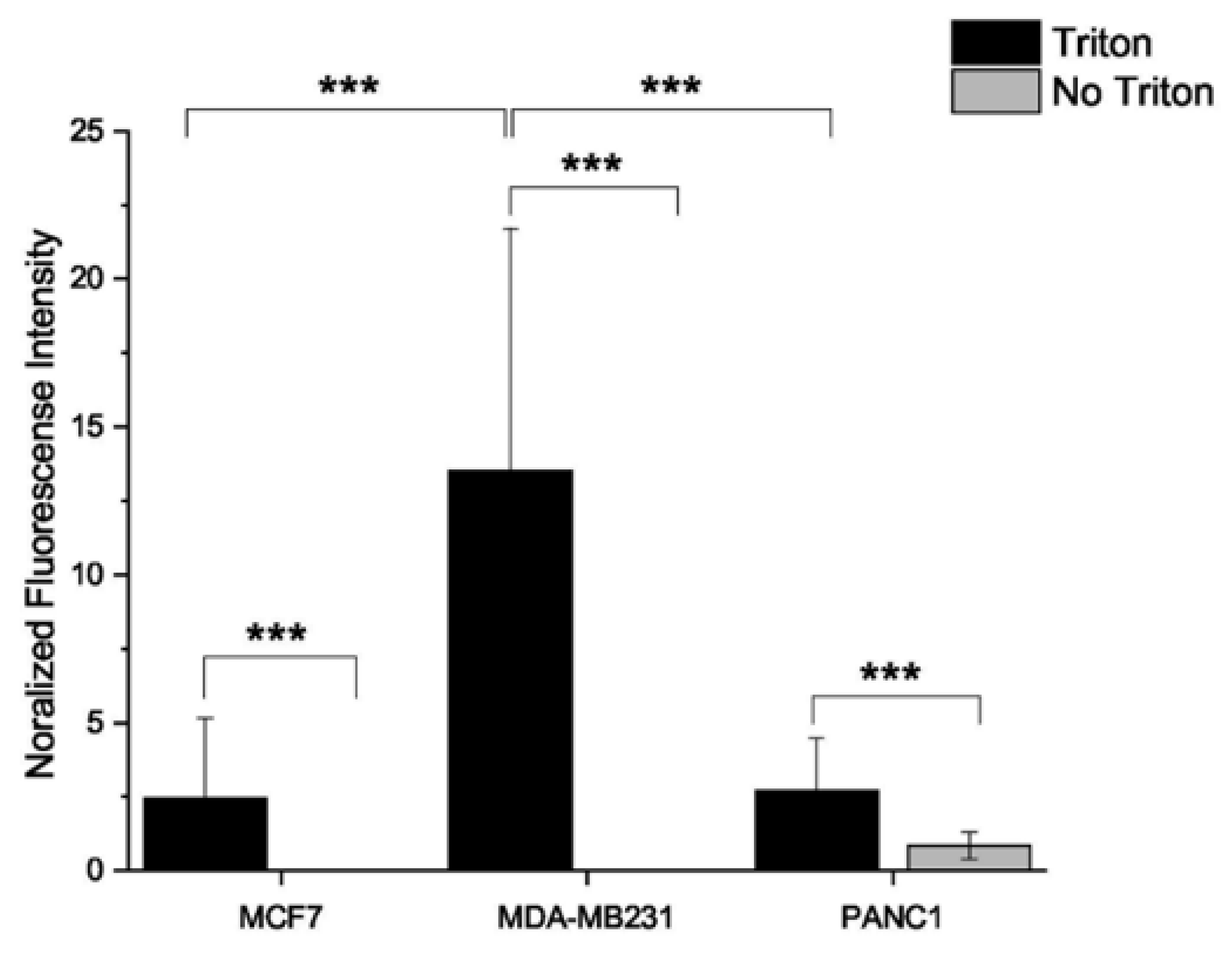
2.3. ROS Production
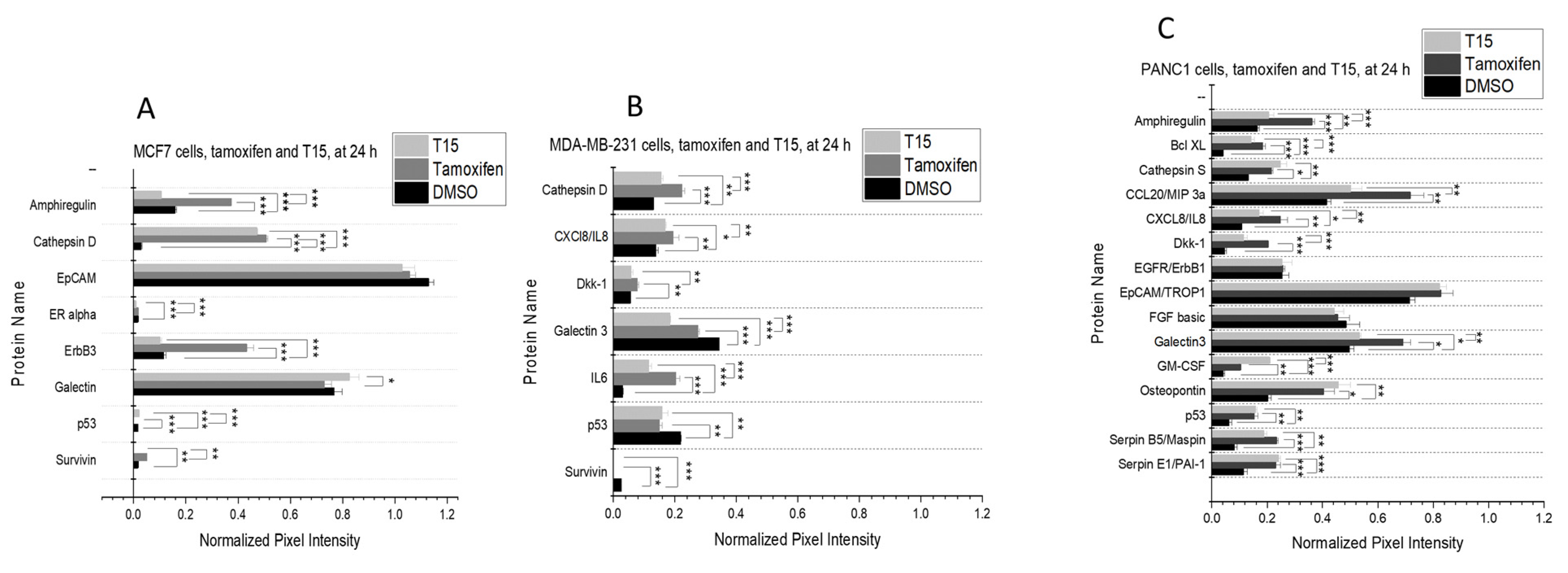
2.4. Proteome Profiler
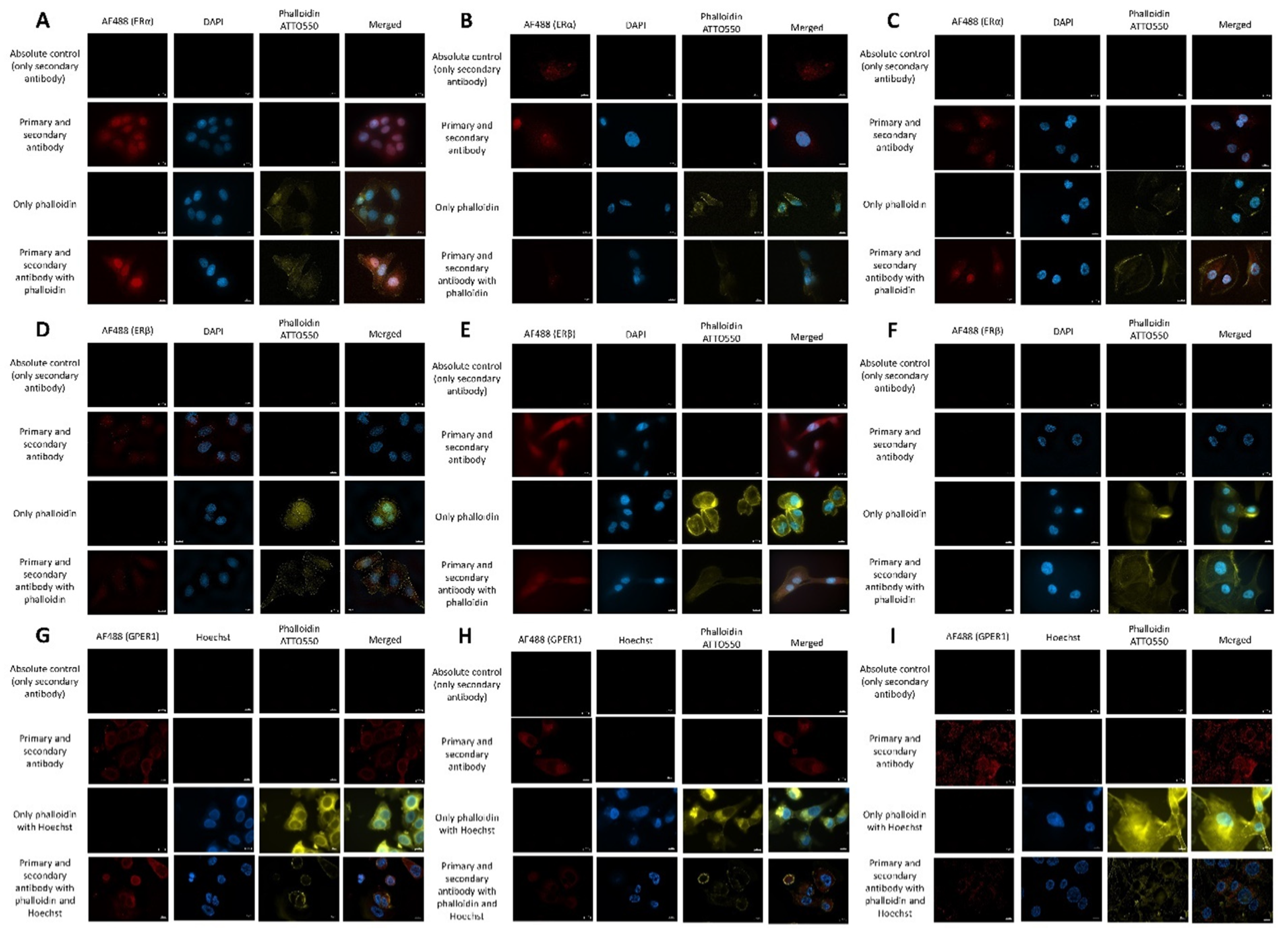
2.5. Estrogen Receptor Expression Profile
3. Discussion
4. Materials and Methods
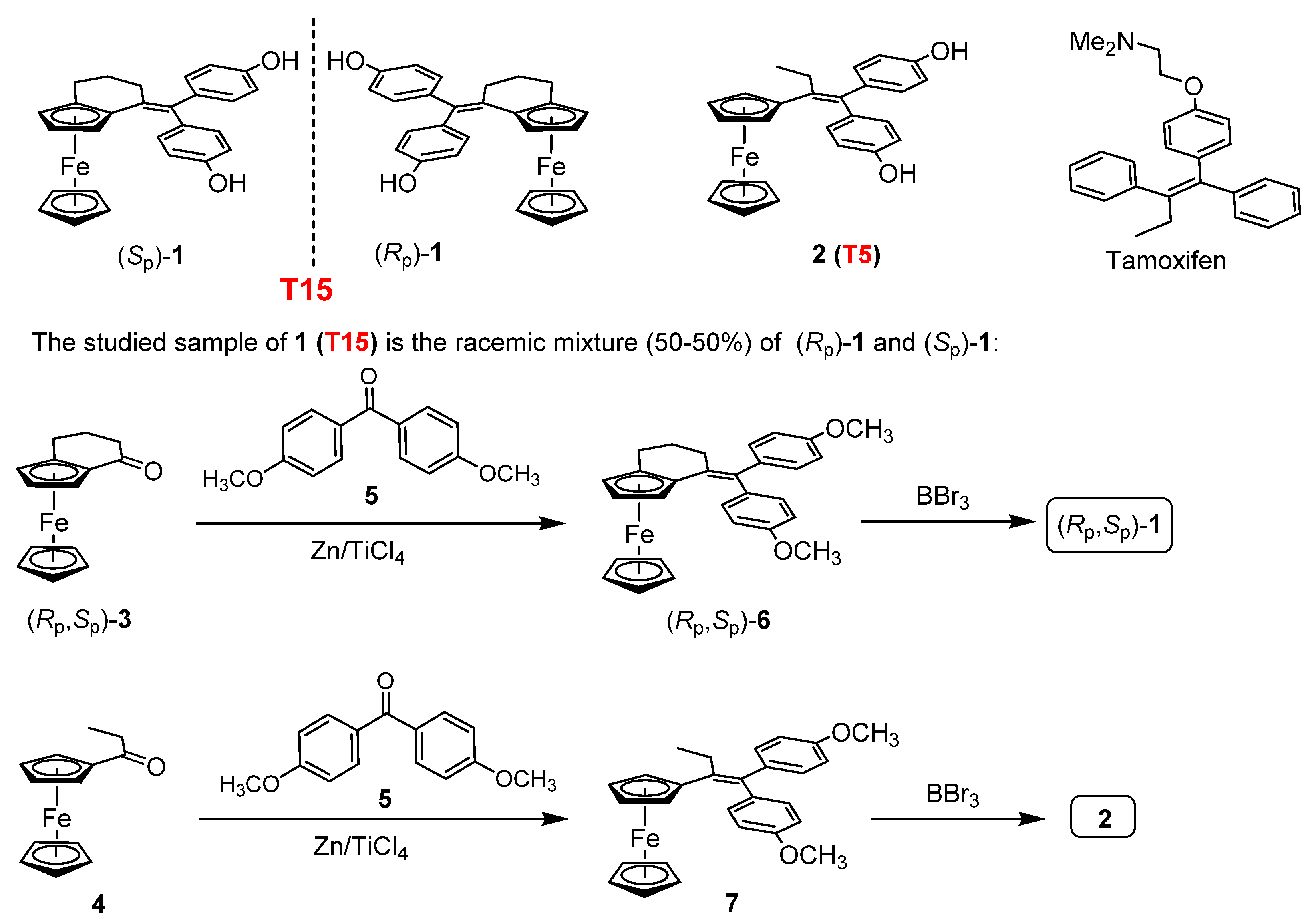
4.1. Synthesis of the Investigated Tamoxifen Derivatives
4.2. Cell Lines and Culturing
4.3. Cytotoxicity Assays
4.4. Cell Cycle Analysis
4.5. ROSGlo Assay
4.6. Proteome Profiler Human Oncology XL Array
4.7. Detection of G-Protein Coupled Estrogen Receptor 1 (GPER1), Estrogen Receptor α (ERα) and Estrogen Receptor β (ERβ) Expression with Indirect Immunohistochemistry
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jordan, V.C. Effect of tamoxifen (ICI 46,474) on initiation and growth of DMBA-induced rat mammary carcinomata. Eur. J. Cancer 1965, 12, 419–424. [Google Scholar] [CrossRef]
- Jordan, V.C. Tamoxifen: A most unlikely pioneering medicine. Nat. Rev. Drug Discov. 2003, 2, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Bogush, T.; Polezhaev, B.B.; Mamichev, I.; Bogush, E.A.; Polotsky, B.E.; Tjulandin, S.A.; Ryabov, A.B. Tamoxifen never ceases to amaze: New findings on non-estrogen receptor molecular targets and mediated effects. Cancer Investig. 2018, 36, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Jie, L.; Li, D.; Yang, C.; Haiying, Z. Tamoxifen versus clomiphene citrate for ovulation induction in infertile women. Eur. J. Obstet. Gynecol. Reprod. Biol. 2018, 228, 57–64. [Google Scholar] [CrossRef]
- Wibowo, E.; Pollock, P.A.; Hollis, N.; Wassersug, R.J. Tamoxifen in men: A review of adverse events. Andrology 2016, 4, 776–788. [Google Scholar] [CrossRef]
- Lapid, O.; van Wingerden, J.J.; Perlemuter, L. Tamoxifen therapy for the management of pubertal gynecomastia: A systematic review. J. Pediatr. Endocrinol. Metab. 2013, 26, 803–807. [Google Scholar] [CrossRef] [PubMed]
- Puce, R.; Porcaro, A.; Curti, P.; Girelli, D.; Pantalena, M.; Malossini, G.; Tallarigo, C. Treatment of retroperitoneal fibrosis with tamoxifen: Case report and review of literature. Arch. Españoles Urol. 2000, 53, 184–190. [Google Scholar]
- Wilcken, N.; Tattersall, M.H.N. Endocrine therapy for desmoid tumors. Cancer 1991, 68, 1384–1388. [Google Scholar] [CrossRef]
- Yang, D.J.; Tewson, T.; Tansey, W.; Kuang, L.-R.; Reger, G.; Cherif, A.; Wright, K.C.; Moult, R.G.; Tilbury, R.S.; Chu, K.; et al. Halogenated analogues of tamoxifen: Synthesis, receptor assay, and inhibition of MCF7 cells. J. Pharm. Sci. 1992, 81, 622–625. [Google Scholar] [CrossRef]
- Jaouen, G.; Vessières, A.; Top, S. Ferrocifen type anti cancer drugs. Chem. Soc. Rev. 2015, 44, 8802–8817. [Google Scholar] [CrossRef]
- Meegan, M.J.; Carr, M.; Knox, A.J.; Zisterer, D.M.; Lloyd, D.G. β-lactam type molecular scaffolds for antiproliferative activity: Synthesis and cytotoxic effects in breast cancer cells. J. Enzyme Inhib. Med. Chem. 2008, 23, 668–685. [Google Scholar] [CrossRef] [PubMed]
- Lainé, A.-L.; Adriaenssens, E.; Vessieres, A.; Jaouen, G.; Corbet, C.; Desruelles, E.; Pigeon, P.; Toillon, R.-A.; Passirani, C. The in vivo performance of ferrocenyl tamoxifen lipid nanocapsules in xenografted triple negative breast cancer. Biomaterials 2013, 34, 6949–6956. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Richard, M.A.; Top, S.; Dansette, P.M.; Pigeon, P.; Vessières, A.; Mansuy, D.; Jaouen, G. Ferrocenyl quinone methide-thiol adducts as new antiproliferative agents: Synthesis, metabolic formation from ferrociphenols, and oxidative transformation. Angew. Chem. Int. Ed. Engl. 2016, 55, 10431–10434. [Google Scholar] [CrossRef] [PubMed]
- Cázares Marinero, J.d.J.; Lapierre, M.; Cavaillès, V.; Saint-Fort, R.; Vessières, A.; Top, S.; Jaouen, G. Efficient new constructs against triple negative breast cancer cells: Synthesis and preliminary biological study of ferrocifen–SAHA hybrids and related species. Dalton Trans. 2013, 42, 15489–15501. [Google Scholar] [CrossRef]
- Resnier, P.; Galopin, N.; Sibiril, Y.; Clavreul, A.; Cayon, J.; Briganti, A.; Legras, P.; Vessières, A.; Montier, T.; Jaouen, G.; et al. Efficient ferrocifen anticancer drug and Bcl-2 gene therapy using lipid nanocapsules on human melanoma xenograft in mouse. Pharmacol. Res. 2017, 126, 54–65. [Google Scholar] [CrossRef]
- Topin-Ruiz, S.; Mellinger, A.; Lepeltier, E.; Bourreau, C.; Fouillet, J.; Riou, J.; Jaouen, G.; Martin, L.; Passirani, C.; Clere, N. P722 ferrocifen loaded lipid nanocapsules improve survival of murine xenografted-melanoma via a potentiation of apoptosis and an activation of CD8+ T lymphocytes. Int. J. Pharm. 2020, 593, 120111. [Google Scholar] [CrossRef]
- Lu, C.; Heldt, J.-M.; Guille-Collignon, M.; Lemaître, F.; Jaouen, G.; Vessières, A.; Amatore, C. Quantitative analyses of ROS and RNS production in breast cancer cell lines incubated with ferrocifens. ChemMedChem 2014, 9, 1286–1293. [Google Scholar] [CrossRef]
- Cázares-Marinero, J.d.J.; Top, S.; Vessières, A.; Jaouen, G. Synthesis and antiproliferative activity of hydroxyferrocifen hybrids against triple-negative breast cancer cells. Dalton Trans. 2014, 43, 817–830. [Google Scholar] [CrossRef]
- Buriez, O.; Heldt, J.M.; Labbé, E.; Vessières, A.; Jaouen, G.; Amatore, C. Reactivity and antiproliferative activity of ferrocenyl-tamoxifen adducts with cyclodextrins against hormone-independent breast-cancer cell lines. Chemistry 2008, 14, 8195–8203. [Google Scholar] [CrossRef]
- Wang, Y.; Heinemann, F.; Top, S.; Dazzi, A.; Policar, C.; Henry, L.; Lambert, F.; Jaouen, G.; Salmain, M.; Vessières, A. Ferrocifens labelled with an infrared rhenium tricarbonyl tag: Synthesis, antiproliferative activity, quantification and nano IR mapping in cancer cells. Dalton Trans. 2018, 47, 9824–9833. [Google Scholar] [CrossRef]
- Cunningham, L.; Wang, Y.; Nottingham, C.; Pagsulingan, J.; Jaouen, G.; Mc Glinchey, M.J.; Guiry, P.J. Enantioselective synthesis of planar chiral ferrocifens that show chiral discrimination in antiproliferative activity on breast cancer cells. ChemBioChem. 2020, 21, 2974–2981. [Google Scholar] [CrossRef] [PubMed]
- Vessieres, A.; Corbet, C.; Heldt, J.M.; Lories, N.; Jouy, N.; Laïos, I.; Leclercq, G.; Jaouen, G.; Toillon, R.-A. A ferrocenyl derivative of hydroxytamoxifen elicits an estrogen receptor-independent mechanism of action in breast cancer cell lines. J. Inorg. Biochem. 2010, 104, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Bertheau, P.; Lehmann-Che, J.; Varna, M.; Dumay, A.; Poirot, B.; Porcher, R.; Turpin, E.; Plassa, L.-F.; de Roquancourt, A.; Bourstyn, E.; et al. P53 in breast cancer subtypes and new insights into response to chemotherapy. Breast 2013, 22, S27–S29. [Google Scholar] [CrossRef] [PubMed]
- Da Veiga, G.L.; da Silva, R.D.M.; Pereira, E.C.; Azzalis, L.A.; Alves, B.D.C.A.; Gehrke, F.D.S.; Gascón, T.M.; Fonseca, F.L.A. The role of Survivin as a biomarker and potential prognostic factor for breast cancer. Rev. Assoc. Méd. Bras. 2019, 65, 893–901. [Google Scholar] [CrossRef]
- Xue, J.; Yao, Y.; Yao, Q.; Tian, X.; Feng, Y.; Su, H.; Kong, D.; Cui, C.; Yan, L.; Hao, C.; et al. Important roles of estrogen receptor alpha in tumor progression and anti-estrogen therapy of pancreatic ductal adenocarcinoma. Life Sci. 2020, 260, 118302. [Google Scholar] [CrossRef] [PubMed]
- Konduri, S.; Schwarz, R.E. Estrogen receptor β/α ratio predicts response of pancreatic cancer cells to estrogens and phytoestrogens. J. Surg. Res. 2007, 140, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Vladusic, E.A.; Hornby, A.E.; Guerra-Vladusic, F.K.; Lupu, R. Expression of estrogen receptor beta messenger RNA variant in breast cancer. Cancer Res. 1998, 58, 210–214. [Google Scholar]
- Woo, H.I.; Lee, S.K.; Kim, J.; Kim, S.W.; Yu, J.; Bae, S.Y.; Lee, J.E.; Nam, S.J.; Lee, S.-Y. Variations in plasma concentrations of tamoxifen metabolites and the effects of genetic polymorphisms on tamoxifen metabolism in Korean patients with breast cancer. Oncotarget 2017, 8, 100296–100311. [Google Scholar] [CrossRef]
- Kim, S.-H.; Katzenellenbogen, J.A. Triarylethylene bisphenols with a novel cycle are ligands for the estrogen receptor. Bioorg. Med. Chem. 2000, 8, 785–793. [Google Scholar] [CrossRef]
- Warner, M.; Huang, B.; Gustafsson, J.A. Estrogen receptor beta as a pharmaceutical target. Trends Pharmacol. Sci. 2017, 38, 92–99. [Google Scholar] [CrossRef]
- Filardo, E.; Quinn, J.; Pang, Y.; Graeber, C.; Shaw, S.; Dong, J.; Thomas, P. Activation of the novel estrogen receptor G protein-coupled receptor 30 (GPR30) at the plasma membrane. Endocrinology 2007, 148, 3236–3245. [Google Scholar] [CrossRef] [PubMed]
- Otto, C.; Rohde-Schulz, B.; Schwarz, G.; Fuchs, I.; Klewer, M.; Brittain, D.; Langer, G.; Bader, B.; Prelle, K.; Nubbemeyer, R.; et al. G protein-coupled receptor 30 localizes to the endoplasmic reticulum and is not activated by estradiol. Endocrinology 2008, 149, 4846–4856. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Quinn, J.A.; Frackelton, A.R., Jr.; Bland, K.I. Estrogen action via the G protein-coupled receptor, GPR30: Stimulation of adenylyl cyclase and c, AMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol. Endocrinol. 2002, 16, 70–84. [Google Scholar] [CrossRef]
- Mesmar, F.; Dai, B.; Ibrahim, A.; Hases, L.; Jafferali, M.H.; Augustine, J.J.; di Lorenzo, S.; Kang, Y.; Zhao, Y.; Wang, J.; et al. Clinical candidate and genistein analogue AXP107-11 has chemoenhancing functions in pancreatic adenocarcinoma through G protein-coupled estrogen receptor signaling. Cancer Med. 2019, 8, 7705–7719. [Google Scholar] [CrossRef]
- Riggs, B.L.; Hartmann, L.C. Selective estrogen-receptor modulators—Mechanisms of action and application to clinical practice. N. Engl. J. Med. 2003, 348, 618–629. [Google Scholar] [CrossRef] [PubMed]
- Emons, G.; Mustea, A.; Tempfer, C. Tamoxifen and endometrial cancer: A janus-headed drug. Cancers 2020, 12, 2535. [Google Scholar] [CrossRef]
- Torres-López, L.; Maycotte, P.; Liñán-Rico, A.; Liñán-Rico, L.; Maturano, L.D.; Delgado-Enciso, I.; Meza-Robles, C.; Vásquez-Jiménez, C.; Hernández-Cruz, A.; Dobrovinskaya, O. Tamoxifen induces toxicity, causes autophagy, and partially reverses dexamethasone resistance in jurkat T cells. J. Leukoc. Biol. 2019, 105, 983–998. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, P.; Nadig, N.; Nambiar, S. Non-canonical estrogen signaling in endocrine resistance. Front. Endocrinol. 2019, 10, 708. [Google Scholar] [CrossRef]
- Dowers, T.S.; Qin, Z.-H.; Thatcher, G.R.J.; Bolton, J.L. Bioactivation of selective estrogen receptor modulators (SERMs). Chem. Res. Toxicol. 2006, 19, 1125–1137. [Google Scholar] [CrossRef]
- Satake, M.; Sawai, H.; Go, V.L.W.; Satake, K.; Reber, H.A.; Hines, O.J.; Eibl, G. Estrogen receptors in pancreatic tumors. Pancreas 2006, 33, 119–127. [Google Scholar] [CrossRef]
- Yip, C.H.; Rhodes, A. Estrogen and progesterone receptors in breast cancer. Future Oncol. 2014, 10, 2293–2301. [Google Scholar] [CrossRef] [PubMed]
- Brian, T.Z.; Zafonte, B.T.; Hulit, J.; Amanatullah, D.F.; Albanese, C.; Wang, C.; Rosen, E.; Reutens, A.; Sparano, J.A.; Lisanti, M.P.; et al. Cell-cycle dysregulation in breast cancer: Breast cancer therapies targeting the cell cycle. Front. Biosci. 2000, 5, D938–D961. [Google Scholar] [CrossRef]
- Bonofiglio, D.; Gabriele, S.; Aquila, S.; Catalano, S.; Gentile, M.; Middea, E.; Giordano, F.; Andò, S. Estrogen receptor alpha binds to peroxisome proliferator-activated receptor response element and negatively interferes with peroxisome proliferator-activated receptor gamma signaling in breast cancer cells. Clin. Cancer Res. 2005, 11, 6139–6147. [Google Scholar] [CrossRef] [PubMed]
- Doisneau-Sixou, S.F.; Sergio, C.M.; Carroll, J.; Hui, R.; Musgrove, E.A.; Sutherland, R.L. Estrogen and antiestrogen regulation of cell cycle progression in breast cancer cells. Endocr. Relat. Cancer 2003, 10, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Prall, O.J.; Sarcevic, B.; Musgrove, E.A.; Watts, C.W.; Sutherland, R.L. Estrogen-induced activation of Cdk4 and Cdk2 during G1-S phase progression is accompanied by increased cyclin D1 expression and decreased cyclin-dependent kinase inhibitor association with cyclin E-Cdk2. J. Biol. Chem. 1997, 272, 10882–10894. [Google Scholar] [CrossRef]
- Hillard, E.; Vessières, A.; Thouin, L.; Jaouen, G.; Amatore, C. Ferrocene-mediated proton-coupled electron transfer in a series of ferrocifen-type breast-cancer drug candidates. Angew. Chem. Int. Ed. 2006, 45, 285–290. [Google Scholar] [CrossRef]
- De Oliveira, A.C.; Hillard, E.A.; Pigeon, P.; Rocha, D.D.; Rodrigues, F.A.; Montenegro, R.C.; Costa-Lotufo, L.V.; Goulart, M.O.; Jaouen, G. Biological evaluation of twenty-eight ferrocenyl tetrasubstituted olefins: Cancer cell growth inhibition, ROS production and hemolytic activity. Eur. J. Med. Chem. 2011, 46, 3778–3787. [Google Scholar] [CrossRef]
- Citta, A.; Folda, A.; Bindoli, A.; Pigeon, P.; Top, S.; Vessieres, A.; Salmain, M.; Jaouen, G.; Rigobello, M.P. Evidence for targeting thioredoxin reductases with ferrocenyl quinone methides. A possible molecular basis for the antiproliferative effect of hydroxyferrocifens on cancer cells. J. Med. Chem. 2014, 57, 8849–8859. [Google Scholar] [CrossRef]
- Wlassoff, W.A.; Albright, C.D.; Sivashinski, M.S.; Ivanova, A.; Appelbaum, J.G.; Salganik, R.I. Hydrogen peroxide overproduced in breast cancer cells can serve as an anticancer prodrug generating apoptosis-stimulating hydroxyl radicals under the effect of tamoxifen-ferrocene conjugate. J. Pharm. Pharmacol. 2007, 59, 1549–1553. [Google Scholar] [CrossRef]
- Michard, Q.; Jaouen, G.; Vessieres, A.; Bernard, B. Evaluation of cytotoxic properties of organometallic ferrocifens on melanocytes, primary and metastatic melanoma cell lines. J. Inorg. Biochem. 2008, 102, 1980–1985. [Google Scholar] [CrossRef]
- Bruyère, C.; Mathieu, V.; Vessieres, A.; Pigeon, P.; Top, S.; Jaouen, G.; Kiss, R. Ferrocifen derivatives that induce senescence in cancer cells: Selected examples. J. Inorg. Biochem. 2014, 141, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wu, H.; Wang, L.; Lu, J.; Duan, H.; Liu, X.; Liang, Z. Expression of amphiregulin predicts poor outcome in patients with pancreatic ductal adenocarcinoma. Diagn. Pathol. 2016, 11, 60. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Hosoda, M.; Nakano, K.; Jia, S.; Hatanaka, K.C.; Takakuwa, E.; Hatanaka, Y.; Matsuno, Y.; Yamashita, H. P53 accumulation is a strong predictor of recurrence in estrogen receptor-positive breast cancer patients treated with aromatase inhibitors. Cancer Sci. 2013, 105, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.Y.; Nam, S.J.; Jung, Y.; Lee, S.B.; Park, B.-W.; Lim, W.; Jung, S.H.; Yang, H.W.; Jung, S.P. Differences in prognosis and efficacy of chemotherapy by p53 expression in triple-negative breast cancer. Breast Cancer Res. Treat. 2018, 172, 437–444. [Google Scholar] [CrossRef]
- Striefler, J.K.; Sinn, M.; Pelzer, U.; Jühling, A.; Wislocka, L.; Bahra, M.; Sinn, B.V.; Denkert, C.; Dörken, B.; Oettle, H.; et al. P53 overexpression and Ki67-index are associated with outcome in ductal pancreatic adenocarcinoma with adjuvant gemcitabine treatment. Pathol. Res. Pr. 2016, 212, 726–734. [Google Scholar] [CrossRef]
- Dutta, S.; Ryan, J.; Chen, T.S.; Kougentakis, C.; Letai, A.; Keating, A.E. Potent and specific peptide inhibitors of human pro-survival protein Bcl-xL. J. Mol. Biol. 2015, 427, 1241–1253. [Google Scholar] [CrossRef]
- Dong, H.; Qian, N.; Wang, Y.; Meng, L.; Chen, D.; Ji, X.; Feng, W. Survivin expression and serum levels in pancreatic cancer. World J. Surg. Oncol. 2015, 13, 189. [Google Scholar] [CrossRef]
- Kuwada, Y.; Sasaki, T.; Morinaka, K.; Kitadai, Y.; Mukaida, N.; Chayama, K. Potential involvement of IL-8 and its receptors in the invasiveness of pancreatic cancer cells. Int. J. Oncol. 2003, 22, 765–771. [Google Scholar] [CrossRef]
- Gao, B.; Chen, P.; Jiang, Q. G protein-coupled estrogen receptor is a critical regulator in metastasis of breast cancer cells. J. Biosci. Med. 2017, 5, 127–140. [Google Scholar] [CrossRef][Green Version]
- Liu, B.; Jia, Y.; Ma, J.; Wu, S.; Jiang, H.; Cao, Y.; Sun, X.; Yin, X.; Yan, S.; Shang, M.; et al. Tumor-associated macrophage-derived CCL20 enhances the growth and metastasis of pancreatic cancer. Acta Biochim. Biophys. Sin. 2016, 48, 1067–1074. [Google Scholar] [CrossRef]
- Masjedi, A.; Hashemi, V.; Hojjat-Farsangi, M.; Ghalamfarsa, G.; Azizi, G.; Yousefi, M.; Jadidi-Niaragh, F. The significant role of interleukin-6 and its signaling pathway in the immunopathogenesis and treatment of breast cancer. Biomed. Pharmacother. 2018, 108, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Baba, T.; Li, Y.Y.; Furukawa, K.; Tanabe, Y.; Matsugo, S.; Sasaki, S.; Mukaida, N. Gemcitabine-induced CXCL8 expression counteracts its actions by inducing tumor neovascularization. Biochem. Biophys. Res. Commun. 2015, 458, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Qin, Y.; Wang, N.; Zhou, L.; Liu, Y.; Chen, S.; Yin, L.; Xiao, Y.; Yao, X.-H.; Yang, X.; et al. CCL20 triggered by chemotherapy hinders the therapeutic efficacy of breast cancer. PLoS Biol. 2018, 16, e2005869. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Luo, M.; Liang, X.; Wang, D.; Gu, X.; Duan, C.; Gu, H.; Chen, G.; Zhao, X.; Zhao, Z.; et al. Correction: Galectin-3 as a marker and potential therapeutic target in breast cancer. PLoS ONE 2020, 15, e0232166. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, R.; Kawahara, K.; Fujii, T.; Takei, H.; Naito, Z. Higher expression of Ep, CAM is associated with poor clinical and pathological responses in breast cancer patients undergoing neoadjuvant chemotherapy. Pathol. Int. 2016, 66, 210–217. [Google Scholar] [CrossRef]
- Liu, D.-J.; Xie, Y.-X.; Liu, X.-X.; Huo, Y.-M.; Yang, M.-W.; Fu, X.-L.; Liu, W.; Yang, J.-Y.; Xue-Liang, F.; Hua, R.; et al. The role of Dickkopf-1 as a potential prognostic marker in pancreatic ductal adenocarcinoma. Cell Cycle 2017, 16, 1622–1629. [Google Scholar] [CrossRef]
- Takahashi, N.; Fukushima, T.; Yorita, K.; Tanaka, H.; Chijiiwa, K.; Kataoka, H. Dickkopf-1 is overexpressed in human pancreatic ductal adenocarcinoma cells and is involved in invasive growth. Int. J. Cancer 2009, 126, 1611–1620. [Google Scholar] [CrossRef]
- Zhou, X.L.; Qin, X.R.; Zhang, X.D.; Ye, L.H. Downregulation of dickkopf-1 is responsible for high proliferation of breast cancer cells via losing control of Wnt/β-catenin signaling. Acta Pharmacol. Sin. 2010, 31, 202–210. [Google Scholar] [CrossRef][Green Version]
- Sun, T.; Jiang, D.; Zhang, L.; Su, Q.; Mao, W.; Jiang, C. Expression profile of cathepsins indicates the potential of cathepsins B and D as prognostic factors in breast cancer patients. Oncol. Lett. 2016, 11, 575–583. [Google Scholar] [CrossRef]
- Dumartin, L.; Whiteman, H.; Weeks, M.; Hariharan, D.; Dmitrovic, B.; Iacobuzio-Donahue, C.A.; Brentnall, T.A.; Bronner, M.P.; Feakins, R.M.; Timms, J.; et al. AGR2 is a novel surface antigen that promotes the dissemination of pancreatic cancer cells through regulation of cathepsins B and D. Cancer Res. 2011, 71, 7091–7102. [Google Scholar] [CrossRef]
- Wilkinson, R.D.A.; Burden, R.E.; Mc Dowell, S.H.; Mc Art, D.G.; Mc Quaid, S.; Bingham, V.; Williams, R.; Cox Órla, T.; O’Connor, R.; Mc Cabe, N.; et al. A novel role for cathepsin S as a potential biomarker in triple negative breast cancer. J. Oncol. 2019, 2019, 3980273. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lu, S.; Xu, R.; Tang, Y.; Liu, J.; Li, C.; Wei, J.; Yao, R.; Zhao, X.; Wei, Q.; et al. Mechanisms of estradiol-induced EGF-like factor expression and oocyte maturation via G protein-coupled estrogen receptor. Endocrinology 2020, 161, bqaa190. [Google Scholar] [CrossRef] [PubMed]
- Stoica, G.E.; Franke, T.F.; Wellstein, A.; Morgan, E.; Czubayko, F.; List, H.J.; Reiter, R.; Martin, M.B.; Stoica, A. Heregulin-β1 regulates the estrogen receptor-alpha gene expression and activity via the Erb, B2/PI 3-K/akt pathway. Oncogene 2003, 22, 2073–2087. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, K.; Helguero, L.A.; Omoto, Y.; Gustafsson, J.; Haldosén, L.A. Estrogen receptor β represses akt signaling in breast cancer cells via downregulation of HER2/HER3 and upregulation of PTEN: Implications for tamoxifen sensitivity. Breast Cancer Res. 2011, 13, R43. [Google Scholar] [CrossRef]
- Lu, W.; Katzenellenbogen, B.S. Estrogen receptor-β modulation of the ERα-p53 loop regulating gene expression, proliferation, and apoptosis in breast cancer. Horm. Cancer 2017, 8, 230–242. [Google Scholar] [CrossRef]
- Alcon, C.; Gómez Tejeda Zañudo, J.; Albert, R.; Wagle, N.; Scaltriti, M.; Letai, A.; Samitier, J.; Montero, J. ER+ breast cancer strongly depends on MCL-1 and BCL-x, L anti-apoptotic proteins. Cells 2021, 10, 1659. [Google Scholar] [CrossRef]
- Wen, K.-C.; Sung, P.-L.; Chou, Y.-T.; Pan, C.-M.; Wang, P.-H.; Lee, O.K.-S.; Wu, C.-W. The role of Ep, CAM in tumor progression and the clinical prognosis of endometrial carcinoma. Gynecol. Oncol. 2018, 148, 383–392. [Google Scholar] [CrossRef]
- Stark, A.M.; Schem, C.; Maass, N.; Hugo, H.H.; Jonat, W.; Mehdorn, H.M.; Held-Feindt, J. Expression of metastasis suppressor gene maspin is reduced in breast cancer brain metastases and correlates with the estrogen receptor status. Neurol. Res. 2010, 32, 303–308. [Google Scholar] [CrossRef]
- Kasoha, M.; Dernektsi, C.; Seibold, A.; Bohle, R.M.; Takacs, Z.; Ioan-Iulian, I.; Solomayer, E.-F.; Juhasz-Böss, I. Crosstalk of estrogen receptors and Wnt/β-catenin signaling in endometrial cancer. J. Cancer Res. Clin. Oncol. 2020, 146, 315–327. [Google Scholar] [CrossRef]
- Chan, H.J.; Li, H.; Liu, Z.; Yuan, Y.C.; Mortimer, J.; Chen, S. SERPINA1 is a direct estrogen receptor target gene and a predictor of survival in breast cancer patients. Oncotarget 2015, 6, 25815–25827. [Google Scholar] [CrossRef]
- Ma, Y.; Ren, Y.; Dai, Z.-J.; Wu, C.-J.; Ji, Y.-H.; Xu, J. IL-6, IL-8 and TNF-α levels correlate with disease stage in breast cancer patients. Adv. Clin. Exp. Med. 2017, 26, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Fuksiewicz, M.; Kaminska, J.; Kotowicz, B.; Kowalska, M.; Rubach, M.; Pienkowski, T. Serum cytokine levels and the expression of estrogen and progesterone receptors in breast cancer patients. Clin. Chem. Lab. Med. CCLM 2006, 44, 1092–1097. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Tang, Z.; Jiang, Y.; Huang, H.; Luo, P.; Qing, B.; Zhang, S.; Tang, R. IL-8 is involved in estrogen-related receptor α-regulated proliferation and migration of colorectal cancer cells. Am. J. Dig. Dis. 2017, 62, 3438–3446. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Guan, X.; Liang, N.; Li, S. Estrogen-related receptor alpha triggers the proliferation and migration of human non-small cell lung cancer via interleukin-6. Cell Biochem. Funct. 2018, 36, 255–262. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Y.; Lan, L.; Liu, R.; Wu, Y.; Qu, Q.; Wen, K. Tamoxifen has a proliferative effect in endometrial carcinoma mediated via the GPER/EGFR/ERK/cyclin D1 pathway: A retrospective study and an in vitro study. Mol. Cell. Endocrinol. 2016, 437, 51–61. [Google Scholar] [CrossRef]
- Molina, L.; Bustamante, F.; Ortloff, A.; Ramos, I.; Ehrenfeld, P.; Figueroa, C.D. Continuous exposure of breast cancer cells to tamoxifen upregulates GPER-1 and increases cell proliferation. Front. Endocrinol. 2020, 11, 563165. [Google Scholar] [CrossRef]
- Li, Y.; Gong, D.; Zhang, L.; Li, H.; Zhang, S.; Zhang, J.; Li, K.; Zheng, Q.; Zhao, G.; Zhang, Y.; et al. Zinc finger protein 32 promotes breast cancer stem cell-like properties through directly promoting GPER transcription. Cell Death Dis. 2018, 9, 1162. [Google Scholar] [CrossRef]
- Ignatov, A.; Ignatov, T.; Roessner, A.; Costa, S.D.; Kalinski, T. Role of GPR30 in the mechanisms of tamoxifen resistance in breast cancer MCF-7 cells. Breast Cancer Res. Treat. 2009, 123, 87–96. [Google Scholar] [CrossRef]
- Wegner, M.-S.; Gruber, L.; Schömel, N.; Trautmann, S.; Brachtendorf, S.; Fuhrmann, D.; Schreiber, Y.; Olesch, C.; Brüne, B.; Geisslinger, G.; et al. GPER1 influences cellular homeostasis and cytostatic drug resistance via influencing long chain ceramide synthesis in breast cancer cells. Int. J. Biochem. Cell Biol. 2019, 112, 95–106. [Google Scholar] [CrossRef]
- Ignatov, T.; Claus, M.; Nass, N.; Haybaeck, J.; Seifert, B.; Kalinski, T.; Ortmann, O.; Ignatov, A. G-protein-coupled estrogen receptor GPER-1 expression in hormone receptor-positive breast cancer is associated with poor benefit of tamoxifen. Breast Cancer Res. Treat. 2019, 174, 121–127. [Google Scholar] [CrossRef]
- Ino, Y.; Akimoto, T.; Takasawa, A.; Takasawa, K.; Aoyama, T.; Ueda, A.; Ota, M.; Magara, K.; Tagami, Y.; Murata, M.; et al. Elevated expression of G protein-coupled receptor 30 (GPR30) is associated with poor prognosis in patients with uterine cervical adenocarcinoma. Histol. Histopathol. 2019, 35, 18157. [Google Scholar]
- Smith, H.O.; Leslie, K.; Singh, M.; Qualls, C.R.; Revankar, C.M.; Joste, N.E.; Prossnitz, E. GPR30: A novel indicator of poor survival for endometrial carcinoma. Am. J. Obstet. Gynecol. 2007, 196, 386.e1–386.e11. [Google Scholar] [CrossRef] [PubMed]
- Plazuk, D.; Top, S.; Vessières, A.; Plamont, M.A.; Huché, M.; Zakrzewski, J.; Makal, A.; Woźniak, K.; Jaouen, G. Organometallic cyclic polyphenols derived from 1,2-(α-keto tri or tetra methylene) ferrocene show strong antiproliferative activity on hormone-independent breast cancer cells. Dalton Trans. 2010, 39, 7444–7450. [Google Scholar] [CrossRef] [PubMed]
- Rinehart, K.L.; Curby, R.J. Ferrocene bridging and homoannular cyclizations. J. Am. Chem. Soc. 1957, 79, 3290–3291. [Google Scholar] [CrossRef]
- Top, S.; Tang, J.; Vessi, A.; Carrez, D.; Provot, C.; Jaouen, G. Ferrocenyl hydroxytamoxifen: A prototype for a new range of oestradiol receptor site-directed cytotoxics. Chem. Commun. 1996, 8, 955–956. [Google Scholar] [CrossRef]
- Top, S.; Dauer, B.; Vaissermann, J.; Jaouen, G. Facile route to ferrocifen, 1-[4-(2-dimethylaminoethoxy)]-1-(phenyl-2-ferrocenyl-but-1-ene), first organometallic analogue of tamoxifen, by the McMurry reaction. J. Organomet. Chem. 1997, 541, 355–361. [Google Scholar] [CrossRef]
- Lajkó, E.; Spring, S.; Hegedüs, R.; Biri-Kovács, B.; Ingebrandt, S.; Mező, G.; Kőhidai, L. Comparative cell biological study of in vitro antitumor and antimetastatic activity on melanoma cells of Gn, RH-III-containing conjugates modified with short-chain fatty acids. Beilstein J. Org. Chem. 2018, 14, 2495–2509. [Google Scholar] [CrossRef]
- Caillot, M.; Zylbersztejn, F.; Maitre, E.; Bourgeais, J.; Hérault, O.; Sola, B. ROS overproduction sensitises myeloma cells to bortezomib-induced apoptosis and alleviates tumour microenvironment-mediated cell resistance. Cells 2020, 9, 2357. [Google Scholar] [CrossRef]
- Li, X.; Liang, M.; Jiang, J.; He, R.; Wang, M.; Guo, X.; Shen, M.; Qin, R. Combined inhibition of autophagy and Nrf2 signaling augments bortezomib-induced apoptosis by increasing ROS production and ER stress in pancreatic cancer cells. Int. J. Biol. Sci. 2018, 14, 1291–1305. [Google Scholar] [CrossRef]
- Mehdizadeh, K.; Ataei, F.; Hosseinkhani, S. Treating MCF7 breast cancer cell with proteasome inhibitor bortezomib restores apoptotic factors and sensitizes cell to docetaxel. Med. Oncol. 2021, 38, 64. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author | Reference | Name(s) of Derivative(s) | IC50 in µM |
|---|---|---|---|
| Kalabay M. et al. | - | T5 T15 | 43.34 (MCF7) 20.2 (MDA-MB-231) 12.46 (PANC1) 23.07 (MCF7), 23.77 (MDA-MB-231), 15.03 (PANC1) |
| Lainé A.-L. | [12] | P5, P15 | 0.5–2 |
| Wang Y. et al. | [13] | FC1 FC2 FC3 | 1.5 (MDA-MB-231) 0.6 (MDA-MB-231) 0.5 (MDA-MB-231) |
| Cázares-Marinero, J. de J et al. | [14] | FcTAM, FcTAM-SAHA | 2.6 (MDA-MB-231), 4.4 (MCF7) 0.7 (MDA-MB-231) 2 (MCF7) |
| Resnier P. et al. | [15] | ansa-FcDiOH FcDiOH | 1 (SKMel28) 3 (SKMel28) |
| Topin-Ruiz S. et al. | [16] | p722 | 168 B16F10 |
| Lu C. et al. | [17] | FcDiOH FcOHTam ansa-Fc-DiOH | 0.6 (MDA-MB-231) 0.089 (MDA-MB-231) 0.5 (MDA-MB-231) |
| Cázares-Marinero, J. de J et al. | [18] | FcOHTam FcOHTam-SAHA FcOHTam-PSA FcOHTam-OPOA | 1.6 (MDA-MB-231) 1.3 (MDA-MB-231) 1.5 (MCF7) 2.1 (MDA-MB-231) 4.4 (MCF7) 4.5 (MDA-MB-231) 6.6 (MCF7) |
| Buriez O. et al. | [19] | 1 (ferrocenyl diphenol) | 0.7 (MCF7) 0.5 (MDA-MB-231) |
| Wang Y. et al. | [20] | 1a 2 | 0.66 (MDA-MB-231) 0.11 (MDA-MB-231) |
| Cunningham L. et al. | [21] | 7 different racemic ferrocifens | 09–16 (MCF7) 05–16.1 (MDA-MB-231) |
| Vessiéres A. et al. | [22] | FcOHTam | Fc-OH-TAM (0.1 and 1 uM) inhibited the growth of breast cancer cell lines MCF7, T-47D, ZR-75-1, MDA-MB-231, SKBR-3 and Hs578-T |
| MCF7 | MDA-MB-231 | PANC1 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Tamoxifen | T5 | T15 | Tamoxifen | T5 | T15 | Tamoxifen | T5 | T15 | |
| 24 h | ND | 42.81 | 24.12 | 25.53 | 29.97 | 25.28 | 33.33 | 21.17 | 24.46 |
| 48 h | 42.73 | 42.95 | 23.24 | 23.05 | 31.6 | 23.6 | 33.68 | 21.29 | 23.80 |
| 72 h | 42.71 | 43.34 | 23.07 | 21.81 | 26.3 | 23.77 | 33.79 | 12.46 | 15.03 |
| 96 h | 44.35 | 42.94 | 12.71 | not measured | not measured | not measured | 46.13 | non-detectable | 5689 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalabay, M.; Szász, Z.; Láng, O.; Lajkó, E.; Pállinger, É.; Duró, C.; Jernei, T.; Csámpai, A.; Takács, A.; Kőhidai, L. Investigation of the Antitumor Effects of Tamoxifen and Its Ferrocene-Linked Derivatives on Pancreatic and Breast Cancer Cell Lines. Pharmaceuticals 2022, 15, 314. https://doi.org/10.3390/ph15030314
Kalabay M, Szász Z, Láng O, Lajkó E, Pállinger É, Duró C, Jernei T, Csámpai A, Takács A, Kőhidai L. Investigation of the Antitumor Effects of Tamoxifen and Its Ferrocene-Linked Derivatives on Pancreatic and Breast Cancer Cell Lines. Pharmaceuticals. 2022; 15(3):314. https://doi.org/10.3390/ph15030314
Chicago/Turabian StyleKalabay, Márton, Zsófia Szász, Orsolya Láng, Eszter Lajkó, Éva Pállinger, Cintia Duró, Tamás Jernei, Antal Csámpai, Angéla Takács, and László Kőhidai. 2022. "Investigation of the Antitumor Effects of Tamoxifen and Its Ferrocene-Linked Derivatives on Pancreatic and Breast Cancer Cell Lines" Pharmaceuticals 15, no. 3: 314. https://doi.org/10.3390/ph15030314
APA StyleKalabay, M., Szász, Z., Láng, O., Lajkó, E., Pállinger, É., Duró, C., Jernei, T., Csámpai, A., Takács, A., & Kőhidai, L. (2022). Investigation of the Antitumor Effects of Tamoxifen and Its Ferrocene-Linked Derivatives on Pancreatic and Breast Cancer Cell Lines. Pharmaceuticals, 15(3), 314. https://doi.org/10.3390/ph15030314