
Rethinking Protein Drug Design with Highly Accurate Structure Prediction of Anti-CRISPR Proteins

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
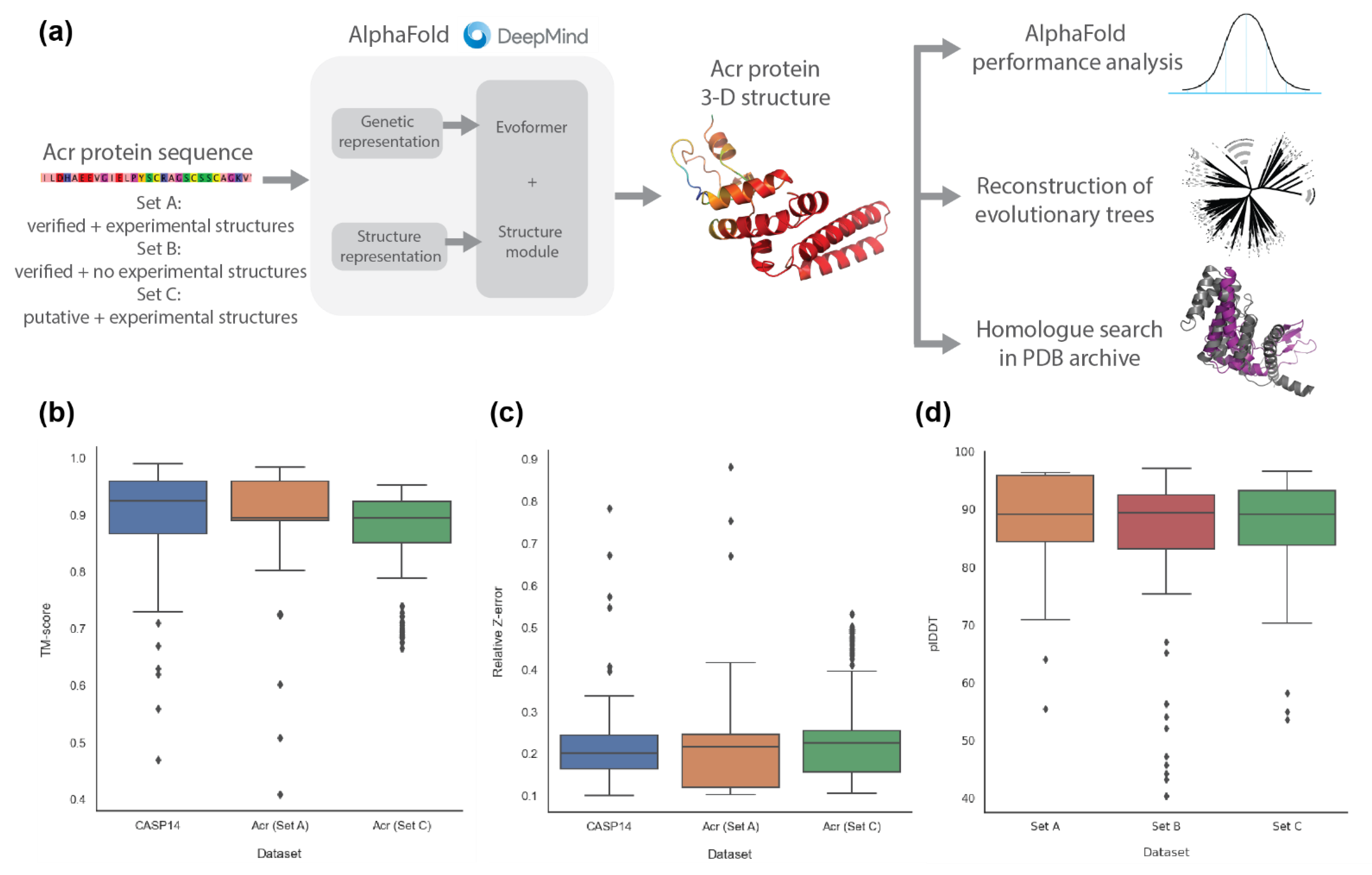
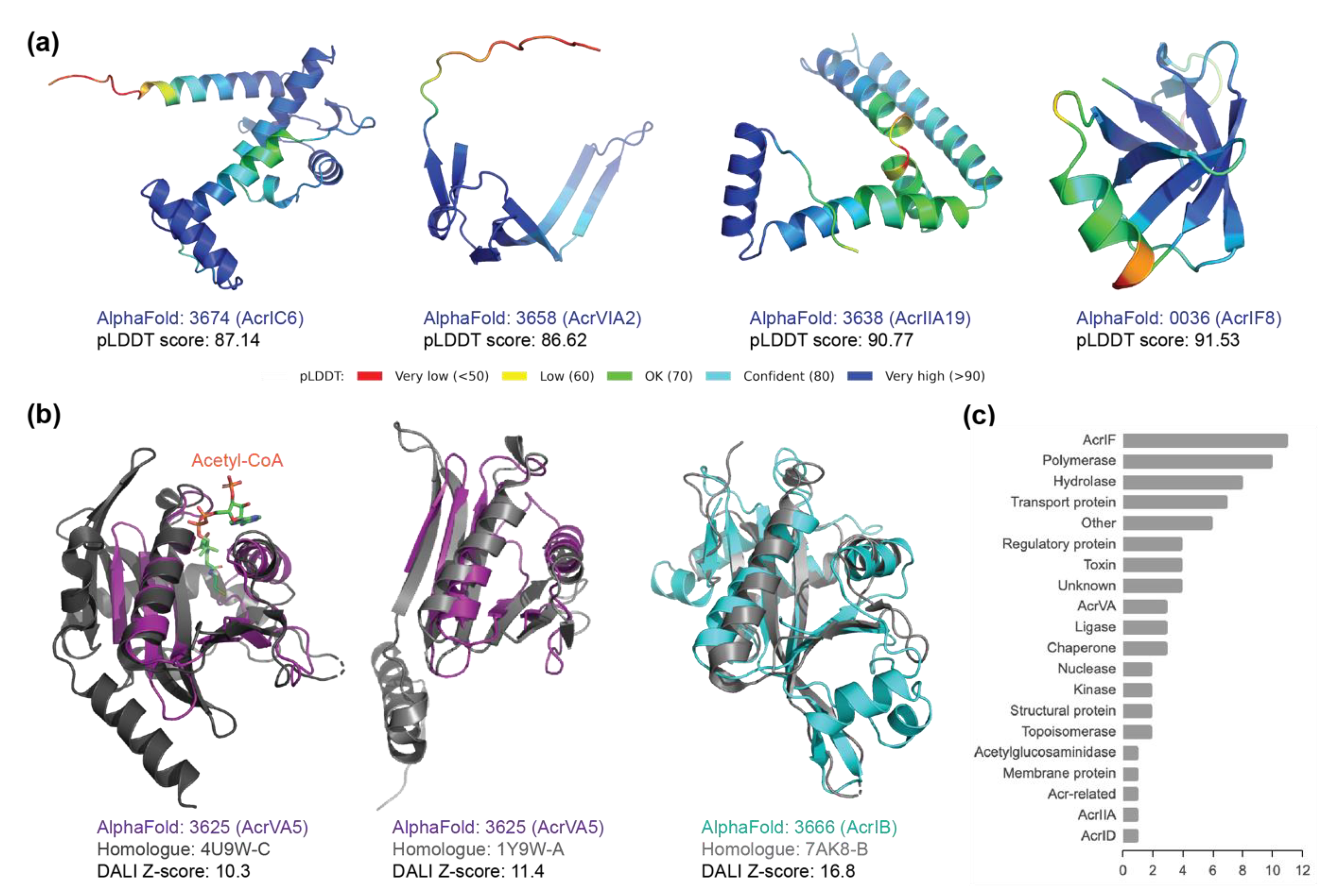
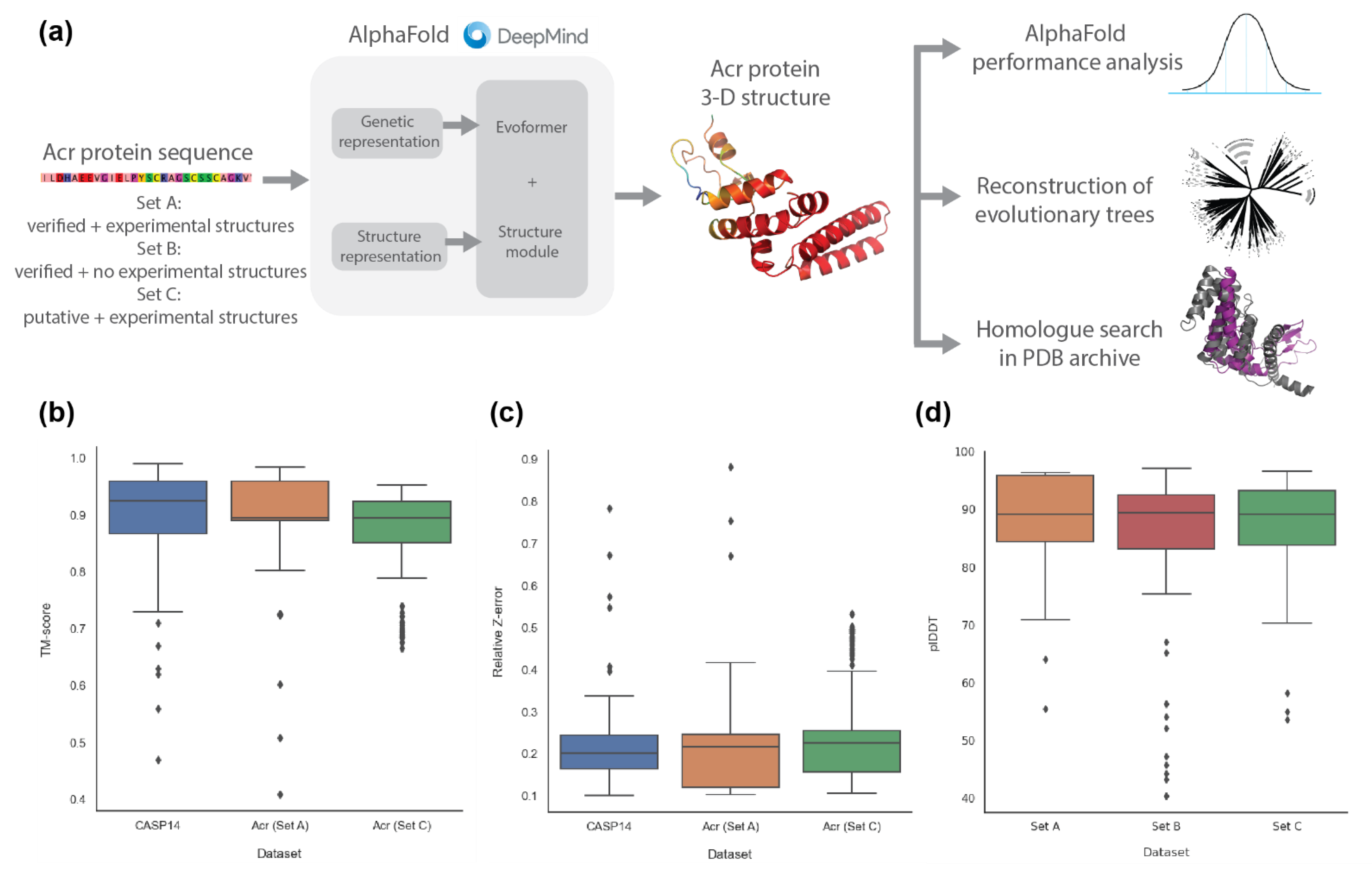
2.1. AlphaFold2 Prediction of Anti-CRISPR Protein Structures
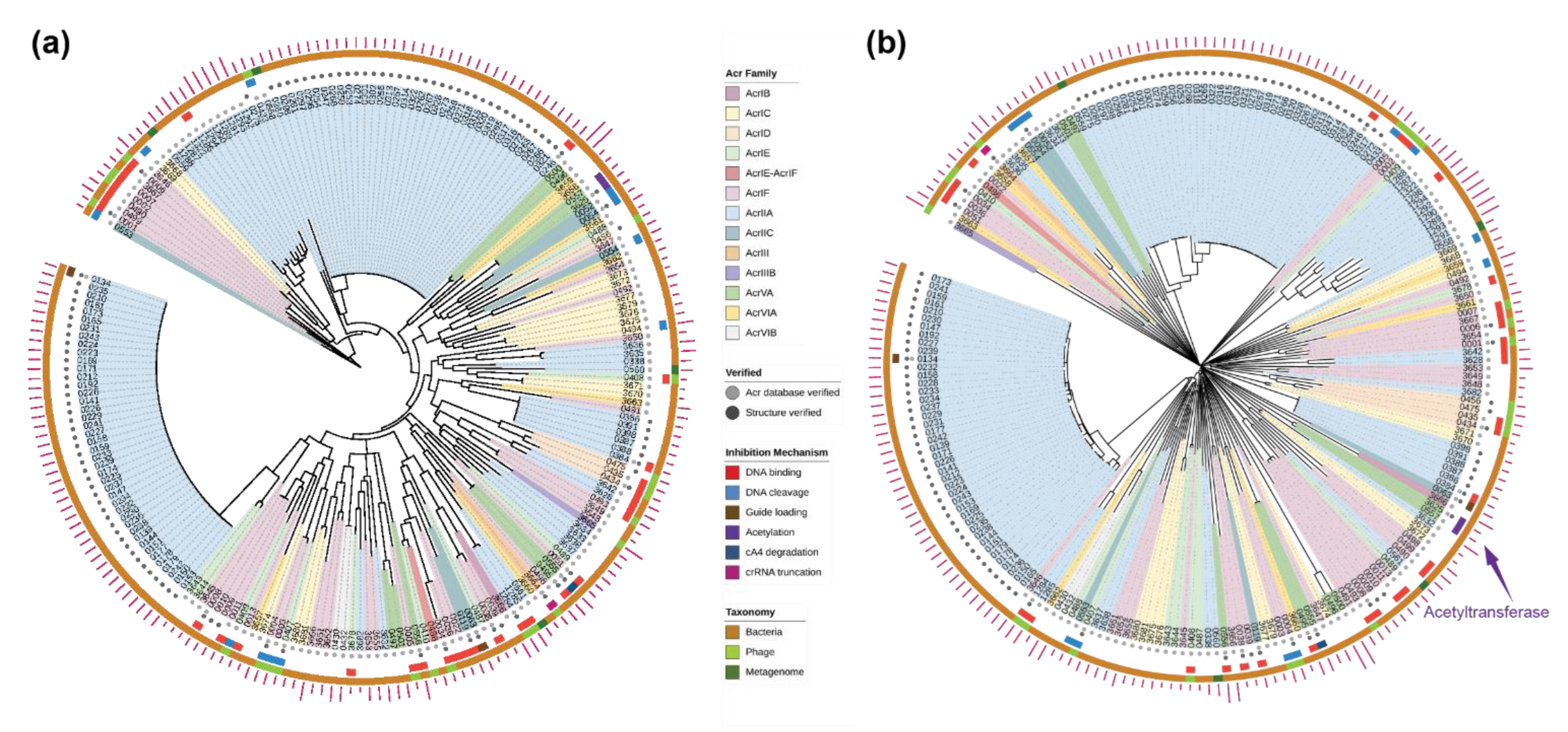
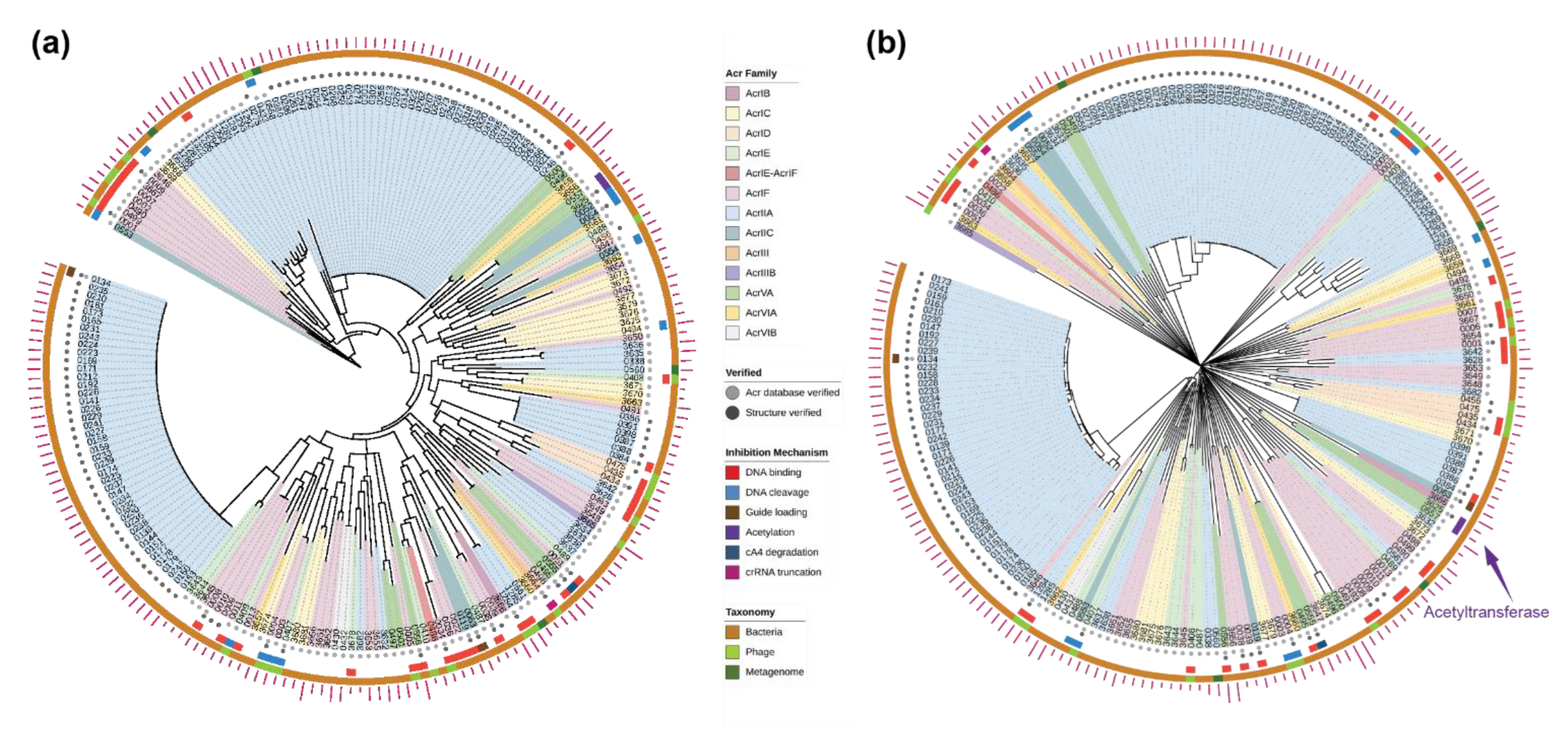
2.2. Evolutionary Trees of Anti-CRISPR Proteins
2.3. Structural Homology to Predicted Anti-CRISPR Structures
2.4. New Anti-CRISPR Family of Acetylation Inhibition
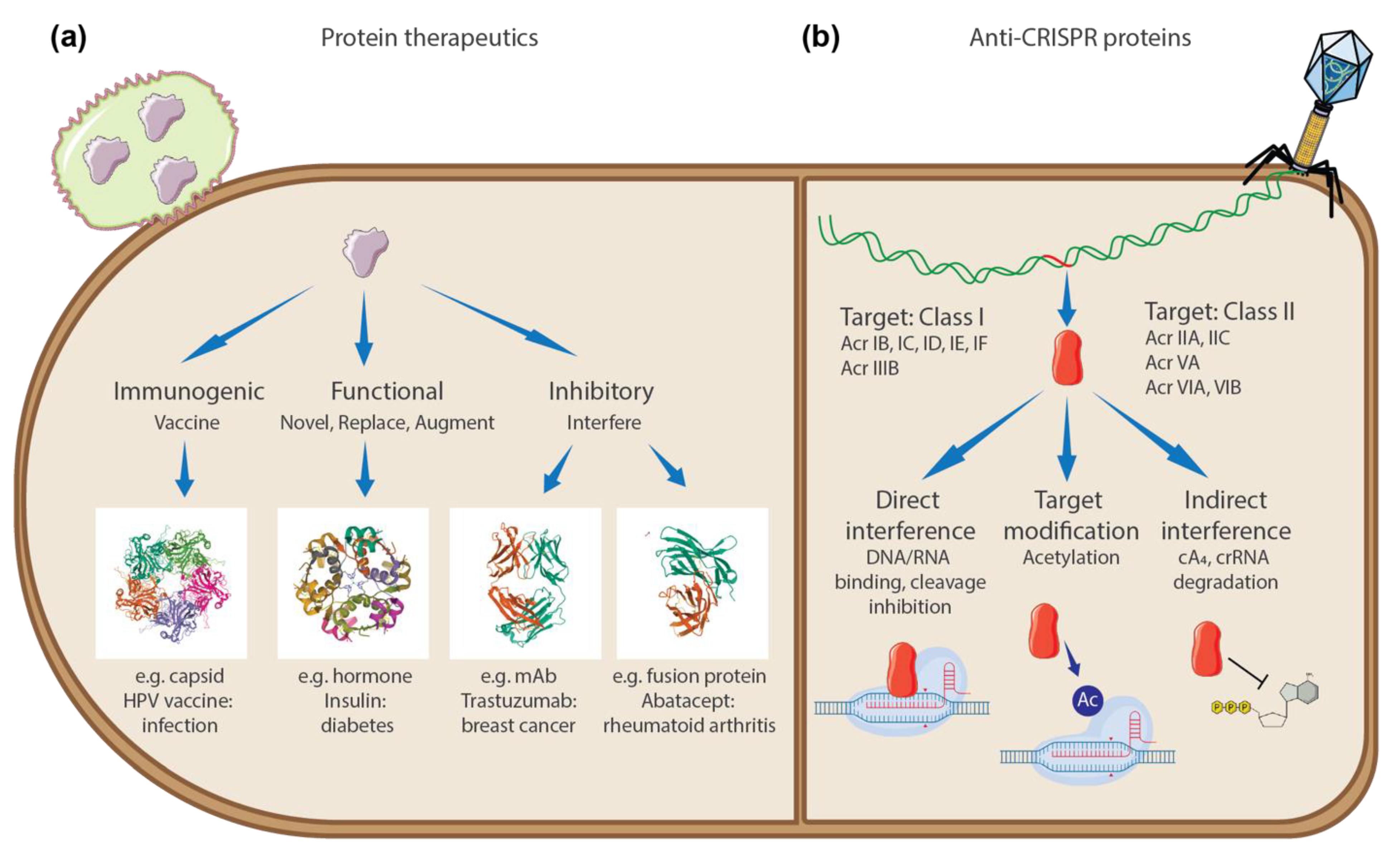
3. Discussion
4. Materials and Methods
4.1. Curation of Anti-CRISPR Datasets
4.2. Prediction of Anti-CRISPR Protein Structure with AlphaFold2
4.3. Comparison of AlphaFold2 Performance on Anti-CRISPR against CASP14
4.4. Reconstruction of Evolutionary Trees of Anti-CRISPR Proteins
4.5. Congruence among Distance Matrices of Sequence-Based and Structure-Based Trees
4.6. Visualization of Protein Structure Superimposition
4.7. Code Availability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [PubMed] [Green Version]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, A.L.; Groom, C.R. The druggable genome. Nat. Rev. Drug Discov. 2002, 1, 727–730. [Google Scholar] [CrossRef]
- Goeddel, D.V.; Kleid, D.G.; Bolivar, F.; Heyneker, H.L.; Yansura, D.G.; Crea, R.; Hirose, T.; Kraszewski, A.; Itakura, K.; Riggs, A.D. Expression in Escherichia coli of chemically synthesized genes for human insulin. Proc. Natl. Acad. Sci. USA 1979, 76, 106–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leader, B.; Baca, Q.J.; Golan, D.E. Protein therapeutics: A summary and pharmacological classification. Nat. Rev. Drug Discov. 2008, 7, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Bishop, B.; Dasgupta, J.; Klein, M.; Garcea, R.L.; Christensen, N.D.; Zhao, R.; Chen, X.S. Crystal structures of four types of human papillomavirus L1 capsid proteins: Understanding the specificity of neutralizing monoclonal antibodies. J. Biol. Chem. 2007, 282, 31803–31811. [Google Scholar] [CrossRef] [Green Version]
- Fávero-Retto, M.P.; Palmieri, L.C.; Souza, T.A.C.B.; Almeida, F.C.L.; Lima, L.M.T.R. Structural meta-analysis of regular human insulin in pharmaceutical formulations. Eur. J. Pharm. Biopharm. 2013, 85, 1112–1121. [Google Scholar] [CrossRef]
- Luthra, A.; Langley, D.B.; Schofield, P.; Jackson, J.; Abdelatti, M.; Rouet, R.; Nevoltris, D.; Mazigi, O.; Crossett, B.; Christie, M.; et al. Human antibody bispecifics through phage display selection. Biochemistry 2019, 58, 1701–1704. [Google Scholar] [CrossRef]
- Ostrov, D.A.; Shi, W.; Schwartz, J.C.; Almo, S.C.; Nathenson, S.G. Structure of murine CTLA-4 and its role in modulating T cell responsiveness. Science 2000, 290, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Dion, M.B.; Oechslin, F.; Moineau, S. Phage diversity, genomics and phylogeny. Nat. Rev. Microbiol. 2020, 18, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Dolja, V.V.; Krupovic, M. Origins and evolution of viruses of eukaryotes: The ultimate modularity. Virology 2015, 479–480, 2–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, B.N.J.; Steens, J.A.; Staals, R.H.J.; Westra, E.R.; van Houte, S. Coevolution between bacterial CRISPR-Cas systems and their bacteriophages. Cell Host Microbe 2021, 29, 715–725. [Google Scholar] [CrossRef]
- Hampton, H.G.; Watson, B.N.J.; Fineran, P.C. The arms race between bacteria and their phage foes. Nature 2020, 577, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.; van Embden, J.D.A.; Gaastra, W.; Schouls, L.M. Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 2002, 43, 1565–1575. [Google Scholar] [CrossRef] [PubMed]
- Mojica, F.J.M.; Díez-Villaseñor, C.; García-Martínez, J.; Soria, E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 2005, 60, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Iranzo, J.; Shmakov, S.A.; Alkhnbashi, O.S.; Brouns, S.J.J.; Charpentier, E.; Cheng, D.; Haft, D.H.; Horvath, P.; et al. Evolutionary classification of CRISPR-Cas systems: A burst of class 2 and derived variants. Nat. Rev. Microbiol. 2020, 18, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Bondy-Denomy, J.; Pawluk, A.; Maxwell, K.L.; Davidson, A.R. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature 2013, 493, 429–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawluk, A.; Bondy-Denomy, J.; Cheung, V.H.W.; Maxwell, K.L.; Davidson, A.R. A new group of phage anti-CRISPR genes inhibits the type I-E CRISPR-Cas system of Pseudomonas aeruginosa. MBio 2014, 5, e00896:1–e00896:7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marino, N.D.; Pinilla-Redondo, R.; Csörgő, B.; Bondy-Denomy, J. Anti-CRISPR protein applications: Natural brakes for CRISPR-Cas technologies. Nat. Methods 2020, 17, 471–479. [Google Scholar] [CrossRef]
- Pawluk, A.; Staals, R.H.J.; Taylor, C.; Watson, B.N.J.; Saha, S.; Fineran, P.C.; Maxwell, K.L.; Davidson, A.R. Inactivation of CRISPR-Cas systems by anti-CRISPR proteins in diverse bacterial species. Nat. Microbiol. 2016, 1, 16085:1–16085:6. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Guan, X.; Li, N.; Zhang, F.; Zhu, Y.; Ren, K.; Yu, L.; Zhou, F.; Han, Z.; Gao, N.; et al. An anti-CRISPR protein disables type V Cas12a by acetylation. Nat. Struct. Mol. Biol. 2019, 26, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Callaway, E. ‘It will change everything’: DeepMind’s AI makes gigantic leap in solving protein structures. Nature 2020, 588, 203–204. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Hao, G.F.; Hua, H.L.; Liu, S.; Labena, A.A.; Chai, G.; Huang, J.; Rao, N.; Guo, F.B. Anti-CRISPRdb: A comprehensive online resource for anti-CRISPR proteins. Nucleic Acids Res. 2018, 46, D393–D398. [Google Scholar] [CrossRef] [PubMed]
- Kufareva, I.; Abagyan, R. Methods of protein structure comparison. Methods Mol. Biol. 2012, 857, 231–257. [Google Scholar] [PubMed] [Green Version]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Balaji, S.; Srinivasan, N. Comparison of sequence-based and structure-based phylogenetic trees of ho-mologous proteins: Inferences on protein evolution. J. Biosci. 2007, 32, 83–96. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watters, K.E.; Shivram, H.; Fellmann, C.; Lew, R.J.; McMahon, B.; Doudna, J.A. Potent CRISPR-Cas9 inhibitors from Staphylococcus genomes. Proc. Natl. Acad. Sci. USA 2020, 117, 6531–6539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athukoralage, J.S.; McMahon, S.A.; Zhang, C.; Grüschow, S.; Graham, S.; Krupovic, M.; Whitaker, R.J.; Gloster, T.M.; White, M.F. An anti-CRISPR viral ring nuclease subverts type III CRISPR immunity. Nature 2020, 577, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Knott, G.J.; Thornton, B.W.; Lobba, M.J.; Liu, J.J.; Al-Shayeb, B.; Watters, K.E.; Doudna, J.A. Broad-spectrum enzymatic inhibition of CRISPR-Cas12a. Nat. Struct. Mol. Biol. 2019, 26, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Shim, H. Feature learning of virus genome evolution with the nucleotide skip-gram neural network. Evol. Bioinform. 2019, 15, 1176934318821072:1–1176934318821072:10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frost, L.S.; Leplae, R.; Summers, A.O.; Toussaint, A. Mobile genetic elements: The agents of open source evolution. Nat. Rev. Microbiol. 2005, 3, 722–732. [Google Scholar] [CrossRef] [PubMed]
- Shim, H.; Shivram, H.; Lei, S.; Doudna, J.A.; Banfield, J.F. Diverse ATPase proteins in mobilomes constitute a large potential sink for prokaryotic host ATP. Front. Microbiol. 2021, 12, 691847:1–691847:11. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics 2010, 26, 889–895. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins 2004, 57, 702–710. [Google Scholar] [CrossRef]
- Holm, L. Using Dali for protein structure comparison. Methods Mol. Biol. 2020, 2112, 29–42. [Google Scholar] [PubMed]
- Kryshtafovych, A.; Schwede, T.; Topf, M.; Fidelis, K.; Moult, J. Critical assessment of methods of pro-tein structure prediction (CASP)-Round XIV. Proteins 2021, 89, 1607–1617. [Google Scholar] [CrossRef] [PubMed]
- Siew, N.; Elofsson, A.; Rychlewski, L.; Fischer, D. MaxSub: An automated measure for the assessment of protein structure prediction quality. Bioinformatics 2000, 16, 776–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Taylor, W.R. Residual colours: A proposal for aminochromography. Protein Eng. 1997, 10, 743–746. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultra-fast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Holm, L. DALI and the persistence of protein shape. Protein Sci. 2019, 29, 128–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics 2007, 23, 127–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legendre, P. Species associations: The Kendall coefficient of concordance revisited. JABES 2005, 10, 226–245. [Google Scholar] [CrossRef]
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef]
- Jackson, N.A.C.; Kester, K.E.; Casimiro, D.; Gurunathan, S.; Derosa, F. The promise of mRNA vaccines: A biotech and industrial perspective. npj Vaccines 2020, 5, 11:1–11:6. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acr ID | Family | Type | Length | Homologue | Dali Z-Score | Annotation | %ID Structure | %ID Sequence |
|---|---|---|---|---|---|---|---|---|
| 3625 | AcrVA5 | V-A | 92 | 4U9W-C | 10.3 | N-Alpha-Acetyltransferase | 14 | 6.8 |
| 3625 | AcrVA5 | V-A | 92 | 1Y9W-A | 11.3 | Acetyltransferase | 22 | 16.8 |
| 3666 | AcrIB | I-B | 193 | 7AK8-B | 16.8 | Acetyltransferase | 21 | 17.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, H.-M.; Park, Y.; Vankerschaver, J.; Van Messem, A.; De Neve, W.; Shim, H. Rethinking Protein Drug Design with Highly Accurate Structure Prediction of Anti-CRISPR Proteins. Pharmaceuticals 2022, 15, 310. https://doi.org/10.3390/ph15030310
Park H-M, Park Y, Vankerschaver J, Van Messem A, De Neve W, Shim H. Rethinking Protein Drug Design with Highly Accurate Structure Prediction of Anti-CRISPR Proteins. Pharmaceuticals. 2022; 15(3):310. https://doi.org/10.3390/ph15030310
Chicago/Turabian StylePark, Ho-Min, Yunseol Park, Joris Vankerschaver, Arnout Van Messem, Wesley De Neve, and Hyunjin Shim. 2022. "Rethinking Protein Drug Design with Highly Accurate Structure Prediction of Anti-CRISPR Proteins" Pharmaceuticals 15, no. 3: 310. https://doi.org/10.3390/ph15030310
APA StylePark, H.-M., Park, Y., Vankerschaver, J., Van Messem, A., De Neve, W., & Shim, H. (2022). Rethinking Protein Drug Design with Highly Accurate Structure Prediction of Anti-CRISPR Proteins. Pharmaceuticals, 15(3), 310. https://doi.org/10.3390/ph15030310