
Hybrids of Imatinib with Quinoline: Synthesis, Antimyeloproliferative Activity Evaluation, and Molecular Docking

 , , ,
, , ,
Abstract
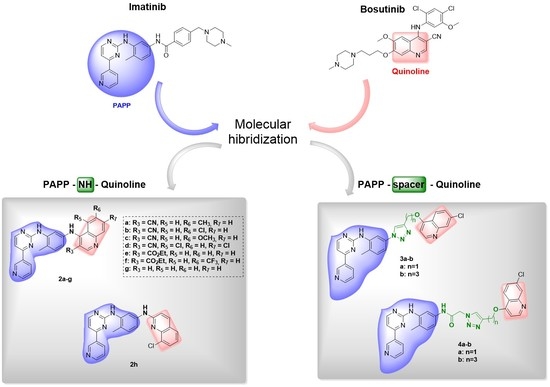
1. Introduction
2. Design and Synthesis of Imatinib Hybrids 2a–h, 3a–b, and 4a–b
3. Results and Discussion
4. Biological Evaluation
5. Molecular Docking
6. Conclusions
7. Experimental Section
7.1. Synthesis
7.2. General Procedure for the Preparation of Ethyl (Z)-3-(Phenylamino)acrylate (6a–f)
7.3. General Procedure for the Preparation of Quinolin-4(1H)-One (9a–f)
7.4. General Procedure for the Preparation of 4-Chloroquinoline (5a–f)
7.5. General Procedure for the Preparation of 4-Methyl-N3-(4-(pyridin-3-yl)pyrimidin-2-yl)-N1-(quinolin-4-yl)benzene-1,3-diamine (2a–h)
7.5.1. 6-Methyl-4-((4-methyl-3-((4-(pyridin-3-yl)pyrimidin-2 yl)amino)phenyl)amino)quinoline-3-carbonitrile (2a)
7.5.2. 6-Chloro-4-((4-methyl-3-((4-(pyridin-3-yl)pyrimidin-2-yl)amino)phenyl)amino)quinoline-3-carbonitrile (2b)
7.5.3. 6-Methoxy-4-((4-methyl-3-((4-pyridin-3-yl)pyrimidin-2-yl)amino)phenyl)amino)quinoline-3-carbonitrile (2c)
7.5.4. 5,7-Dichloro-4-((4-methyl-3-((4-(pyridin-3-yl)pyrimidin-2-yl)amino)phenyl)amino)quinoline-3-carbonitrile (2d)
7.5.5. Ethyl 4-((4-methyl-3-((4-(pyridin-3-yl)pyrimidin-2-yl)amino)phenyl)amino)quinoline-3-carboxylate (2e)
7.5.6. Ethyl 4-((4-methyl-3-((4-(pyridin-3-yl)pyrimidin-2-yl)amino)phenyl)amino)-6-(trifluoromethyl)quinoline-3-carboxylate (2f)
7.5.7. 4-Methyl-N3-(4-(pyridin-3-yl)pyrimidin-2-yl)-N1-(quinolin-4-yl)benzene-1,3-diamine (2g)
7.5.8. N1-(8-Chloroquinolin-2-yl)-4-methyl-N3-(4-(pyridin-3-yl)pyrimidin-2-yl)benzene-1,3-diamine (2h)
7.6. General Procedure for the Preparation of N-(5-Azido-2-methylphenyl)-4-(pyridin-3-yl)pyrimidin-2-amine (10)
7.7. General Procedure for the Preparation of Alcohol (4-Methyl-3-((4-(pyridin-3-yl)pyrimidin-2-yl)amino)phenyl)-1H-1,2,3-triazol-4-yl) (11a–b)
7.7.1. 1-(4-Methyl-3-((4-(pyridin-3-yl)pyrimidin-2-yl)amino)phenil)-1H-1,2,3-triazol-4-yl)methanol (11a)
7.7.2. 3-(1-(4-Methyl-3-((4-(pyridin-3-yl)pyrimidin-2-yl)amino)phenyl)-1H-1,2,3-triazol-4-yl)propan-1-ol (11b)
7.8. General Procedure for the Preparation of N-(5-(4-(((7-Chloroquinolin-4-yl)oxy))-1H-1,2,3-triazol-1-yl)-2-methylphenyl)-4-(pyridin-3-yl)pyrimidin-2-amine (3a–b)
7.8.1. N-(5-(4-(((7-Chloroquinolin-4-yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)-2-methylphenyl)-4-(pyridin-3-yl)pyrimidin-2-amine (3a)
7.8.2. N-(5-(4-(3-((7-Chloroquinolin-4-yl)oxy)propyl)-1H-1,2,3-triazol-1-yl)-2-methylphenyl)-4-(pyridin-3-yl)pyrimidin-2-amine (3b)
7.9. General Procedure for the Preparation of 2-Chloro-N-(4-methyl-3-((4-(pyridin-3-yl)pyrimidin-2-yl)amino)phenyl)acetamide (13)
7.10. General Procedure for the Preparation of 2-Azido-N-(4-methyl-3-((4-(pyridin-3-yl)pyrimidin-2-yl)amino)phenyl)acetamide (14)
7.11. General Procedure for the Preparation of 2-(4-(Hydroxy)-1H-1,2,3-triazol-1-yl)-N-(4-methyl-3-((4-(pyridin-3-yl)pyrimidin-2-yl)amino)phenyl)acetamide (15a–b)
7.11.1. 2-(4-(Hydroxymethyl)-1H-1,2,3-triazol-1-yl)-N-(4-methyl-3-((4-(pyridin-3-yl)pyrimidin-2-yl)amino)phenyl)acetamide (15a)
7.11.2. 2-(4-(3-Hydroxypropyl)-1H-1,2,3-triazol-1-yl)-N-(4-methyl-3-((4-(pyridin-3-yl)pyrimidin-2-yl)amino)phenyl)acetamide (15b)
7.12. General Procedure for the Preparation of 2-(4-(((7-Chloroquinolin-4-yl)oxy))-1H-1,2,3-triazol-1-yl)-N-(4-methyl-3-((4-(pyridin-3-yl)pyrimidin-2-yl)amino)phenyl)acetamide (4a–b)
7.12.1. 2-(4-(((7-Chloroquinolin-4-yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(4-methyl-3-((4-(pyridin-3-yl)pyrimidin-2-yl)amino)phenyl)acetamide (4a)
7.12.2. 2-(4-(3-((7-Chloroquinolin-4-yl)oxy)propyl)-1H-1,2,3-triazol-1-yl)-N-(4-methyl-3-((4-(pyridin-3-yl)pyrimidin-2-yl)amino)phenyl)acetamide (4b)
8. Biological Evaluation
8.1. Cell Viability Analysis
8.2. WSS-1 Cell Assay
8.3. K562 Cell Assay
8.4. Data Analysis and Graph Construction
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mercy, M.S. Targeted Therapies: A Life Saving Approach for Cancer. World J. Pharm. Pharm. Sci. 2016, 5, 612–626. [Google Scholar]
- Burnmeister, T.; Schwartz, S.; Bartram, C.R.; Gökbuget, N.; Hoelzer, D.; Thiel, E. Patients’ age and BCR-ABL frequency in adult B-precursor ALL: A retrospective analysis from the GMALL study group. Blood 2008, 112, 918–919. [Google Scholar] [CrossRef] [PubMed]
- Panjarian, S.; Iacob, R.E.; Chen, S.; Engen, J.R.; Smithgall, T.E.J. Structure and Dynamic Regulation of Abl Kinases. Biol. Chem. 2013, 288, 5443–5450. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Ohno, S.; Buchdunger, E.; Tamura, S.; Zimmermann, J.; Lydon, N.B. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat. Med. 1996, 2, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Yaish, P.; Gazit, A.; Gilon, C.; Levitzki, A. Blocking of EGF-dependent cell proliferation by EGF receptor kinase inhibitors. Science 1988, 242, 933–935. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, X.; You, Q. Lead Discovery of Type II BRAF V600E Inhibitors Targeting the Structurally Validated DFG-Out Conformation Based upon Selected Fragments. Molecules 2016, 21, 879. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Cardona, A.F.; Enciso, L.; Ruiz, G.; Yepes, A.; Ospina, V.; Gálvez, K.; García, J.; Rosales, J.; Rosales, M.; et al. Leucemia mieloide crónica en crisis blástica: Perspectiva terapéutica según la evidencia (parte II). Rev. Colomb. Cancerol. 2006, 10, 267–281. [Google Scholar]
- Kannaiayan, R.; Mahadevan, D.A. A comprehensive review of protein kinase inhibitors for cancer therapy. Expert Rev. Anticancer Ther. 2018, 18, 1249–1270. [Google Scholar] [CrossRef]
- Padula, W.V.; Larson, R.A.; Dusetzina, S.B.; Apperley, J.F.; Hehlmann, R.; Baccarani, M.; Eigendorff, E.; Guilhot, J.; Guilhot, F.; Hehlmann, R.; et al. Cost-effectiveness of Tyrosine Kinase Inhibitor Treatment Strategies for Chronic Myeloid Leukemia in Chronic Phase after Generic Entry of Imatinib in the United States. J. Natl. Cancer Inst. 2016, 108, djw003. [Google Scholar] [CrossRef] [PubMed]
- Hochhaus, A.; Saussele, S.; Rosti, G.; Mahon, F.-X.; Janssen, J.J.W.M.; Hansen, H.-H.; Richter, J.; Buske, C. Chronic myeloid leukemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2017, 28 (Suppl. S4), 41–51. [Google Scholar] [CrossRef]
- Rossari, F.; Minutolo, F.; Orciuolo, E. Past, present, and future of Bcr-Abl inhibitors: From chemical development to clinical efficacy. J. Hematol. Oncol. 2018, 11, 1–14. [Google Scholar] [CrossRef]
- Rosti, G.; Castagnetti, F.; Gugliotta, G.; Palandri, F.; Baccarani, M. Phisician’s guide to the clinical management of adverce events on nilotinib therapy for the treatment of CML. Cancer Treat. Rev. 2012, 38, 241–248. [Google Scholar] [CrossRef]
- Azevedo, L.D.; Bastos, M.M.; Vasconcelos, F.C.; Hoelz, L.V.B.; Junior, F.P.S.; Dantas, R.F.; Almeida, A.C.M.; Oliveira, A.P.; Gomes, L.C.; Maia, R.C.; et al. Imatinib derivatives as inhibitors of K562 cells in chronic myeloid leukemia. Med. Chem. Res. 2017, 26, 2929–2941. [Google Scholar] [CrossRef]
- Selvam, T.P.; James, C.R.; Dniandev, P.V.; Valzita, S.K. A mini review of pyrimidine and fused pyrimidine marketed drugs. Res. Pharm. 2012, 2, 1–9. [Google Scholar]
- Cocco, M.T.; Congiu, C.; Onnis, V.; Piras, R. Synthesis and antitumor evaluation of 6-thioxo-, -6-oxo- and 2,4-dioxopyrimidine derivatives. Farmaco 2001, 56, 741–748. [Google Scholar] [CrossRef]
- Ashour, H.M.; Shaaban, O.G.; Rizk, O.H.; El-Ashmawy, I.M. Synthesis and biological evaluation of thieno[2’,3’:4,5]pyrimido[1,2-b][1,2,4]triazines and thieno[2,3-d][1,2,4]triazolo[1,5-a]pyrimidines as anti-inflamatory and analgesic agents. Eur. J. Med. Chem. 2013, 62, 341–351. [Google Scholar] [CrossRef]
- Kumar, S.; Narasimhan, B. Therapeutic potential of heterocyclic pyrimidine scaffolds. Chem. Cent. J. 2018, 12, 38. [Google Scholar] [CrossRef]
- Pimentel, L.C.F.; Cunha, A.C.; Hoelz, L.V.B.; Canzian, H.F.; Marinho, D.I.L.F.; Boechat, N.; Bastos, M.M. Phenylamino-pyrimidine (PAP) Privileged Structure: Synthesis and Medicinal Applications. Curr. Top. Med. Chem. 2020, 20, 227–243. [Google Scholar] [CrossRef]
- Matada, B.S.; Pattanashettar, R.; Yernale, N.G. A comprehensive review on the biological interest of quinoline and its derivatives. Bioorg. Med. Chem. 2021, 32, 115973. [Google Scholar] [CrossRef]
- Yadav, P.; Shah, K. Quinolines, a perpetual, multipurpose scaffold in medicinal chemistry. Bioorg. Chem. 2021, 109, 104639. [Google Scholar] [CrossRef]
- Levinson, N.M.; Boxer, S.G. Structural and Spectroscopic Analysis of the Kinase Inhibitor Bosutinib and an Isomer of Bosutinib Binding to the Abl Tyrosine Kinase Domain. PLoS ONE 2012, 7, e29828. [Google Scholar] [CrossRef] [PubMed]
- Maračić, S.; Lapić, J.; Djaković, S.; Bernardi, T.-O.; Obrovac, L.-G.; Vrček, V.; Malić, S.-R. Quinoline and ferrocene conjugates: Synthesis, computational study and biological evaluations. Appl. Organomet. Chem. 2019, 33, e4628. [Google Scholar] [CrossRef]
- Abbas, S.M.; Hafeez, A.-E.; Shoman, M.E.; Montano, M.M.; Hassan, H.A. New quinolone/chalcone hybrids as anti-cancer agents: Design, synthesis and evaluations of cytotoxicity and PI3K inhibitory activity. Bioorg. Chem. 2019, 82, 360–377. [Google Scholar] [CrossRef] [PubMed]
- González, S.-M.; Arístegui, S.-R.; Oliva, C.A.G.; Hernández, A.I.; Cantalapiedra, E.G.; Varela, C.; García, A.B.; Rabal, O.; Oyarzabal, J.; Bischoff, J.R.; et al. Discovery of novel triazolo[4,3-b]pyridazin-3-il-quinoline derivatives as PIM inhibitors. Eur. J. Med. Chem. 2019, 168, 87–109. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Hou, X.; Wang, G.; Pan, W.; Yang, X.; Zhang, Y.; Fang, H. Design, synthesis and biological evaluation of quinolone derivatives as HDAC class I inhibitors. Eur. J. Med. Chem. 2017, 133, 11–23. [Google Scholar] [CrossRef]
- Orfi, Z.; Waczek, F.; Baska, F.; Szabadkai, I.; Torka, R.; Hartmann, J.; Orfi, L.; Ullrich, A. Novel members of quinolone compound family enhance insulin secretion in RIN-5AH beta cells and in rat pancreatic islet microtissue. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef]
- Ghorab, M.M.; Alsaid, M.S. Anti-breast cancer activityof some novel quinolone derivatives. Acta Pharm. 2015, 65, 271–283. [Google Scholar] [CrossRef]
- Köprülü, T.K.; Ökten, S.; Tekin, Ş.; Çakmak, O. Biological evaluation of some quinolone derivatives with different funcitional groups as anticancer agents. J. Biochem. Mol. Toxicol. 2019, 33, e22260. [Google Scholar] [CrossRef]
- Tang, Y.; Wang, L.; Xiang, J.; Liu, Y. An Application of 4-Substituted-quinoline-2-amine Compound in Preparation of Anticancer Drugs. CN 104027335, 10 September 2014. [Google Scholar]
- Korcz, M.; Saczewski, F.; Bednarski, P.J.; Kornicka, A. Synthesis, Structure, Chemical Stability, and In Vitro Cytotoxic Properties of Novel Quinoline—3-Carbaldehyde Hydrazones Bearing a 1,2,4-triazole or Benzotriazole Moiety. Molecules 2018, 23, 1497. [Google Scholar] [CrossRef]
- Boechat, N.; Bastos, M.M. Fluorinated Compounds Against Mycobacterium tuberculosis. Curr. Top. Med. Chem. 2013, 13, 2885. [Google Scholar] [CrossRef]
- Li, Y.-T.; Wang, J.-H.; Pan, C.-W.; Meng, F.-F.; Chu, X.-Q.; Ding, Y.-H.; Qu, W.-Z.; Li, H.-Y.; Yang, C.; Zhang, Q.; et al. Synthesis and biological evaluation of 1,2,3-triazole and 1,3,4-oxadiazole derivatives of imatinib. Bioorg. Med. Chem. Lett. 2016, 26, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Sen, R.; Natarajan, K.; Bhullar, J.; Shukla, S.; Fang, H.B.; Cai, L.; Chen, Z.S.; Ambudkar, S.V.; Baer, M.R. The Novel BCR-ABL and FLT3 Inhibitor of the MDR-Associated ATP-Binding Cassette Transporter ABCG2. Mol. Cancer Ther. 2012, 11, 2033–2044. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, G.A.; Hauser, C.R. Ethyl β-anilinocrotonate. Org. Synth. 1949, 29, 42. [Google Scholar] [CrossRef]
- Davis, T.A.; Dobish, M.C.; Schwieter, K.E.; Chun, A.C.; Johnston, J.N. Preparation of H, 4PyrrolidineQuin-BAM (PBAM). Org. Synth. 2012, 89, 380–393. [Google Scholar] [CrossRef] [PubMed]
- Arioli, F.; Borrelli, S.; Colombo, F.; Falchi, F.; Filippi, I.; Crespan, E.; Naldini, A.; Scalia, G.; Silvani, A.; Maga, G.; et al. N—[2-Methyl-5-(triazol-1-yl)phenyl]pyrimidin-2-amine as a scaffold for the synthesis of inhibitors of Bcr-Abl. ChemMedChem 2011, 6, 2009–2018. [Google Scholar] [CrossRef]
- Moorhouse, A.D.; Moses, J.E. Microwave Enhancement of a “One-pot” Tandem Azidation-“Click” Cycloaddition of Anilines. Synlett 2008, 14, 2089–2092. [Google Scholar] [CrossRef]
- Chen, Y.; Rao, Z.; Yang, C.; Bai, C.; Pan, C.; Meng, F.; Zhang, W.; Wang, T.; Wang, S.; Liu, H.; et al. 1,4-Dissubstituted-1,2,3-triazole Compounds and Their Preparation, Pharmaceutical Compositions and Use in the Treatment of Leukemia. CN 103159739, 19 June 2013. [Google Scholar]
- Yeon, K.D.; Jim, C.D.; Yeol, L.G.; Yeop, K.H.; Hun, W.S.; Fon, L.H.; Mu, K.S.; Am, C. Isothiocyanate Compound Containin N-Phenylpyrimidin-2-amine Moiety and Method for the Preparation Thereof. KR20120052095A, 23 May 2012. [Google Scholar]
- Pimentel, L.C.F.; Hoelz, L.V.B.; Canzian, H.F.; Branco, F.S.C.; Oliveira, A.P.; Campos, V.R.; Júnior, F.P.S.; Dantas, R.F.; Resende, J.A.L.C.; Cunha, A.C.; et al. (Phenylamino)pyrimidine-1,2,3-triazole derivatives as analogs of imatinib: Searching for novel compounds against chronic myeloid leukemia. Beilstein J. Org. Chem. 2021, 17, 2260–2269. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Wilson, I.; Orton, T.; Pognan, F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 2000, 267, 5421–5426. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.R.; Churas, C.P.; Shuai, L.; Swift, R.V.; Chiu, M.; Shao, C.; Feher, V.A.; Burley, S.K.; Gilson, M.K.; Rommie, E.; et al. Continuous Evaluation of Ligand Protein Predictions: A Weekly Community Challenge for Drug Docking. Structure 2019, 27, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Campobasso, N.; Biju, M.P.; Fisher, K.; Pan, X.-Q.; Cottom, J.; Galbraith, S.; Ho, T.; Zhang, H.; Hong, X.; et al. Discovery and Characterization of a Cell-Permeable, Small-Molecule c-Abl Kinase Activator that Binds to the Myristoyl Binding Site. Chem. Biol. 2011, 18, 177–186. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Golas, J.M.; Arndt, K.; Etienne, C.; Lucas, J.; Nardin, D.; Gibbons, J.; Frost, P.; Ye, F.; Boschelli, D.H.; Boschelli, F. SKI-606, a 4-anilino-3-quinolinocarbonitrile dual inhibitor of Src and Abl kinases, is a potent antiproliferative agent against chronic myelogenous leukemia cells in culture and causes regression of K562 xenografts in nude mice. Cancer Res. 2003, 63, 375–381. Available online: https://pubmed.ncbi.nlm.nih.gov/12543790/ (accessed on 9 January 2022). [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CC50 (μM) | |||||
|---|---|---|---|---|---|
| Compounds | K562 | CI * | WSS-1 | CI | SI ** WSS-1/K562 |
| IMT | 0.08 | 0.05 to 0.10 | 8.9 | 7.8 to 10.0 | 111.2 |
| 2g | 0.9 | 0.7 to 1.1 | 0.2 | 0.2 to 0.3 | 0.2 |
| 2h | 6.2 | 5.3 to 7.3 | 4.9 | 4.4 to 5.6 | 0.8 |
| Inhibitors | H-Bond Energy (a.u.) a | Residues (H-Bond Interaction) | Steric Interaction Energy by PLP (a.u.) b | Residues (Steric Interactions) | MolDock Score (a.u.) |
|---|---|---|---|---|---|
| IMT | −8.97 | Asp400, Glu305, Met 337, Thr334 | −193.65 | Asp400, Glu305, Ile379 | −206.02 |
| 2g | −7.34 | Asp400, Met 337, Thr334 | −169.73 | Glu305, Leu389, Met 337, Phe336, Val308, | −177.07 |
| 2h | −5.31 | Asp400, Thr334 | −172.11 | Glu305, Ile312, Tyr272, Val318 | −176.04 |
| Inhibitors | H-Bond Energy (a.u.) a | Residues (H-Bond Interaction) | Steric Interaction Energy by PLP (a.u.) b | Residues (Steric Interactions) | MolDock Score (a.u.) |
|---|---|---|---|---|---|
| BST | −5.20 | Met318, Thr315 | −308.41 | Asn322, Lys271, Phe382, Tyr320 | −332.36 |
| 2g | −5.00 | Thr315 | −282.51 | Ala269, Phe336 | −287.51 |
| 2h | −8.99 | Glu316, Thr315 | −291.88 | Ala308, Leu370, Met318, Phe382, Thr 315 | −176.04 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, C.; Pimentel, L.; Canzian, H.; Oliveira, A.; Junior, F.; Dantas, R.; Hoelz, L.; Marinho, D.; Cunha, A.; Bastos, M.; et al. Hybrids of Imatinib with Quinoline: Synthesis, Antimyeloproliferative Activity Evaluation, and Molecular Docking. Pharmaceuticals 2022, 15, 309. https://doi.org/10.3390/ph15030309
Santos C, Pimentel L, Canzian H, Oliveira A, Junior F, Dantas R, Hoelz L, Marinho D, Cunha A, Bastos M, et al. Hybrids of Imatinib with Quinoline: Synthesis, Antimyeloproliferative Activity Evaluation, and Molecular Docking. Pharmaceuticals. 2022; 15(3):309. https://doi.org/10.3390/ph15030309
Chicago/Turabian StyleSantos, Carine, Luiz Pimentel, Henayle Canzian, Andressa Oliveira, Floriano Junior, Rafael Dantas, Lucas Hoelz, Debora Marinho, Anna Cunha, Monica Bastos, and et al. 2022. "Hybrids of Imatinib with Quinoline: Synthesis, Antimyeloproliferative Activity Evaluation, and Molecular Docking" Pharmaceuticals 15, no. 3: 309. https://doi.org/10.3390/ph15030309
APA StyleSantos, C., Pimentel, L., Canzian, H., Oliveira, A., Junior, F., Dantas, R., Hoelz, L., Marinho, D., Cunha, A., Bastos, M., & Boechat, N. (2022). Hybrids of Imatinib with Quinoline: Synthesis, Antimyeloproliferative Activity Evaluation, and Molecular Docking. Pharmaceuticals, 15(3), 309. https://doi.org/10.3390/ph15030309