
Chiral Switch: Between Therapeutical Benefit and Marketing Strategy



Abstract
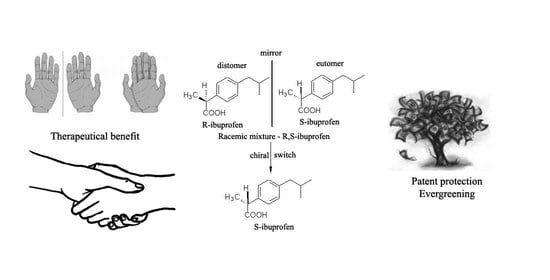
:
1. Introduction
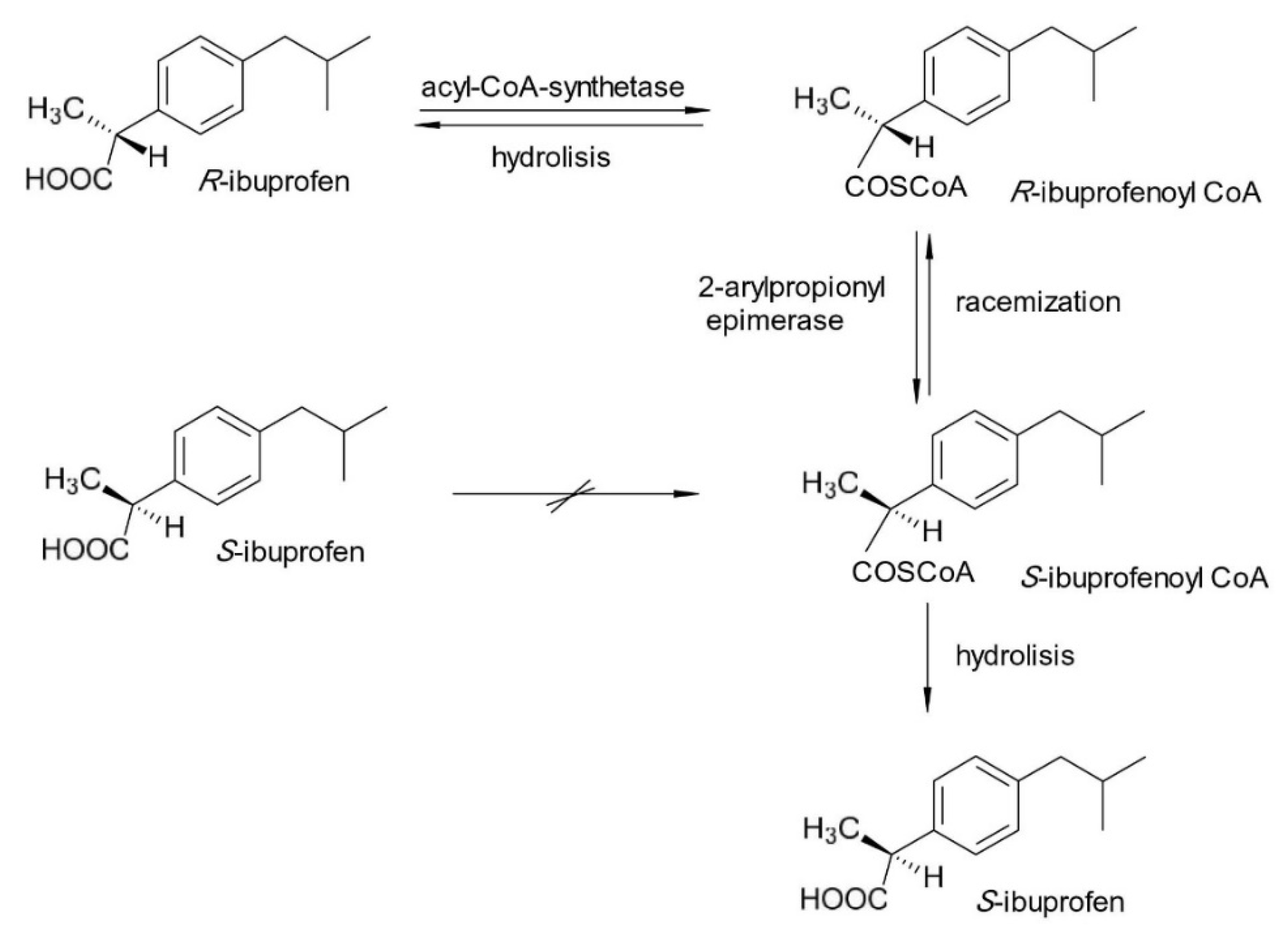
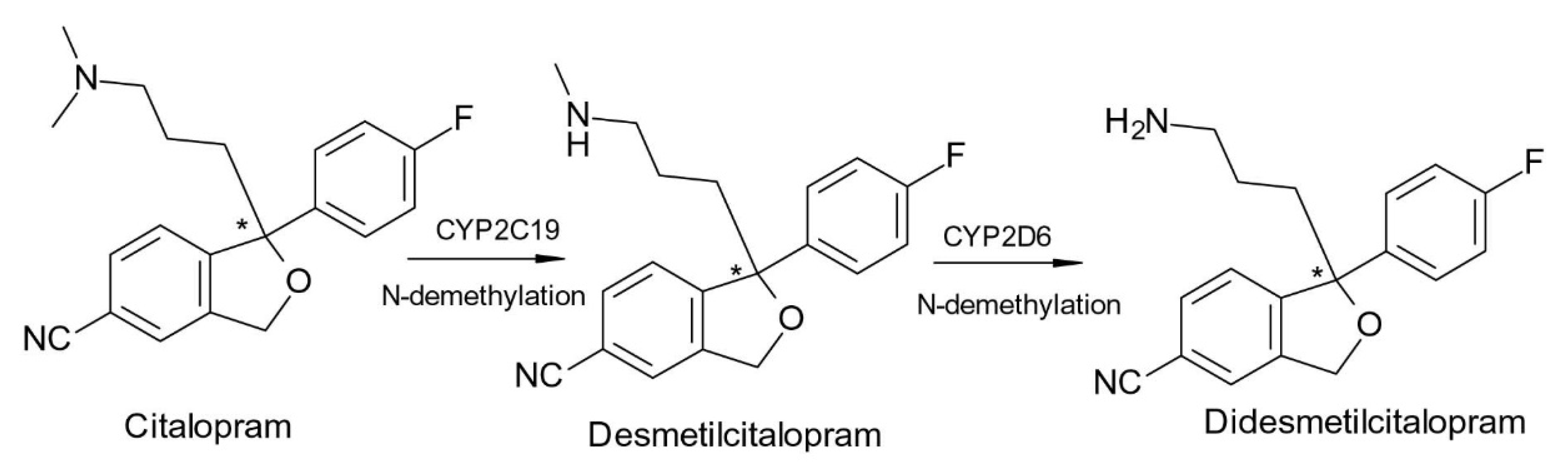
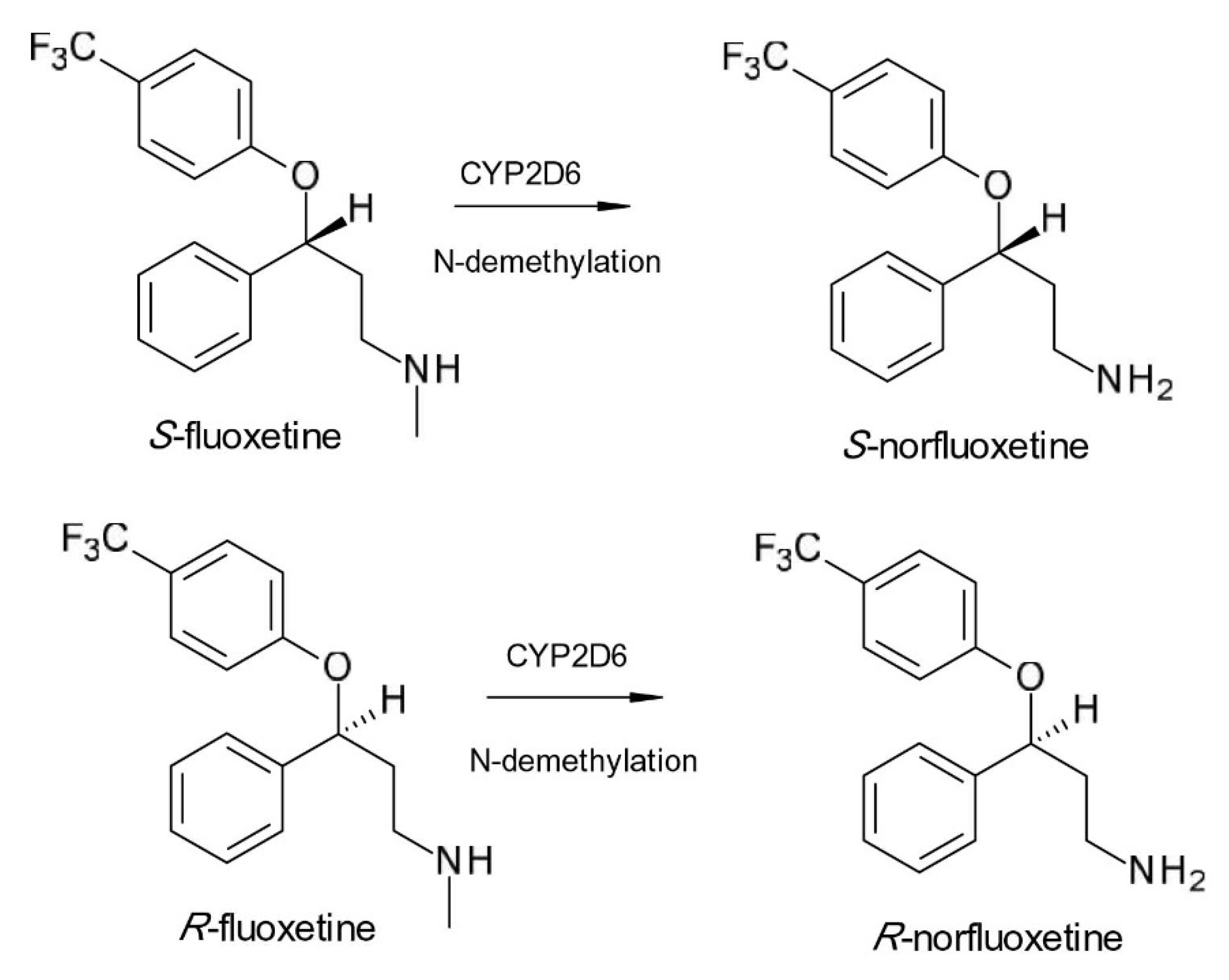
2. Chiral Switches in Therapy
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nguyen, L.A.; He, H.; Pham-Huy, C. Chiral drugs: An overview. Int. J. Biomed. Sci. 2006, 2, 85–100. [Google Scholar]
- Salam, A. The role of chirality in the origin of life. J. Mol. Evol. 1991, 33, 105–113. [Google Scholar] [CrossRef]
- De Camp, W.H. Chiral drugs: The FDA perspective on manufacturing and control. J. Pharm. Biomed. Anal. 1993, 11, 1167–1172. [Google Scholar] [CrossRef]
- Kumkumian, C.S. Chirality and drug development. Science 1992, 257, 145. [Google Scholar] [CrossRef]
- Sekhon, B.S. Exploiting the power of stereochemistry in drugs: An overview of racemic and enantiopure drugs. J. Mod. Med. Chem. 2013, 1, 10–36. [Google Scholar] [CrossRef] [Green Version]
- Hutt, A.J.; Tan, S.C. Drug chirality and its clinical significance. Drugs 1996, 52, 1–12. [Google Scholar] [CrossRef]
- Brooks, H.; Guida, C.W.; Daniel, K.G. The significance of chirality in drug design and development. Curr. Top. Med. Chem. 2011, 11, 760–770. [Google Scholar] [CrossRef]
- Murakami, H. From racemates to single enantiomers–chiral synthetic drugs over the last 20 years. Top. Curr. Chem. 2007, 269, 273–299. [Google Scholar]
- Agranat, I.; Caner, H.; Caldwell, J. Putting chirality to work: The strategy of chiral switches. Nat. Rev. Drug Discov. 2002, 1, 753–768. [Google Scholar] [CrossRef]
- Hutt, A.J.; Valentova, J. The chiral switch: The development of single enantiomer drugs from racemates. Acta Fac. Pharm. Univ. Comen. 2003, 50, 7–23. [Google Scholar]
- Tucker, G.T. Chiral switches. Lancet 2000, 355, 1085–1087. [Google Scholar] [CrossRef]
- Gellad, W.F.; Choi, P.; Mizah, M.; Good, C.B.; Kesselheim, A.S. Assessing the chiral switch: Approval and use of single-enantiomer drugs, 2001 to 2011. Am. J. Manag. Care 2014, 20, e90–e97. [Google Scholar]
- Agranat, I.; Wainschtein, S.R. The strategy of enantiomer patents of drugs. Drug Discov. Today 2010, 15, 163–170. [Google Scholar] [CrossRef]
- Eriksson, T.; Björkman, S.; Höglund, P. Clinical pharmacology of thalidomide. Eur. J. Clin. Pharmacol. 2001, 57, 365–376. [Google Scholar] [CrossRef]
- Tokunaga, E.; Yamamoto, T.; Ito, E.; Shibata, N. Understanding the thalidomide chirality in biological processes by the self-disproportionation of enantiomers. Sci. Rep. 2018, 8, 17131. [Google Scholar] [CrossRef]
- Evans, A.M. Pharmacodynamics and pharmacokinetics of the profens: Enantioselectivity, clinical implications, and special reference to S-(+)-ibuprofen. J. Clin. Pharmacol. 1996, 36, 7–15. [Google Scholar]
- Davies, N.M. Clinical pharmacokinetics of ibuprofen. Clin. Pharmacokinet. 1998, 34, 101–154. [Google Scholar] [CrossRef]
- Hao, H.; Wang, G.; Sun, J. Enantioselective pharmacokinetics of ibuprofen and involved mechanisms. Drug Metab. Rev. 2005, 37, 215–234. [Google Scholar] [CrossRef]
- Jamali, F.; Brocks, D.R. Clinical pharmacokinetics of ketoprofen and its enantiomers. Clin. Pharmacokinet. 1990, 19, 197–217. [Google Scholar] [CrossRef]
- Foster, R.T.; Jamali, F.; Russell, A.S.; Alballa, S.R. Pharmacokinetics of ketoprofen enantiomers in healthy subjects following single and multiple doses. J. Pharm. Sci. 1998, 77, 70–73. [Google Scholar] [CrossRef]
- Andersson, T.; Weidolf, L. Stereoselective disposition of proton pump inhibitors. Clin. Drug Investig. 2008, 28, 263–279. [Google Scholar] [CrossRef]
- Shi, S.; Klotz, U. Proton pump inhibitors: An update of their clinical use and pharmacokinetics. Eur. J. Clin. Pharmacol. 2008, 64, 935–951. [Google Scholar] [CrossRef]
- Olbe, L.; Carlsson, E.; Lindberg, P. A proton-pump inhibitor expedition: The case histories of omeprazole and esomeprazole. Nat. Rev. Drug Discov. 2003, 2, 132–139. [Google Scholar] [CrossRef]
- Asghar, W.; Pittman, E.; Jamali, F. Comparative efficacy of esomeprazole and omeprazole: Racemate to single enantiomer switch. DARU J. Pharm. Sci. 2015, 23, 50. [Google Scholar] [CrossRef] [Green Version]
- Katsuki, H.; Hamada, A.; Nakamura, C.; Arimori, K.; Nakano, M. Role of CYP3A4 and CYP2C19 in the stereoselective metabolism of lansoprazole by human liver microsomes. Eur. J. Clin. Pharmacol. 2001, 57, 709–715. [Google Scholar] [CrossRef]
- Baumann, P.; Zullino, D.F.; Eap, C.B. Enantiomers’ potential in psychopharmacology- a critical analysis with special emphasis on the antidepressant escitalopram. Eur. Neuropsychopharm. 2002, 12, 433–444. [Google Scholar] [CrossRef]
- Gorman, J.M.; Korotzer, A.; Su, G. Efficacy comparison of escitalopram and citalopram in the treatment of major depressive disorder: Pooled analysis of placebo-controlled trials. CNS Spectr. 2002, 7, 40–44. [Google Scholar] [CrossRef]
- Wong, D.T.; Fuller, R.W.; Robertson, D.W. Fluoxetine and its two enantiomers as selective serotonin uptake inhibitors. Acta Pharm. Nord. 1990, 2, 171–180. [Google Scholar]
- Eap, C.B.; Bondolfi, G.; Zullino, D.; Savary-Cosendai, L.; Powell-Golay, K.; Kosel, M.; Baumann, P. Concentrations of the enantiomers of fluoxetine and norfluoxetine after multiple doses of fluoxetine in cytochrome P4502D6 poor and extensive metabolizers. J. Clin. Psychopharmacol. 2001, 21, 330–334. [Google Scholar] [CrossRef]
- Johansson, F.; Rydberg, I.; Aberg, G.; Andersson, R.G. Effects of albuterol enantiomers on in vitro bronchial reactivity. Clin. Rev. Allergy Immunol. 1996, 14, 57–64. [Google Scholar] [CrossRef]
- Blake, K.; Raissy, H. Chiral switch drugs for asthma and allergies: True benefit or marketing hype. Pediatr. Allergy Immunol. 2013, 26, 157–160. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, G.A.; Hostrup, M.; Narkowicz, C.K.; Nichols, D.S.; Walters, E.H. Enantioselective disposition of (R,R)-formoterol, (S,S)-formoterol and their respective glucuronides in urine following single inhaled dosing and application to doping control. Drug Test. Anal. 2019, 11, 950–956. [Google Scholar] [CrossRef]
- Bardsley, H.; Gristwood, R.; Baker, H.; Watson, N.; Nimmo, W. A comparison of the cardiovascular effects of levobupivacaine and rac-bupivacaine following intravenous administration to healthy volunteers. Br. J. Clin. Pharmacol. 1998, 46, 245–249. [Google Scholar] [CrossRef] [Green Version]
- Gristwood, R.W. Cardiac and CNS toxicity of levobupivacaine: Strengths of evidence for advantage over bupivacaine. Drug Saf. 2002, 25, 153–163. [Google Scholar] [CrossRef]
- Heppolette, C.A.; Brunnen, D.; Bampoe, S.; Odor, P.M. Clinical pharmacokinetics and pharmacodynamics of levobupivacaine. Clin. Pharmacokinet. 2020, 59, 715–745. [Google Scholar] [CrossRef]
- Scott, D.B.; Lee, A.; Fagan, D.; Bowler, G.M.; Bloomfield, P.; Lundh, R. Acute toxicity of ropivacaine compared with that of bupivacaine. Anesth. Analg. 1989, 69, 563–569. [Google Scholar] [CrossRef]
- Tillement, J.P.; Testa, B.; Brée, F. Compared pharmacological characteristics in humans of racemic cetirizine and levocetirizine, two histamine H1-receptor antagonists. Biochem. Pharmacol. 2003, 66, 1123–1126. [Google Scholar] [CrossRef]
- Swainson, J.; Thomas, R.K.; Archer, S.; Chrenek, C.; MacKay, M.A.; Baker, G.; Demas, M.L. Esketamine for treatment resistant depression. Expert Rev. Neurother. 2019, 19, 899–911. [Google Scholar] [CrossRef]
- Quinn, D. Does chirality matter? Pharmacodynamics of enantiomers of methylphenidate in patients with attention-deficit/hyperactivity disorder. J. Clin. Psychopharmacol. 2008, 28, S62–S66. [Google Scholar] [CrossRef]
- Fish, D.N.; Chow, A.T. The clinical pharmacokinetics of levofloxacin. Clin. Pharmacokinet. 1997, 32, 101–119. [Google Scholar] [CrossRef]
- Preston, S.L.; Drusano, G.L.; Berman, A.L.; Fowler, C.L.; Chow, A.T.; Dornseif, B.; Corrado, M. Pharmacodynamics of levofloxacin: A new paradigm for early clinical trials. JAMA 1998, 279, 125–129. [Google Scholar] [CrossRef] [Green Version]
- Gardin, J.M.; Schumacher, D.; Constantine, G.; Davis, K.D.; Leung, C.; Reid, C.L. Valvular abnormalities and cardiovascular status following exposure to dexfenfluramine or phentermine/fenfluramine. JAMA 2000, 283, 1703–1709. [Google Scholar] [CrossRef] [Green Version]
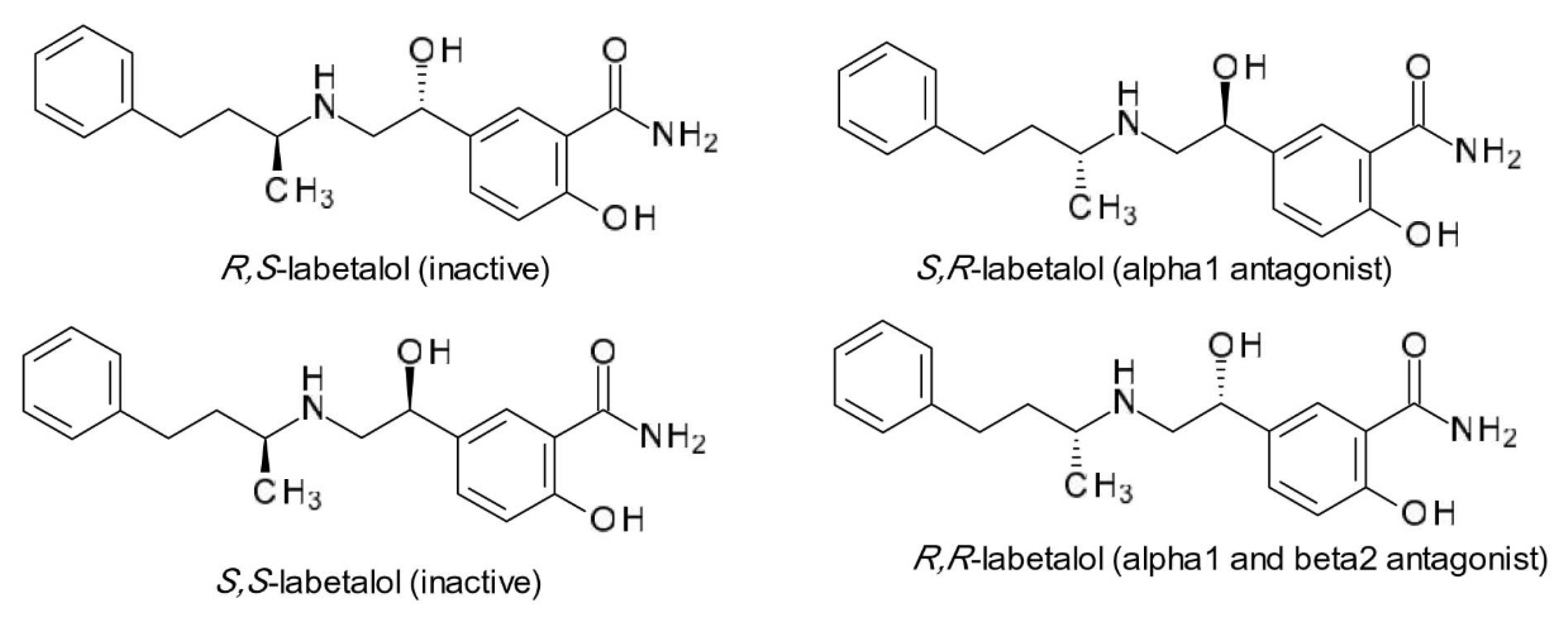
- Vashistha, V.K.; Kumar, A. Stereochemical facets of clinical β-blockers: An overview. Chirality 2020, 32, 722–735. [Google Scholar] [CrossRef]
- Kelly, R.; Daley, J.; Avolio, A.; O’Rourke, M. Arterial dilation and reduced wave reflection. Benefit of dilevalol in hypertension. Hypertension 1989, 14, 14–21. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, R.; Macphee, G.J. Clinical pharmacokinetics and kinetic-dynamic relationships of dilevalol and labetalol. Clin. Pharmacokinet. 1991, 21, 95–109. [Google Scholar] [CrossRef]
- Waldo, A.L.; Camm, A.J.; deRuyter, H.; Friedman, P.L.; MacNeil, D.J.; Pauls, J.F.; Pitt, B.; Pratt, C.M.; Schwartz, P.J.; Veltri, E.P. Effect of d-sotalol on mortality in patients with left ventricular dysfunction after recent and remote myocardial infarction. Lancet 1996, 348, 7–12. [Google Scholar] [CrossRef]
- Pratt, C.M.; Camm, A.J.; Cooper, W.; Friedman, P.L.; MacNeil, D.J.; Moulton, K.M.; Waldo, A.L. Mortality in the Survival with ORal D-sotalol (SWORD) trial: Why did patients die? Am. J. Cardiol. 1998, 81, 869–876. [Google Scholar] [CrossRef]
- FDA. Development of New Stereoisomeric Drugs. Available online: www.fda.gov/regulatory-information/search-fda-guidance-documents/development-new-stereoisomeric-drugs (accessed on 14 January 2022).
- Calcaterra, A.; D’Acquarica, I. The market of chiral drugs: Chiral switches versus de novo enantiomerically pure compounds. J. Pharm. Biomed. Anal. 2018, 147, 323–340. [Google Scholar] [CrossRef]
- FDA. New Drugs at FDA: CDER’s New Molecular Entities and New Therapeutic Biological Products. Available online: www.fda.gov/drugs/development-approval-process-drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products (accessed on 14 January 2022).
- Nunez, M.C.; Garcia-Rubino, M.E.; Conejo-Garcia, A.; Cruz-Lopez, O.; Kimatrai, M.; Gallo, M.A.; Espinosa, A.; Campos, J.M. Homochiral drugs: A demanding tendency of the pharmaceutical industry. Curr. Med. Chem. 2009, 16, 2064–2074. [Google Scholar] [CrossRef]
- Long, A.S.; Zhang, A.D.; Meyer, C.E.; Egilman, A.C.; Ross, J.S.; Wallach, J.D. Evaluation of trials comparing single-enantiomer drugs to their racemic precursors: A systematic review. JAMA Network Open 2021, 4, e215731. [Google Scholar] [CrossRef]
- Gupta, H.; Kumar, S.; Roy, S.K.; Gaud, R.S. Patent protection strategies. J. Pharm. Bioallied. Sci. 2010, 2, 2–7. [Google Scholar] [CrossRef]
- Branch, S.K.; Agranat, I. “New drug” designations for new therapeutic entities: New active substance, new chemical entity, new biological entity, new molecular entity. J. Med. Chem. 2014, 57, 8729–8765. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Racemate | Active Enantiomer | Chemical Structure | Pharmacological Activity |
|---|---|---|---|---|
| 1 | R,S-Albuterol | R-(−)-Albuterol (levalbuterol) | 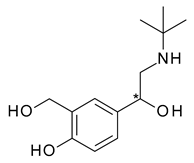 | β2 adrenergic receptor agonist antiasthmatic |
| 2 | R,S-Bupivacaine | S-(−)-Bupivacaine (levobupivacaine) | 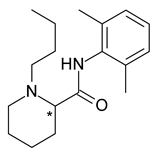 | Local anesthetic |
| 3 | R,S-Cetirizine | R-(−)-Cetirizine (levocetirizine) | 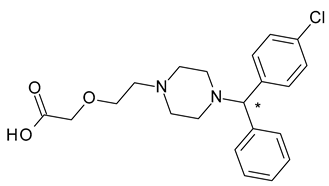 | H1 antihistaminic antiallergic |
| 4 | R,S-Citalopram | S-(+)-Citalopram (escitalopram) |  | Selective serotonin reuptake inhibitor (SSRI) antidepressant |
| 5 | R,S-Fenfluramine | S-(+)-Fenfluramine (dexfenfluramine) | 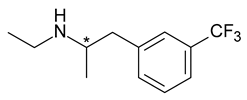 | Anorectic—withdrawn from the market due to cardiovascular effects |
| 6 | R,R,S,S-Formoterol | R,R-(−) Formoterol (arformoterol) |  | β2 adrenergic receptor agonist antiasthmatic |
| 7 | R,S-Ibuprofen | S-(+)-Ibuprofen (dexibuprofen) | 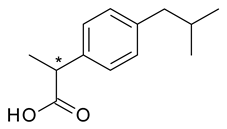 | Nonsteroidal anti-inflammatory |
| 8 | R,S-Ketamine | S-(+)-Ketamine (esketamine) |  | General anaesthetic |
| 9 | R,S-Ketoprofen | S-(+)-Ketoprofen (dexketoprofen) |  | Nonsteroidal anti-inflammatory |
| 10 | R,S-Lansoprazole | R-(+)-Lansoprazole (dexlansoprazole) | 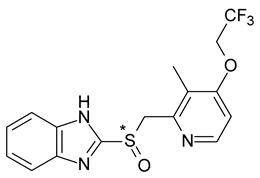 | Proton pump inhibitor (PPI) antacid |
| 11 | R,S-Leucovorin (Folinic acid) | S-(−)-Leucovorin (levoleucovorin) | 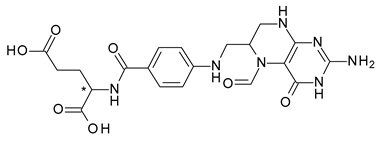 | Folate deficiency, decreases the toxic effects of methotrexate and pyrimethamine treatments |
| 12 | R,R,S,S-Methylphenidate | R,R-(+)-Methylphenidate (dexmethylphenidate) |  | Stimulant in ADHD and narcolepsy |
| 13 | R,S-Milnacipran | S-(−)-Milnacipran (levomilnacipran) |  | Serotonin and norepinephrine reuptake inhibitor (SNRI) antidepressant |
| 14 | R,S-Modafinil | R-(−)-Modafinil (armodafinil) | 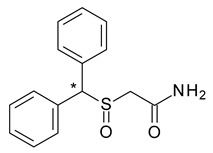 | Eugeroic (wakefulness-promoter) in narcolepsy |
| 15 | R,S-Ofloxacin | S-(−)-Ofloxacin (levofloxacin) |  | Fluoroquinolone antibacterial |
| 16 | R,S-Omeprazole | S-(+)-Omeprazole (esomeprazole) |  | Proton pump inhibitor (PPI) antacid |
| 17 | R,S-Zopiclone | S-(+)-Zopiclone (eszopiclone) | 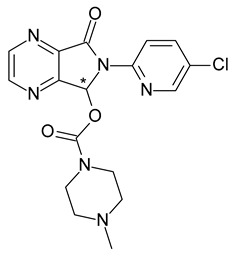 | Sedative-hypnotic |
| No. | Racemate | Active Enantiomer | Potential Therapeutic Advantage |
|---|---|---|---|
| 1 | Albuterol | Levalbuterol | Increased potency, decreased development of airway hyperreactivity |
| 2 | Bupivacaine | Levobupivacaine | Decreased risk of cardiotoxicity |
| 3 | Cetirizine | Levocetirizine | Increased potency |
| 4 | Citalopram | Escitalopram | Increased potency, faster onset of action, improved tolerability profile |
| 5 | Formoterol | Arformoterol | Increased potency, decreased development of airway hyperreactivity |
| 6 | Ibuprofen | Dexibuprofen | Faster onset of action |
| 7 | Ketamine | Esketamine | Increased tolerance, shorter recovery time, decrease incidence of side-effects |
| 8 | Ketoprofen | Dexketoprofen | Increased potency, faster onset of action, decrease incidence of gastrointestinal side-effects (trometamol salt) |
| 9 | Lansoprazole | Dexlansoprazole | Less variable pharmacokinetic profile in some patients |
| 10 | Methylphenidate | Dexmethylphenidate | Increased potency |
| 11 | Ofloxacin | Levofloxacin | Increased potency |
| 12 | Omeprazole | Esomeprazole | Less variable pharmacokinetic profile in some patients |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hancu, G.; Modroiu, A. Chiral Switch: Between Therapeutical Benefit and Marketing Strategy. Pharmaceuticals 2022, 15, 240. https://doi.org/10.3390/ph15020240
Hancu G, Modroiu A. Chiral Switch: Between Therapeutical Benefit and Marketing Strategy. Pharmaceuticals. 2022; 15(2):240. https://doi.org/10.3390/ph15020240
Chicago/Turabian StyleHancu, Gabriel, and Adriana Modroiu. 2022. "Chiral Switch: Between Therapeutical Benefit and Marketing Strategy" Pharmaceuticals 15, no. 2: 240. https://doi.org/10.3390/ph15020240
APA StyleHancu, G., & Modroiu, A. (2022). Chiral Switch: Between Therapeutical Benefit and Marketing Strategy. Pharmaceuticals, 15(2), 240. https://doi.org/10.3390/ph15020240