
Increasing Inhibition of the Rat Brain 2-Oxoglutarate Dehydrogenase Decreases Glutathione Redox State, Elevating Anxiety and Perturbing Stress Adaptation

 , , , ,
, , , , 
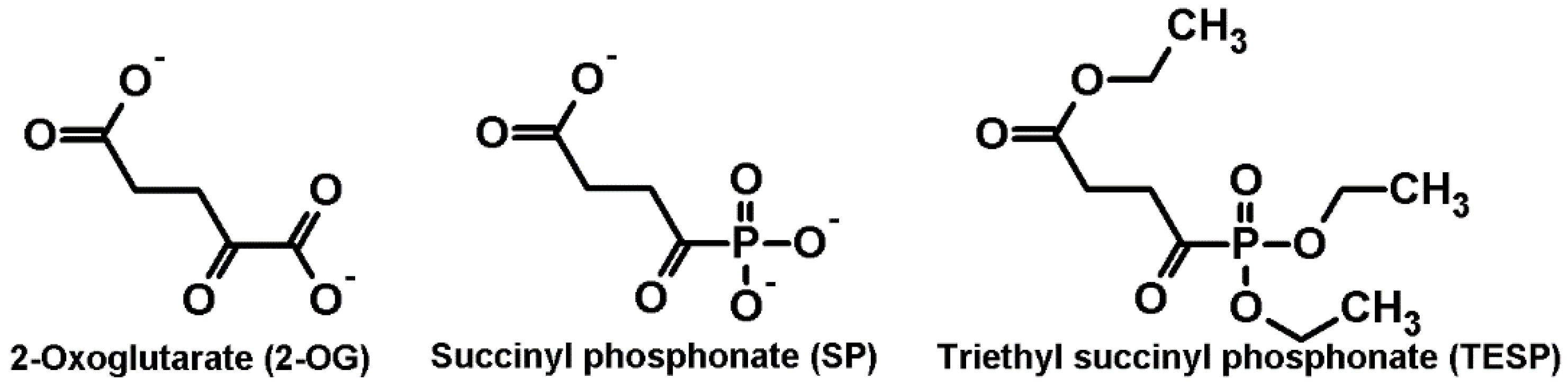{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
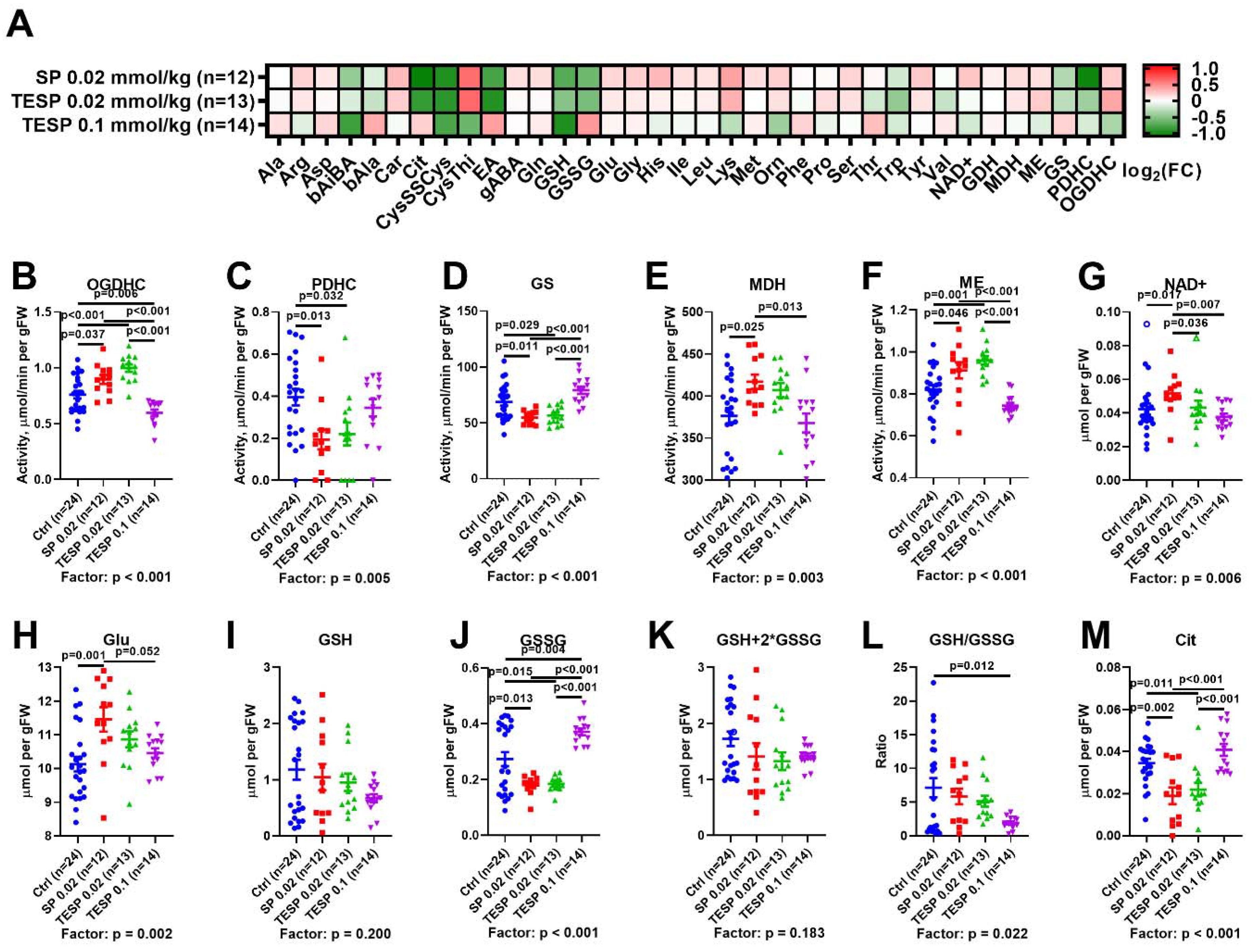
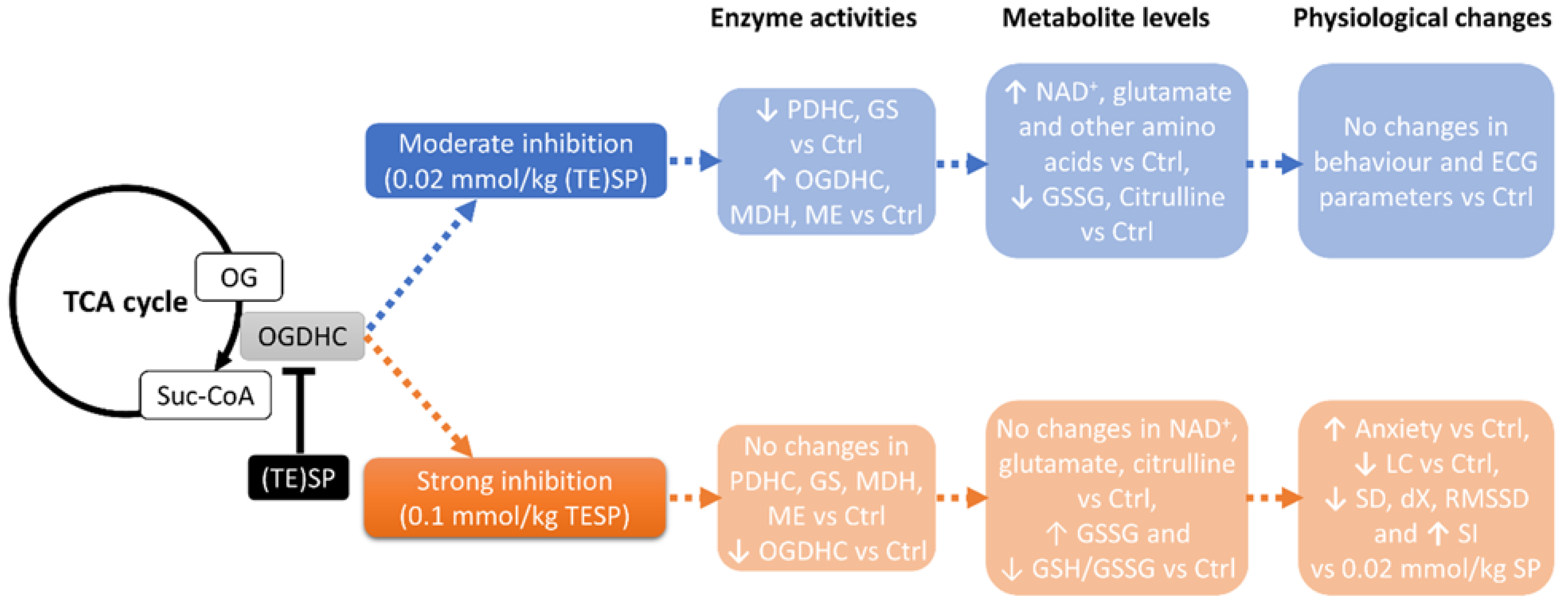
2.1. Biphasic Changes in Biochemical Parameters of the Rat Cerebral Cortex in Response to Increasing Dosage of 2-Oxo Phosphonate Inhibitors of OGDH
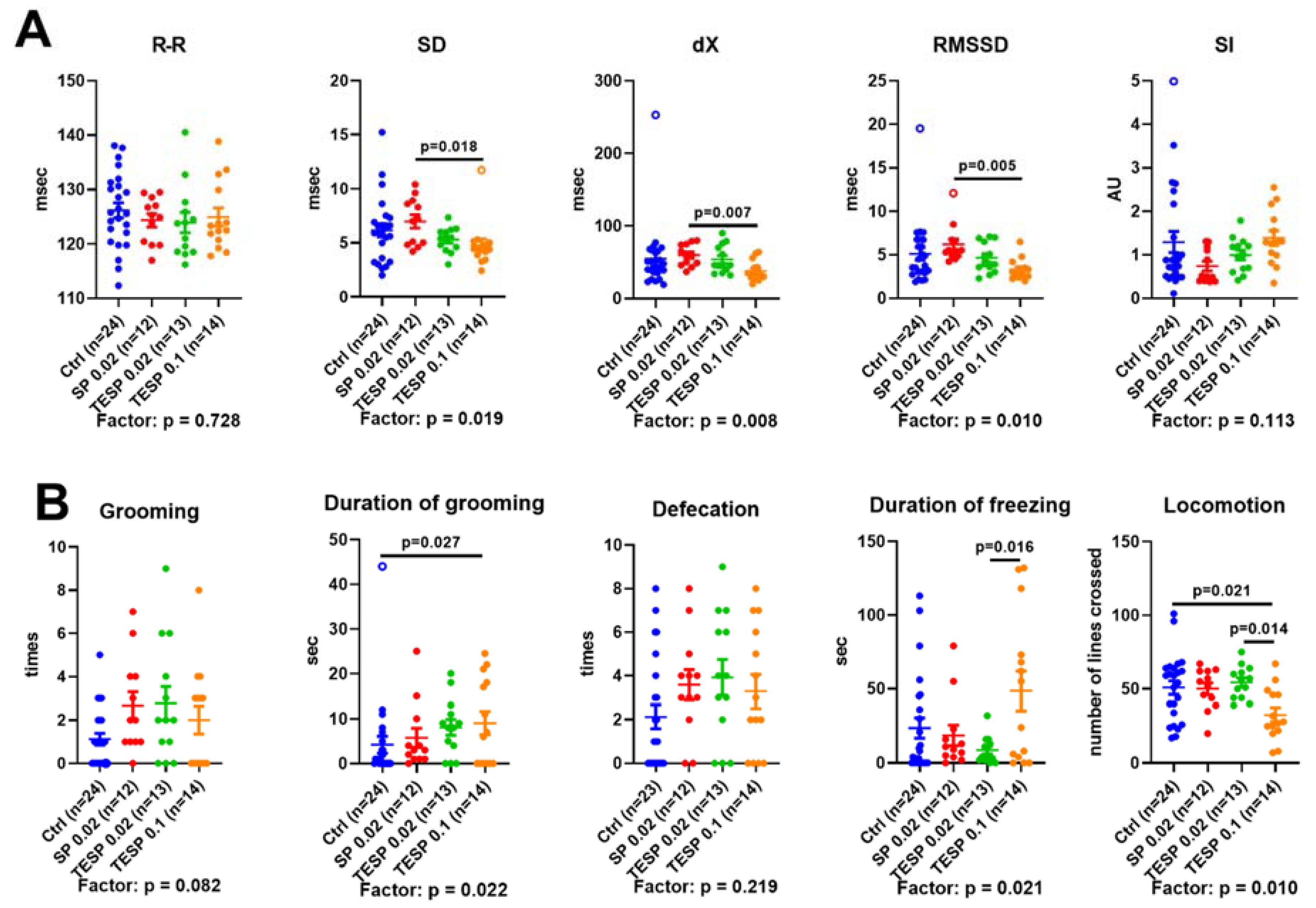
2.2. Effects of OGDH Inhibition on the ECG Parameters and Animal Behavior
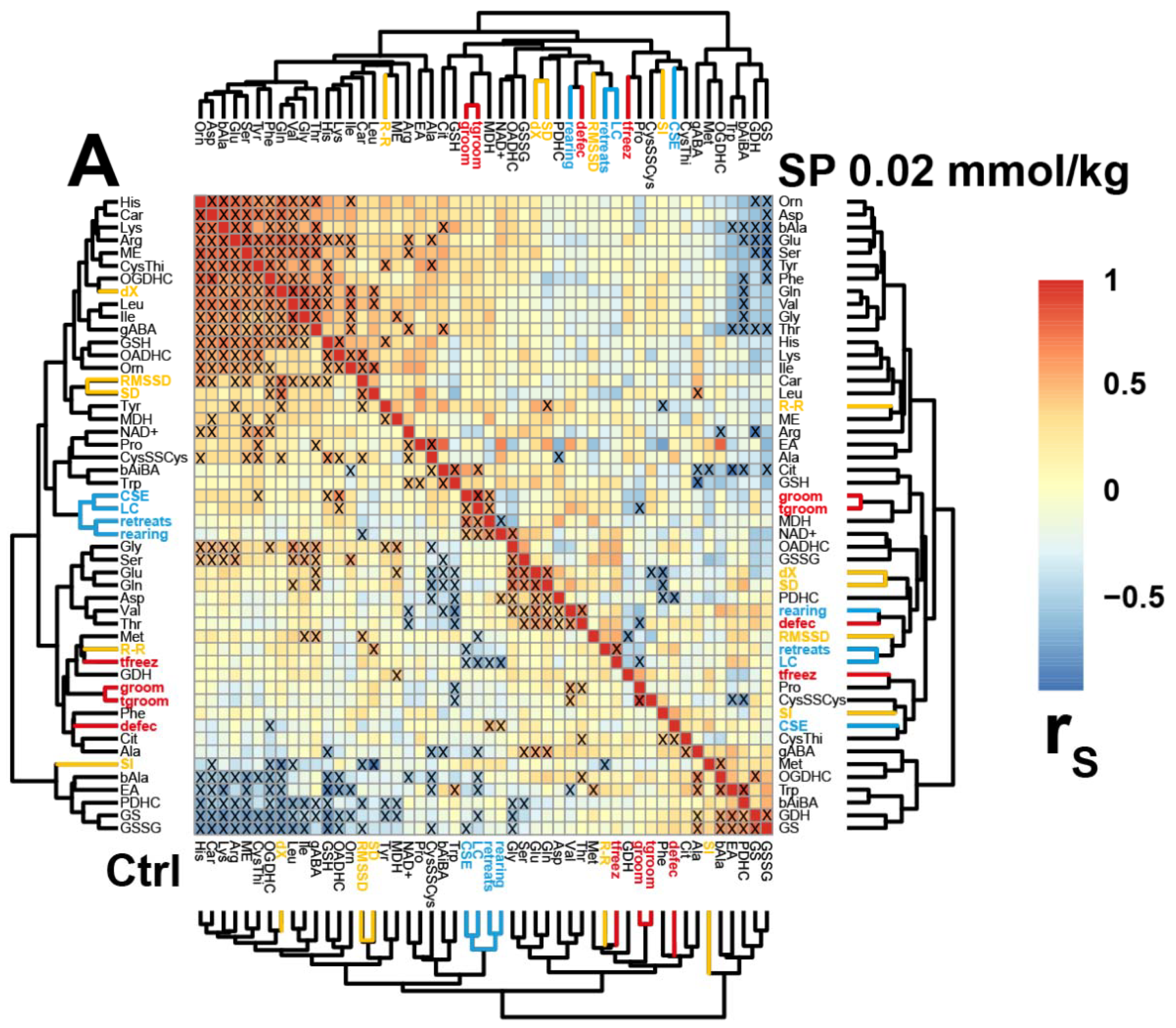
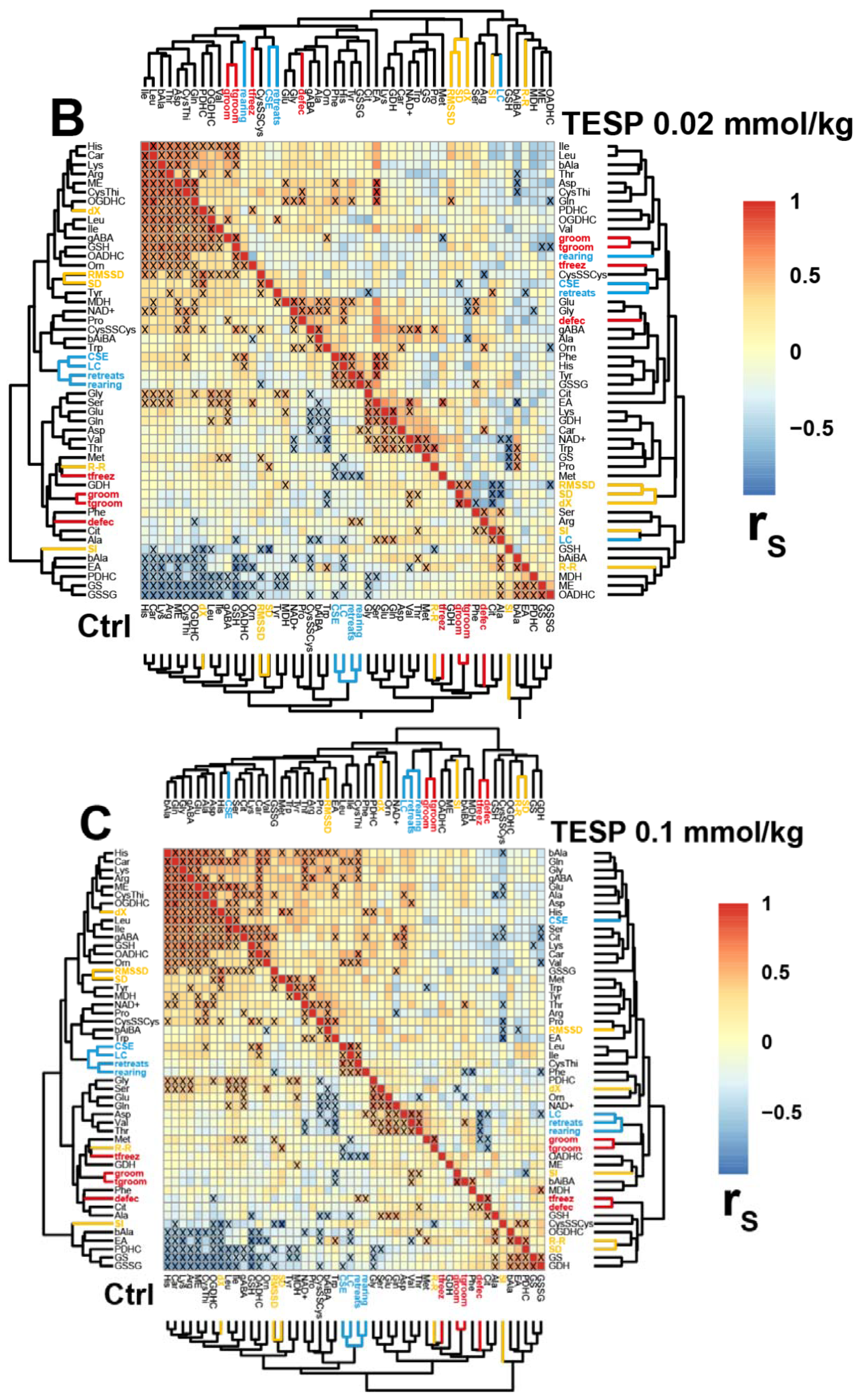
2.3. Correlations between the Brain Biochemical Markers and Physiological Changes
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Animal Experiments
4.3. Physiological Tests
4.4. Metabolite and Enzyme Activity Assays
4.5. Statistics and Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jha, S.K.; Jha, N.K.; Kumar, D.; Ambasta, R.K.; Kumar, P. Linking mitochondrial dysfunction, metabolic syndrome and stress signaling in Neurodegeneration. Biochem. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1132–1146. [Google Scholar] [CrossRef] [PubMed]
- Muddapu, V.R.; Dharshini, S.A.P.; Chakravarthy, V.S.; Gromiha, M.M. Neurodegenerative diseases—Is metabolic deficiency the root cause? Front. Neurosci. 2020, 14, 213. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Wang, X.; Zheng, L.; Perry, G.; Smith, M.A.; Zhu, X. Abnormal mitochondrial dynamics and neurodegenerative diseases. Biochim. Biophys. Acta 2010, 1802, 135–142. [Google Scholar] [CrossRef]
- Kim, Y.; Vadodaria, K.C.; Lenkei, Z.; Kato, T.; Gage, F.H.; Marchetto, M.C.; Santos, R. Mitochondria, metabolism, and redox mechanisms in psychiatric disorders. Antioxid. Redox Signal. 2019, 31, 275–317. [Google Scholar] [CrossRef]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef]
- Patel, R.P.; McAndrew, J.; Sellak, H.; White, C.R.; Jo, H.; Freeman, B.A.; Darley-Usmar, V.M. Biological aspects of reactive nitrogen species. Biochim. Biophys. Acta 1999, 1411, 385–400. [Google Scholar] [CrossRef]
- Seo, A.Y.; Joseph, A.M.; Dutta, D.; Hwang, J.C.; Aris, J.P.; Leeuwenburgh, C. New insights into the role of mitochondria in aging: Mitochondrial dynamics and more. J. Cell Sci. 2010, 123, 2533–2542. [Google Scholar] [CrossRef]
- Procaccini, C.; Santopaolo, M.; Faicchia, D.; Colamatteo, A.; Formisano, L.; de Candia, P.; Galgani, M.; De Rosa, V.; Matarese, G. Role of metabolism in neurodegenerative disorders. Metab. Clin. Exp. 2016, 65, 1376–1390. [Google Scholar] [CrossRef]
- Blandini, F.; Braunewell, K.H.; Manahan-Vaughan, D.; Orzi, F.; Sarti, P. Neurodegeneration and energy metabolism: From chemistry to clinics. Cell Death Differ. 2004, 11, 479–484. [Google Scholar] [CrossRef][Green Version]
- Zuccoli, G.S.; Saia-Cereda, V.M.; Nascimento, J.M.; Martins-de-Souza, D. The energy metabolism dysfunction in psychiatric disorders postmortem brains: Focus on proteomic evidence. Front. Neurosci. 2017, 11, 493. [Google Scholar] [CrossRef]
- Chen, X.; Pan, W. The treatment strategies for neurodegenerative diseases by integrative medicine. Integr. Med. Int. 2014, 1, 223–225. [Google Scholar] [CrossRef]
- Reynolds, G.P.; Kirk, S.L. Metabolic side effects of antipsychotic drug treatment-pharmacological mechanisms. Pharmacol. Ther. 2010, 125, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Siafis, S.; Tzachanis, D.; Samara, M.; Papazisis, G. Antipsychotic drugs: From receptor-binding profiles to metabolic side effects. Curr. Neuropharmacol. 2018, 16, 1210–1223. [Google Scholar] [CrossRef]
- Atti, A.R.; Ferrari Gozzi, B.; Zuliani, G.; Bernabei, V.; Scudellari, P.; Berardi, D.; De Ronchi, D.; Tarricone, I.; Menchetti, M. A systematic review of metabolic side effects related to the use of antipsychotic drugs in dementia. Int. Psychogeriatr. 2014, 26, 19–37. [Google Scholar] [CrossRef] [PubMed]
- Mintzer, S.; Skidmore, C.T.; Abidin, C.J.; Morales, M.C.; Chervoneva, I.; Capuzzi, D.M.; Sperling, M.R. Effects of antiepileptic drugs on lipids, homocysteine, and C-reactive protein. Ann. Neurol. 2009, 65, 448–456. [Google Scholar] [CrossRef]
- Bunik, V.I.; Fernie, A.R. Metabolic control exerted by the 2-oxoglutarate dehydrogenase reaction: A cross-kingdom comparison of the crossroad between energy production and nitrogen assimilation. Biochem. J. 2009, 422, 405–421. [Google Scholar] [CrossRef] [PubMed]
- Mkrtchyan, G.V.; Graf, A.; Trofimova, L.; Ksenofontov, A.; Baratova, L.; Bunik, V. Positive correlation between rat brain glutamate concentrations and mitochondrial 2-oxoglutarate dehydrogenase activity. Anal. Biochem. 2018, 552, 100–109. [Google Scholar] [CrossRef]
- Artiukhov, A.V.; Graf, A.V.; Bunik, V.I. Directed regulation of multienzyme complexes of 2-Oxo acid dehydrogenases using phosphonate and phosphinate analogs of 2-Oxo acids. Biochemistry 2016, 81, 1498–1521. [Google Scholar] [CrossRef]
- Graf, A.; Trofimova, L.; Loshinskaja, A.; Mkrtchyan, G.; Strokina, A.; Lovat, M.; Tylicky, A.; Strumilo, S.; Bettendorff, L.; Bunik, V.I. Up-regulation of 2-oxoglutarate dehydrogenase as a stress response. Int. J. Biochem. Cell Biol. 2013, 45, 175–189. [Google Scholar] [CrossRef]
- Mkrtchyan, G.V.; Ucal, M.; Mullebner, A.; Dumitrescu, S.; Kames, M.; Moldzio, R.; Molcanyi, M.; Schaefer, S.; Weidinger, A.; Schaefer, U.; et al. Thiamine preserves mitochondrial function in a rat model of traumatic brain injury, preventing inactivation of the 2-oxoglutarate dehydrogenase complex. Biochim. Biophys. Acta 2018, 1859, 925–931. [Google Scholar] [CrossRef]
- Boyko, A.; Tsepkova, P.; Aleshin, V.; Artiukhov, A.; Mkrtchyan, G.; Ksenofontov, A.; Baratova, L.; Ryabov, S.; Graf, A.; Bunik, V. Severe spinal cord injury in rats induces chronic changes in the spinal cord and cerebral cortex metabolism, adjusted by thiamine that improves locomotor performance. Front. Mol. Neurosci. 2021, 14, 620593. [Google Scholar] [CrossRef] [PubMed]
- Parkhomenko, Y.M.; Kudryavtsev, P.A.; Pylypchuk, S.Y.; Chekhivska, L.I.; Stepanenko, S.P.; Sergiichuk, A.A.; Bunik, V.I. Chronic alcoholism in rats induces a compensatory response, preserving brain thiamine diphosphate, but the brain 2-oxo acid dehydrogenases are inactivated despite unchanged coenzyme levels. J. Neurochem. 2011, 117, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Sheu, K.F.; Blass, J.P.; Baker, A.; Carlson, K.C.; Harding, B.; Perrino, P. Reduced activities of thiamine-dependent enzymes in the brains and peripheral tissues of patients with Alzheimer’s disease. Arch. Neurol. 1988, 45, 836–840. [Google Scholar] [CrossRef] [PubMed]
- Yap, Z.Y.; Strucinska, K.; Matsuzaki, S.; Lee, S.; Si, Y.; Humphries, K.; Tarnopolsky, M.A.; Yoon, W.H. A biallelic pathogenic variant in the OGDH gene results in a neurological disorder with features of a mitochondrial disease. J. Inherit. Metab. Dis. 2021, 44, 388–400. [Google Scholar] [CrossRef]
- Csaban, D.; Pentelenyi, K.; Toth-Bencsik, R.; Illes, A.; Grosz, Z.; Gezsi, A.; Molnar, M.J. The role of the rare variants in the genes encoding the alpha-ketoglutarate dehydrogenase in Alzheimer’s disease. Life 2021, 11, 321. [Google Scholar] [CrossRef]
- Yoon, W.H.; Sandoval, H.; Nagarkar-Jaiswal, S.; Jaiswal, M.; Yamamoto, S.; Haelterman, N.A.; Putluri, N.; Putluri, V.; Sreekumar, A.; Tos, T.; et al. Loss of nardilysin, a mitochondrial co-chaperone for alpha-ketoglutarate dehydrogenase, Promotes mTORC1 activation and neurodegeneration. Neuron 2017, 93, 115–131. [Google Scholar] [CrossRef]
- Sherrill, J.D.; Kc, K.; Wang, X.; Wen, T.; Chamberlin, A.; Stucke, E.M.; Collins, M.H.; Abonia, J.P.; Peng, Y.; Wu, Q.; et al. Whole-exome sequencing uncovers oxidoreductases DHTKD1 and OGDHL as linkers between mitochondrial dysfunction and eosinophilic esophagitis. JCI Insight 2018, 3, 3. [Google Scholar] [CrossRef]
- Kessler, M.; Berger, I.M.; Just, S.; Rottbauer, W. Loss of dihydrolipoyl succinyltransferase (DLST) leads to reduced resting heart rate in the zebrafish. Basic Res. Cardiol. 2015, 110, 14. [Google Scholar] [CrossRef]
- Amsterdam, A.; Nissen, R.M.; Sun, Z.; Swindell, E.C.; Farrington, S.; Hopkins, N. Identification of 315 genes essential for early zebrafish development. Proc. Natl. Acad. Sci. USA 2004, 101, 12792–12797. [Google Scholar] [CrossRef]
- Johnson, M.T.; Yang, H.S.; Magnuson, T.; Patel, M.S. Targeted disruption of the murine dihydrolipoamide dehydrogenase gene (Dld) results in perigastrulation lethality. Proc. Natl. Acad. Sci. USA 1997, 94, 14512–14517. [Google Scholar] [CrossRef]
- Bubber, P.; Haroutunian, V.; Fisch, G.; Blass, J.P.; Gibson, G.E. Mitochondrial abnormalities in Alzheimer brain: Mechanistic implications. Ann. Neurol. 2005, 57, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Park, L.C.; Albers, D.S.; Xu, H.; Lindsay, J.G.; Beal, M.F.; Gibson, G.E. Mitochondrial impairment in the cerebellum of the patients with progressive supranuclear palsy. J. Neurosci. Res. 2001, 66, 1028–1034. [Google Scholar] [CrossRef] [PubMed]
- Albers, D.S.; Augood, S.J.; Park, L.C.; Browne, S.E.; Martin, D.M.; Adamson, J.; Hutton, M.; Standaert, D.G.; Vonsattel, J.P.; Gibson, G.E.; et al. Frontal lobe dysfunction in progressive supranuclear palsy: Evidence for oxidative stress and mitochondrial impairment. J. Neurochem. 2000, 74, 878–881. [Google Scholar] [CrossRef] [PubMed]
- Heroux, M.; Raghavendra Rao, V.L.; Lavoie, J.; Richardson, J.S.; Butterworth, R.F. Alterations of thiamine phosphorylation and of thiamine-dependent enzymes in Alzheimer’s disease. Metab. Brain Dis. 1996, 11, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, R.F.; Besnard, A.M. Thiamine-dependent enzyme changes in temporal cortex of patients with Alzheimer’s disease. Metab. Brain Dis. 1990, 5, 179–184. [Google Scholar] [CrossRef]
- Mastrogiacomo, F.; LaMarche, J.; Dozic, S.; Lindsay, G.; Bettendorff, L.; Robitaille, Y.; Schut, L.; Kish, S.J. Immunoreactive levels of alpha-ketoglutarate dehydrogenase subunits in Friedreich’s ataxia and spinocerebellar ataxia type 1. J. Neurodegenerati. Disord. Neuroprot. Neuroreg. 1996, 5, 27–33. [Google Scholar]
- Mastrogiacomo, F.; Bergeron, C.; Kish, S.J. Brain alpha-ketoglutarate dehydrogenase complex activity in Alzheimer’s disease. J. Neurochem. 1993, 61, 2007–2014. [Google Scholar] [CrossRef]
- Mizuno, Y.; Matuda, S.; Yoshino, H.; Mori, H.; Hattori, N.; Ikebe, S. An immunohistochemical study on alpha-ketoglutarate dehydrogenase complex in Parkinson’s disease. Ann. Neurol. 1994, 35, 204–210. [Google Scholar] [CrossRef]
- Terwel, D.; Bothmer, J.; Wolf, E.; Meng, F.; Jolles, J. Affected enzyme activities in Alzheimer’s disease are sensitive to antemortem hypoxia. J. Neurol. Sci. 1998, 161, 47–56. [Google Scholar] [CrossRef]
- Bunik, V.I.; Denton, T.T.; Xu, H.; Thompson, C.M.; Cooper, A.J.; Gibson, G.E. Phosphonate analogues of alpha-ketoglutarate inhibit the activity of the alpha-ketoglutarate dehydrogenase complex isolated from brain and in cultured cells. Biochemistry 2005, 44, 10552–10561. [Google Scholar] [CrossRef]
- Zundorf, G.; Kahlert, S.; Bunik, V.I.; Reiser, G. Aalpha-ketoglutarate dehydrogenase contributes to production of reactive oxygen species in glutamate-stimulated hippocampal neurons in situ. Neuroscience 2009, 158, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Araujo, W.L.; Trofimova, L.; Mkrtchyan, G.; Steinhauser, D.; Krall, L.; Graf, A.; Fernie, A.R.; Bunik, V.I. On the role of the mitochondrial 2-oxoglutarate dehydrogenase complex in amino acid metabolism. Amino Acids 2013, 44, 683–700. [Google Scholar] [CrossRef] [PubMed]
- Artiukhov, A.V.; Grabarska, A.; Gumbarewicz, E.; Aleshin, V.A.; Kahne, T.; Obata, T.; Kazantsev, A.V.; Lukashev, N.V.; Stepulak, A.; Fernie, A.R.; et al. Synthetic analogues of 2-oxo acids discriminate metabolic contribution of the 2-oxoglutarate and 2-oxoadipate dehydrogenases in mammalian cells and tissues. Sci. Rep. 2020, 10, 1886. [Google Scholar] [CrossRef]
- Artiukhov, A.V.; Kazantsev, A.V.; Lukashev, N.V.; Bellinzoni, M.; Bunik, V.I. Selective Inhibition of 2-Oxoglutarate and 2-Oxoadipate Dehydrogenases by the Phosphonate Analogs of Their 2-Oxo Acid Substrates. Front. Chem. 2021, 8, 596187. [Google Scholar] [CrossRef] [PubMed]
- Trofimova, L.; Lovat, M.; Groznaya, A.; Efimova, E.; Dunaeva, T.; Maslova, M.; Graf, A.; Bunik, V. Behavioral impact of the regulation of the brain 2-oxoglutarate dehydrogenase complex by synthetic phosphonate analog of 2-oxoglutarate: Implications into the role of the complex in neurodegenerative diseases. Int. J. Alzheimer’s Dis. 2010, 2010, 749061. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Munoz, M.D.; Bell, S.E.; Fairfax, K.; Monzon-Casanova, E.; Cunningham, A.F.; Gonzalez-Porta, M.; Andrews, S.R.; Bunik, V.I.; Zarnack, K.; Curk, T.; et al. The RNA-binding protein HuR is essential for the B cell antibody response. Nat. Immunol. 2015, 16, 415–425. [Google Scholar] [CrossRef]
- Bunik, V.I.; Artiukhov, A.; Kazantsev, A.; Goncalves, R.; Daloso, D.; Oppermann, H.; Kulakovskaya, E.; Lukashev, N.; Fernie, A.; Brand, M.; et al. Specific inhibition by synthetic analogs of pyruvate reveals that the pyruvate dehydrogenase reaction is essential for metabolism and viability of glioblastoma cells. Oncotarget 2015, 6, 40036–40052. [Google Scholar] [CrossRef]
- Boyko, A.; Ksenofontov, A.; Ryabov, S.; Baratova, L.; Graf, A.; Bunik, V. Delayed Influence of Spinal Cord Injury on the Amino Acids of NO(*) Metabolism in Rat Cerebral Cortex Is Attenuated by Thiamine. Front. Med. 2017, 4, 249. [Google Scholar] [CrossRef]
- Wanchu, A.; Khullar, M.; Sud, A.; Bambery, P. Nitric oxide production is increased in patients with inflammatory myositis. Nitric Oxide 1999, 3, 454–458. [Google Scholar] [CrossRef]
- Graf, A.; Ksenofontov, A.; Bunik, V. Inhibition of 2-oxoglutarate dehydrogenase as a chemical model of acute hypobaric hypoxia. Front. Med. 2021, 8, 751639. [Google Scholar] [CrossRef]
- Tsepkova, P.M.; Artiukhov, A.V.; Boyko, A.I.; Aleshin, V.A.; Mkrtchyan, G.V.; Zvyagintseva, M.A.; Ryabov, S.I.; Ksenofontov, A.L.; Baratova, L.A.; Graf, A.V.; et al. Thiamine induces long-term changes in amino acid profiles and activities of 2-xoglutarate and 2-Oxoadipate Dehydrogenases in rat brain. Biochemistry 2017, 82, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Bunik, V.; Mkrtchyan, G.; Grabarska, A.; Oppermann, H.; Daloso, D.; Araujo, W.L.; Juszczak, M.; Rzeski, W.; Bettendorff, L.; Fernie, A.R.; et al. Inhibition of mitochondrial 2-oxoglutarate dehydrogenase impairs viability of cancer cells in a cell-specific metabolism-dependent manner. Oncotarget 2016, 7, 26400–26421. [Google Scholar] [CrossRef] [PubMed]
- Trofimova, L.K.; Araujo, W.L.; Strokina, A.A.; Fernie, A.R.; Bettendorff, L.; Bunik, V.I. Consequences of the alpha-ketoglutarate dehydrogenase inhibition for neuronal metabolism and survival: Implications for neurodegenerative diseases. Curr. Med. Chem. 2012, 19, 5895–5906. [Google Scholar] [CrossRef]
- Martin, A.; David, V.; Malaval, L.; Lafage-Proust, M.H.; Vico, L.; Thomas, T. Opposite effects of leptin on bone metabolism: A dose-dependent balance related to energy intake and insulin-like growth factor-I pathway. Endocrinology 2007, 148, 3419–3425. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ravera, S.; Cossu, V.; Tappino, B.; Nicchia, E.; Dufour, C.; Cavani, S.; Sciutto, A.; Bolognesi, C.; Columbaro, M.; Degan, P.; et al. Concentration-dependent metabolic effects of metformin in healthy and Fanconi anemia lymphoblast cells. J. Cell. Physiol. 2018, 233, 1736–1751. [Google Scholar] [CrossRef] [PubMed]
- Wasik, A.; Kajta, M.; Lenda, T.; Antkiewicz-Michaluk, L. Concentration-dependent opposite effects of 1-benzyl-1,2,3,4-tetrahydroisoquinoline on markers of apoptosis: In vitro and ex vivo studies. Neurotox. Res. 2014, 25, 90–99. [Google Scholar] [CrossRef]
- Kazmierczak-Baranska, J.; Boguszewska, K.; Adamus-Grabicka, A.; Karwowski, B.T. Two faces of vitamin C-antioxidative and pro-oxidative agent. Nutrients 2020, 12, 1501. [Google Scholar] [CrossRef]
- Dintcheva, N.T.; D’Anna, F. Anti-/pro-oxidant behavior of naturally occurring molecules in polymers and biopolymers: A brief review. ACS Sustain. Chem. Eng. 2019, 7, 12656–12670. [Google Scholar] [CrossRef]
- Sharifi-Rad, M.; Anil Kumar, N.V.; Zucca, P.; Varoni, E.M.; Dini, L.; Panzarini, E.; Rajkovic, J.; Tsouh Fokou, P.V.; Azzini, E.; Peluso, I.; et al. Lifestyle, oxidative stress, and antioxidants: Back and forth in the pathophysiology of chronic diseases. Front. Physiol. 2020, 11, 694. [Google Scholar] [CrossRef]
- Chen, H.; Denton, T.T.; Xu, H.; Calingasan, N.; Beal, M.F.; Gibson, G.E. Reductions in the mitochondrial enzyme alpha-ketoglutarate dehydrogenase complex in neurodegenerative disease—Beneficial or detrimental? J. Neurochem. 2016, 139, 823–838. [Google Scholar] [CrossRef]
- Bunik, V.I. Redox-driven signaling: 2-Oxo acid dehydrogenase complexes as sensors and transmitters of metabolic imbalance. Antioxid. Redox Signal. 2019, 30, 1911–1947. [Google Scholar] [CrossRef] [PubMed]
- Zalachoras, I.; Hollis, F.; Ramos-Fernandez, E.; Trovo, L.; Sonnay, S.; Geiser, E.; Preitner, N.; Steiner, P.; Sandi, C.; Morato, L. Therapeutic potential of glutathione-enhancers in stress-related psychopathologies. Neurosci. Biobehav. Rev. 2020, 114, 134–155. [Google Scholar] [CrossRef] [PubMed]
- Hassan, W.; Silva, C.E.; Mohammadzai, I.U.; da Rocha, J.B.; J, L.F. Association of oxidative stress to the genesis of anxiety: Implications for possible therapeutic interventions. Curr. Neuropharmacol. 2014, 12, 120–139. [Google Scholar] [CrossRef] [PubMed]
- Krolow, R.; Arcego, D.M.; Noschang, C.; Weis, S.N.; Dalmaz, C. Oxidative imbalance and anxiety disorders. Curr. Neuropharmacol. 2014, 12, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.; Stomrud, E.; Lindberg, O.; Westman, E.; Johansson, P.M.; van Westen, D.; Mattsson, N.; Hansson, O. Apathy and anxiety are early markers of Alzheimer’s disease. Neurobiol. Aging 2020, 85, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Santabarbara, J.; Lipnicki, D.M.; Bueno-Notivol, J.; Olaya-Guzman, B.; Villagrasa, B.; Lopez-Anton, R. Updating the evidence for an association between anxiety and risk of Alzheimer’s disease: A meta-analysis of prospective cohort studies. J. Affect. Disord. 2020, 262, 397–404. [Google Scholar] [CrossRef]
- Pentkowski, N.S.; Rogge-Obando, K.K.; Donaldson, T.N.; Bouquin, S.J.; Clark, B.J. Anxiety and Alzheimer’s disease: Behavioral analysis and neural basis in rodent models of Alzheimer’s-related neuropathology. Neurosci. Biobehav. Rev. 2021, 127, 647–658. [Google Scholar] [CrossRef]
- Gibson, G.E.; Blass, J.P.; Beal, M.F.; Bunik, V. The alpha-ketoglutarate-dehydrogenase complex: A mediator between mitochondria and oxidative stress in neurodegeneration. Mol. Neurobiol. 2005, 31, 43–63. [Google Scholar] [CrossRef]
- Bubber, P.; Hartounian, V.; Gibson, G.E.; Blass, J.P. Abnormalities in the tricarboxylic acid (TCA) cycle in the brains of schizophrenia patients. Eur. Neuropsychopharmacol. 2011, 21, 254–260. [Google Scholar] [CrossRef]
- Bubber, P.; Tang, J.; Haroutunian, V.; Xu, H.; Davis, K.L.; Blass, J.P.; Gibson, G.E. Mitochondrial enzymes in schizophrenia. J. Mol. Neurosci. 2004, 24, 315–321. [Google Scholar] [CrossRef]
- Yao, J.K.; Leonard, S.; Reddy, R. Altered glutathione redox state in schizophrenia. Dis. Markers 2006, 22, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Luchsinger, J.A.; Cirio, R.; Chen, H.; Franchino-Elder, J.; Hirsch, J.A.; Bettendorff, L.; Chen, Z.; Flowers, S.A.; Gerber, L.M.; et al. Benfotiamine and cognitive decline in Alzheimer’s Disease: Results of a randomized placebo-controlled Phase IIa clinical trial. J. Alzheimer’s Dis. 2020, 78, 989–1010. [Google Scholar] [CrossRef]
- Pan, X.; Chen, Z.; Fei, G.; Pan, S.; Bao, W.; Ren, S.; Guan, Y.; Zhong, C. Long-term cognitive improvement after benfotiamine administration in patients with Alzheimer’s disease. Neurosci. Bull. 2016, 32, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Tapias, V.; Jainuddin, S.; Ahuja, M.; Stack, C.; Elipenahli, C.; Vignisse, J.; Gerges, M.; Starkova, N.; Xu, H.; Starkov, A.A.; et al. Benfotiamine treatment activates the Nrf2/ARE pathway and is neuroprotective in a transgenic mouse model of tauopathy. Hum. Mol. Genet. 2018, 27, 2874–2892. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Gong, N.; Zhao, J.; Yu, Z.; Gu, F.; Chen, J.; Sun, X.; Zhao, L.; Yu, M.; Xu, Z.; et al. Powerful beneficial effects of benfotiamine on cognitive impairment and beta-amyloid deposition in amyloid precursor protein/presenilin-1 transgenic mice. Brain J. Neurol. 2010, 133, 1342–1351. [Google Scholar] [CrossRef] [PubMed]
- Sambon, M.; Gorlova, A.; Demelenne, A.; Alhama-Riba, J.; Coumans, B.; Lakaye, B.; Wins, P.; Fillet, M.; Anthony, D.C.; Strekalova, T.; et al. Dibenzoylthiamine has powerful antioxidant and anti-inflammatory properties in cultured cells and in mouse models of stress and neurodegeneration. Biomedicines 2020, 8, 361. [Google Scholar] [CrossRef]
- Markova, N.; Bazhenova, N.; Anthony, D.C.; Vignisse, J.; Svistunov, A.; Lesch, K.P.; Bettendorff, L.; Strekalova, T. Thiamine and benfotiamine improve cognition and ameliorate GSK-3beta-associated stress-induced behaviours in mice. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2017, 75, 148–156. [Google Scholar] [CrossRef]
- Pavlov, D.; Gorlova, A.; Bettendorff, L.; Kalueff, A.A.; Umriukhin, A.; Proshin, A.; Lysko, A.; Landgraf, R.; Anthony, D.C.; Strekalova, T. Enhanced conditioning of adverse memories in the mouse modified swim test is associated with neuroinflammatory changes—Effects that are susceptible to antidepressants. Neurobiol. Learn. Mem. 2020, 172, 107227. [Google Scholar] [CrossRef]
- Gorlova, A.; Pavlov, D.; Anthony, D.C.; Ponomarev, E.D.; Sambon, M.; Proshin, A.; Shafarevich, I.; Babaevskaya, D.; Lessmall es, C.K.P.; Bettendorff, L.; et al. Thiamine and benfotiamine counteract ultrasound-induced aggression, normalize AMPA receptor expression and plasticity markers, and reduce oxidative stress in mice. Neuropharmacology 2019, 156, 107543. [Google Scholar] [CrossRef]
- Pavlov, D.; Markova, N.; Bettendorff, L.; Chekhonin, V.; Pomytkin, I.; Lioudyno, V.; Svistunov, A.; Ponomarev, E.; Lesch, K.P.; Strekalova, T. Elucidating the functions of brain GSK3alpha: Possible synergy with GSK3beta upregulation and reversal by antidepressant treatment in a mouse model of depressive-like behaviour. Behav. Brain. Res. 2017, 335, 122–127. [Google Scholar] [CrossRef]
- Vignisse, J.; Sambon, M.; Gorlova, A.; Pavlov, D.; Caron, N.; Malgrange, B.; Shevtsova, E.; Svistunov, A.; Anthony, D.C.; Markova, N.; et al. Thiamine and benfotiamine prevent stress-induced suppression of hippocampal neurogenesis in mice exposed to predation without affecting brain thiamine diphosphate levels. Mol. Cell. Neurosci. 2017, 82, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Aleshin, V.A.; Graf, A.V.; Artiukhov, A.V.; Boyko, A.I.; Ksenofontov, A.L.; Maslova, M.V.; Nogues, I.; di Salvo, M.L.; Bunik, V.I. Physiological and biochemical markers of the sex-specific sensitivity to epileptogenic factors, delayed consequences of seizures and their response to vitamins B1 and B6 in a rat model. Pharmaceuticals 2021, 14, 737. [Google Scholar] [CrossRef] [PubMed]
- Costantini, A.; Laureti, T.; Pala, M.I.; Colangeli, M.; Cavalieri, S.; Pozzi, E.; Brusco, A.; Salvarani, S.; Serrati, C.; Fancellu, R. Long-term treatment with thiamine as possible medical therapy for Friedreich ataxia. J. Neurol. 2016, 263, 2170–2178. [Google Scholar] [CrossRef] [PubMed]
- Costantini, A.; Giorgi, R.; D’Agostino, S.; Pala, M.I. High-dose thiamine improves the symptoms of Friedreich’s ataxia. BMJ Case Rep. 2013, 2013, bcr2013009424. [Google Scholar] [CrossRef]
- Song, X.S.; Huang, Z.J.; Song, X.J. Thiamine suppresses thermal hyperalgesia, inhibits hyperexcitability, and lessens alterations of sodium currents in injured, dorsal root ganglion neurons in rats. Anesthesiology 2009, 110, 387–400. [Google Scholar] [CrossRef]
- Botez, M.I.; Botez, T.; Ross-Chouinard, A.; Lalonde, R. Thiamine and folate treatment of chronic epileptic patients: A controlled study with the Wechsler IQ scale. Epilepsy Res. 1993, 16, 157–163. [Google Scholar] [CrossRef]
- Costantini, A.; Nappo, A.; Pala, M.I.; Zappone, A. High dose thiamine improves fatigue in multiple sclerosis. BMJ Case Rep. 2013, 2013, bcr2013009144. [Google Scholar] [CrossRef]
- Leary, S.; Underwood, W.; Anthony, R.; Cartner, S.; Grandin, T.; Greenacre, C.; Gwaltney-Brant, S.; McCrackin, M.A.; Meyer, R.; Miller, D.; et al. AVMA Guidelines for the Euthanasia of Animals, 2020 ed.; American Veterinary Medical Association: Schaumburg, IL, USA, 2020. [Google Scholar]
- Aleshin, V.A.; Mkrtchyan, G.V.; Kaehne, T.; Graf, A.V.; Maslova, M.V.; Bunik, V.I. Diurnal regulation of the function of the rat brain glutamate dehydrogenase by acetylation and its dependence on thiamine administration. J. Neurochem. 2020, 153, 80–102. [Google Scholar] [CrossRef]
- Graff, C.L.; Pollack, G.M. Drug transport at the blood-brain barrier and the choroid plexus. Curr. Drug Metab. 2004, 5, 95–108. [Google Scholar] [CrossRef]
- Badhan, R.K.; Kaur, M.; Lungare, S.; Obuobi, S. Improving brain drug targeting through exploitation of the nose-to-brain route: A physiological and pharmacokinetic perspective. Curr. Drug Deliv. 2014, 11, 458–471. [Google Scholar] [CrossRef]
- Djupesland, P.G.; Messina, J.C.; Mahmoud, R.A. The nasal approach to delivering treatment for brain diseases: An anatomic, physiologic, and delivery technology overview. Ther. Deliv. 2014, 5, 709–733. [Google Scholar] [CrossRef]
- La Monaca, E.; Fodale, V. Effects of anesthetics on mitochondrial signaling and function. Curr. Drug. Saf. 2012, 7, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Brunner, E.A.; Cheng, S.C.; Berman, M.L. Effects of anesthesia on intermediary metabolism. Annu. Rev. Med. 1975, 26, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.J. Effect of anesthetics on mitochondrial function. Anesthesiology 1973, 39, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.P.; Pum, M.E.; Amato, D.; Schuttler, J.; Huston, J.P.; Silva, M.A. The in vivo neurochemistry of the brain during general anesthesia. J. Neurochem. 2011, 119, 419–446. [Google Scholar] [CrossRef] [PubMed]
- Karmarkar, S.W.; Bottum, K.M.; Tischkau, S.A. Considerations for the use of anesthetics in neurotoxicity studies. Comp. Med. 2010, 60, 256–262. [Google Scholar]
- Zhang, Y.M.; Yu, D.X.; Yin, B.S.; Li, X.R.; Li, L.N.; Li, Y.N.; Wang, Y.X.; Chen, Y.; Liu, W.H.; Gao, L. Xylazine regulates the release of glycine and aspartic acid in rat brain. J. Vet. Res. 2018, 62, 121–128. [Google Scholar] [CrossRef]
- Pierozan, P.; Jerneren, F.; Ransome, Y.; Karlsson, O. The choice of euthanasia method affects metabolic serum biomarkers. Basic Clin. Pharmacol. Toxicol. 2017, 121, 113–118. [Google Scholar] [CrossRef]
- Jackson, H.F.; Broadhurst, P.L. The effects of parachlorophenylalanine and stimulus intensity on open-field test measures in rats. Neuropharmacology 1982, 21, 1279–1282. [Google Scholar] [CrossRef]
- Baevsky, R.M.; Chernikova, A.G. Heart rate variability analysis: Physiological foundations and main methods. Cardiometry 2017, 10, 66–76. [Google Scholar] [CrossRef]
- Shaffer, F.; Ginsberg, J.P. An Overview of Heart Rate Variability Metrics and Norms. Front. Public Health 2017, 5, 258. [Google Scholar] [CrossRef] [PubMed]
- Ksenofontov, A.L.; Boyko, A.I.; Mkrtchyan, G.V.; Tashlitsky, V.N.; Timofeeva, A.V.; Graf, A.V.; Bunik, V.I.; Baratova, L.A. Analysis of free amino acids in mammalian brain homogenates. Biochemistry 2017, 82, 1183–1192. [Google Scholar] [PubMed]
- Artiukhov, A.V.; Pometun, A.A.; Zubanova, S.A.; Tishkov, V.I.; Bunik, V.I. Advantages of formate dehydrogenase reaction for efficient NAD(+) quantification in biological samples. Anal. Biochem. 2020, 603, 113797. [Google Scholar] [CrossRef] [PubMed]
- Hissin, P.J.; Hilf, R. A fluorometric method for determination of oxidized and reduced glutathione in tissues. Anal. Biochem. 1976, 74, 214–226. [Google Scholar] [CrossRef]
- Senft, A.P.; Dalton, T.P.; Shertzer, H.G. Determining glutathione and glutathione disulfide using the fluorescence probe o-phthalaldehyde. Anal. Biochem. 2000, 280, 80–86. [Google Scholar] [CrossRef]
- Levintow, L. The glutamyltransferase activity of normal and neoplastic tissues. J. Natl. Cancer Inst. 1954, 15, 347–352. [Google Scholar]
- Hinman, L.M.; Blass, J.P. An NADH-linked spectrophotometric assay for pyruvate dehydrogenase complex in crude tissue homogenates. J. Biol. Chem. 1981, 256, 6583–6586. [Google Scholar] [CrossRef]
- Schwab, M.A.; Kolker, S.; van den Heuvel, L.P.; Sauer, S.; Wolf, N.I.; Rating, D.; Hoffmann, G.F.; Smeitink, J.A.; Okun, J.G. Optimized spectrophotometric assay for the completely activated pyruvate dehydrogenase complex in fibroblasts. Clin. Chem. 2005, 51, 151–160. [Google Scholar] [CrossRef]
- Owens, T.G.; King, F.D. The measurement of respiratory electron-transport-system activity in marine zooplankton. Mar. Biol. 1975, 30, 27–36. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Artiukhov, A.V.; Graf, A.V.; Kazantsev, A.V.; Boyko, A.I.; Aleshin, V.A.; Ksenofontov, A.L.; Bunik, V.I. Increasing Inhibition of the Rat Brain 2-Oxoglutarate Dehydrogenase Decreases Glutathione Redox State, Elevating Anxiety and Perturbing Stress Adaptation. Pharmaceuticals 2022, 15, 182. https://doi.org/10.3390/ph15020182
Artiukhov AV, Graf AV, Kazantsev AV, Boyko AI, Aleshin VA, Ksenofontov AL, Bunik VI. Increasing Inhibition of the Rat Brain 2-Oxoglutarate Dehydrogenase Decreases Glutathione Redox State, Elevating Anxiety and Perturbing Stress Adaptation. Pharmaceuticals. 2022; 15(2):182. https://doi.org/10.3390/ph15020182
Chicago/Turabian StyleArtiukhov, Artem V., Anastasia V. Graf, Alexey V. Kazantsev, Alexandra I. Boyko, Vasily A. Aleshin, Alexander L. Ksenofontov, and Victoria I. Bunik. 2022. "Increasing Inhibition of the Rat Brain 2-Oxoglutarate Dehydrogenase Decreases Glutathione Redox State, Elevating Anxiety and Perturbing Stress Adaptation" Pharmaceuticals 15, no. 2: 182. https://doi.org/10.3390/ph15020182
APA StyleArtiukhov, A. V., Graf, A. V., Kazantsev, A. V., Boyko, A. I., Aleshin, V. A., Ksenofontov, A. L., & Bunik, V. I. (2022). Increasing Inhibition of the Rat Brain 2-Oxoglutarate Dehydrogenase Decreases Glutathione Redox State, Elevating Anxiety and Perturbing Stress Adaptation. Pharmaceuticals, 15(2), 182. https://doi.org/10.3390/ph15020182