
Therapies Targeting Epigenetic Alterations in Acute Kidney Injury-to-Chronic Kidney Disease Transition


Abstract
1. Introduction
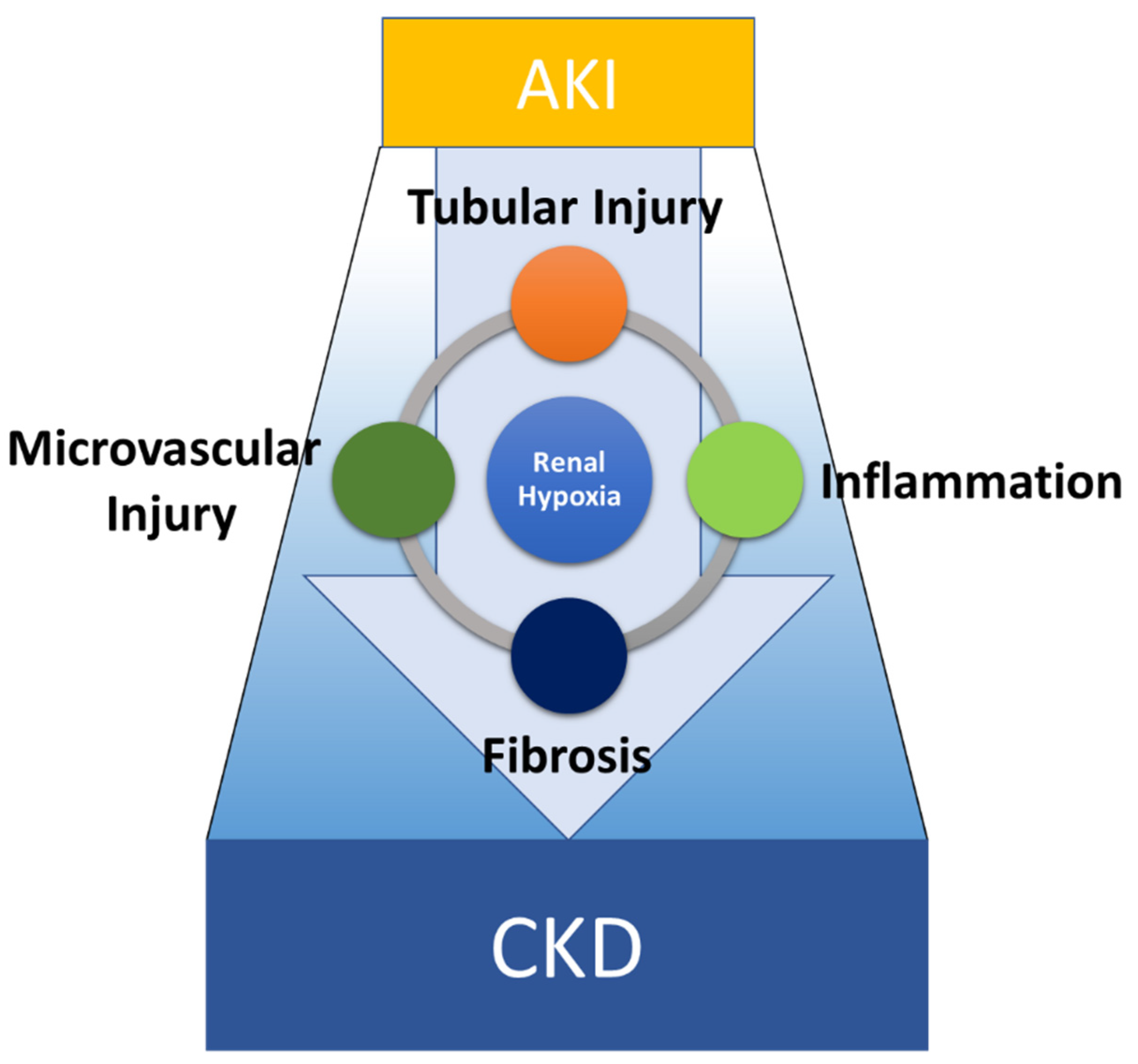
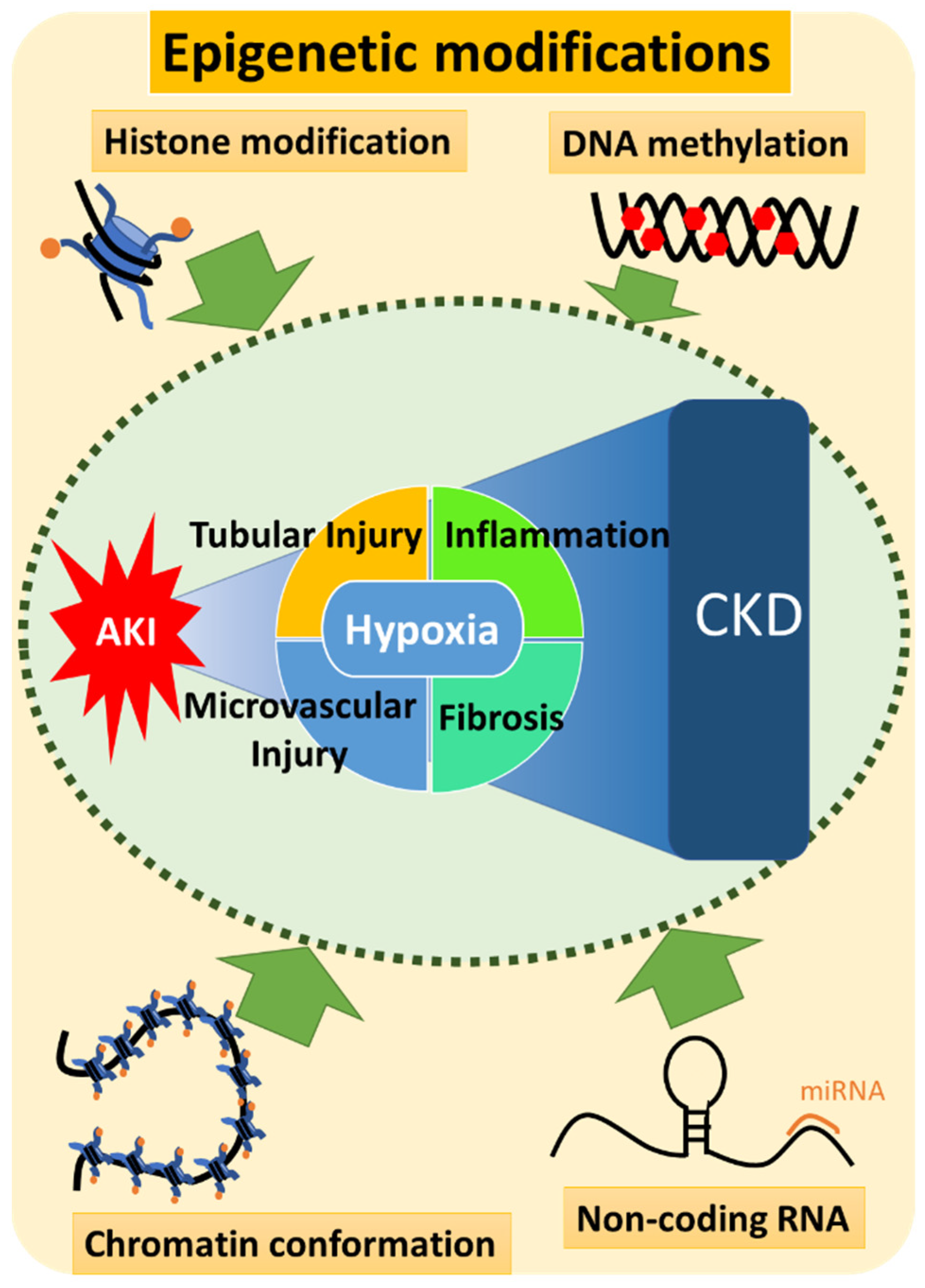
2. Current Overview of the Mechanisms Involved in AKI-to-CKD Transition
2.1. The Pathophysiology of AKI-to-CKD Transition
2.2. TECs Are a Primary Target of AKI and a Driver of Inflammation and Fibrosis
2.3. Molecular Mechanisms of AKI-to-CKD Transition: Insights from Omics Approaches
3. Epigenetics of AKI-to-CKD Transition
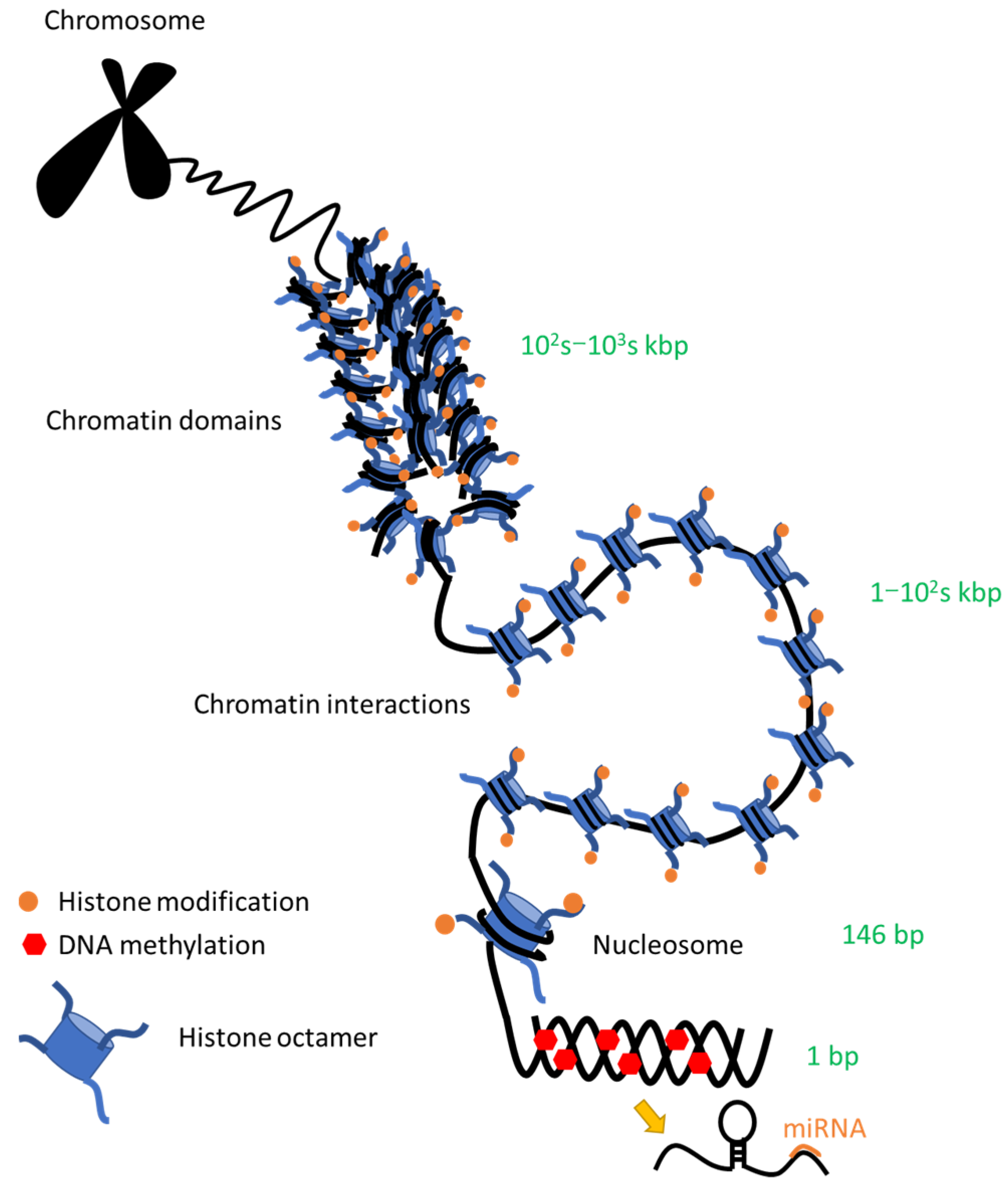
3.1. Epigenetics Overview
3.2. Involvement of Epigenetics in the Pathogenesis of AKI-to-CKD Transition
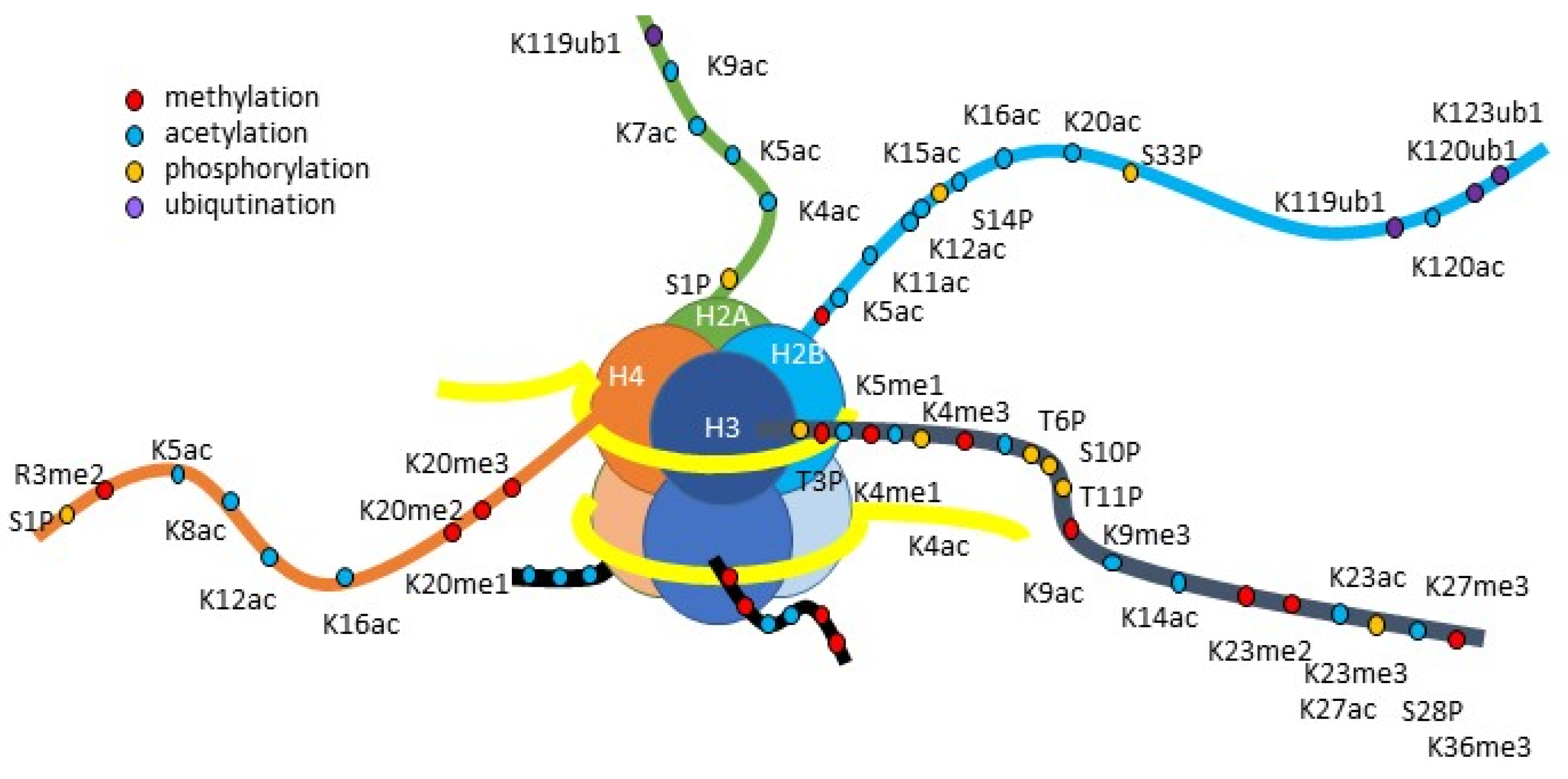
3.3. Histone Modification
3.3.1. Histone Acetylation
- Basic Mechanisms of Histone Acetylation

- 2.
- Histone Acetylation in AKI-to-CKD Transition
- HDAC Inhibitors in AKI-to-CKD Transition
- HDAC Activators in AKI-to-CKD Transition
- HAT Inhibitors in AKI-to-CKD Transition
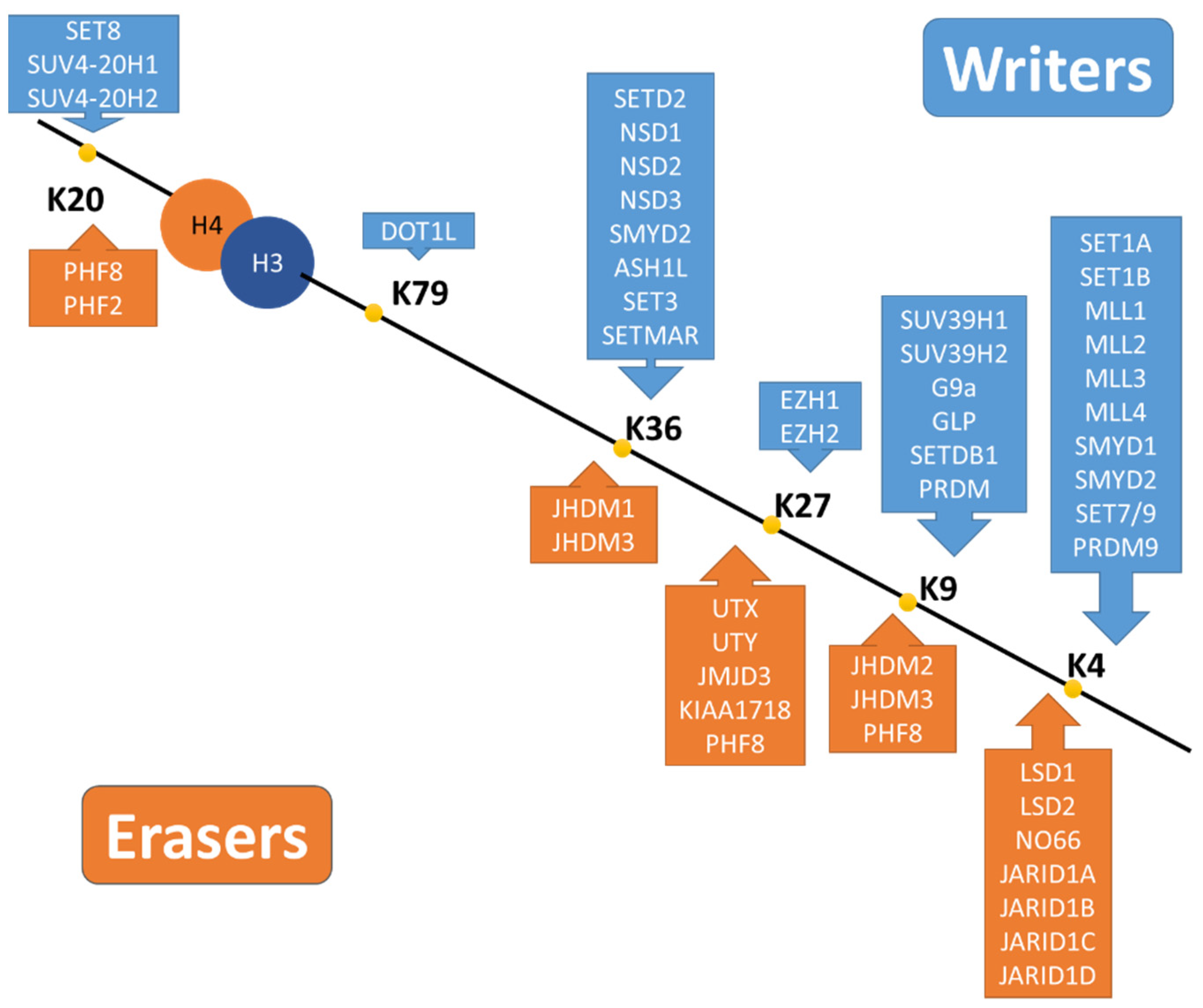
3.3.2. Histone Methylation
- Basic Mechanisms of Histone Methylation
- 2.
- Histone Methylation in AKI-to-CKD Transition
3.3.3. Other Histone Modifications
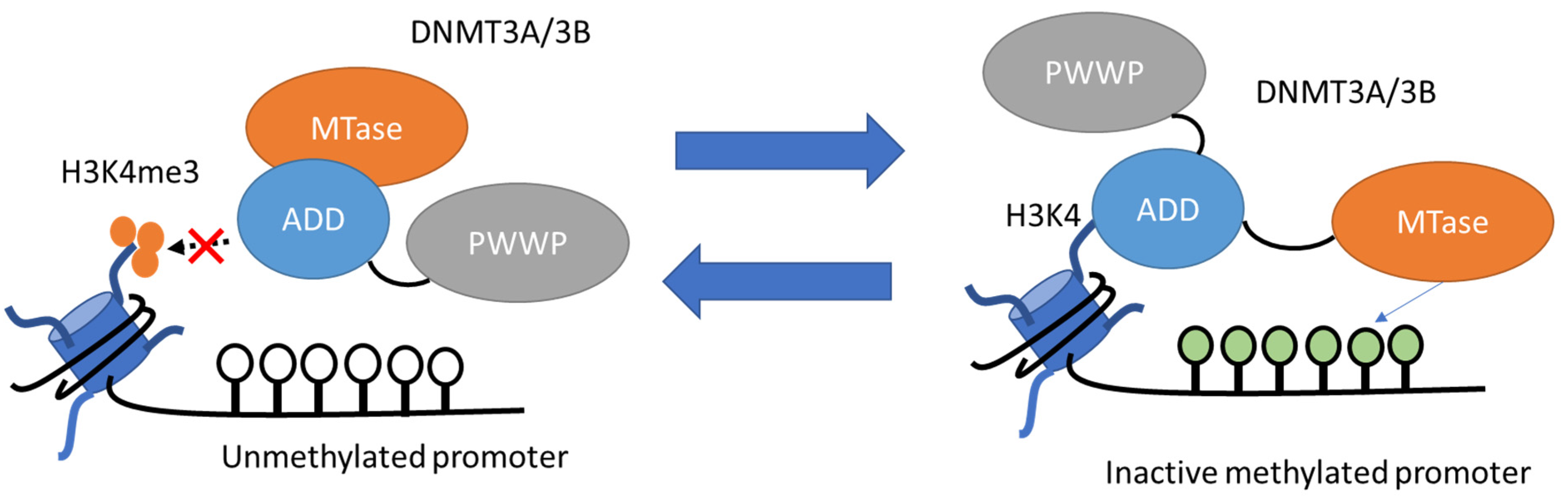
3.4. DNA Methylation
3.4.1. Basic Mechanisms of DNA Methylation
- Writers and Erasers of DNA Methylation
3.4.2. DNA Methylation in AKI-to-CKD Transition
3.5. Non-Coding RNAs
3.5.1. Basic Mechanisms of Non-Coding RNAs
3.5.2. Non-Coding RNAs in AKI-to-CKD Transition
3.6. Chromatin Conformational Changes
3.6.1. High-Order Chromatin Architecture
3.6.2. Chromatin Conformational Changes in AKI-to-CKD Transition
4. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Romagnani, P.; Remuzzi, G.; Glassock, R.; Levin, A.; Jager, K.J.; Tonelli, M.; Massy, Z.; Wanner, C.; Anders, H.J. Chronic kidney disease. Nat. Rev. Dis. Primers 2017, 3, 17088. [Google Scholar] [CrossRef]
- Kuppe, C.; Ibrahim, M.M.; Kranz, J.; Zhang, X.; Ziegler, S.; Perales-Paton, J.; Jansen, J.; Reimer, K.C.; Smith, J.R.; Dobie, R.; et al. Decoding myofibroblast origins in human kidney fibrosis. Nature 2021, 589, 281–286. [Google Scholar] [CrossRef]
- Nangaku, M. Mechanisms of tubulointerstitial injury in the kidney: Final common pathways to end-stage renal failure. Intern. Med. 2004, 43, 9–17. [Google Scholar] [CrossRef]
- Nangaku, M. Chronic hypoxia and tubulointerstitial injury: A final common pathway to end-stage renal failure. J. Am. Soc. Nephrol. 2006, 17, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.I.; Lau, L.F. Resolution of organ fibrosis. J. Clin. Investig. 2018, 128, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Mimura, I.; Nangaku, M. The suffocating kidney: Tubulointerstitial hypoxia in end-stage renal disease. Nat. Rev. Nephrol. 2010, 6, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Kellum, J.A.; Romagnani, P.; Ashuntantang, G.; Ronco, C.; Zarbock, A.; Anders, H.J. Acute kidney injury. Nat. Rev. Dis. Primers 2021, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.L.; Burdmann, E.A.; Cerdá, J.; Feehally, J.; Finkelstein, F.; García-García, G.; Godin, M.; Jha, V.; Lameire, N.H.; Levin, N.W.; et al. Recognition and management of acute kidney injury in the International Society of Nephrology 0by25 Global Snapshot: A multinational cross-sectional study. Lancet 2016, 387, 2017–2025. [Google Scholar] [CrossRef]
- Ullah, M.M.; Basile, D.P. Role of Renal Hypoxia in the Progression from Acute Kidney Injury to Chronic Kidney Disease. Semin. Nephrol. 2019, 39, 567–580. [Google Scholar] [CrossRef]
- Shu, S.; Wang, Y.; Zheng, M.; Liu, Z.; Cai, J.; Tang, C.; Dong, Z. Hypoxia and Hypoxia-Inducible Factors in Kidney Injury and Repair. Cells 2019, 8, 207. [Google Scholar] [CrossRef]
- Loren, P.; Saavedra, N.; Saavedra, K.; Zambrano, T.; Moriel, P.; Salazar, L.A. Epigenetic Mechanisms Involved in Cisplatin-Induced Nephrotoxicity: An Update. Pharmaceuticals 2021, 14, 491. [Google Scholar] [CrossRef]
- Nangaku, M.; Hirakawa, Y.; Mimura, I.; Inagi, R.; Tanaka, T. Epigenetic Changes in the Acute Kidney Injury-to-Chronic Kidney Disease Transition. Nephron 2017, 137, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Tanaka, T.; Nangaku, M. Hypoxia as a key player in the AKI-to-CKD transition. Am. J. Physiol. Ren. Physiol. 2014, 307, F1187–F1195. [Google Scholar] [CrossRef]
- Coca, S.G.; Singanamala, S.; Parikh, C.R. Chronic kidney disease after acute kidney injury: A systematic review and meta-analysis. Kidney Int. 2012, 81, 442–448. [Google Scholar] [CrossRef]
- See, E.J.; Jayasinghe, K.; Glassford, N.; Bailey, M.; Johnson, D.W.; Polkinghorne, K.R.; Toussaint, N.D.; Bellomo, R. Long-term risk of adverse outcomes after acute kidney injury: A systematic review and meta-analysis of cohort studies using consensus definitions of exposure. Kidney Int. 2019, 95, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.P.; Bonventre, J.V.; Mehta, R.; Nangaku, M.; Unwin, R.; Rosner, M.H.; Kellum, J.A.; Ronco, C.; Group, A.X.W. Progression after AKI: Understanding Maladaptive Repair Processes to Predict and Identify Therapeutic Treatments. J. Am. Soc. Nephrol. 2016, 27, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Guzzi, F.; Cirillo, L.; Roperto, R.M.; Romagnani, P.; Lazzeri, E. Molecular Mechanisms of the Acute Kidney Injury to Chronic Kidney Disease Transition: An Updated View. Int. J. Mol. Sci. 2019, 20, 4941. [Google Scholar] [CrossRef]
- Morgado-Pascual, J.L.; Marchant, V.; Rodrigues-Diez, R.; Dolade, N.; Suarez-Alvarez, B.; Kerr, B.; Valdivielso, J.M.; Ruiz-Ortega, M.; Rayego-Mateos, S. Epigenetic Modification Mechanisms Involved in Inflammation and Fibrosis in Renal Pathology. Mediat. Inflamm. 2018, 2018, 2931049. [Google Scholar] [CrossRef]
- Chevalier, R.L. The proximal tubule is the primary target of injury and progression of kidney disease: Role of the glomerulotubular junction. Am. J. Physiol. Ren. Physiol. 2016, 311, F145–F161. [Google Scholar] [CrossRef]
- Sato, Y.; Takahashi, M.; Yanagita, M. Pathophysiology of AKI to CKD progression. Semin. Nephrol. 2020, 40, 206–215. [Google Scholar] [CrossRef]
- Kumar, S. Cellular and molecular pathways of renal repair after acute kidney injury. Kidney Int. 2018, 93, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Anders, H.J. Immune system modulation of kidney regeneration—Mechanisms and implications. Nat. Rev. Nephrol. 2014, 10, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From mechanisms to medicines. Nature 2020, 587, 555–566. [Google Scholar] [CrossRef]
- Kramann, R.; Wongboonsin, J.; Chang-Panesso, M.; Machado, F.G.; Humphreys, B.D. Gli1+ Pericyte Loss Induces Capillary Rarefaction and Proximal Tubular Injury. J. Am. Soc. Nephrol. 2017, 28, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Sharfuddin, A.A.; Molitoris, B.A. Pathophysiology of ischemic acute kidney injury. Nat. Rev. Nephrol. 2011, 7, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Stockwell, B.R.; Krautwald, S.; Anders, H.J. Regulated cell death and inflammation: An auto-amplification loop causes organ failure. Nat. Rev. Immunol. 2014, 14, 759–767. [Google Scholar] [CrossRef]
- Mulay, S.R.; Linkermann, A.; Anders, H.J. Necroinflammation in Kidney Disease. J. Am. Soc. Nephrol. 2016, 27, 27–39. [Google Scholar] [CrossRef]
- Lazzeri, E.; Angelotti, M.L.; Conte, C.; Anders, H.J.; Romagnani, P. Surviving Acute Organ Failure: Cell Polyploidization and Progenitor Proliferation. Trends Mol. Med. 2019, 25, 366–381. [Google Scholar] [CrossRef]
- Lombardi, D.; Becherucci, F.; Romagnani, P. How much can the tubule regenerate and who does it? An open question. Nephrol. Dial. Transplant. 2016, 31, 1243–1250. [Google Scholar] [CrossRef]
- Kumar, S.; Liu, J.; Pang, P.; Krautzberger, A.M.; Reginensi, A.; Akiyama, H.; Schedl, A.; Humphreys, B.D.; McMahon, A.P. Sox9 Activation Highlights a Cellular Pathway of Renal Repair in the Acutely Injured Mammalian Kidney. Cell Rep. 2015, 12, 1325–1338. [Google Scholar] [CrossRef]
- Lazzeri, E.; Angelotti, M.L.; Peired, A.; Conte, C.; Marschner, J.A.; Maggi, L.; Mazzinghi, B.; Lombardi, D.; Melica, M.E.; Nardi, S.; et al. Endocycle-related tubular cell hypertrophy and progenitor proliferation recover renal function after acute kidney injury. Nat. Commun. 2018, 9, 1344. [Google Scholar] [CrossRef]
- Shu, Z.; Row, S.; Deng, W.M. Endoreplication: The Good, the Bad, and the Ugly. Trends Cell Biol. 2018, 28, 465–474. [Google Scholar] [CrossRef]
- Liu, B.C.; Tang, T.T.; Lv, L.L.; Lan, H.Y. Renal tubule injury: A driving force toward chronic kidney disease. Kidney Int. 2018, 93, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Canaud, G.; Bonventre, J.V. Cell cycle arrest and the evolution of chronic kidney disease from acute kidney injury. Nephrol. Dial. Transplant. 2015, 30, 575–583. [Google Scholar] [CrossRef]
- Liu, J.; Kumar, S.; Dolzhenko, E.; Alvarado, G.F.; Guo, J.; Lu, C.; Chen, Y.; Li, M.; Dessing, M.C.; Parvez, R.K.; et al. Molecular characterization of the transition from acute to chronic kidney injury following ischemia/reperfusion. JCI Insight 2017, 2, e94716. [Google Scholar] [CrossRef] [PubMed]
- Sharifian, R.; Okamura, D.M.; Denisenko, O.; Zager, R.A.; Johnson, A.; Gharib, S.A.; Bomsztyk, K. Distinct patterns of transcriptional and epigenetic alterations characterize acute and chronic kidney injury. Sci. Rep. 2018, 8, 17870. [Google Scholar] [CrossRef] [PubMed]
- Cippa, P.E.; Sun, B.; Liu, J.; Chen, L.; Naesens, M.; McMahon, A.P. Transcriptional trajectories of human kidney injury progression. JCI Insight 2018, 3, e123151. [Google Scholar] [CrossRef] [PubMed]
- Kirita, Y.; Wu, H.; Uchimura, K.; Wilson, P.C.; Humphreys, B.D. Cell profiling of mouse acute kidney injury reveals conserved cellular responses to injury. Proc. Natl. Acad. Sci. USA 2020, 117, 15874–15883. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, L.M.S.; Liu, J.; Koppitch, K.; Cippa, P.E.; McMahon, A.P. Single-nuclear transcriptomics reveals diversity of proximal tubule cell states in a dynamic response to acute kidney injury. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Cao, J.; Cusanovich, D.A.; Ramani, V.; Aghamirzaie, D.; Pliner, H.A.; Hill, A.J.; Daza, R.M.; McFaline-Figueroa, J.L.; Packer, J.S.; Christiansen, L.; et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 2018, 361, 1380–1385. [Google Scholar] [CrossRef]
- Muto, Y.; Wilson, P.C.; Ledru, N.; Wu, H.; Dimke, H.; Waikar, S.S.; Humphreys, B.D. Single cell transcriptional and chromatin accessibility profiling redefine cellular heterogeneity in the adult human kidney. Nat. Commun. 2021, 12, 2190. [Google Scholar] [CrossRef] [PubMed]
- Govender, M.A.; Brandenburg, J.T.; Fabian, J.; Ramsay, M. The Use of Omics for Diagnosing and Predicting Progression of Chronic Kidney Disease: A Scoping Review. Front. Genet. 2021, 12, 682929. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, Y.; Wang, X.; Huang, H.; Bao, J.; Zhong, W.; Li, A. PTEN alleviates maladaptive repair of renal tubular epithelial cells by restoring CHMP2A-mediated phagosome closure. Cell Death Dis. 2021, 12, 1087. [Google Scholar] [CrossRef] [PubMed]
- Tran, M.T.; Zsengeller, Z.K.; Berg, A.H.; Khankin, E.V.; Bhasin, M.K.; Kim, W.; Clish, C.B.; Stillman, I.E.; Karumanchi, S.A.; Rhee, E.P.; et al. PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 2016, 531, 528–532. [Google Scholar] [CrossRef]
- Dubin, R.F.; Rhee, E.P. Proteomics and Metabolomics in Kidney Disease, including Insights into Etiology, Treatment, and Prevention. Clin. J. Am. Soc. Nephrol. 2020, 15, 404–411. [Google Scholar] [CrossRef]
- Li, Z.; Lu, S.; Li, X. The role of metabolic reprogramming in tubular epithelial cells during the progression of acute kidney injury. Cell. Mol. Life Sci. 2021, 78, 5731–5741. [Google Scholar] [CrossRef]
- Morigi, M.; Perico, L.; Benigni, A. Sirtuins in Renal Health and Disease. J. Am. Soc. Nephrol. 2018, 29, 1799–1809. [Google Scholar] [CrossRef]
- Wang, Q.; Xu, J.; Li, X.; Liu, Z.; Han, Y.; Xu, X.; Li, X.; Tang, Y.; Liu, Y.; Yu, T.; et al. Sirt3 modulate renal ischemia-reperfusion injury through enhancing mitochondrial fusion and activating the ERK-OPA1 signaling pathway. J. Cell. Physiol. 2019, 234, 23495–23506. [Google Scholar] [CrossRef]
- Li, W.; Sun, Z. Mechanism of Action for HDAC Inhibitors-Insights from Omics Approaches. Int. J. Mol. Sci. 2019, 20, 1616. [Google Scholar] [CrossRef]
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004, 429, 457–463. [Google Scholar] [CrossRef]
- Guo, C.; Dong, G.; Liang, X.; Dong, Z. Epigenetic regulation in AKI and kidney repair: Mechanisms and therapeutic implications. Nat. Rev. Nephrol. 2019, 15, 220–239. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Zhang, L.; Yang, Q.; Zhang, X.; Li, X. Epigenetics in kidney diseases. Adv. Clin. Chem. 2021, 104, 233–297. [Google Scholar] [CrossRef] [PubMed]
- Mimura, I.; Nangaku, M.; Kanki, Y.; Tsutsumi, S.; Inoue, T.; Kohro, T.; Yamamoto, S.; Fujita, T.; Shimamura, T.; Suehiro, J.; et al. Dynamic change of chromatin conformation in response to hypoxia enhances the expression of GLUT3 (SLC2A3) by cooperative interaction of hypoxia-inducible factor 1 and KDM3A. Mol. Cell. Biol. 2012, 32, 3018–3032. [Google Scholar] [CrossRef]
- Wanner, N.; Bechtel-Walz, W. Epigenetics of kidney disease. Cell Tissue Res. 2017, 369, 75–92. [Google Scholar] [CrossRef] [PubMed]
- Prachayasittikul, V.; Prathipati, P.; Pratiwi, R.; Phanus-Umporn, C.; Malik, A.A.; Schaduangrat, N.; Seenprachawong, K.; Wongchitrat, P.; Supokawej, A.; Prachayasittikul, V.; et al. Exploring the epigenetic drug discovery landscape. Expert Opin. Drug Discov. 2017, 12, 345–362. [Google Scholar] [CrossRef]
- Wilson, P.C.; Ledru, N.; Humphreys, B.D. Epigenomics and the kidney. Curr. Opin. Nephrol. Hypertens. 2020, 29, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Tampe, B.; Zeisberg, M. Chromatin dynamics at the core of kidney fibrosis. Matrix Biol. 2018, 68–69, 194–229. [Google Scholar] [CrossRef]
- Yamanaka, S.; Nishihara, H.; Toh, H.; Eijy Nagai, L.A.; Hashimoto, K.; Park, S.J.; Shibuya, A.; Suzuki, A.M.; Tanaka, Y.; Nakai, K.; et al. Broad Heterochromatic Domains Open in Gonocyte Development Prior to De Novo DNA Methylation. Dev. Cell 2019, 51, 21–34.e5. [Google Scholar] [CrossRef]
- Stricker, S.H.; Koferle, A.; Beck, S. From profiles to function in epigenomics. Nat. Rev. Genet. 2017, 18, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T. Epigenetic changes mediating transition to chronic kidney disease: Hypoxic memory. Acta Physiol. 2018, 222, e13023. [Google Scholar] [CrossRef] [PubMed]
- Mimura, I.; Tanaka, T.; Nangaku, M. Novel therapeutic strategy with hypoxia-inducible factors via reversible epigenetic regulation mechanisms in progressive tubulointerstitial fibrosis. Semin. Nephrol. 2013, 33, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Mimura, I.; Tanaka, T.; Wada, Y.; Kodama, T.; Nangaku, M. Pathophysiological response to hypoxia—From the molecular mechanisms of malady to drug discovery: Epigenetic regulation of the hypoxic response via hypoxia-inducible factor and histone modifying enzymes. J. Pharmacol. Sci. 2011, 115, 453–458. [Google Scholar] [CrossRef]
- Mimura, I.; Tanaka, T.; Nangaku, M. New insights into molecular mechanisms of epigenetic regulation in kidney disease. Clin. Exp. Pharmacol. Physiol. 2016, 43, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Mimura, I.; Hirakawa, Y.; Kanki, Y.; Kushida, N.; Nakaki, R.; Suzuki, Y.; Tanaka, T.; Aburatani, H.; Nangaku, M. Novel lnc RNA regulated by HIF-1 inhibits apoptotic cell death in the renal tubular epithelial cells under hypoxia. Physiol. Rep. 2017, 5, e13203. [Google Scholar] [CrossRef] [PubMed]
- Mimura, I.; Kanki, Y.; Kodama, T.; Nangaku, M. Revolution of nephrology research by deep sequencing: ChIP-seq and RNA-seq. Kidney Int. 2014, 85, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Shahbazian, M.D.; Grunstein, M. Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 2007, 76, 75–100. [Google Scholar] [CrossRef]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef]
- Hodawadekar, S.C.; Marmorstein, R. Chemistry of acetyl transfer by histone modifying enzymes: Structure, mechanism and implications for effector design. Oncogene 2007, 26, 5528–5540. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Knapp, S. Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356. [Google Scholar] [CrossRef] [PubMed]
- Belkina, A.C.; Denis, G.V. BET domain co-regulators in obesity, inflammation and cancer. Nat. Rev. Cancer 2012, 12, 465–477. [Google Scholar] [CrossRef]
- Fontecha-Barriuso, M.; Martin-Sanchez, D.; Ruiz-Andres, O.; Poveda, J.; Sanchez-Nino, M.D.; Valino-Rivas, L.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. Targeting epigenetic DNA and histone modifications to treat kidney disease. Nephrol. Dial. Transplant. 2018, 33, 1875–1886. [Google Scholar] [CrossRef]
- Fu, L.L.; Tian, M.; Li, X.; Li, J.J.; Huang, J.; Ouyang, L.; Zhang, Y.; Liu, B. Inhibition of BET bromodomains as a therapeutic strategy for cancer drug discovery. Oncotarget 2015, 6, 5501–5516. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Marumo, T.; Hishikawa, K.; Yoshikawa, M.; Fujita, T. Epigenetic regulation of BMP7 in the regenerative response to ischemia. J. Am. Soc. Nephrol. 2008, 19, 1311–1320. [Google Scholar] [CrossRef]
- Ruiz-Andres, O.; Suarez-Alvarez, B.; Sánchez-Ramos, C.; Monsalve, M.; Sanchez-Niño, M.D.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A.; Sanz, A.B. The inflammatory cytokine TWEAK decreases PGC-1α expression and mitochondrial function in acute kidney injury. Kidney Int. 2016, 89, 399–410. [Google Scholar] [CrossRef]
- Moreno, J.A.; Izquierdo, M.C.; Sanchez-Niño, M.D.; Suárez-Alvarez, B.; Lopez-Larrea, C.; Jakubowski, A.; Blanco, J.; Ramirez, R.; Selgas, R.; Ruiz-Ortega, M.; et al. The inflammatory cytokines TWEAK and TNFα reduce renal klotho expression through NFκB. J. Am. Soc. Nephrol. 2011, 22, 1315–1325. [Google Scholar] [CrossRef]
- Zager, R.A.; Johnson, A.C.; Becker, K. Acute unilateral ischemic renal injury induces progressive renal inflammation, lipid accumulation, histone modification, and “end-stage” kidney disease. Am. J. Physiol. Ren. Physiol. 2011, 301, F1334–F1345. [Google Scholar] [CrossRef] [PubMed]
- Hewitson, T.D.; Holt, S.G.; Tan, S.J.; Wigg, B.; Samuel, C.S.; Smith, E.R. Epigenetic Modifications to H3K9 in Renal Tubulointerstitial Cells after Unilateral Ureteric Obstruction and TGF-β1 Stimulation. Front. Pharmacol. 2017, 8, 307. [Google Scholar] [CrossRef]
- Huang, J.; Wan, D.; Li, J.; Chen, H.; Huang, K.; Zheng, L. Histone acetyltransferase PCAF regulates inflammatory molecules in the development of renal injury. Epigenetics 2015, 10, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Mar, D.; Gharib, S.A.; Zager, R.A.; Johnson, A.; Denisenko, O.; Bomsztyk, K. Heterogeneity of epigenetic changes at ischemia/reperfusion- and endotoxin-induced acute kidney injury genes. Kidney Int. 2015, 88, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Chen, H.; Shi, Y.; Ma, X.; Zhuang, S.; Liu, N. The Role and Mechanism of Histone Deacetylases in Acute Kidney Injury. Front. Pharmacol. 2021, 12, 695237. [Google Scholar] [CrossRef]
- Kim, J.I.; Jung, K.J.; Jang, H.S.; Park, K.M. Gender-specific role of HDAC11 in kidney ischemia- and reperfusion-induced PAI-1 expression and injury. Am. J. Physiol. Ren. Physiol. 2013, 305, F61–F70. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zhuang, S. Treatment of chronic kidney diseases with histone deacetylase inhibitors. Front. Physiol. 2015, 6, 121. [Google Scholar] [CrossRef] [PubMed]
- Hyndman, K.A.; Speed, J.S.; Mendoza, L.D.; Allan, J.M.; Colson, J.; Sedaka, R.; Jin, C.; Jung, H.J.; El-Dahr, S.; Pollock, D.M.; et al. Fluid-electrolyte homeostasis requires histone deacetylase function. JCI Insight 2020, 5, e137792. [Google Scholar] [CrossRef]
- Do Amaral, C.L.; Francescato, H.D.; Coimbra, T.M.; Costa, R.S.; Darin, J.D.; Antunes, L.M.; Bianchi Mde, L. Resveratrol attenuates cisplatin-induced nephrotoxicity in rats. Arch. Toxicol. 2008, 82, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Gao, Y.; Zhang, Q.; Wei, S.; Chen, Z.; Dai, X.; Zeng, Z.; Zhao, K.S. SIRT1/3 Activation by Resveratrol Attenuates Acute Kidney Injury in a Septic Rat Model. Oxid. Med. Cell. Longev. 2016, 2016, 7296092. [Google Scholar] [CrossRef]
- Fan, H.; Yang, H.C.; You, L.; Wang, Y.Y.; He, W.J.; Hao, C.M. The histone deacetylase, SIRT1, contributes to the resistance of young mice to ischemia/reperfusion-induced acute kidney injury. Kidney Int. 2013, 83, 404–413. [Google Scholar] [CrossRef]
- Bayrak, O.; Uz, E.; Bayrak, R.; Turgut, F.; Atmaca, A.F.; Sahin, S.; Yildirim, M.E.; Kaya, A.; Cimentepe, E.; Akcay, A. Curcumin protects against ischemia/reperfusion injury in rat kidneys. World J. Urol. 2008, 26, 285–291. [Google Scholar] [CrossRef]
- Kasi, P.D.; Tamilselvam, R.; Skalicka-Woźniak, K.; Nabavi, S.F.; Daglia, M.; Bishayee, A.; Pazoki-Toroudi, H.; Nabavi, S.M. Molecular targets of curcumin for cancer therapy: An updated review. Tumour Biol. 2016, 37, 13017–13028. [Google Scholar] [CrossRef]
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Mol. Cell 2012, 48, 491–507. [Google Scholar] [CrossRef]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef] [PubMed]
- Rea, S.; Eisenhaber, F.; O’Carroll, D.; Strahl, B.D.; Sun, Z.W.; Schmid, M.; Opravil, S.; Mechtler, K.; Ponting, C.P.; Allis, C.D.; et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 2000, 406, 593–599. [Google Scholar] [CrossRef]
- Dambacher, S.; Hahn, M.; Schotta, G. Epigenetic regulation of development by histone lysine methylation. Heredity 2010, 105, 24–37. [Google Scholar] [CrossRef]
- Zhou, X.; Zang, X.; Ponnusamy, M.; Masucci, M.V.; Tolbert, E.; Gong, R.; Zhao, T.C.; Liu, N.; Bayliss, G.; Dworkin, L.D.; et al. Enhancer of Zeste Homolog 2 Inhibition Attenuates Renal Fibrosis by Maintaining Smad7 and Phosphatase and Tensin Homolog Expression. J. Am. Soc. Nephrol. 2016, 27, 2092–2108. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, N. Epigenetic Modification Drives Acute Kidney Injury-to-Chronic Kidney Disease Progression. Nephron 2021, 145, 737–747. [Google Scholar] [CrossRef]
- Naito, M.; Bomsztyk, K.; Zager, R.A. Endotoxin mediates recruitment of RNA polymerase II to target genes in acute renal failure. J. Am. Soc. Nephrol. 2008, 19, 1321–1330. [Google Scholar] [CrossRef]
- Zager, R.A.; Johnson, A.C. Renal ischemia-reperfusion injury upregulates histone-modifying enzyme systems and alters histone expression at proinflammatory/profibrotic genes. Am. J. Physiol. Ren. Physiol. 2009, 296, F1032–F1041. [Google Scholar] [CrossRef]
- Naito, M.; Bomsztyk, K.; Zager, R.A. Renal ischemia-induced cholesterol loading: Transcription factor recruitment and chromatin remodeling along the HMG CoA reductase gene. Am. J. Pathol. 2009, 174, 54–62. [Google Scholar] [CrossRef]
- Johnson, A.C.; Ware, L.B.; Himmelfarb, J.; Zager, R.A. HMG-CoA reductase activation and urinary pellet cholesterol elevations in acute kidney injury. Clin. J. Am. Soc. Nephrol. 2011, 6, 2108–2113. [Google Scholar] [CrossRef]
- Mimura, I.; Hirakawa, Y.; Kanki, Y.; Nakaki, R.; Suzuki, Y.; Tanaka, T.; Aburatani, H.; Nangaku, M. Genome-wide analysis revealed that DZNep reduces tubulointerstitial fibrosis via down-regulation of pro-fibrotic genes. Sci. Rep. 2018, 8, 3779. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Doi, S.; Nakashima, A.; Irifuku, T.; Yamada, K.; Kokoroishi, K.; Ueno, T.; Doi, T.; Hida, E.; Arihiro, K.; et al. Inhibition of SET Domain-Containing Lysine Methyltransferase 7/9 Ameliorates Renal Fibrosis. J. Am. Soc. Nephrol. 2016, 27, 203–215. [Google Scholar] [CrossRef]
- Irifuku, T.; Doi, S.; Sasaki, K.; Doi, T.; Nakashima, A.; Ueno, T.; Yamada, K.; Arihiro, K.; Kohno, N.; Masaki, T. Inhibition of H3K9 histone methyltransferase G9a attenuates renal fibrosis and retains klotho expression. Kidney Int. 2016, 89, 147–157. [Google Scholar] [CrossRef]
- Xu, H.; Wu, M.; Ma, X.; Huang, W.; Xu, Y. Function and Mechanism of Novel Histone Posttranslational Modifications in Health and Disease. Biomed. Res. Int. 2021, 2021, 6635225. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Luo, H.; Lee, S.; Jin, F.; Yang, J.S.; Montellier, E.; Buchou, T.; Cheng, Z.; Rousseaux, S.; Rajagopal, N.; et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 2011, 146, 1016–1028. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Andres, O.; Sanchez-Niño, M.D.; Cannata-Ortiz, P.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A.; Sanz, A.B. Histone lysine crotonylation during acute kidney injury in mice. Dis. Model. Mech. 2016, 9, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Liu, H.; Chu, J.; Zhang, H. Functions and mechanisms of lysine crotonylation. J. Cell. Mol. Med. 2019, 23, 7163–7169. [Google Scholar] [CrossRef] [PubMed]
- Hellman, A.; Chess, A. Gene body-specific methylation on the active X chromosome. Science 2007, 315, 1141–1143. [Google Scholar] [CrossRef]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef]
- Robertson, K.D. DNA methylation, methyltransferases, and cancer. Oncogene 2001, 20, 3139–3155. [Google Scholar] [CrossRef]
- Guo, X.; Wang, L.; Li, J.; Ding, Z.; Xiao, J.; Yin, X.; He, S.; Shi, P.; Dong, L.; Li, G.; et al. Structural insight into autoinhibition and histone H3-induced activation of DNMT3A. Nature 2015, 517, 640–644. [Google Scholar] [CrossRef]
- Piunti, A.; Shilatifard, A. Epigenetic balance of gene expression by Polycomb and COMPASS families. Science 2016, 352, aad9780. [Google Scholar] [CrossRef]
- Ishiyama, S.; Nishiyama, A.; Saeki, Y.; Moritsugu, K.; Morimoto, D.; Yamaguchi, L.; Arai, N.; Matsumura, R.; Kawakami, T.; Mishima, Y.; et al. Structure of the Dnmt1 Reader Module Complexed with a Unique Two-Mono-Ubiquitin Mark on Histone H3 Reveals the Basis for DNA Methylation Maintenance. Mol. Cell 2017, 68, 350–360.e357. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Tan, L.; Xue, Z.; Cang, J.; Wang, H. Reduction of DNA hydroxymethylation in the mouse kidney insulted by ischemia reperfusion. Biochem. Biophys. Res. Commun. 2012, 422, 697–702. [Google Scholar] [CrossRef]
- Zhao, Y.; Ding, C.; Xue, W.; Ding, X.; Zheng, J.; Gao, Y.; Xia, X.; Li, S.; Liu, J.; Han, F.; et al. Genome-wide DNA methylation analysis in renal ischemia reperfusion injury. Gene 2017, 610, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.T.; Yang, C.C.; Pan, S.Y.; Chou, Y.H.; Chang, F.C.; Lai, C.F.; Tsai, M.H.; Hsu, H.L.; Lin, C.H.; Chiang, W.C.; et al. DNA methyltransferase inhibition restores erythropoietin production in fibrotic murine kidneys. J. Clin. Investig. 2016, 126, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liang, Y.; Lei, L.; Zhu, G.; Chen, X.; Jin, T.; Wu, Q. Hypermethylations of RASAL1 and KLOTHO is associated with renal dysfunction in a Chinese population environmentally exposed to cadmium. Toxicol. Appl. Pharmacol. 2013, 271, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Tan, X.; Tampe, B.; Wilhelmi, T.; Hulshoff, M.S.; Saito, S.; Moser, T.; Kalluri, R.; Hasenfuss, G.; Zeisberg, E.M.; et al. High-fidelity CRISPR/Cas9- based gene-specific hydroxymethylation rescues gene expression and attenuates renal fibrosis. Nat. Commun. 2018, 9, 3509. [Google Scholar] [CrossRef]
- Tampe, B.; Steinle, U.; Tampe, D.; Carstens, J.L.; Korsten, P.; Zeisberg, E.M.; Muller, G.A.; Kalluri, R.; Zeisberg, M. Low-dose hydralazine prevents fibrosis in a murine model of acute kidney injury-to-chronic kidney disease progression. Kidney Int. 2017, 91, 157–176. [Google Scholar] [CrossRef]
- Bechtel, W.; McGoohan, S.; Zeisberg, E.M.; Müller, G.A.; Kalbacher, H.; Salant, D.J.; Müller, C.A.; Kalluri, R.; Zeisberg, M. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat. Med. 2010, 16, 544–550. [Google Scholar] [CrossRef]
- Kandler, M.R.; Mah, G.T.; Tejani, A.M.; Stabler, S.N.; Salzwedel, D.M. Hydralazine for essential hypertension. Cochrane Database Syst. Rev. 2011, Cd004934. [Google Scholar] [CrossRef]
- Larkin, B.P.; Glastras, S.J.; Chen, H.; Pollock, C.A.; Saad, S. DNA methylation and the potential role of demethylating agents in prevention of progressive chronic kidney disease. FASEB J. 2018, 32, 5215–5226. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.H.; Wang, Z.; Bhatti, T.R.; Wang, Y.; Aufhauser, D.D.; McNeal, S.; Liu, Y.; Cheraghlou, S.; Han, R.; Wang, L.; et al. Class-specific histone/protein deacetylase inhibition protects against renal ischemia reperfusion injury and fibrosis formation. Am. J. Transplant. 2015, 15, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Manson, S.R.; Song, J.B.; Hruska, K.A.; Austin, P.F. HDAC dependent transcriptional repression of Bmp-7 potentiates TGF-β mediated renal fibrosis in obstructive uropathy. J. Urol. 2014, 191, 242–252. [Google Scholar] [CrossRef]
- Novitskaya, T.; McDermott, L.; Zhang, K.X.; Chiba, T.; Paueksakon, P.; Hukriede, N.A.; de Caestecker, M.P. A PTBA small molecule enhances recovery and reduces postinjury fibrosis after aristolochic acid-induced kidney injury. Am. J. Physiol. Ren. Physiol. 2014, 306, F496–F504. [Google Scholar] [CrossRef] [PubMed]
- Skrypnyk, N.I.; Sanker, S.; Skvarca, L.B.; Novitskaya, T.; Woods, C.; Chiba, T.; Patel, K.; Goldberg, N.D.; McDermott, L.; Vinson, P.N.; et al. Delayed treatment with PTBA analogs reduces postinjury renal fibrosis after kidney injury. Am. J. Physiol. Ren. Physiol. 2016, 310, F705–F716. [Google Scholar] [CrossRef]
- Kinugasa, F.; Noto, T.; Matsuoka, H.; Urano, Y.; Sudo, Y.; Takakura, S.; Mutoh, S. Prevention of renal interstitial fibrosis via histone deacetylase inhibition in rats with unilateral ureteral obstruction. Transpl. Immunol. 2010, 23, 18–23. [Google Scholar] [CrossRef]
- Costalonga, E.C.; Silva, F.M.; Noronha, I.L. Valproic Acid Prevents Renal Dysfunction and Inflammation in the Ischemia-Reperfusion Injury Model. Biomed. Res. Int. 2016, 2016, 5985903. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, W.; Jiao, F.; Li, X.; Zhang, H.; Wang, L.; Gong, Z. The Nephroprotective Effect of MS-275 on Lipopolysaccharide (LPS)-Induced Acute Kidney Injury by Inhibiting Reactive Oxygen Species (ROS)-Oxidative Stress and Endoplasmic Reticulum Stress. Med. Sci. Monit. 2018, 24, 2620–2630. [Google Scholar] [CrossRef]
- Liu, N.; He, S.; Ma, L.; Ponnusamy, M.; Tang, J.; Tolbert, E.; Bayliss, G.; Zhao, T.C.; Yan, H.; Zhuang, S. Blocking the class I histone deacetylase ameliorates renal fibrosis and inhibits renal fibroblast activation via modulating TGF-beta and EGFR signaling. PLoS ONE 2013, 8, e54001. [Google Scholar] [CrossRef]
- Tang, J.; Yan, Y.; Zhao, T.C.; Gong, R.; Bayliss, G.; Yan, H.; Zhuang, S. Class I HDAC activity is required for renal protection and regeneration after acute kidney injury. Am. J. Physiol. Ren. Physiol. 2014, 307, F303–F316. [Google Scholar] [CrossRef]
- Xiong, C.; Guan, Y.; Zhou, X.; Liu, L.; Zhuang, M.A.; Zhang, W.; Zhang, Y.; Masucci, M.V.; Bayliss, G.; Zhao, T.C.; et al. Selective inhibition of class IIa histone deacetylases alleviates renal fibrosis. FASEB J. 2019, 33, 8249–8262. [Google Scholar] [CrossRef]
- Zhang, W.; Guan, Y.; Bayliss, G.; Zhuang, S. Class IIa HDAC inhibitor TMP195 alleviates lipopolysaccharide-induced acute kidney injury. Am. J. Physiol. Ren. Physiol. 2020, 319, F1015–F1026. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Gao, Q.; Wei, A.; Chen, X.; Shi, Y.; Wang, H.; Cao, W. Histone deacetylase 3 aberration inhibits Klotho transcription and promotes renal fibrosis. Cell Death Differ. 2021, 28, 1001–1012. [Google Scholar] [CrossRef]
- Chen, X.; Yu, C.; Hou, X.; Li, J.; Li, T.; Qiu, A.; Liu, N.; Zhuang, S. Histone deacetylase 6 inhibition mitigates renal fibrosis by suppressing TGF-β and EGFR signaling pathways in obstructive nephropathy. Am. J. Physiol. Ren. Physiol. 2020, 319, F1003–F1014. [Google Scholar] [CrossRef] [PubMed]
- Cianciolo Cosentino, C.; Skrypnyk, N.I.; Brilli, L.L.; Chiba, T.; Novitskaya, T.; Woods, C.; West, J.; Korotchenko, V.N.; McDermott, L.; Day, B.W.; et al. Histone deacetylase inhibitor enhances recovery after AKI. J. Am. Soc. Nephrol. 2013, 24, 943–953. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Wang, Y.; Zhang, M.Z.; You, L.; Davis, L.S.; Fan, H.; Yang, H.C.; Fogo, A.B.; Zent, R.; Harris, R.C.; et al. Sirt1 activation protects the mouse renal medulla from oxidative injury. J. Clin. Investig. 2010, 120, 1056–1068. [Google Scholar] [CrossRef] [PubMed]
- Kuhad, A.; Pilkhwal, S.; Sharma, S.; Tirkey, N.; Chopra, K. Effect of curcumin on inflammation and oxidative stress in cisplatin-induced experimental nephrotoxicity. J. Agric. Food Chem. 2007, 55, 10150–10155. [Google Scholar] [CrossRef]
- Memis, D.; Hekimoglu, S.; Sezer, A.; Altaner, S.; Sut, N.; Usta, U. Curcumin attenuates the organ dysfunction caused by endotoxemia in the rat. Nutrition 2008, 24, 1133–1138. [Google Scholar] [CrossRef]
- Liang, H.; Huang, Q.; Liao, M.J.; Xu, F.; Zhang, T.; He, J.; Zhang, L.; Liu, H.Z. EZH2 plays a crucial role in ischemia/reperfusion-induced acute kidney injury by regulating p38 signaling. Inflamm. Res. 2019, 68, 325–336. [Google Scholar] [CrossRef]
- Yin, S.; Zhang, Q.; Yang, J.; Lin, W.; Li, Y.; Chen, F.; Cao, W. TGFβ-incurred epigenetic aberrations of miRNA and DNA methyltransferase suppress Klotho and potentiate renal fibrosis. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1207–1216. [Google Scholar] [CrossRef]
- Kota, S.K.; Kota, S.B. Noncoding RNA and epigenetic gene regulation in renal diseases. Drug Discov. Today 2017, 22, 1112–1122. [Google Scholar] [CrossRef]
- Fan, Y.; Chen, H.; Huang, Z.; Zheng, H.; Zhou, J. Emerging role of miRNAs in renal fibrosis. RNA Biol. 2020, 17, 1–12. [Google Scholar] [CrossRef]
- Carthew, R.W.; Sontheimer, E.J. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [PubMed]
- Brennecke, J.; Malone, C.D.; Aravin, A.A.; Sachidanandam, R.; Stark, A.; Hannon, G.J. An epigenetic role for maternally inherited piRNAs in transposon silencing. Science 2008, 322, 1387–1392. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Mattick, J.S. Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Biol. 2013, 20, 300–307. [Google Scholar] [CrossRef]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef]
- Jiang, X.; Li, D.; Shen, W.; Shen, X.; Liu, Y. LncRNA NEAT1 promotes hypoxia-induced renal tubular epithelial apoptosis through downregulating miR-27a-3p. J. Cell. Biochem. 2019, 120, 16273–16282. [Google Scholar] [CrossRef]
- Bijkerk, R.; van Solingen, C.; de Boer, H.C.; van der Pol, P.; Khairoun, M.; de Bruin, R.G.; van Oeveren-Rietdijk, A.M.; Lievers, E.; Schlagwein, N.; van Gijlswijk, D.J.; et al. Hematopoietic microRNA-126 protects against renal ischemia/reperfusion injury by promoting vascular integrity. J. Am. Soc. Nephrol. 2014, 25, 1710–1722. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Wei, Q.; Mei, S.; Li, L.; Su, Y.; Mei, C.; Dong, Z. Induction of microRNA-17-5p by p53 protects against renal ischemia-reperfusion injury by targeting death receptor 6. Kidney Int. 2017, 91, 106–118. [Google Scholar] [CrossRef]
- Wei, Q.; Sun, H.; Song, S.; Liu, Y.; Liu, P.; Livingston, M.J.; Wang, J.; Liang, M.; Mi, Q.S.; Huo, Y.; et al. MicroRNA-668 represses MTP18 to preserve mitochondrial dynamics in ischemic acute kidney injury. J. Clin. Investig. 2018, 128, 5448–5464. [Google Scholar] [CrossRef]
- Chen, W.; Ruan, Y.; Zhao, S.; Ning, J.; Rao, T.; Yu, W.; Zhou, X.; Liu, C.; Qi, Y.; Cheng, F. MicroRNA-205 inhibits the apoptosis of renal tubular epithelial cells via the PTEN/Akt pathway in renal ischemia-reperfusion injury. Am. J. Transl. Res. 2019, 11, 7364–7375. [Google Scholar]
- Lorenzen, J.M.; Kaucsar, T.; Schauerte, C.; Schmitt, R.; Rong, S.; Hübner, A.; Scherf, K.; Fiedler, J.; Martino, F.; Kumarswamy, R.; et al. MicroRNA-24 antagonism prevents renal ischemia reperfusion injury. J. Am. Soc. Nephrol. 2014, 25, 2717–2729. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, K.; Wei, Q.; Pabla, N.; Dong, G.; Mi, Q.S.; Liang, M.; Mei, C.; Dong, Z. MicroRNA-687 Induced by Hypoxia-Inducible Factor-1 Targets Phosphatase and Tensin Homolog in Renal Ischemia-Reperfusion Injury. J. Am. Soc. Nephrol. 2015, 26, 1588–1596. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Benway, C.J.; Bagley, J.; Iacomini, J. MicroRNA-494 promotes cyclosporine-induced nephrotoxicity and epithelial to mesenchymal transition by inhibiting PTEN. Am. J. Transplant. 2015, 15, 1682–1691. [Google Scholar] [CrossRef] [PubMed]
- Guan, H.; Peng, R.; Mao, L.; Fang, F.; Xu, B.; Chen, M. Injured tubular epithelial cells activate fibroblasts to promote kidney fibrosis through miR-150-containing exosomes. Exp. Cell Res. 2020, 392, 112007. [Google Scholar] [CrossRef]
- Huang, S.J.; Huang, J.; Yan, Y.B.; Qiu, J.; Tan, R.Q.; Liu, Y.; Tian, Q.; Guan, L.; Niu, S.S.; Zhang, Y.; et al. The renoprotective effect of curcumin against cisplatin-induced acute kidney injury in mice: Involvement of miR-181a/PTEN axis. Ren. Fail. 2020, 42, 350–357. [Google Scholar] [CrossRef]
- Lv, W.; Fan, F.; Wang, Y.; Gonzalez-Fernandez, E.; Wang, C.; Yang, L.; Booz, G.W.; Roman, R.J. Therapeutic potential of microRNAs for the treatment of renal fibrosis and CKD. Physiol. Genom. 2018, 50, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Fan, Y.; Jing, H.; Tang, S.; Zhou, J. Emerging role of lncRNAs in renal fibrosis. Arch. Biochem. Biophys. 2020, 692, 108530. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Wu, M.; Zhou, P.; Huang, C.; Ye, C.; Wang, L. The long non-coding RNA MALAT1 is increased in renal ischemia-reperfusion injury and inhibits hypoxia-induced inflammation. Ren. Fail. 2018, 40, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Bijkerk, R.; Au, Y.W.; Stam, W.; Duijs, J.; Koudijs, A.; Lievers, E.; Rabelink, T.J.; van Zonneveld, A.J. Long Non-coding RNAs Rian and Miat Mediate Myofibroblast Formation in Kidney Fibrosis. Front. Pharmacol. 2019, 10, 215. [Google Scholar] [CrossRef]
- Yu, T.M.; Palanisamy, K.; Sun, K.T.; Day, Y.J.; Shu, K.H.; Wang, I.K.; Shyu, W.C.; Chen, P.; Chen, Y.L.; Li, C.Y. RANTES mediates kidney ischemia reperfusion injury through a possible role of HIF-1α and LncRNA PRINS. Sci. Rep. 2016, 6, 18424. [Google Scholar] [CrossRef]
- Tian, X.; Ji, Y.; Liang, Y.; Zhang, J.; Guan, L.; Wang, C. LINC00520 targeting miR-27b-3p regulates OSMR expression level to promote acute kidney injury development through the PI3K/AKT signaling pathway. J. Cell. Physiol. 2019, 234, 14221–14233. [Google Scholar] [CrossRef]
- Vermunt, M.W.; Zhang, D.; Blobel, G.A. The interdependence of gene-regulatory elements and the 3D genome. J. Cell Biol. 2019, 218, 12–26. [Google Scholar] [CrossRef]
- van Steensel, B.; Belmont, A.S. Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin, and Gene Repression. Cell 2017, 169, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Yanez-Cuna, J.O.; van Steensel, B. Genome-nuclear lamina interactions: From cell populations to single cells. Curr. Opin. Genet. Dev. 2017, 43, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Guelen, L.; Pagie, L.; Brasset, E.; Meuleman, W.; Faza, M.B.; Talhout, W.; Eussen, B.H.; de Klein, A.; Wessels, L.; de Laat, W.; et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 2008, 453, 948–951. [Google Scholar] [CrossRef] [PubMed]
- Wijchers, P.J.; Geeven, G.; Eyres, M.; Bergsma, A.J.; Janssen, M.; Verstegen, M.; Zhu, Y.; Schell, Y.; Vermeulen, C.; de Wit, E.; et al. Characterization and dynamics of pericentromere-associated domains in mice. Genome Res. 2015, 25, 958–969. [Google Scholar] [CrossRef] [PubMed]
- van Koningsbruggen, S.; Gierlinski, M.; Schofield, P.; Martin, D.; Barton, G.J.; Ariyurek, Y.; den Dunnen, J.T.; Lamond, A.I. High-resolution whole-genome sequencing reveals that specific chromatin domains from most human chromosomes associate with nucleoli. Mol. Biol. Cell 2010, 21, 3735–3748. [Google Scholar] [CrossRef]
- Schoenfelder, S.; Fraser, P. Long-range enhancer-promoter contacts in gene expression control. Nat. Rev. Genet. 2019, 20, 437–455. [Google Scholar] [CrossRef]
- Lupianez, D.G.; Spielmann, M.; Mundlos, S. Breaking TADs: How Alterations of Chromatin Domains Result in Disease. Trends Genet. 2016, 32, 225–237. [Google Scholar] [CrossRef]
- Fudenberg, G.; Imakaev, M.; Lu, C.; Goloborodko, A.; Abdennur, N.; Mirny, L.A. Formation of Chromosomal Domains by Loop Extrusion. Cell Rep. 2016, 15, 2038–2049. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Brandt, M.M.; Meddens, C.A.; Louzao-Martinez, L.; van den Dungen, N.A.M.; Lansu, N.R.; Nieuwenhuis, E.E.S.; Duncker, D.J.; Verhaar, M.C.; Joles, J.A.; Mokry, M.; et al. Chromatin Conformation Links Distal Target Genes to CKD Loci. J. Am. Soc. Nephrol. 2018, 29, 462–476. [Google Scholar] [CrossRef]
- Wilflingseder, J.; Willi, M.; Lee, H.K.; Olauson, H.; Jankowski, J.; Ichimura, T.; Erben, R.; Valerius, M.T.; Hennighausen, L.; Bonventre, J.V. Enhancer and super-enhancer dynamics in repair after ischemic acute kidney injury. Nat. Commun. 2020, 11, 3383. [Google Scholar] [CrossRef] [PubMed]
- De Raedt, T.; Beert, E.; Pasmant, E.; Luscan, A.; Brems, H.; Ortonne, N.; Helin, K.; Hornick, J.L.; Mautner, V.; Kehrer-Sawatzki, H.; et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 2014, 514, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Philpott, M.; Muller, S.; Schulze, J.; Badock, V.; Eberspacher, U.; Moosmayer, D.; Bader, B.; Schmees, N.; Fernandez-Montalvan, A.; et al. Affinity map of bromodomain protein 4 (BRD4) interactions with the histone H4 tail and the small molecule inhibitor JQ1. J. Biol. Chem. 2014, 289, 9304–9319. [Google Scholar] [CrossRef] [PubMed]
- Crump, N.T.; Ballabio, E.; Godfrey, L.; Thorne, R.; Repapi, E.; Kerry, J.; Tapia, M.; Hua, P.; Lagerholm, C.; Filippakopoulos, P.; et al. BET inhibition disrupts transcription but retains enhancer-promoter contact. Nat. Commun. 2021, 12, 223. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Target | Model | Effects on Kidney | Ref. |
|---|---|---|---|---|
| Histone acetylation | ||||
| HDAC inhibitors | ||||
| TSA | Class Ι and ΙΙ HDAC inhibitor | IRI | ↓ fibrosis | [123] |
| UUO | ↓ fibrosis | [124] | ||
| PTBA | Class Ι HDAC inhibitor | AA | ↓ fibrosis, ↓ inflammation | [125] |
| PTBA analogs, UPHD 25 | Class Ι HDAC inhibitor | IRI | ↓ fibrosis, ↓ inflammation | [126] |
| FR276457 | Class Ι and ΙΙ HDAC inhibitor | UUO | ↓ fibrosis | [127] |
| Valproic acid | Class Ι HDAC inhibitor | IRI | ↓ fibrosis, ↓ inflammation | [128] |
| MS-275 | Class Ι HDAC inhibitor | LPS | ↓ inflammation, ↑ renal function | [129] |
| UUO | ↓ fibrosis | [130] | ||
| IRI | ↓ fibrosis | [123] | ||
| FA | ↑ renal injury | [131] | ||
| TMP195 | Class ΙΙ HDAC inhibitor | LPS | ↓ inflammation, ↑ renal function | [132] |
| MC1568 | Class ΙΙ HDAC inhibitor | UUO | ↓ fibrosis, ↓ inflammation | [133] |
| RGFP966 | Class Ι HDAC inhibitor | UUO/AA | ↑ Klotho, ↓ fibrosis | [134] |
| ACY-1215 | Class ΙΙ HDAC inhibitor | UUO | ↓ fibrosis | [135] |
| Entinostat | Class Ι HDAC inhibitor | IRI | ↑ renal function | [123,130] |
| UUO | ↓ fibrosis | |||
| M4-PTB | Class Ι HDAC inhibitor | AA | ↑ proliferation | [125,136] |
| IRI | ↓ fibrosis, ↓ inflammation | |||
| HDAC (SIRT1) activators | ||||
| Resveratrol | SIRT1 activator | Cisplatin | ↓ inflammation | [85] |
| LPS | ↓ inflammation | [86] | ||
| SRT-1720 | SIRT1 activator | UUO | ↓ fibrosis | [137] |
| IRI | ↑ proliferation, ↓ fibrosis | [87] | ||
| HAT inhibitors | ||||
| Curcumin | HAT p300/CBP inhibitor | Cisplatin | ↓ inflammation | [138] |
| LPS | ↓ inflammation | [139] | ||
| IRI | ↓ fibrosis | [88] | ||
| Histone methylation | ||||
| HMT inhibitors | ||||
| DZNeP | HMT EZH2 inhibitor | UUO | ↓ fibrosis | [140] |
| IRI | ↓ fibrosis | [100] | ||
| Sinefungin | HMT SET7/9 inhibitor | UUO | ↓ fibrosis | [101] |
| BIX01294I | HMT G9a inhibitor | UUO | ↓ fibrosis, ↑ Klotho | [102] |
| DNA methylation | ||||
| DNMT inhibitors | ||||
| 5-azacytidine | DNMT inhibitor | UUO | ↓ fibrosis | [116] |
| Decitabine | DNMT inhibitor | UUO | ↓ fibrosis, ↑ Klotho | [141] |
| Hydralazine | induction of TET3 | IRI | ↓ fibrosis, ↑ renal function | [119] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanemoto, F.; Mimura, I. Therapies Targeting Epigenetic Alterations in Acute Kidney Injury-to-Chronic Kidney Disease Transition. Pharmaceuticals 2022, 15, 123. https://doi.org/10.3390/ph15020123
Tanemoto F, Mimura I. Therapies Targeting Epigenetic Alterations in Acute Kidney Injury-to-Chronic Kidney Disease Transition. Pharmaceuticals. 2022; 15(2):123. https://doi.org/10.3390/ph15020123
Chicago/Turabian StyleTanemoto, Fumiaki, and Imari Mimura. 2022. "Therapies Targeting Epigenetic Alterations in Acute Kidney Injury-to-Chronic Kidney Disease Transition" Pharmaceuticals 15, no. 2: 123. https://doi.org/10.3390/ph15020123
APA StyleTanemoto, F., & Mimura, I. (2022). Therapies Targeting Epigenetic Alterations in Acute Kidney Injury-to-Chronic Kidney Disease Transition. Pharmaceuticals, 15(2), 123. https://doi.org/10.3390/ph15020123