
Toosendanin Induces Hepatocyte Damage by Inhibiting Autophagic Flux via TFEB-Mediated Lysosomal Dysfunction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
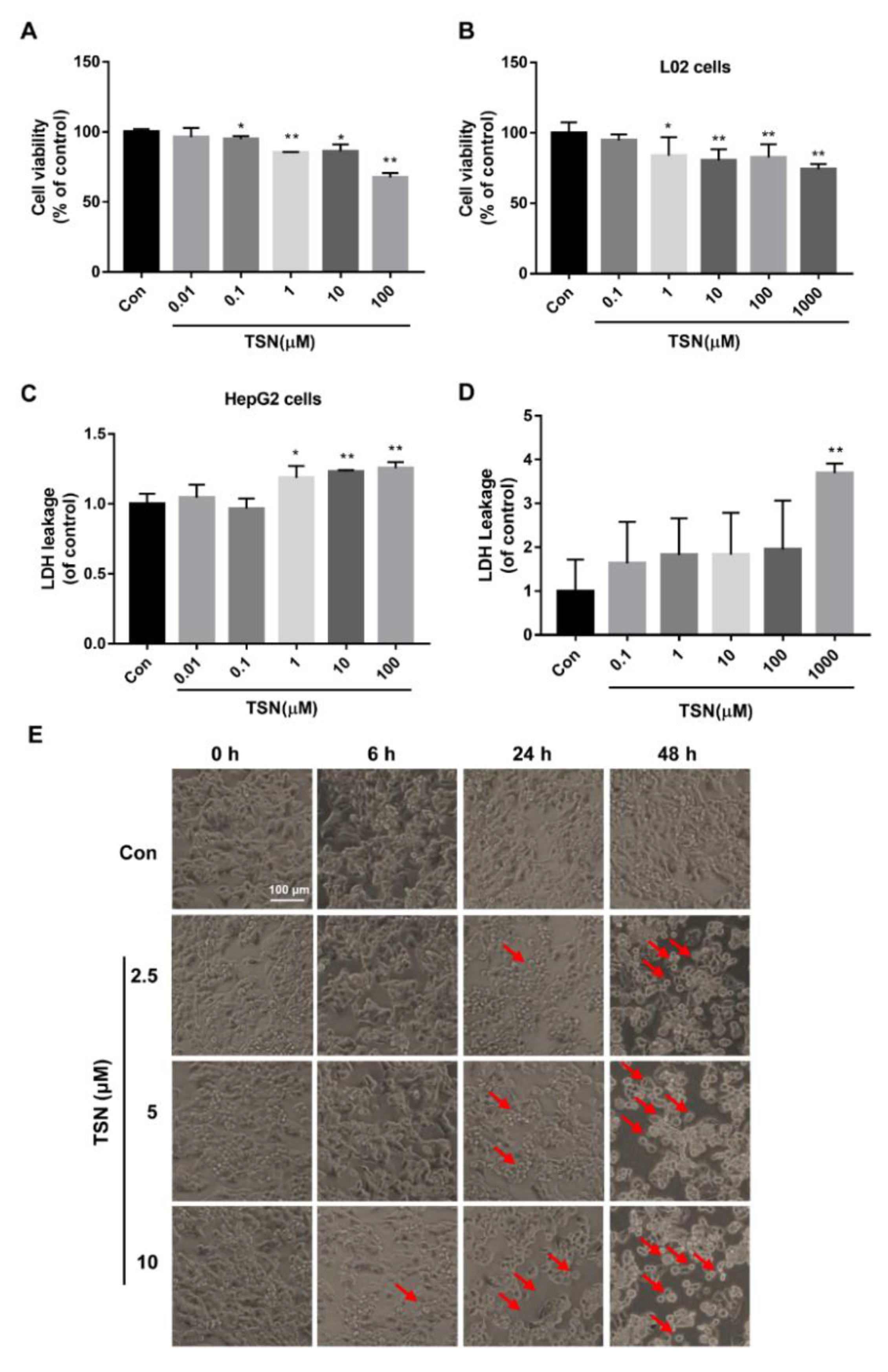
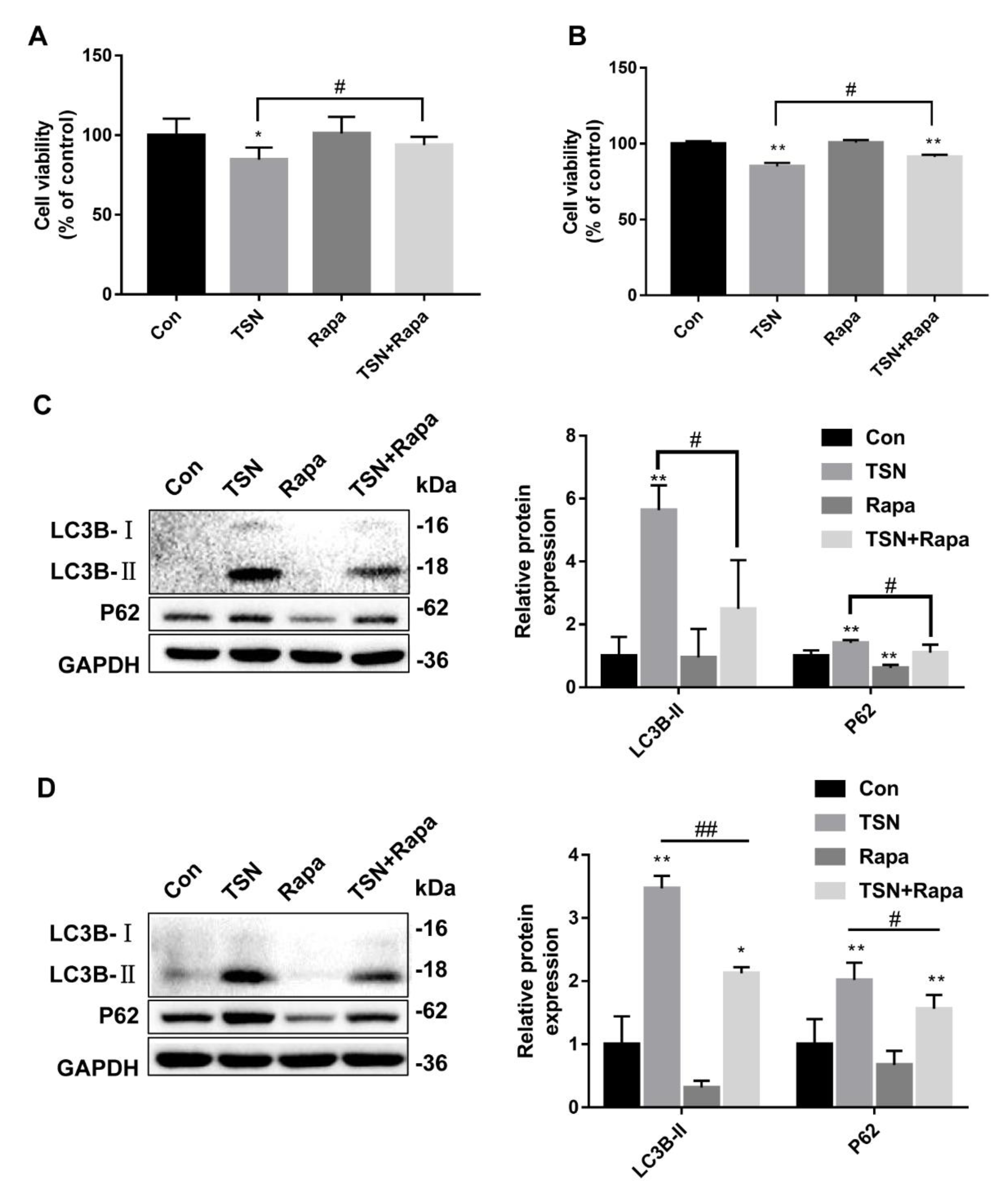
2.1. TSN Induces Cell Damage in HepG2 Cells and L02 Cells
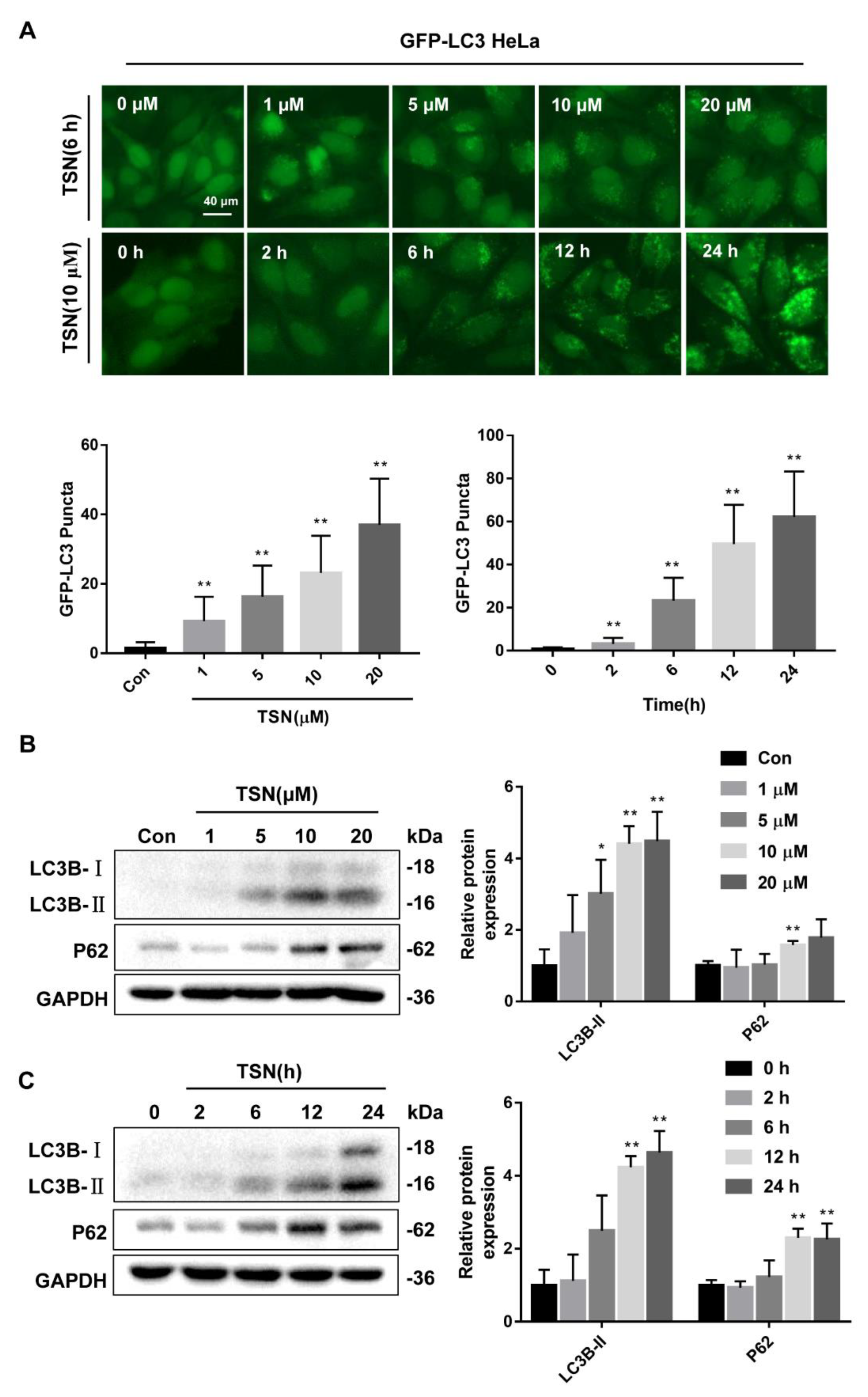
2.2. TSN Inhibits Autophagic Flux in HepG2 Cells
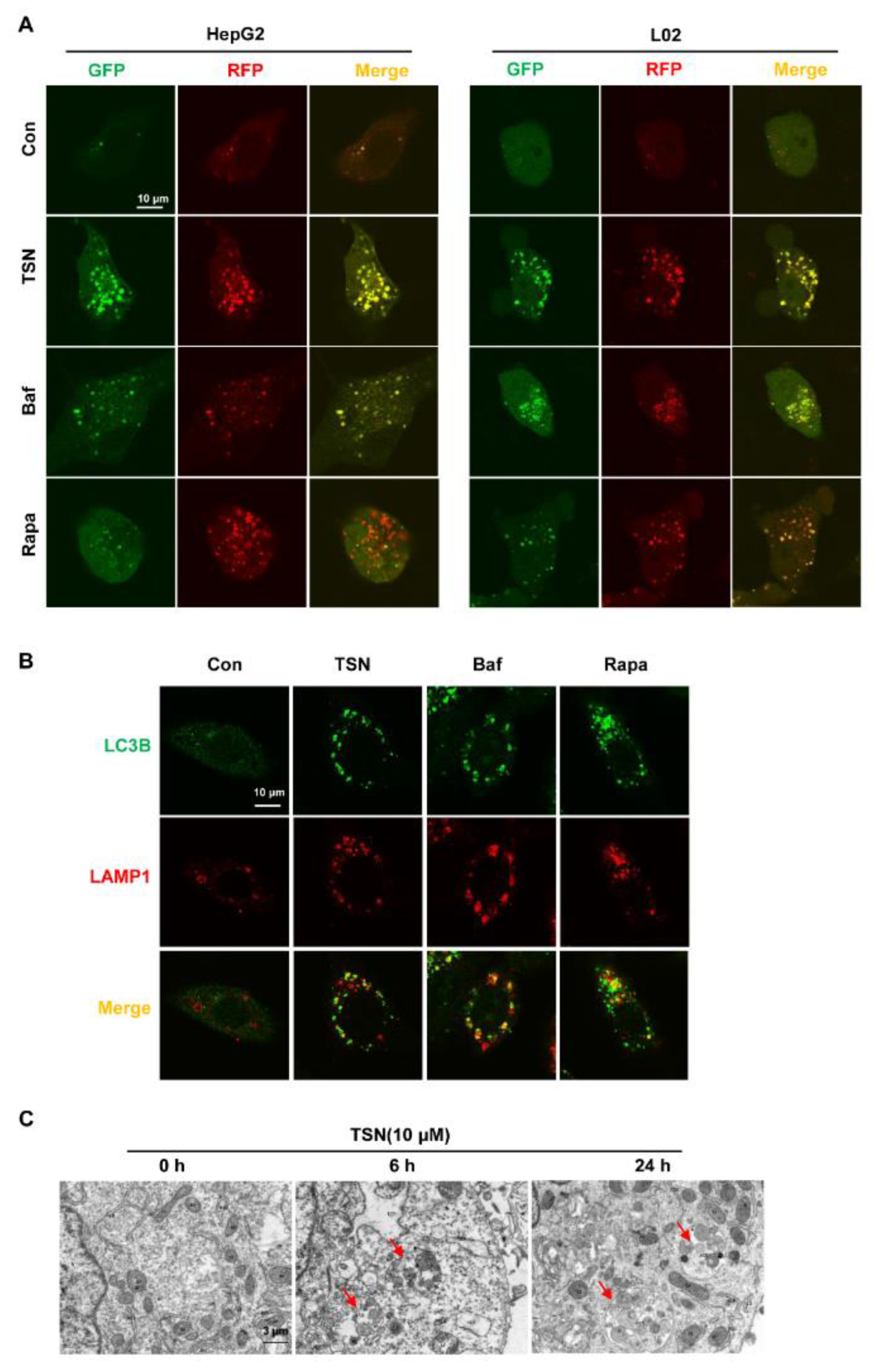
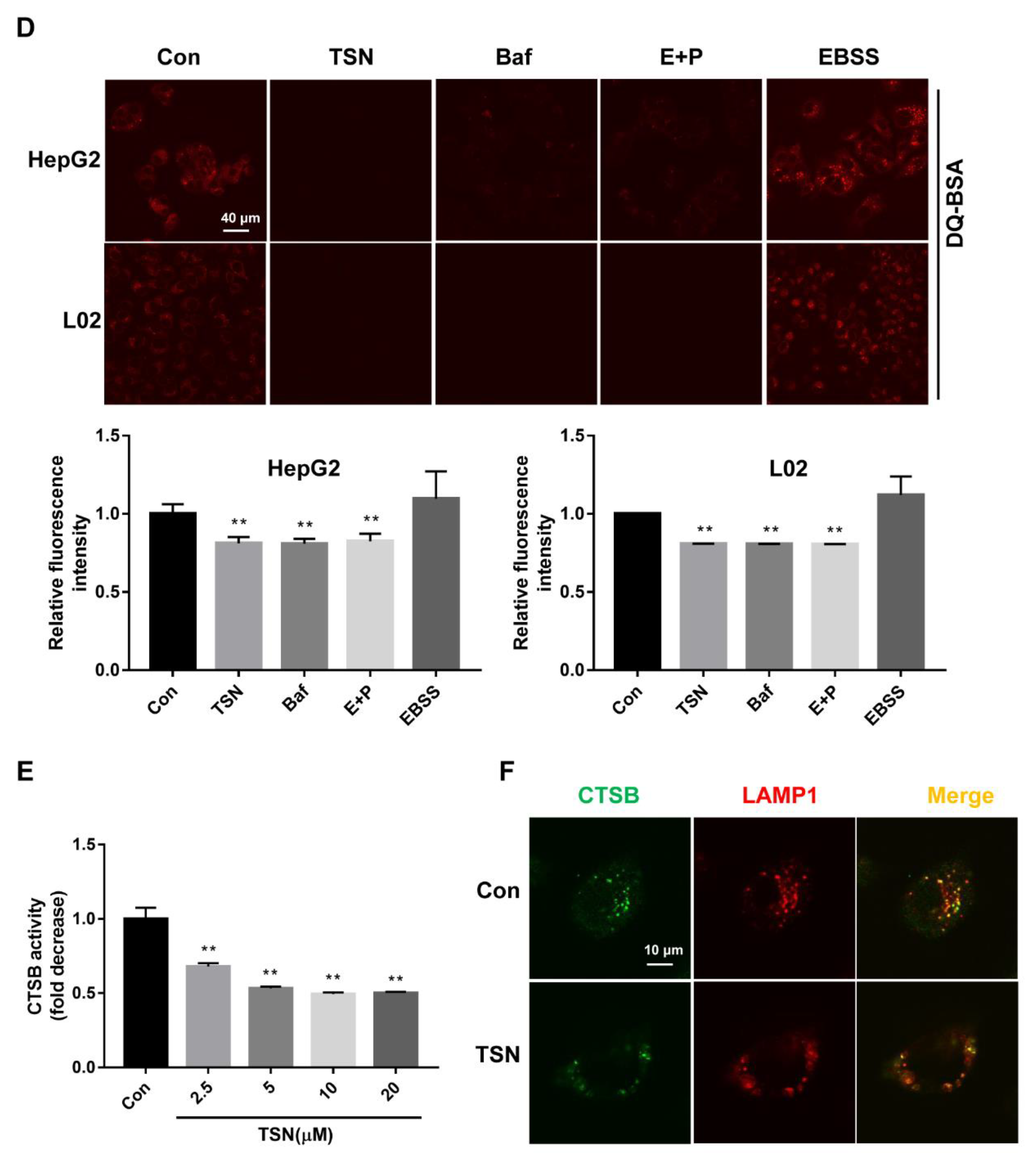
2.3. TSN Increases the Accumulation of Autolysosomes in HepG2 Cells and L02 Cells
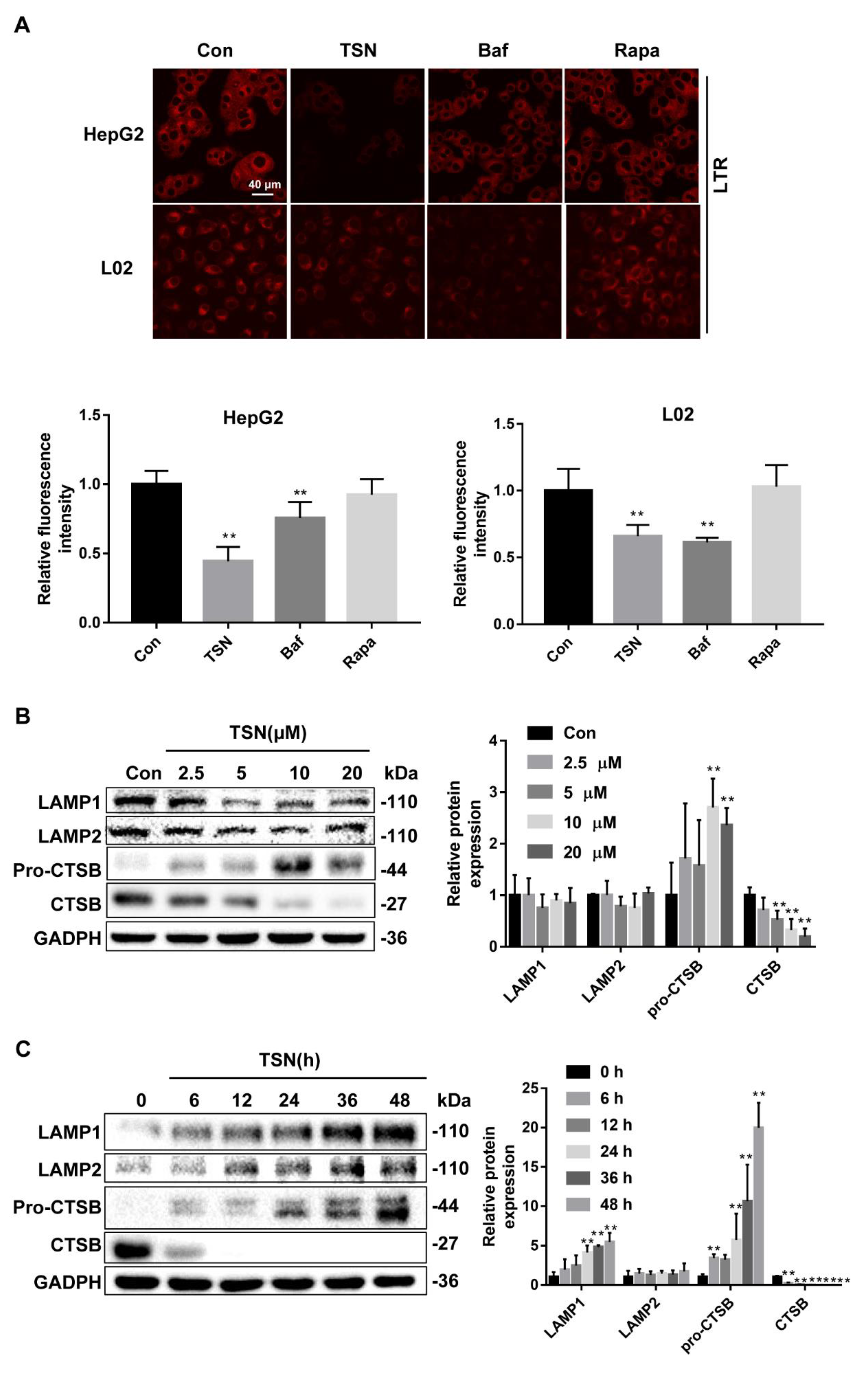
2.4. TSN Induces Lysosome Dysfunction in Vitro
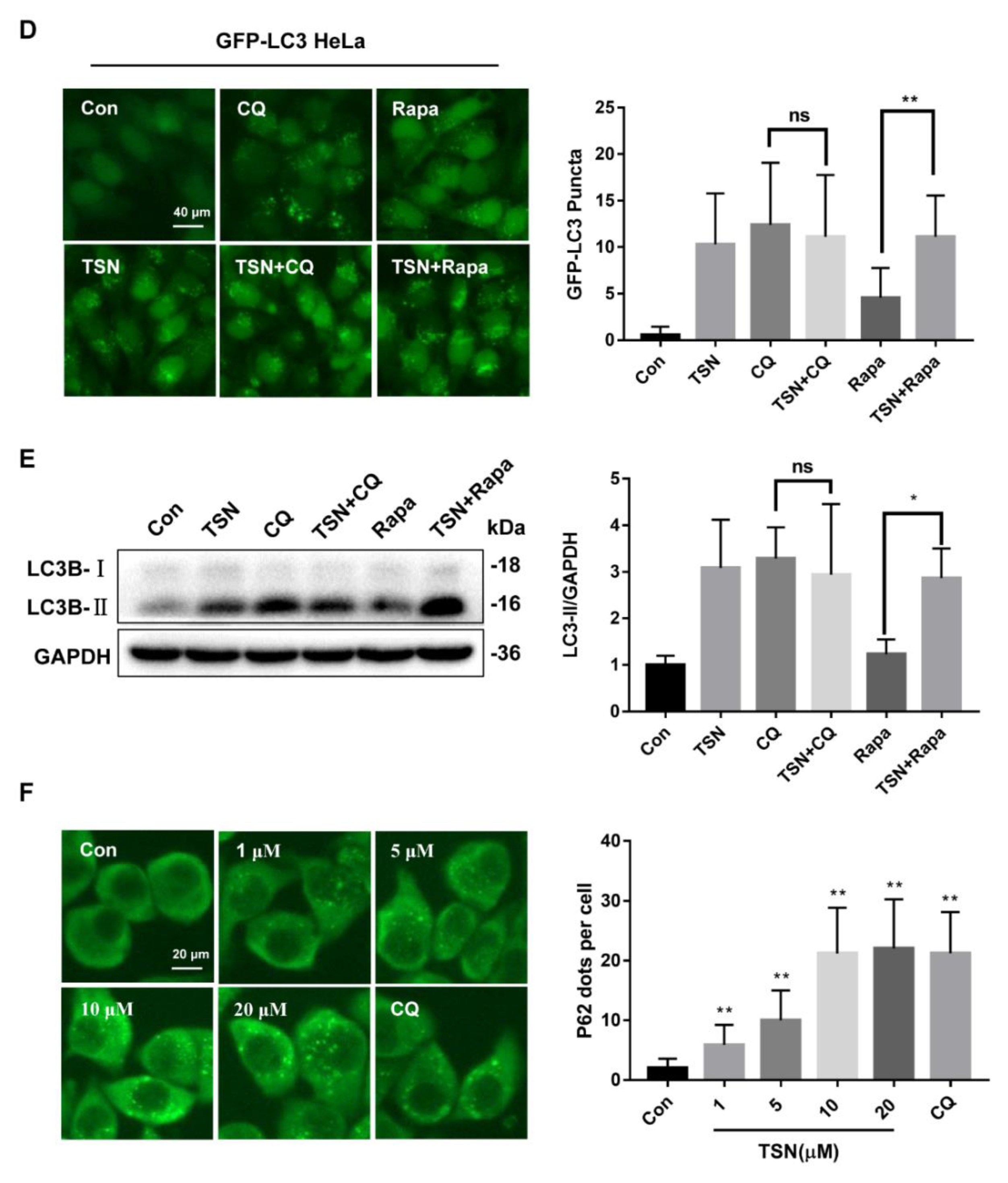
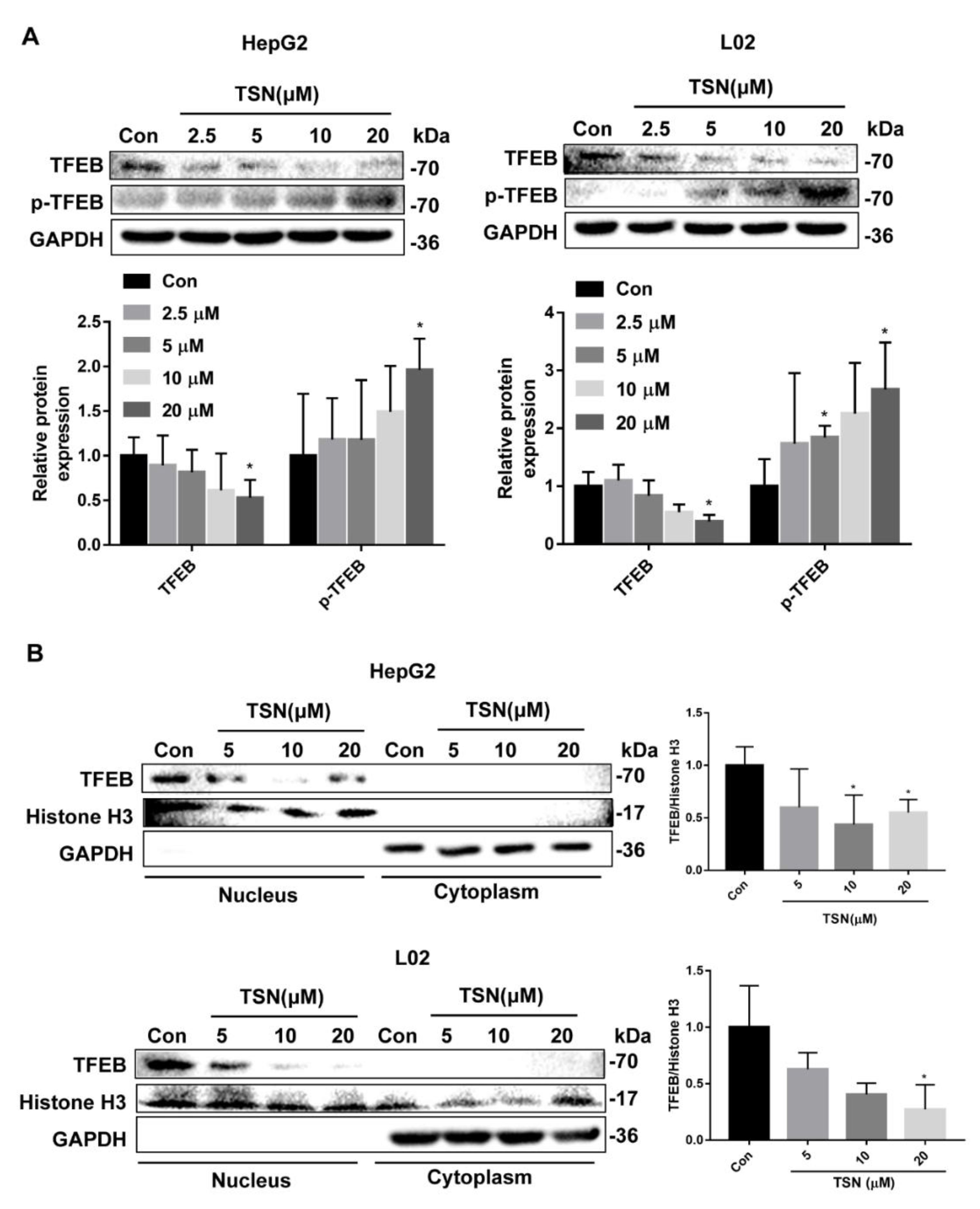
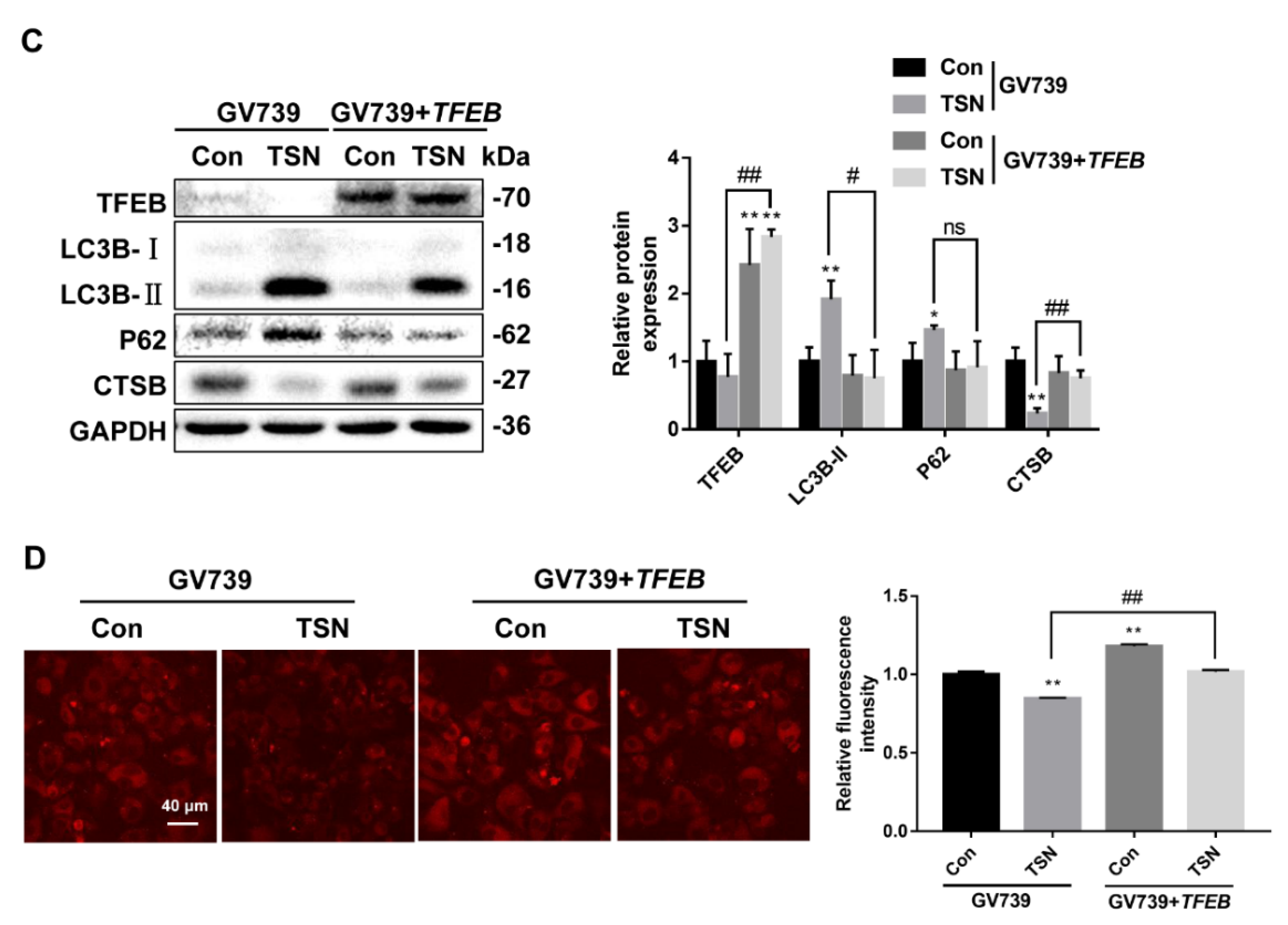
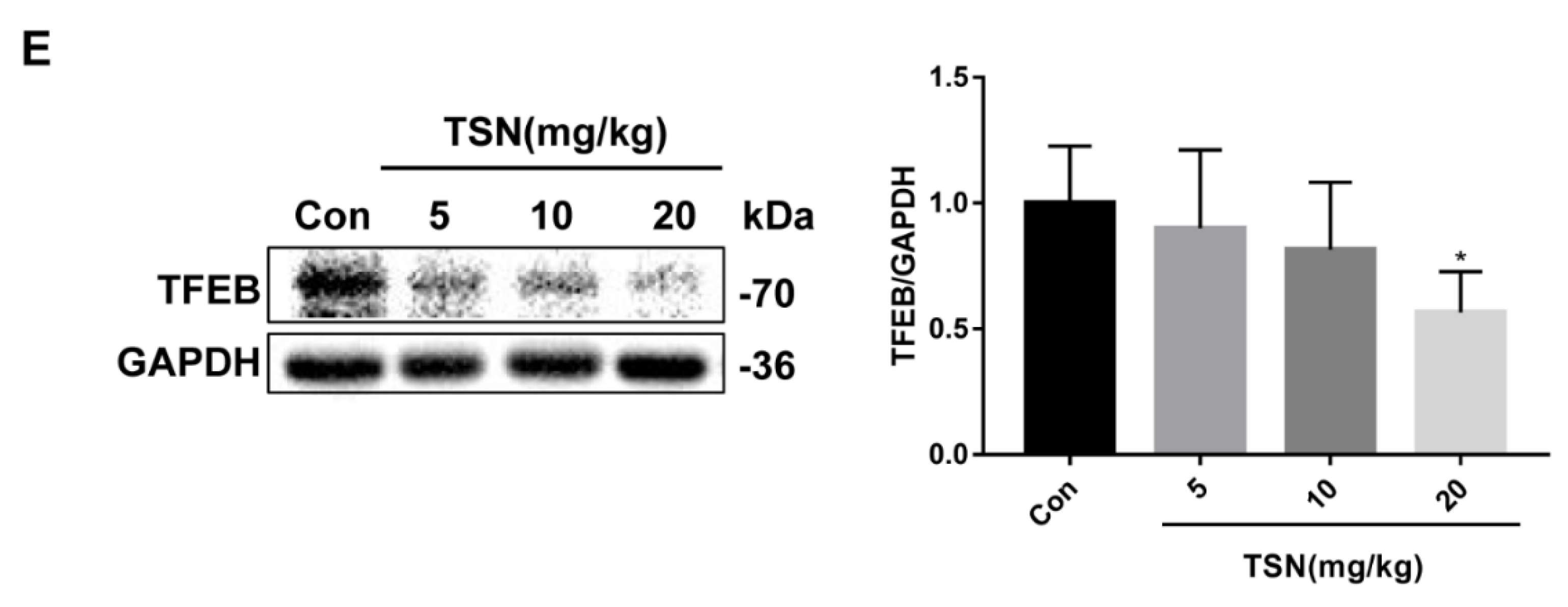
2.5. TSN Blocks Autophagic Flux by Inhibiting TFEB in Vitro
2.6. Activation of Autophagy Alleviates TSN-Induced Cell Damage
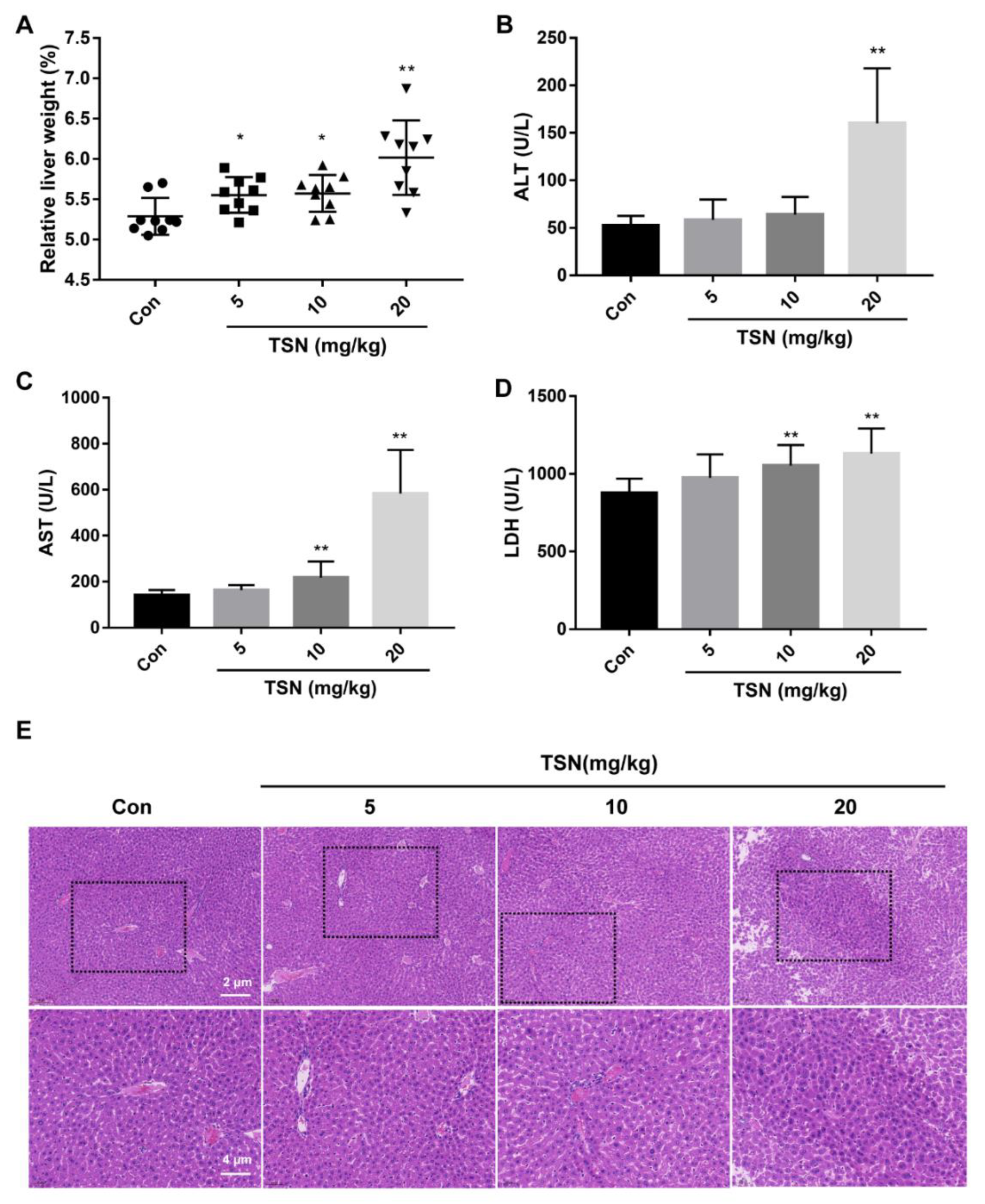
2.7. TSN Induces Liver Injury in Balb/c Mice
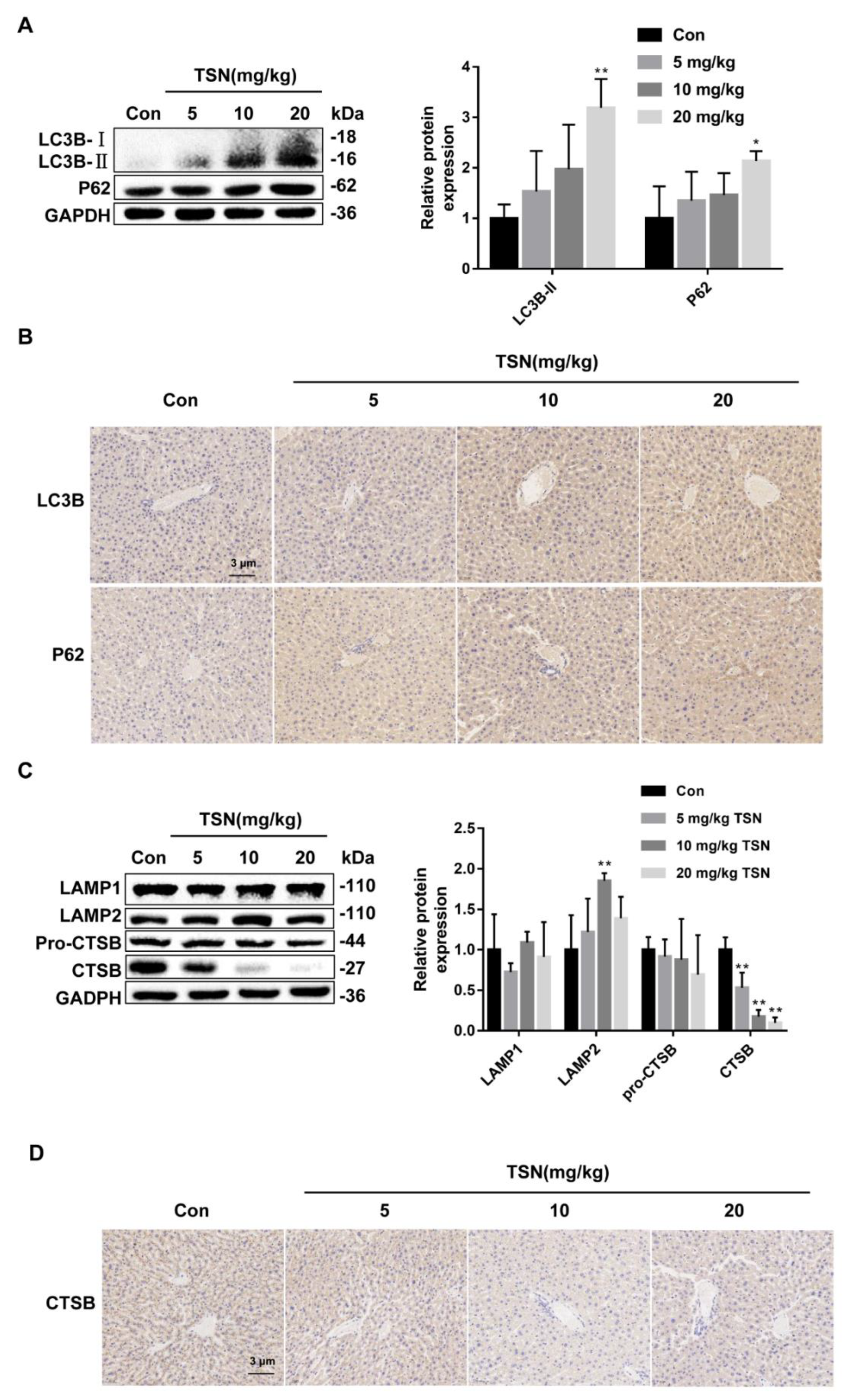
2.8. TSN Inhibits Autophagic Flux and Induces Lysosome Dysfunction in Balb/c Mice
3. Discussion
4. Material and Methods
4.1. Materials
4.2. Cell Culture
4.3. Cell Transfection
4.4. Cell Viability Assay
4.5. LDH Leakage Assay
4.6. Analysis of Cell Morphology
4.7. Western Blot Analysis
4.8. Quantitative Real-Time PCR Analysis
4.9. Immunofluorescence Staining
4.10. RFP–GFP–LC3 Assay
4.11. Transmission Electron Microscopy
4.12. LTR Staining
4.13. DQ-BSA Assay
4.14. CTSB Activity Assay
4.15. Animal Experiment
4.16. Serum Biochemical Assay
4.17. Histopathological Examinations (HE)
4.18. IHC
4.19. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ALT | serum alanine aminotransferase |
| AST | aspartate aminotransferase |
| ATG | autophagy-related gene |
| Baf | bafilomycin A1 |
| CLEAR | coordinated lysosomal enhancement and regulation |
| CQ | chloroquine |
| CTSB | cathepsin B |
| CTSD | cathepsin D |
| CYP | cytochromes |
| DILI | drug-induced liver injury |
| DQ-BSA | Dye Quenched-Bovine Serum Albumin |
| GFP | green fluorescent protein |
| GSH | glutathione |
| HDS | herbal medicine and dietary supplements |
| LAMP1 | lysosomal membrane proteins 1 |
| LAMP2 | lysosomal membrane proteins 2 |
| LC3 | microtubule-associated proteins 1A/1B light chain 3 |
| LDH | lactate dehydrogenase |
| LTR | LysoTracker Red |
| MTORC1 | mechanistic target of rapamycin kinase complex 1 |
| Rapa | rapamycin |
| RFP | red fluorescent protein |
| TFEB | transcription factor EB |
| TSN | toosendanin |
References
- Li, X.; Tang, J.; Mao, Y. Incidence and Risk Factors of Drug-Induced Liver Injury. Liver Int. 2022, 42, 1999–2014. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Cortes, M.; Robles-Diaz, M.; Stephens, C.; Ortega-Alonso, A.; Lucena, M.I.; Andrade, R.J. Drug Induced Liver Injury: An Update. Arch. Toxicol. 2020, 94, 3381–3407. [Google Scholar] [CrossRef] [PubMed]
- Sgro, C.; Clinard, F.; Ouazir, K.; Chanay, H.; Allard, C.; Guilleminet, C.; Lenoir, C.; Lemoine, A.; Hillon, P. Incidence of Drug-Induced Hepatic Injuries: A French Population-Based Study. Hepatology 2002, 36, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Björnsson, E.S.; Bergmann, O.M.; Björnsson, H.K.; Kvaran, R.B.; Olafsson, S. Incidence, Presentation, and Outcomes in Patients with Drug-Induced Liver Injury in the General Population of Iceland. Gastroenterology 2013, 144, 1419–1425.e3. [Google Scholar] [CrossRef] [PubMed]
- De Abajo, F.J.; Montero, D.; Madurga, M.; Rodríguez, L.A.G. Acute and Clinically Relevant Drug-Induced Liver Injury: A Population Based Case-Control Study. Br. J. Clin. Pharmacol. 2004, 58, 71–80. [Google Scholar] [CrossRef]
- De Valle, M.B.; Av Klinteberg, V.; Alem, N.; Olsson, R.; Björnsson, E. Drug-Induced Liver Injury in a Swedish University Hospital out-Patient Hepatology Clinic. Aliment. Pharmacol. Ther. 2006, 24, 1187–1195. [Google Scholar] [CrossRef]
- Andrade, R.J.; Lucena, M.I.; Fernández, M.C.; Pelaez, G.; Pachkoria, K.; García-Ruiz, E.; García-Muñoz, B.; González-Grande, R.; Pizarro, A.; Durán, J.A.; et al. Drug-Induced Liver Injury: An Analysis of 461 Incidences Submitted to the Spanish Registry Over a 10-Year Period. Gastroenterology 2005, 129, 512–521. [Google Scholar] [CrossRef]
- Suk, K.T.; Kim, D.J.; Kim, C.H.; Park, S.H.; Yoon, J.H.; Kim, Y.S.; Baik, G.H.; Kim, J.B.; Kweon, Y.O.; Kim, B.I.; et al. A Prospective Nationwide Study of Drug-Induced Liver Injury in Korea. Off. J. Am. Coll. Gastroenterol. ACG 2012, 107, 1380–1387. [Google Scholar] [CrossRef]
- Shen, T.; Liu, Y.; Shang, J.; Xie, Q.; Li, J.; Yan, M.; Xu, J.; Niu, J.; Liu, J.; Watkins, P.B.; et al. Incidence and Etiology of Drug-Induced Liver Injury in Mainland China. Gastroenterology 2019, 156, 2230–2241.e11. [Google Scholar] [CrossRef]
- Zhang, S.; Cao, L.; Wang, Z.-R.; Li, Z.; Ma, J. Anti-Cancer Effect of Toosendanin and Its Underlying Mechanisms. J. Asian Nat. Prod. Res. 2019, 21, 270–283. [Google Scholar] [CrossRef]
- Li, H.; Zhang, J.; Ma, T.; Li, C.; Ma, Z.; Zhang, X. Acting Target of Toosendanin Locates in the Midgut Epithelium Cells of Mythimna Separate Walker Larvae (Lepidoptera: Noctuidae). Ecotoxicol. Environ. Saf. 2020, 201, 110828. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, F.; Mei, X.; Yang, R.; Lu, B.; Wang, Z.; Ji, L. Toosendanin and Isotoosendanin Suppress Triple-Negative Breast Cancer Growth via Inducing Necrosis, Apoptosis and Autophagy. Chem. Biol. Interact. 2022, 351, 109739. [Google Scholar] [CrossRef]
- Yang, T.; Xu, R.; Huo, J.; Wang, B.; Du, X.; Dai, B.; Zhu, M.; Zhan, Y.; Zhang, D.; Zhang, Y. WWOX Activation by Toosendanin Suppresses Hepatocellular Carcinoma Metastasis through JAK2/Stat3 and Wnt/β-Catenin Signaling. Cancer Lett. 2021, 513, 50–62. [Google Scholar] [CrossRef]
- Jin, Y.-H.; Kwon, S.; Choi, J.-G.; Cho, W.-K.; Lee, B.; Ma, J.Y. Toosendanin From Melia Fructus Suppresses Influenza A Virus Infection by Altering Nuclear Localization of Viral Polymerase PA Protein. Front. Pharmacol. 2019, 10, 1025. [Google Scholar] [CrossRef]
- Nakai, Y.; Pellett, S.; Tepp, W.H.; Johnson, E.A.; Janda, K.D. Toosendanin: Synthesis of the AB-Ring and Investigations of Its Anti-Botulinum Properties (Part II). Bioorg. Med. Chem. 2010, 18, 1280. [Google Scholar] [CrossRef]
- Jin, Y.; Huang, Z.; Li, L.; Yang, Y.; Wang, C.; Wang, Z.; Ji, L. Quercetin Attenuates Toosendanin-Induced Hepatotoxicity through Inducing the Nrf2/GCL/GSH Antioxidant Signaling Pathway. Acta Pharmacol. Sin. 2019, 40, 75–85. [Google Scholar] [CrossRef]
- Yang, F.; Li, L.; Yang, R.; Wei, M.; Sheng, Y.; Ji, L. Identification of Serum MicroRNAs as Potential Toxicological Biomarkers for Toosendanin-Induced Liver Injury in Mice. Phytomedicine 2019, 58, 152867. [Google Scholar] [CrossRef]
- Yan, X.; Zhuo, Y.; Bian, X.; Li, J.; Zhang, Y.; Ma, L.; Lu, G.; Guo, M.-Q.; Wu, J.-L.; Li, N. Integrated Proteomics, Biological Functional Assessments, and Metabolomics Reveal Toosendanin-Induced Hepatic Energy Metabolic Disorders. Chem. Res. Toxicol. 2019, 32, 668–680. [Google Scholar] [CrossRef]
- Behrends, C.; Sowa, M.E.; Gygi, S.P.; Harper, J.W. Network Organization of the Human Autophagy System. Nature 2010, 466, 68–76. [Google Scholar] [CrossRef]
- Qian, H.; Chao, X.; Williams, J.; Fulte, S.; Li, T.; Yang, L.; Ding, W.-X. Autophagy in Liver Diseases: A Review. Mol. Aspects Med. 2021, 82, 100973. [Google Scholar] [CrossRef]
- Zhou, J.; Li, Y.; Liu, X.; Long, Y.; Chen, J. LncRNA-Regulated Autophagy and Its Potential Role in Drug-Induced Liver Injury. Ann. Hepatol. 2018, 17, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Ding, W.-X. Role of Autophagy in Alcohol and Drug-Induced Liver Injury. Food Chem. Toxicol. 2020, 136, 111075. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.-Y.; Shin, J.-K.; Lee, S.-M. Pterostilbene Protects against Acetaminophen-Induced Liver Injury by Restoring Impaired Autophagic Flux. Food Chem. Toxicol. 2019, 123, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Apostolova, N.; Gomez-Sucerquia, L.; Moran, A.; Alvarez, A.; Blas-Garcia, A.; Esplugues, J. Enhanced Oxidative Stress and Increased Mitochondrial Mass during Efavirenz-Induced Apoptosis in Human Hepatic Cells. Br. J. Pharmacol. 2010, 160, 2069–2084. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.-X.; Li, M.; Chen, X.; Ni, H.-M.; Lin, C.-W.; Gao, W.; Lu, B.; Stolz, D.B.; Clemens, D.L.; Yin, X.-M. Autophagy Reduces Acute Ethanol-Induced Hepatotoxicity and Steatosis in Mice. Gastroenterology 2010, 139, 1740–1752. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Liu, Q.; Liang, Q.; Gao, S.; Zhuang, K.; Zhang, Y.; Zhang, H.; Liu, K.; She, G.; Xia, Q. Toosendanin Triggered Hepatotoxicity in Zebrafish via Inflammation, Autophagy, and Apoptosis Pathways. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2021, 250, 109171. [Google Scholar] [CrossRef]
- Zou, H.; Wang, T.; Yuan, J.; Sun, J.; Yuan, Y.; Gu, J.; Liu, X.; Bian, J.; Liu, Z. Cadmium-Induced Cytotoxicity in Mouse Liver Cells Is Associated with the Disruption of Autophagic Flux via Inhibiting the Fusion of Autophagosomes and Lysosomes. Toxicol. Lett. 2020, 321, 32–43. [Google Scholar] [CrossRef]
- Lu, J.; Fan, Y.; Liu, M.; Zhang, Q.; Guan, S. 1,3-Dichloro-2-Propanol Induced Lipid Accumulation by Blocking Autophagy Flux in HepG2 Cells. Toxicology 2021, 454, 152716. [Google Scholar] [CrossRef]
- Mohamud, Y.; Tang, H.; Xue, Y.C.; Liu, H.; Ng, C.S.; Bahreyni, A.; Luo, H. Coxsackievirus B3 Targets TFEB to Disrupt Lysosomal Function. Autophagy 2021, 17, 3924–3938. [Google Scholar] [CrossRef]
- Zhu, L.; Yuan, Y.; Yuan, L.; Li, L.; Liu, F.; Liu, J.; Chen, Y.; Lu, Y.; Cheng, J. Activation of TFEB-Mediated Autophagy by Trehalose Attenuates Mitochondrial Dysfunction in Cisplatin-Induced Acute Kidney Injury. Theranostics 2020, 10, 5829–5844. [Google Scholar] [CrossRef]
- Zhao, S.; Feng, J.; Li, J.; Cao, R.; Zhang, Y.; Yang, S.; Yin, L. The RNA Binding Protein HnRNPK Protects against Adriamycin-Induced Podocyte Injury. Ann. Transl. Med. 2021, 9, 1303. [Google Scholar] [CrossRef]
- Wang, Y.; Hao, C.-L.; Zhang, Z.-H.; Wang, L.-H.; Yan, L.-N.; Zhang, R.-J.; Lin, L.; Yang, Y. Valproic Acid Increased Autophagic Flux in Human Multiple Myeloma Cells in Vitro. Biomed. Pharmacother. 2020, 127, 110167. [Google Scholar] [CrossRef]
- Lu, J.; Yang, Y.; Zhu, L.; Li, M.; Xu, W.; Zhang, C.; Cheng, J.; Tao, L.; Li, Z.; Zhang, Y. Exposure to Environmental Concentrations of Natural Pyrethrins Induces Hepatotoxicity: Assessment in HepG2 Cell Lines and Zebrafish Models. Chemosphere 2022, 288, 132565. [Google Scholar] [CrossRef]
- Arzumanian, V.A.; Kiseleva, O.I.; Poverennaya, E.V. The Curious Case of the HepG2 Cell Line: 40 Years of Expertise. Int. J. Mol. Sci. 2021, 22, 13135. [Google Scholar] [CrossRef]
- Gerets, H.H.J.; Tilmant, K.; Gerin, B.; Chanteux, H.; Depelchin, B.O.; Dhalluin, S.; Atienzar, F.A. Characterization of Primary Human Hepatocytes, HepG2 Cells, and HepaRG Cells at the MRNA Level and CYP Activity in Response to Inducers and Their Predictivity for the Detection of Human Hepatotoxins. Cell Biol. Toxicol. 2012, 28, 69–87. [Google Scholar] [CrossRef]
- McGill, M.R.; Jaeschke, H. Metabolism and Disposition of Acetaminophen: Recent Advances in Relation to Hepatotoxicity and Diagnosis. Pharm. Res. 2013, 30, 2174–2187. [Google Scholar] [CrossRef]
- Shi, C.; Xue, W.; Han, B.; Yang, F.; Yin, Y.; Hu, C. Acetaminophen Aggravates Fat Accumulation in NAFLD by Inhibiting Autophagy via the AMPK/MTOR Pathway. Eur. J. Pharmacol. 2019, 850, 15–22. [Google Scholar] [CrossRef]
- Ni, H.-M.; Bockus, A.; Boggess, N.; Jaeschke, H.; Ding, W.-X. Activation of Autophagy Protects against Acetaminophen-Induced Hepatotoxicity. Hepatology 2012, 55, 222–232. [Google Scholar] [CrossRef]
- Peng, S.; Yang, Q.; Li, H.; Pan, Y.; Wang, J.; Hu, P.; Zhang, N. CTSB Knockdown Inhibits Proliferation and Tumorigenesis in HL-60 Cells. Int. J. Med. Sci. 2021, 18, 1484–1491. [Google Scholar] [CrossRef]
- Tao, Y.; Qiu, T.; Yao, X.; Jiang, L.; Wang, N.; Jia, X.; Wei, S.; Wang, Z.; Pei, P.; Zhang, J.; et al. Autophagic-CTSB-Inflammasome Axis Modulates Hepatic Stellate Cells Activation in Arsenic-Induced Liver Fibrosis. Chemosphere 2020, 242, 124959. [Google Scholar] [CrossRef]
- Araujo, T.F.; Cordeiro, A.V.; Vasconcelos, D.A.A.; Vitzel, K.F.; Silva, V.R.R. The Role of Cathepsin B in Autophagy during Obesity: A Systematic Review. Life Sci. 2018, 209, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Chitranshi, N.; Kumar, A.; Sheriff, S.; Gupta, V.; Godinez, A.; Saks, D.; Sarkar, S.; Shen, T.; Mirzaei, M.; Basavarajappa, D.; et al. Identification of Novel Cathepsin B Inhibitors with Implications in Alzheimer’s Disease: Computational Refining and Biochemical Evaluation. Cells 2021, 10, 1946. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, Z.-F.; Zhang, D.; Wang, Z.-Y.; Wang, L. Quercetin Alleviates Cadmium-Induced Autophagy Inhibition via TFEB-Dependent Lysosomal Restoration in Primary Proximal Tubular Cells. Ecotoxicol. Environ. Saf. 2021, 208, 111743. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Yuan, Y.; Liu, F.; Li, L.; Liu, J.; Chen, Y.; Cheng, J.; Lu, Y. PGC-1α Alleviates Mitochondrial Dysfunction via TFEB-Mediated Autophagy in Cisplatin-Induced Acute Kidney Injury. Aging 2021, 13, 8421–8439. [Google Scholar] [CrossRef]
- Fu, Y.; Hong, L.; Xu, J.; Zhong, G.; Gu, Q.; Gu, Q.; Guan, Y.; Zheng, X.; Dai, Q.; Luo, X.; et al. Discovery of a Small Molecule Targeting Autophagy via ATG4B Inhibition and Cell Death of Colorectal Cancer Cells in Vitro and in Vivo. Autophagy 2018, 15, 295–311. [Google Scholar] [CrossRef]
- Liu, Y.; Luo, X.; Shan, H.; Fu, Y.; Gu, Q.; Zheng, X.; Dai, Q.; Xia, F.; Zheng, Z.; Liu, P.; et al. Niclosamide Triggers Non-Canonical LC3 Lipidation. Cells 2019, 8, 248. [Google Scholar] [CrossRef]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, L.; Liang, Y.; Fu, Y.; Liang, Z.; Zheng, J.; Lan, J.; Shen, F.; Huang, Z. Toosendanin Induces Hepatocyte Damage by Inhibiting Autophagic Flux via TFEB-Mediated Lysosomal Dysfunction. Pharmaceuticals 2022, 15, 1509. https://doi.org/10.3390/ph15121509
Luo L, Liang Y, Fu Y, Liang Z, Zheng J, Lan J, Shen F, Huang Z. Toosendanin Induces Hepatocyte Damage by Inhibiting Autophagic Flux via TFEB-Mediated Lysosomal Dysfunction. Pharmaceuticals. 2022; 15(12):1509. https://doi.org/10.3390/ph15121509
Chicago/Turabian StyleLuo, Li, Yonghong Liang, Yuanyuan Fu, Zhiyuan Liang, Jinfen Zheng, Jie Lan, Feihai Shen, and Zhiying Huang. 2022. "Toosendanin Induces Hepatocyte Damage by Inhibiting Autophagic Flux via TFEB-Mediated Lysosomal Dysfunction" Pharmaceuticals 15, no. 12: 1509. https://doi.org/10.3390/ph15121509
APA StyleLuo, L., Liang, Y., Fu, Y., Liang, Z., Zheng, J., Lan, J., Shen, F., & Huang, Z. (2022). Toosendanin Induces Hepatocyte Damage by Inhibiting Autophagic Flux via TFEB-Mediated Lysosomal Dysfunction. Pharmaceuticals, 15(12), 1509. https://doi.org/10.3390/ph15121509