


Cytoskeletal and Cytoskeleton-Associated Proteins: Key Regulators of Cancer Stem Cell Properties
 , ,
, ,
Abstract
1. Introduction
2. Cancer Stem Cells
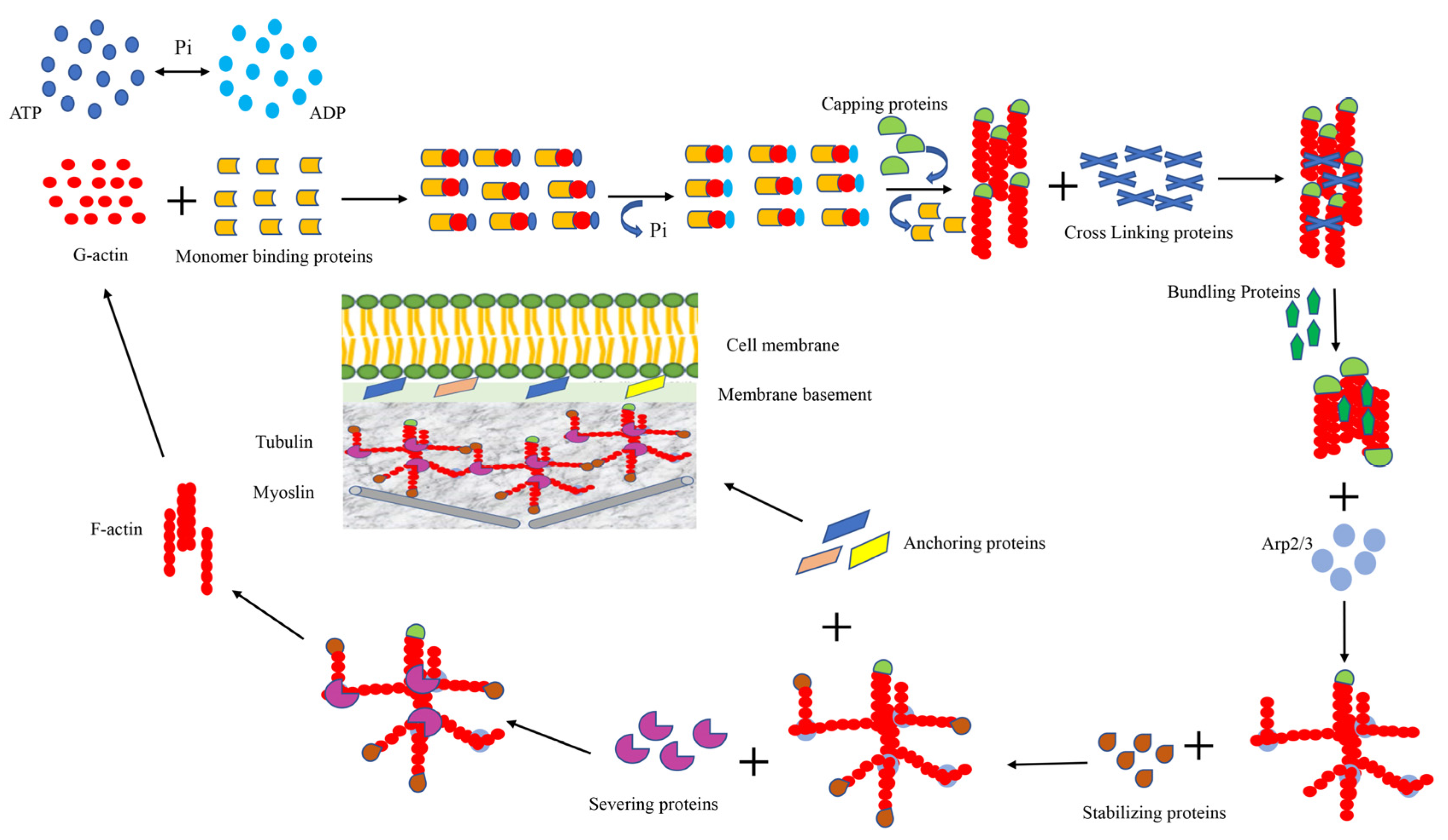
3. Cytoskeleton of the Cell
3.1. Monomer-Binding Proteins
3.2. Cross-Linking and Bundling Proteins
3.3. Anchoring Proteins
3.4. Capping and Severing Proteins
3.5. Stabilizing Protein and Signaling Protein
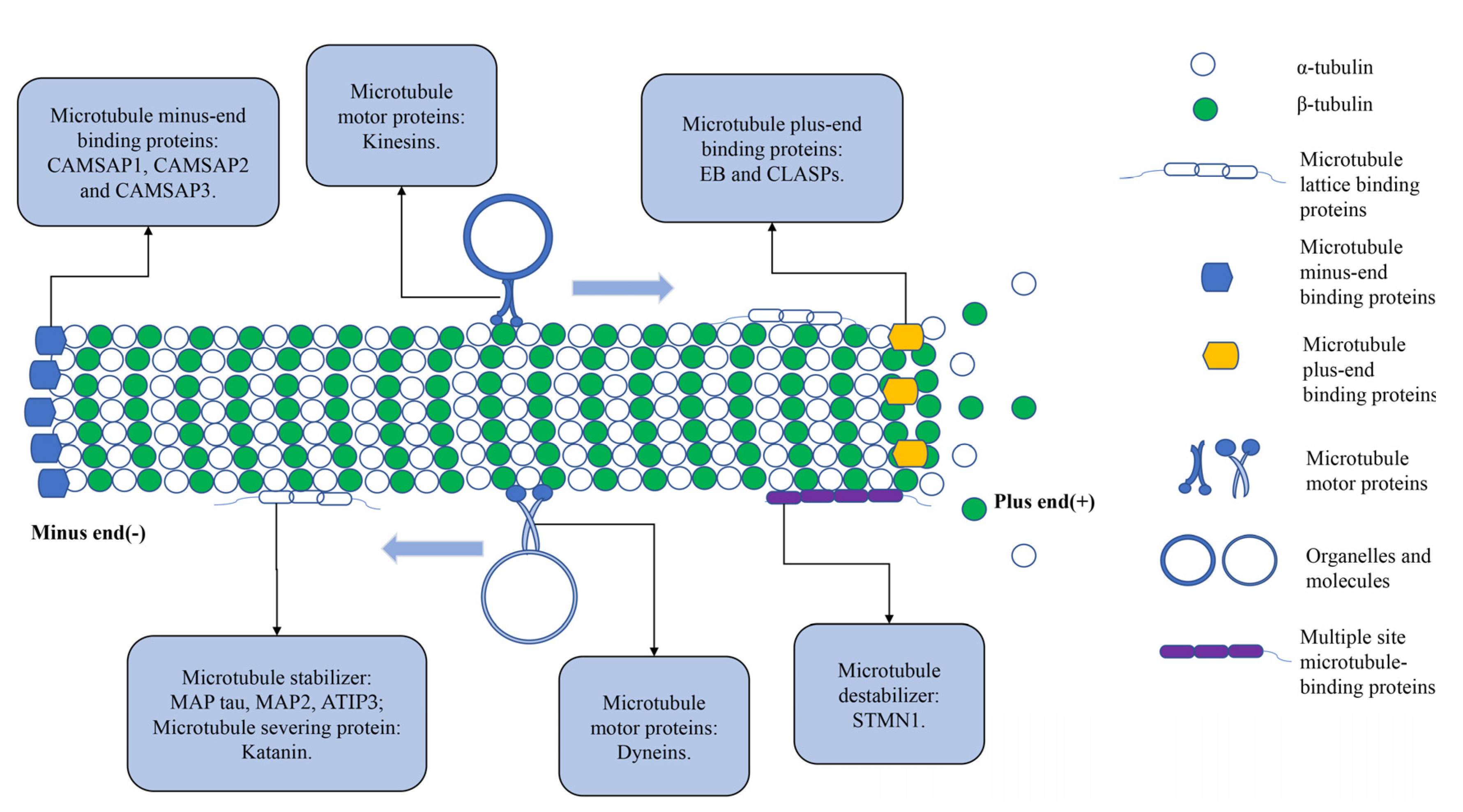
3.6. Microtubule-Associated Proteins
3.7. Other Components of the Cytoskeleton
4. Cytoskeleton and CSCs
4.1. Actin and CSCs
4.2. Monomer-Binding Proteins and CSCs
4.3. Cross-Linking and Bundling Proteins Interact with CSCs
4.4. Anchoring Proteins and CSCs
4.5. Capping and Severing Proteins Interact with CSCs
4.6. Stabilizing and Signaling Proteins Interact with CSCs
4.7. Microtubule-Associated Proteins Interact with CSCs
4.8. Other Components of the Cytoskeleton on CSCs
4.9. Mitochondria-Cytoskeleton Interactions and CSCs
5. Therapeutic Strategies Targeting Cytoskeleton
5.1. Therapeutic Strategies Targeting Actin
5.2. Therapeutic Strategies Targeting ABPs
5.3. Therapeutic Strategies Targeting Microtubules and IFs
5.4. Therapeutic Strategies Targeting MAPs
5.5. Therapeutic Strategies Targeting the Metabolism of CSCs
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- D’Alterio, C.; Scala, S.; Sozzi, G.; Roz, L.; Bertolini, G. Paradoxical effects of chemotherapy on tumor relapse and metastasis promotion. Semin. Cancer Biol. 2020, 60, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Mayea, Y.; Mir, C.; Masson, F.; Paciucci, R.; ME, L.L. Insights into new mechanisms and models of cancer stem cell multidrug resistance. Semin. Cancer Biol. 2020, 60, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Walcher, L.; Kistenmacher, A.K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauss, A.; Blaudszun, A.R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells-Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Li, Y.; Ge, H.; Ghadban, T.; Reeh, M.; Gungor, C. The Extracellular Matrix: A Key Accomplice of Cancer Stem Cell Migration, Metastasis Formation, and Drug Resistance in PDAC. Cancers 2022, 14, 3998. [Google Scholar] [CrossRef]
- Voog, J.; Jones, D.L. Stem cells and the niche: A dynamic duo. Cell Stem Cell 2010, 6, 103–115. [Google Scholar] [CrossRef]
- Jin, J.; Bakker, A.D.; Wu, G.; Klein-Nulend, J.; Jaspers, R.T. Physicochemical Niche Conditions and Mechanosensing by Osteocytes and Myocytes. Curr. Osteoporos. Rep. 2019, 17, 235–249. [Google Scholar] [CrossRef]
- Morrison, S.J.; Kimble, J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature 2006, 441, 1068–1074. [Google Scholar] [CrossRef]
- Li, X.; Wang, J. Mechanical tumor microenvironment and transduction: Cytoskeleton mediates cancer cell invasion and metastasis. Int. J. Biol. Sci. 2020, 16, 2014–2028. [Google Scholar] [CrossRef] [PubMed]
- Fife, C.M.; McCarroll, J.A.; Kavallaris, M. Movers and shakers: Cell cytoskeleton in cancer metastasis. Br. J. Pharmacol. 2014, 171, 5507–5523. [Google Scholar] [CrossRef]
- Strube, F.; Infanger, M.; Wehland, M.; Delvinioti, X.; Romswinkel, A.; Dietz, C.; Kraus, A. Alteration of Cytoskeleton Morphology and Gene Expression in Human Breast Cancer Cells under Simulated Microgravity. Cell J. 2020, 22, 106–114. [Google Scholar] [CrossRef]
- Samardzija, C.; Greening, D.W.; Escalona, R.; Chen, M.; Bilandzic, M.; Luwor, R.; Kannourakis, G.; Findlay, J.K.; Ahmed, N. Knockdown of stem cell regulator Oct4A in ovarian cancer reveals cellular reprogramming associated with key regulators of cytoskeleton-extracellular matrix remodelling. Sci. Rep. 2017, 7, 46312. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Cheong, J.H. Role of Mitochondria-Cytoskeleton Interactions in the Regulation of Mitochondrial Structure and Function in Cancer Stem Cells. Cells 2020, 9, 1691. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- So, J.Y.; Suh, N. Targeting cancer stem cells in solid tumors by vitamin D. J. Steroid Biochem. Mol. Biol. 2015, 148, 79–85. [Google Scholar] [CrossRef]
- Sulaiman, A.; McGarry, S.; Han, X.; Liu, S.; Wang, L. CSCs in Breast Cancer-One Size Does Not Fit All: Therapeutic Advances in Targeting Heterogeneous Epithelial and Mesenchymal CSCs. Cancers 2019, 11, 1128. [Google Scholar] [CrossRef]
- Shibata, M.; Hoque, M.O. Targeting Cancer Stem Cells: A Strategy for Effective Eradication of Cancer. Cancers 2019, 11, 732. [Google Scholar] [CrossRef]
- Matsui, W.; Huff, C.A.; Wang, Q.; Malehorn, M.T.; Barber, J.; Tanhehco, Y.; Smith, B.D.; Civin, C.I.; Jones, R.J. Characterization of clonogenic multiple myeloma cells. Blood 2004, 103, 2332–2336. [Google Scholar] [CrossRef]
- Bjerkvig, R.; Tysnes, B.B.; Aboody, K.S.; Najbauer, J.; Terzis, A.J. Opinion: The origin of the cancer stem cell: Current controversies and new insights. Nat. Rev. Cancer 2005, 5, 899–904. [Google Scholar] [CrossRef]
- Hill, R.P. Identifying cancer stem cells in solid tumors: Case not proven. Cancer Res. 2006, 66, 1891–1895. [Google Scholar] [CrossRef]
- Kanwar, S.S.; Yu, Y.; Nautiyal, J.; Patel, B.B.; Majumdar, A.P. The Wnt/beta-catenin pathway regulates growth and maintenance of colonospheres. Mol. Cancer 2010, 9, 212. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wakeman, T.P.; Lathia, J.D.; Hjelmeland, A.B.; Wang, X.F.; White, R.R.; Rich, J.N.; Sullenger, B.A. Notch promotes radioresistance of glioma stem cells. Stem Cells 2010, 28, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef]
- Nguyen, L.V.; Vanner, R.; Dirks, P.; Eaves, C.J. Cancer stem cells: An evolving concept. Nat. Rev. Cancer 2012, 12, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef]
- Jin, X.; Jin, X.; Kim, H. Cancer stem cells and differentiation therapy. Tumour Biol. 2017, 39, 1010428317729933. [Google Scholar] [CrossRef]
- Talukdar, S.; Bhoopathi, P.; Emdad, L.; Das, S.; Sarkar, D.; Fisher, P.B. Dormancy and cancer stem cells: An enigma for cancer therapeutic targeting. Adv. Cancer Res. 2019, 141, 43–84. [Google Scholar] [CrossRef]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef]
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 2019, 18, 157. [Google Scholar] [CrossRef]
- Lee, E.; Yang, J.; Ku, M.; Kim, N.H.; Park, Y.; Park, C.B.; Suh, J.S.; Park, E.S.; Yook, J.I.; Mills, G.B.; et al. Metabolic stress induces a Wnt-dependent cancer stem cell-like state transition. Cell Death Dis. 2015, 6, e1805. [Google Scholar] [CrossRef]
- Peiris-Pages, M.; Martinez-Outschoorn, U.E.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer stem cell metabolism. Breast Cancer Res. 2016, 18, 55. [Google Scholar] [CrossRef] [PubMed]
- Obre, E.; Rossignol, R. Emerging concepts in bioenergetics and cancer research: Metabolic flexibility, coupling, symbiosis, switch, oxidative tumors, metabolic remodeling, signaling and bioenergetic therapy. Int. J. Biochem. Cell Biol. 2015, 59, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y. Targeting cancer energy metabolism: A potential systemic cure for cancer. Arch. Pharm. Res. 2019, 42, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef]
- Araujo, E.P.; Carvalheira, J.B.; Velloso, L.A. Disruption of metabolic pathways--perspectives for the treatment of cancer. Curr. Cancer Drug Targets 2006, 6, 77–87. [Google Scholar] [CrossRef]
- Janiszewska, M.; Suva, M.L.; Riggi, N.; Houtkooper, R.H.; Auwerx, J.; Clement-Schatlo, V.; Radovanovic, I.; Rheinbay, E.; Provero, P.; Stamenkovic, I. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012, 26, 1926–1944. [Google Scholar] [CrossRef]
- Pasto, A.; Bellio, C.; Pilotto, G.; Ciminale, V.; Silic-Benussi, M.; Guzzo, G.; Rasola, A.; Frasson, C.; Nardo, G.; Zulato, E.; et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget 2014, 5, 4305–4319. [Google Scholar] [CrossRef]
- Shen, Y.A.; Lin, C.H.; Chi, W.H.; Wang, C.Y.; Hsieh, Y.T.; Wei, Y.H.; Chen, Y.J. Resveratrol Impedes the Stemness, Epithelial-Mesenchymal Transition, and Metabolic Reprogramming of Cancer Stem Cells in Nasopharyngeal Carcinoma through p53 Activation. Evid. Based Complement. Alternat. Med. 2013, 2013, 590393. [Google Scholar] [CrossRef]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef]
- Ye, X.Q.; Li, Q.; Wang, G.H.; Sun, F.F.; Huang, G.J.; Bian, X.W.; Yu, S.C.; Qian, G.S. Mitochondrial and energy metabolism-related properties as novel indicators of lung cancer stem cells. Int. J. Cancer 2011, 129, 820–831. [Google Scholar] [CrossRef]
- Farnie, G.; Sotgia, F.; Lisanti, M.P. High mitochondrial mass identifies a sub-population of stem-like cancer cells that are chemo-resistant. Oncotarget 2015, 6, 30472–30486. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, D.; Wang, D.; Liu, X.; Yin, N.; Song, Y.; Lu, S.H.; Ju, Z.; Zhan, Q. Quiescence and attenuated DNA damage response promote survival of esophageal cancer stem cells. J. Cell. Biochem. 2012, 113, 3643–3652. [Google Scholar] [CrossRef] [PubMed]
- Kreso, A.; O’Brien, C.A.; van Galen, P.; Gan, O.I.; Notta, F.; Brown, A.M.; Ng, K.; Ma, J.; Wienholds, E.; Dunant, C.; et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science 2013, 339, 543–548. [Google Scholar] [CrossRef]
- Erin, N.; Grahovac, J.; Brozovic, A.; Efferth, T. Tumor microenvironment and epithelial mesenchymal transition as targets to overcome tumor multidrug resistance. Drug Resist. Updates 2020, 53, 100715. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Nogales, E. Structural insight into microtubule function. Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 397–420. [Google Scholar] [CrossRef]
- Heald, R.; Nogales, E. Microtubule dynamics. J. Cell Sci. 2002, 115, 3–4. [Google Scholar] [CrossRef]
- Popowicz, G.M.; Schleicher, M.; Noegel, A.A.; Holak, T.A. Filamins: Promiscuous organizers of the cytoskeleton. Trends Biochem. Sci. 2006, 31, 411–419. [Google Scholar] [CrossRef]
- Grintsevich, E.E.; Ahmed, G.; Ginosyan, A.A.; Wu, H.; Rich, S.K.; Reisler, E.; Terman, J.R. Profilin and Mical combine to impair F-actin assembly and promote disassembly and remodeling. Nat. Commun. 2021, 12, 5542. [Google Scholar] [CrossRef]
- Merino, F.; Pospich, S.; Raunser, S. Towards a structural understanding of the remodeling of the actin cytoskeleton. Semin. Cell Dev. Biol. 2020, 102, 51–64. [Google Scholar] [CrossRef]
- Ofer, N.; Abu Shah, E.; Keren, K. Differential mapping of the free barbed and pointed ends of actin filaments in cells. Cytoskeleton 2014, 71, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Bisaria, A.; Hayer, A.; Garbett, D.; Cohen, D.; Meyer, T. Membrane-proximal F-actin restricts local membrane protrusions and directs cell migration. Science 2020, 368, 1205–1210. [Google Scholar] [CrossRef] [PubMed]
- Manor, U.; Bartholomew, S.; Golani, G.; Christenson, E.; Kozlov, M.; Higgs, H.; Spudich, J.; Lippincott-Schwartz, J. A mitochondria-anchored isoform of the actin-nucleating spire protein regulates mitochondrial division. Elife 2015, 4, e08828. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.S.; Wong, Y.C.; Simpson, C.L.; Holzbaur, E.L. Dynamic actin cycling through mitochondrial subpopulations locally regulates the fission-fusion balance within mitochondrial networks. Nat. Commun. 2016, 7, 12886. [Google Scholar] [CrossRef] [PubMed]
- Kruppa, A.J.; Kishi-Itakura, C.; Masters, T.A.; Rorbach, J.E.; Grice, G.L.; Kendrick-Jones, J.; Nathan, J.A.; Minczuk, M.; Buss, F. Myosin VI-Dependent Actin Cages Encapsulate Parkin-Positive Damaged Mitochondria. Dev. Cell 2018, 44, 484–499.e486. [Google Scholar] [CrossRef]
- Valencia, D.A.; Quinlan, M.E. Formins. Curr. Biol. 2021, 31, R517–R522. [Google Scholar] [CrossRef]
- Izdebska, M.; Zielinska, W.; Halas-Wisniewska, M.; Grzanka, A. Involvement of Actin and Actin-Binding Proteins in Carcinogenesis. Cells 2020, 9, 2245. [Google Scholar] [CrossRef]
- Izdebska, M.; Zielinska, W.; Grzanka, D.; Gagat, M. The Role of Actin Dynamics and Actin-Binding Proteins Expression in Epithelial-to-Mesenchymal Transition and Its Association with Cancer Progression and Evaluation of Possible Therapeutic Targets. Biomed. Res. Int. 2018, 2018, 4578373. [Google Scholar] [CrossRef]
- Namgoong, S.; Kim, N.H. Roles of actin binding proteins in mammalian oocyte maturation and beyond. Cell Cycle 2016, 15, 1830–1843. [Google Scholar] [CrossRef]
- Lee, C.W.; Vitriol, E.A.; Shim, S.; Wise, A.L.; Velayutham, R.P.; Zheng, J.Q. Dynamic localization of G-actin during membrane protrusion in neuronal motility. Curr. Biol. 2013, 23, 1046–1056. [Google Scholar] [CrossRef]
- Pinto-Costa, R.; Sousa, M.M. Profilin as a dual regulator of actin and microtubule dynamics. Cytoskeleton 2020, 77, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.J.; Funes, S.; McKeon, J.E.; Morgan, B.R.; Boopathy, S.; O’Connor, L.C.; Bilsel, O.; Massi, F.; Jegou, A.; Bosco, D.A. ALS-linked PFN1 variants exhibit loss and gain of functions in the context of formin-induced actin polymerization. Proc. Natl. Acad. Sci. USA 2021, 118, e2024605118. [Google Scholar] [CrossRef] [PubMed]
- Skruber, K.; Read, T.A.; Vitriol, E.A. Reconsidering an active role for G-actin in cytoskeletal regulation. J. Cell Sci. 2018, 131, jcs203760. [Google Scholar] [CrossRef] [PubMed]
- Coumans, J.V.F.; Davey, R.J.; Moens, P.D.J. Cofilin and profilin: Partners in cancer aggressiveness. Biophys. Rev. 2018, 10, 1323–1335. [Google Scholar] [CrossRef]
- Vartiainen, M.K.; Sarkkinen, E.M.; Matilainen, T.; Salminen, M.; Lappalainen, P. Mammals have two twinfilin isoforms whose subcellular localizations and tissue distributions are differentially regulated. J. Biol. Chem. 2003, 278, 34347–34355. [Google Scholar] [CrossRef] [PubMed]
- Poukkula, M.; Kremneva, E.; Serlachius, M.; Lappalainen, P. Actin-depolymerizing factor homology domain: A conserved fold performing diverse roles in cytoskeletal dynamics. Cytoskeleton 2011, 68, 471–490. [Google Scholar] [CrossRef]
- Becker, I.C.; Scheller, I.; Wackerbarth, L.M.; Beck, S.; Heib, T.; Aurbach, K.; Manukjan, G.; Gross, C.; Spindler, M.; Nagy, Z.; et al. Actin/microtubule crosstalk during platelet biogenesis in mice is critically regulated by Twinfilin1 and Cofilin1. Blood Adv. 2020, 4, 2124–2134. [Google Scholar] [CrossRef]
- Johnston, A.B.; Hilton, D.M.; McConnell, P.; Johnson, B.; Harris, M.T.; Simone, A.; Amarasinghe, G.K.; Cooper, J.A.; Goode, B.L. A novel mode of capping protein-regulation by twinfilin. Elife 2018, 7, e41313. [Google Scholar] [CrossRef]
- Kaishang, Z.; Xue, P.; Shaozhong, Z.; Yingying, F.; Yan, Z.; Chanjun, S.; Zhenzhen, L.; Xiangnan, L. Elevated expression of Twinfilin-1 is correlated with inferior prognosis of lung adenocarcinoma. Life Sci. 2018, 215, 159–169. [Google Scholar] [CrossRef]
- Husson, C.; Cantrelle, F.X.; Roblin, P.; Didry, D.; Le, K.H.; Perez, J.; Guittet, E.; Van Heijenoort, C.; Renault, L.; Carlier, M.F. Multifunctionality of the beta-thymosin/WH2 module: G-actin sequestration, actin filament growth, nucleation, and severing. Ann. N. Y. Acad. Sci. 2010, 1194, 44–52. [Google Scholar] [CrossRef]
- Bjorklund, G.; Dadar, M.; Aaseth, J.; Chirumbolo, S. Thymosin beta4: A Multi-Faceted Tissue Repair Stimulating Protein in Heart Injury. Curr. Med. Chem. 2020, 27, 6294–6305. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.N.; Masci, P.P.; Lavin, M.F. Disruption of spectrin-like cytoskeleton in differentiating keratinocytes by PKCdelta activation is associated with phosphorylated adducin. PLoS ONE 2011, 6, e28267. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.M.; Moon, E.Y. Actin-sequestering protein, thymosin beta-4, induces paclitaxel resistance through ROS/HIF-1alpha stabilization in HeLa human cervical tumor cells. Life Sci. 2010, 87, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.O.; Lee, J.I.; Hong, S.P.; Hong, S.D. Thymosin beta4 induces proliferation, invasion, and epithelial-to-mesenchymal transition of oral squamous cell carcinoma. Amino Acids 2016, 48, 117–127. [Google Scholar] [CrossRef]
- Lv, S.; Cai, H.; Xu, Y.; Dai, J.; Rong, X.; Zheng, L. Thymosinbeta 4 induces angiogenesis in critical limb ischemia mice via regulating Notch/NFkappaB pathway. Int. J. Mol. Med. 2020, 46, 1347–1358. [Google Scholar] [CrossRef]
- Kudryashova, E.; Heisler, D.B.; Williams, B.; Harker, A.J.; Shafer, K.; Quinlan, M.E.; Kovar, D.R.; Vavylonis, D.; Kudryashov, D.S. Actin Cross-Linking Toxin Is a Universal Inhibitor of Tandem-Organized and Oligomeric G-Actin Binding Proteins. Curr. Biol. 2018, 28, 1536–1547.e1539. [Google Scholar] [CrossRef]
- Jayo, A.; Malboubi, M.; Antoku, S.; Chang, W.; Ortiz-Zapater, E.; Groen, C.; Pfisterer, K.; Tootle, T.; Charras, G.; Gundersen, G.G.; et al. Fascin Regulates Nuclear Movement and Deformation in Migrating Cells. Dev. Cell 2016, 38, 371–383. [Google Scholar] [CrossRef]
- Gallop, J.L. Filopodia and their links with membrane traffic and cell adhesion. Semin. Cell Dev. Biol. 2020, 102, 81–89. [Google Scholar] [CrossRef]
- Lin, S.; Taylor, M.D.; Singh, P.K.; Yang, S. How does fascin promote cancer metastasis? FEBS J. 2021, 288, 1434–1446. [Google Scholar] [CrossRef]
- Cardama, G.A.; Gonzalez, N.; Maggio, J.; Menna, P.L.; Gomez, D.E. Rho GTPases as therapeutic targets in cancer (Review). Int. J. Oncol. 2017, 51, 1025–1034. [Google Scholar] [CrossRef]
- Parsons, M.; Adams, J.C. Rac regulates the interaction of fascin with protein kinase C in cell migration. J. Cell Sci. 2008, 121, 2805–2813. [Google Scholar] [CrossRef] [PubMed]
- Jayo, A.; Parsons, M.; Adams, J.C. A novel Rho-dependent pathway that drives interaction of fascin-1 with p-Lin-11/Isl-1/Mec-3 kinase (LIMK) 1/2 to promote fascin-1/actin binding and filopodia stability. BMC Biol. 2012, 10, 72. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Morales, N.; Holenka, T.K.; Schock, F. Filamin actin-binding and titin-binding fulfill distinct functions in Z-disc cohesion. PLoS Genet. 2017, 13, e1006880. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhu, H.; Lu, Y.; Wang, Z.; Kennedy, D. The elastic properties and deformation mechanisms of actin filament networks crosslinked by filamins. J. Mech. Behav. Biomed. Mater. 2020, 112, 104075. [Google Scholar] [CrossRef] [PubMed]
- Lad, Y.; Kiema, T.; Jiang, P.; Pentikainen, O.T.; Coles, C.H.; Campbell, I.D.; Calderwood, D.A.; Ylanne, J. Structure of three tandem filamin domains reveals auto-inhibition of ligand binding. EMBO J. 2007, 26, 3993–4004. [Google Scholar] [CrossRef]
- Svitkina, T. The Actin Cytoskeleton and Actin-Based Motility. Cold Spring Harb. Perspect. Biol. 2018, 10, a018267. [Google Scholar] [CrossRef]
- Iwamoto, D.V.; Huehn, A.; Simon, B.; Huet-Calderwood, C.; Baldassarre, M.; Sindelar, C.V.; Calderwood, D.A. Structural basis of the filamin A actin-binding domain interaction with F-actin. Nat. Struct. Mol. Biol. 2018, 25, 918–927. [Google Scholar] [CrossRef]
- Tirupula, K.C.; Ithychanda, S.S.; Mohan, M.L.; Naga Prasad, S.V.; Qin, J.; Karnik, S.S. G protein-coupled receptors directly bind filamin A with high affinity and promote filamin phosphorylation. Biochemistry 2015, 54, 6673–6683. [Google Scholar] [CrossRef]
- Liem, R.K. Cytoskeletal Integrators: The Spectrin Superfamily. Cold Spring Harb. Perspect. Biol. 2016, 8, a018259. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, C.; Zhao, Q.; Li, D. Spectrin: Structure, function and disease. Sci. China Life Sci. 2013, 56, 1076–1085. [Google Scholar] [CrossRef]
- Unudurthi, S.D.; Greer-Short, A.; Patel, N.; Nassal, D.; Hund, T.J. Spectrin-based pathways underlying electrical and mechanical dysfunction in cardiac disease. Expert Rev. Cardiovasc. Ther. 2018, 16, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yago, T.; Zhang, N.; Abdisalaam, S.; Alexandrakis, G.; Rodgers, W.; McEver, R.P. Cytoskeletal regulation of CD44 membrane organization and interactions with E-selectin. J. Biol. Chem. 2014, 289, 35159–35171. [Google Scholar] [CrossRef] [PubMed]
- Aseervatham, J. Cytoskeletal Remodeling in Cancer. Biology 2020, 9, 385. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Song, Y.; Tu, W.; He, X.; Lin, J.; Liu, F. beta-2 spectrin is involved in hepatocyte proliferation through the interaction of TGFbeta/Smad and PI3K/AKT signalling. Liver Int. 2012, 32, 1103–1111. [Google Scholar] [CrossRef]
- Wu, X.T.; Sun, L.W.; Yang, X.; Ding, D.; Han, D.; Fan, Y.B. The potential role of spectrin network in the mechanotransduction of MLO-Y4 osteocytes. Sci. Rep. 2017, 7, 40940. [Google Scholar] [CrossRef]
- Murphy, A.C.; Young, P.W. The actinin family of actin cross-linking proteins—A genetic perspective. Cell Biosci. 2015, 5, 49. [Google Scholar] [CrossRef]
- Thomas, D.G.; Robinson, D.N. The fifth sense: Mechanosensory regulation of alpha-actinin-4 and its relevance for cancer metastasis. Semin. Cell Dev. Biol. 2017, 71, 68–74. [Google Scholar] [CrossRef]
- Nagano, M.; Hoshino, D.; Koshikawa, N.; Akizawa, T.; Seiki, M. Turnover of focal adhesions and cancer cell migration. Int. J. Cell Biol. 2012, 2012, 310616. [Google Scholar] [CrossRef]
- Castellano, E.; Downward, J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef]
- Deming, P.B.; Campbell, S.L.; Stone, J.B.; Rivard, R.L.; Mercier, A.L.; Howe, A.K. Anchoring of protein kinase A by ERM (ezrin-radixin-moesin) proteins is required for proper netrin signaling through DCC (deleted in colorectal cancer). J. Biol. Chem. 2015, 290, 5783–5796. [Google Scholar] [CrossRef]
- Gao, S.; Dai, Y.; Yin, M.; Ye, J.; Li, G.; Yu, J. Potential transcriptional regulatory regions exist upstream of the human ezrin gene promoter in esophageal carcinoma cells. Acta Biochim. Biophys. Sin. 2011, 43, 455–464. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Biri-Kovacs, B.; Kiss, B.; Vadaszi, H.; Gogl, G.; Palfy, G.; Torok, G.; Homolya, L.; Bodor, A.; Nyitray, L. Ezrin interacts with S100A4 via both its N- and C-terminal domains. PLoS ONE 2017, 12, e0177489. [Google Scholar] [CrossRef]
- Lema, B.E.; Patricio, G.M.; Kreimann, E.L. Nuclear expression of NHERF1/EBP50 in Clear Cell Renal Cell Carcinoma. Acta Histochem. 2021, 123, 151717. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, R.P.; Veltman, D.; Machesky, L.M. Actin-bundling proteins in cancer progression at a glance. J. Cell Sci. 2012, 125, 1073–1079. [Google Scholar] [CrossRef]
- Shi, K.; Yang, L.; Du, X.; Guo, D.; Xue, L. Molecular chaperone Hsp90 protects KCBP from degradation by proteasome in Dunaliella salina cells. Folia Microbiol. 2021, 66, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Kamioka, H.; Tomono, T.; Fujita, A.; Onozato, R.; Iijima, M.; Tsuchida, S.; Arai, T.; Fujita, Y.; Zhang, X.; Yano, K.; et al. Moesin-Mediated P-Glycoprotein Activation During Snail-Induced Epithelial-Mesenchymal Transition in Lung Cancer Cells. J. Pharm. Sci. 2020, 109, 2302–2308. [Google Scholar] [CrossRef]
- Rahimi, N.; Ho, R.X.Y.; Chandler, K.B.; De La Cena, K.O.C.; Amraei, R.; Mitchel, A.J.; Engblom, N.; Costello, C.E. The cell adhesion molecule TMIGD1 binds to moesin and regulates tubulin acetylation and cell migration. J. Biomed. Sci. 2021, 28, 61. [Google Scholar] [CrossRef]
- Karvar, S.; Ansa-Addo, E.A.; Suda, J.; Singh, S.; Zhu, L.; Li, Z.; Rockey, D.C. Moesin, an Ezrin/Radixin/Moesin Family Member, Regulates Hepatic Fibrosis. Hepatology 2020, 72, 1073–1084. [Google Scholar] [CrossRef]
- Jiang, Q.H.; Wang, A.X.; Chen, Y. Radixin enhances colon cancer cell invasion by increasing MMP-7 production via Rac1-ERK pathway. Sci. World J. 2014, 2014, 340271. [Google Scholar] [CrossRef]
- Hoeflich, K.P.; Ikura, M. Radixin: Cytoskeletal adopter and signaling protein. Int. J. Biochem. Cell Biol. 2004, 36, 2131–2136. [Google Scholar] [CrossRef]
- Michie, K.A.; Bermeister, A.; Robertson, N.O.; Goodchild, S.C.; Curmi, P.M.G. Two Sides of the Coin: Ezrin/Radixin/Moesin and Merlin Control Membrane Structure and Contact Inhibition. Int. J. Mol. Sci. 2019, 20, 1996. [Google Scholar] [CrossRef] [PubMed]
- Chinthalapudi, K.; Mandati, V.; Zheng, J.; Sharff, A.J.; Bricogne, G.; Griffin, P.R.; Kissil, J.; Izard, T. Lipid binding promotes the open conformation and tumor-suppressive activity of neurofibromin 2. Nat. Commun. 2018, 9, 1338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Luo, X.; Lin, J.; Fu, S.; Feng, P.; Su, H.; He, X.; Liang, X.; Liu, K.; Deng, W. Gelsolin Promotes Cancer Progression by Regulating Epithelial-Mesenchymal Transition in Hepatocellular Carcinoma and Correlates with a Poor Prognosis. J. Oncol. 2020, 2020, 1980368. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.; Larsson, M.; Robinson, R.C.; Burtnick, L.D. Gelsolin: The tail of a molecular gymnast. Cytoskeleton 2013, 70, 360–384. [Google Scholar] [CrossRef] [PubMed]
- Morley, S.C.; Sung, J.; Sun, G.P.; Martelli, M.P.; Bunnell, S.C.; Bierer, B.E. Gelsolin overexpression alters actin dynamics and tyrosine phosphorylation of lipid raft-associated proteins in Jurkat T cells. Mol. Immunol. 2007, 44, 2469–2480. [Google Scholar] [CrossRef][Green Version]
- Narita, A. ADF/cofilin regulation from a structural viewpoint. J. Muscle Res. Cell Motil. 2020, 41, 141–151. [Google Scholar] [CrossRef]
- Ostrowska, Z.; Moraczewska, J. Cofilin—A protein controlling dynamics of actin filaments. Postepy Hig. Med. Dosw. (Online) 2017, 71, 339–351. [Google Scholar] [CrossRef]
- Hamill, S.; Lou, H.J.; Turk, B.E.; Boggon, T.J. Structural Basis for Noncanonical Substrate Recognition of Cofilin/ADF Proteins by LIM Kinases. Mol. Cell 2016, 62, 397–408. [Google Scholar] [CrossRef]
- Kaushik, V.; Brunnert, D.; Hanschmann, E.M.; Sharma, P.K.; Anand, B.G.; Kar, K.; Kateriya, S.; Goyal, P. The intrinsic amyloidogenic propensity of cofilin-1 is aggravated by Cys-80 oxidation: A possible link with neurodegenerative diseases. Biochem. Biophys. Res. Commun. 2021, 569, 187–192. [Google Scholar] [CrossRef]
- Ubelmann, F.; Chamaillard, M.; El-Marjou, F.; Simon, A.; Netter, J.; Vignjevic, D.; Nichols, B.L.; Quezada-Calvillo, R.; Grandjean, T.; Louvard, D.; et al. Enterocyte loss of polarity and gut wound healing rely upon the F-actin-severing function of villin. Proc. Natl. Acad. Sci. USA 2013, 110, E1380–E1389. [Google Scholar] [CrossRef]
- Hampton, C.M.; Liu, J.; Taylor, D.W.; DeRosier, D.J.; Taylor, K.A. The 3D structure of villin as an unusual F-Actin crosslinker. Structure 2008, 16, 1882–1891. [Google Scholar] [CrossRef] [PubMed]
- Ghoshdastider, U.; Popp, D.; Burtnick, L.D.; Robinson, R.C. The expanding superfamily of gelsolin homology domain proteins. Cytoskeleton 2013, 70, 775–795. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Kumar, M.; Sharma, N.R.; Sarkar, D.P. Site-specific phosphorylation of villin remodels the actin cytoskeleton to regulate Sendai viral glycoprotein-mediated membrane fusion. FEBS Lett. 2019, 593, 1927–1943. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Zhao, P.; Tomar, A.; Galea, C.A.; Khurana, S. Association of villin with phosphatidylinositol 4,5-bisphosphate regulates the actin cytoskeleton. J. Biol. Chem. 2004, 279, 3096–3110. [Google Scholar] [CrossRef]
- Fowler, V.M.; Dominguez, R. Tropomodulins and Leiomodins: Actin Pointed End Caps and Nucleators in Muscles. Biophys. J. 2017, 112, 1742–1760. [Google Scholar] [CrossRef]
- Ghosh, A.; Fowler, V.M. Tropomodulins. Curr. Biol. 2021, 31, R501–R503. [Google Scholar] [CrossRef]
- Ostrowska, Z.; Robaszkiewicz, K.; Moraczewska, J. Regulation of actin filament turnover by cofilin-1 and cytoplasmic tropomyosin isoforms. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 88–98. [Google Scholar] [CrossRef]
- Gateva, G.; Tojkander, S.; Koho, S.; Carpen, O.; Lappalainen, P. Palladin promotes assembly of non-contractile dorsal stress fibers through VASP recruitment. J. Cell Sci. 2014, 127, 1887–1898. [Google Scholar] [CrossRef]
- Salazar, M.A.; Kwiatkowski, A.V.; Pellegrini, L.; Cestra, G.; Butler, M.H.; Rossman, K.L.; Serna, D.M.; Sondek, J.; Gertler, F.B.; De Camilli, P. Tuba, a novel protein containing bin/amphiphysin/Rvs and Dbl homology domains, links dynamin to regulation of the actin cytoskeleton. J. Biol. Chem. 2003, 278, 49031–49043. [Google Scholar] [CrossRef]
- Bear, J.E.; Gertler, F.B. Ena/VASP: Towards resolving a pointed controversy at the barbed end. J. Cell Sci. 2009, 122, 1947–1953. [Google Scholar] [CrossRef]
- Zhang, Y.T.; Xu, L.H.; Lu, Q.; Liu, K.P.; Liu, P.Y.; Ji, F.; Liu, X.M.; Ouyang, D.Y.; He, X.H. VASP activation via the Galpha13/RhoA/PKA pathway mediates cucurbitacin-B-induced actin aggregation and cofilin-actin rod formation. PLoS ONE 2014, 9, e93547. [Google Scholar] [CrossRef]
- Goodson, H.V.; Jonasson, E.M. Microtubules and Microtubule-Associated Proteins. Cold Spring Harb. Perspect. Biol. 2018, 10, a022608. [Google Scholar] [CrossRef] [PubMed]
- Janke, C.; Magiera, M.M. The tubulin code and its role in controlling microtubule properties and functions. Nat. Rev. Mol. Cell Biol. 2020, 21, 307–326. [Google Scholar] [CrossRef]
- Chaudhary, N.; Nagaraj, R. Tau fibrillogenesis. Subcell. Biochem. 2012, 65, 75–90. [Google Scholar] [CrossRef]
- Breuzard, G.; Pagano, A.; Bastonero, S.; Malesinski, S.; Parat, F.; Barbier, P.; Peyrot, V.; Kovacic, H. Tau regulates the microtubule-dependent migration of glioblastoma cells via the Rho-ROCK signaling pathway. J. Cell Sci. 2019, 132, jcs222851. [Google Scholar] [CrossRef]
- Teng, J.; Takei, Y.; Harada, A.; Nakata, T.; Chen, J.; Hirokawa, N. Synergistic effects of MAP2 and MAP1B knockout in neuronal migration, dendritic outgrowth, and microtubule organization. J. Cell Biol. 2001, 155, 65–76. [Google Scholar] [CrossRef]
- Zehr, E.; Szyk, A.; Piszczek, G.; Szczesna, E.; Zuo, X.; Roll-Mecak, A. Katanin spiral and ring structures shed light on power stroke for microtubule severing. Nat. Struct. Mol. Biol. 2017, 24, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Nehlig, A.; Seiler, C.; Steblyanko, Y.; Dingli, F.; Arras, G.; Loew, D.; Welburn, J.; Prigent, C.; Barisic, M.; Nahmias, C. Reciprocal regulation of Aurora kinase A and ATIP3 in the control of metaphase spindle length. Cell Mol. Life Sci. 2021, 78, 1765–1779. [Google Scholar] [CrossRef]
- Lu, W.; Gelfand, V.I. Moonlighting Motors: Kinesin, Dynein, and Cell Polarity. Trends Cell Biol. 2017, 27, 505–514. [Google Scholar] [CrossRef]
- Gigant, B.; Wang, W.; Dreier, B.; Jiang, Q.; Pecqueur, L.; Pluckthun, A.; Wang, C.; Knossow, M. Structure of a kinesin-tubulin complex and implications for kinesin motility. Nat. Struct. Mol. Biol. 2013, 20, 1001–1007. [Google Scholar] [CrossRef]
- Arora, K.; Talje, L.; Asenjo, A.B.; Andersen, P.; Atchia, K.; Joshi, M.; Sosa, H.; Allingham, J.S.; Kwok, B.H. KIF14 binds tightly to microtubules and adopts a rigor-like conformation. J. Mol. Biol. 2014, 426, 2997–3015. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.J.; Kon, T.; Knight, P.J.; Sutoh, K.; Burgess, S.A. Functions and mechanics of dynein motor proteins. Nat. Rev. Mol. Cell Biol. 2013, 14, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Wattanathamsan, O.; Pongrakhananon, V. Emerging role of microtubule-associated proteins on cancer metastasis. Front. Pharmacol. 2022, 13, 935493. [Google Scholar] [CrossRef] [PubMed]
- Zeitz, M.; Kierfeld, J. Feedback mechanism for microtubule length regulation by stathmin gradients. Biophys. J. 2014, 107, 2860–2871. [Google Scholar] [CrossRef][Green Version]
- Gupta, K.K.; Li, C.; Duan, A.; Alberico, E.O.; Kim, O.V.; Alber, M.S.; Goodson, H.V. Mechanism for the catastrophe-promoting activity of the microtubule destabilizer Op18/stathmin. Proc. Natl. Acad. Sci. USA 2013, 110, 20449–20454. [Google Scholar] [CrossRef]
- Matov, A.; Applegate, K.; Kumar, P.; Thoma, C.; Krek, W.; Danuser, G.; Wittmann, T. Analysis of microtubule dynamic instability using a plus-end growth marker. NaMethods 2010, 7, 761–768. [Google Scholar] [CrossRef]
- Nehlig, A.; Molina, A.; Rodrigues-Ferreira, S.; Honore, S.; Nahmias, C. Regulation of end-binding protein EB1 in the control of microtubule dynamics. Cell Mol. Life Sci. 2017, 74, 2381–2393. [Google Scholar] [CrossRef]
- Lindeboom, J.J.; Nakamura, M.; Saltini, M.; Hibbel, A.; Walia, A.; Ketelaar, T.; Emons, A.M.C.; Sedbrook, J.C.; Kirik, V.; Mulder, B.M.; et al. CLASP stabilization of plus ends created by severing promotes microtubule creation and reorientation. J. Cell Biol. 2019, 218, 190–205. [Google Scholar] [CrossRef]
- Akhmanova, A.; Hoogenraad, C.C. Microtubule minus-end-targeting proteins. Curr. Biol. 2015, 25, R162–R171. [Google Scholar] [CrossRef]
- Atherton, J.; Jiang, K.; Stangier, M.M.; Luo, Y.; Hua, S.; Houben, K.; van Hooff, J.J.E.; Joseph, A.P.; Scarabelli, G.; Grant, B.J.; et al. A structural model for microtubule minus-end recognition and protection by CAMSAP proteins. Nat. Struct. Mol. Biol. 2017, 24, 931–943. [Google Scholar] [CrossRef]
- Guhathakurta, P.; Prochniewicz, E.; Thomas, D.D. Actin-Myosin Interaction: Structure, Function and Drug Discovery. Int. J. Mol. Sci. 2018, 19, 2628. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.C.; Walczak, C.E.; Cochran, J.C. Switch-1 instability at the active site decouples ATP hydrolysis from force generation in myosin II. Cytoskeleton 2021, 78, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Cross, J.A.; Dodding, M.P. Motor-cargo adaptors at the organelle-cytoskeleton interface. Curr. Opin. Cell Biol. 2019, 59, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, H.; Aebi, U. Intermediate Filaments: Structure and Assembly. Cold Spring Harb. Perspect. Biol. 2016, 8, a018242. [Google Scholar] [CrossRef] [PubMed]
- Chung, B.M.; Rotty, J.D.; Coulombe, P.A. Networking galore: Intermediate filaments and cell migration. Curr. Opin. Cell Biol. 2013, 25, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Etienne-Manneville, S. Cytoplasmic Intermediate Filaments in Cell Biology. Annu. Rev. Cell Dev. Biol. 2018, 34, 1–28. [Google Scholar] [CrossRef]
- Hyder, C.L.; Pallari, H.M.; Kochin, V.; Eriksson, J.E. Providing cellular signposts—Post-translational modifications of intermediate filaments. FEBS Lett. 2008, 582, 2140–2148. [Google Scholar] [CrossRef]
- Leduc, C.; Etienne-Manneville, S. Intermediate filaments in cell migration and invasion: The unusual suspects. Curr. Opin. Cell Biol. 2015, 32, 102–112. [Google Scholar] [CrossRef]
- de Pereda, J.M.; Lillo, M.P.; Sonnenberg, A. Structural basis of the interaction between integrin alpha6beta4 and plectin at the hemidesmosomes. EMBO J. 2009, 28, 1180–1190. [Google Scholar] [CrossRef]
- Satelli, A.; Li, S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol. Life Sci. 2011, 68, 3033–3046. [Google Scholar] [CrossRef]
- Ambriz, X.; de Lanerolle, P.; Ambrosio, J.R. The Mechanobiology of the Actin Cytoskeleton in Stem Cells during Differentiation and Interaction with Biomaterials. Stem Cells Int. 2018, 2018, 2891957. [Google Scholar] [CrossRef] [PubMed]
- Low, B.C.; Pan, C.Q.; Shivashankar, G.V.; Bershadsky, A.; Sudol, M.; Sheetz, M. YAP/TAZ as mechanosensors and mechanotransducers in regulating organ size and tumor growth. FEBS Lett. 2014, 588, 2663–2670. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.M.; Chang, J.W. Current status and issues in cancer stem cell study. Cancer Investig. 2008, 26, 741–755. [Google Scholar] [CrossRef] [PubMed]
- Jabbari, E.; Sarvestani, S.K.; Daneshian, L.; Moeinzadeh, S. Optimum 3D Matrix Stiffness for Maintenance of Cancer Stem Cells Is Dependent on Tissue Origin of Cancer Cells. PLoS ONE 2015, 10, e0132377. [Google Scholar] [CrossRef]
- Bourguignon, L.Y.; Shiina, M.; Li, J.J. Hyaluronan-CD44 interaction promotes oncogenic signaling, microRNA functions, chemoresistance, and radiation resistance in cancer stem cells leading to tumor progression. Adv. Cancer Res. 2014, 123, 255–275. [Google Scholar] [CrossRef]
- Witke, W. The role of profilin complexes in cell motility and other cellular processes. Trends Cell Biol. 2004, 14, 461–469. [Google Scholar] [CrossRef]
- Kim, M.J.; Lee, Y.S.; Han, G.Y.; Lee, H.N.; Ahn, C.; Kim, C.W. Profilin 2 promotes migration, invasion, and stemness of HT29 human colorectal cancer stem cells. Biosci. Biotechnol. Biochem. 2015, 79, 1438–1446. [Google Scholar] [CrossRef][Green Version]
- Pouremamali, F.; Vahedian, V.; Hassani, N.; Mirzaei, S.; Pouremamali, A.; Kazemzadeh, H.; Faridvand, Y.; Jafari-Gharabaghlou, D.; Nouri, M.; Maroufi, N.F. The role of SOX family in cancer stem cell maintenance: With a focus on SOX2. Pathol. Res. Pract. 2022, 231, 153783. [Google Scholar] [CrossRef]
- Jiang, C.; Ding, Z.; Joy, M.; Chakraborty, S.; Kim, S.H.; Bottcher, R.; Condeelis, J.; Singh, S.; Roy, P. A balanced level of profilin-1 promotes stemness and tumor-initiating potential of breast cancer cells. Cell Cycle 2017, 16, 2366–2373. [Google Scholar] [CrossRef]
- Kobayashi, T.; Okada, F.; Fujii, N.; Tomita, N.; Ito, S.; Tazawa, H.; Aoyama, T.; Choi, S.K.; Shibata, T.; Fujita, H.; et al. Thymosin-beta4 regulates motility and metastasis of malignant mouse fibrosarcoma cells. Am. J. Pathol. 2002, 160, 869–882. [Google Scholar] [CrossRef]
- Nemolato, S.; Restivo, A.; Cabras, T.; Coni, P.; Zorcolo, L.; Orru, G.; Fanari, M.; Cau, F.; Gerosa, C.; Fanni, D.; et al. Thymosin beta 4 in colorectal cancer is localized predominantly at the invasion front in tumor cells undergoing epithelial mesenchymal transition. Cancer Biol. Ther. 2012, 13, 191–197. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ricci-Vitiani, L.; Mollinari, C.; di Martino, S.; Biffoni, M.; Pilozzi, E.; Pagliuca, A.; de Stefano, M.C.; Circo, R.; Merlo, D.; De Maria, R.; et al. Thymosin beta4 targeting impairs tumorigenic activity of colon cancer stem cells. FASEB J. 2010, 24, 4291–4301. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.I.; Lee, B.Y.; Kang, Y.J.; Jo, J.O.; Lee, S.H.; Kim, H.Y.; Kim, Y.O.; Lee, C.; Koh, S.B.; Kim, A.; et al. Expression patterns of Thymosin beta4 and cancer stem cell marker CD133 in ovarian cancers. Pathol. Oncol. Res. 2013, 19, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Feurino, L.W.; Zhai, Q.; Wang, H.; Fisher, W.E.; Chen, C.; Yao, Q.; Li, M. Thymosin Beta 4 is overexpressed in human pancreatic cancer cells and stimulates proinflammatory cytokine secretion and JNK activation. Cancer Biol. Ther. 2008, 7, 419–423. [Google Scholar] [CrossRef]
- Lee, S.I.; Kim, D.S.; Lee, H.J.; Cha, H.J.; Kim, E.C. The role of thymosin beta 4 on odontogenic differentiation in human dental pulp cells. PLoS ONE 2013, 8, e61960. [Google Scholar] [CrossRef]
- Bragdon, B.; Moseychuk, O.; Saldanha, S.; King, D.; Julian, J.; Nohe, A. Bone morphogenetic proteins: A critical review. Cell Signal 2011, 23, 609–620. [Google Scholar] [CrossRef]
- Tang, M.C.; Chan, L.C.; Yeh, Y.C.; Chen, C.Y.; Chou, T.Y.; Wang, W.S.; Su, Y. Thymosin beta 4 induces colon cancer cell migration and clinical metastasis via enhancing ILK/IQGAP1/Rac1 signal transduction pathway. Cancer Lett. 2011, 308, 162–171. [Google Scholar] [CrossRef]
- Samaeekia, R.; Adorno-Cruz, V.; Bockhorn, J.; Chang, Y.F.; Huang, S.; Prat, A.; Ha, N.; Kibria, G.; Huo, D.; Zheng, H.; et al. miR-206 Inhibits Stemness and Metastasis of Breast Cancer by Targeting MKL1/IL11 Pathway. Clin. Cancer Res. 2017, 23, 1091–1103. [Google Scholar] [CrossRef]
- Meacham, C.E.; Ho, E.E.; Dubrovsky, E.; Gertler, F.B.; Hemann, M.T. In vivo RNAi screening identifies regulators of actin dynamics as key determinants of lymphoma progression. Nat. Genet. 2009, 41, 1133–1137. [Google Scholar] [CrossRef]
- Barnawi, R.; Al-Khaldi, S.; Colak, D.; Tulbah, A.; Al-Tweigeri, T.; Fallatah, M.; Monies, D.; Ghebeh, H.; Al-Alwan, M. beta1 Integrin is essential for fascin-mediated breast cancer stem cell function and disease progression. Int. J. Cancer 2019, 145, 830–841. [Google Scholar] [CrossRef]
- Barnawi, R.; Al-Khaldi, S.; Bakheet, T.; Fallatah, M.; Alaiya, A.; Ghebeh, H.; Al-Alwan, M. Fascin Activates beta-Catenin Signaling and Promotes Breast Cancer Stem Cell Function Mainly Through Focal Adhesion Kinase (FAK): Relation with Disease Progression. Front. Oncol. 2020, 10, 440. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Gao, J.; Wu, J.; Liu, Q.H.; Wang, Z.G.; Li, H.L.; Xing, L.H. Expression of Fascin-1 on human lung cancer and paracarcinoma tissue and its relation to clinicopathological characteristics in patients with lung cancer. Onco Targets Ther. 2015, 8, 2571–2576. [Google Scholar] [CrossRef] [PubMed]
- McGuire, S.; Kara, B.; Hart, P.C.; Montag, A.; Wroblewski, K.; Fazal, S.; Huang, X.Y.; Lengyel, E.; Kenny, H.A. Inhibition of fascin in cancer and stromal cells blocks ovarian cancer metastasis. Gynecol. Oncol. 2019, 153, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Leung, R.; Wang, Y.; Cuddy, K.; Sun, C.; Magalhaes, J.; Grynpas, M.; Glogauer, M. Filamin A regulates monocyte migration through Rho small GTPases during osteoclastogenesis. J. Bone Miner Res. 2010, 25, 1077–1091. [Google Scholar] [CrossRef]
- Yue, J.; Lu, H.; Liu, J.; Berwick, M.; Shen, Z. Filamin-A as a marker and target for DNA damage based cancer therapy. DNA Repair 2012, 11, 192–200. [Google Scholar] [CrossRef]
- Savoy, R.M.; Ghosh, P.M. The dual role of filamin A in cancer: Can’t live with (too much of) it, can’t live without it. Endocr. Relat. Cancer 2013, 20, R341–R356. [Google Scholar] [CrossRef]
- Bourguignon, L.Y.W.; Earle, C.; Shiina, M. Activation of Matrix Hyaluronan-Mediated CD44 Signaling, Epigenetic Regulation and Chemoresistance in Head and Neck Cancer Stem Cells. Int. J. Mol. Sci. 2017, 18, 1849. [Google Scholar] [CrossRef]
- Zhang, R.; Liu, C.; Niu, Y.; Jing, Y.; Zhang, H.; Wang, J.; Yang, J.; Zen, K.; Zhang, J.; Zhang, C.Y.; et al. MicroRNA-128-3p regulates mitomycin C-induced DNA damage response in lung cancer cells through repressing SPTAN1. Oncotarget 2017, 8, 58098–58107. [Google Scholar] [CrossRef]
- Schrecker, C.; Behrens, S.; Schonherr, R.; Ackermann, A.; Pauli, D.; Plotz, G.; Zeuzem, S.; Brieger, A. SPTAN1 Expression Predicts Treatment and Survival Outcomes in Colorectal Cancer. Cancers 2021, 13, 3638. [Google Scholar] [CrossRef]
- Ackermann, A.; Schrecker, C.; Bon, D.; Friedrichs, N.; Bankov, K.; Wild, P.; Plotz, G.; Zeuzem, S.; Herrmann, E.; Hansmann, M.L.; et al. Downregulation of SPTAN1 is related to MLH1 deficiency and metastasis in colorectal cancer. PLoS ONE 2019, 14, e0213411. [Google Scholar] [CrossRef]
- Chen, Y.; Meng, L.; Shang, H.; Dou, Q.; Lu, Z.; Liu, L.; Wang, Z.; He, X.; Song, Y. beta2 spectrin-mediated differentiation repressed the properties of liver cancer stem cells through beta-catenin. Cell Death Dis. 2018, 9, 424. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Kim, S.; An, H.T.; Ko, J. alpha-Actinin-4 regulates cancer stem cell properties and chemoresistance in cervical cancer. Carcinogenesis 2020, 41, 940–949. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Wang, Q.; Tang, H.; Zhang, F.; Zheng, Y.; Wang, S.; Zhang, J.; Wang, Z.; Xie, X. Direct inhibition of ACTN4 by ellagic acid limits breast cancer metastasis via regulation of beta-catenin stabilization in cancer stem cells. J. Exp. Clin. Cancer Res. 2017, 36, 172. [Google Scholar] [CrossRef]
- Kikuchi, S.; Honda, K.; Tsuda, H.; Hiraoka, N.; Imoto, I.; Kosuge, T.; Umaki, T.; Onozato, K.; Shitashige, M.; Yamaguchi, U.; et al. Expression and gene amplification of actinin-4 in invasive ductal carcinoma of the pancreas. Clin. Cancer Res. 2008, 14, 5348–5356. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Hong, S.H.; Chen, Q.R.; Briggs, J.; Cassavaugh, J.; Srinivasan, S.; Lizardo, M.M.; Mendoza, A.; Xia, A.Y.; Avadhani, N.; et al. Dysregulation of ezrin phosphorylation prevents metastasis and alters cellular metabolism in osteosarcoma. Cancer Res. 2012, 72, 1001–1012. [Google Scholar] [CrossRef][Green Version]
- Sarrio, D.; Rodriguez-Pinilla, S.M.; Dotor, A.; Calero, F.; Hardisson, D.; Palacios, J. Abnormal ezrin localization is associated with clinicopathological features in invasive breast carcinomas. Breast Cancer Res. Treat. 2006, 98, 71–79. [Google Scholar] [CrossRef]
- Barik, G.K.; Sahay, O.; Paul, D.; Santra, M.K. Ezrin gone rogue in cancer progression and metastasis: An enticing therapeutic target. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188753. [Google Scholar] [CrossRef]
- Ma, L.; Jiang, T. Clinical implications of Ezrin and CD44 coexpression in breast cancer. Oncol. Rep. 2013, 30, 1899–1905. [Google Scholar] [CrossRef]
- Penchev, V.R.; Chang, Y.T.; Begum, A.; Ewachiw, T.; Gocke, C.; Li, J.; McMillan, R.H.; Wang, Q.; Anders, R.; Marchionni, L.; et al. Ezrin Promotes Stem Cell Properties in Pancreatic Ductal Adenocarcinoma. Mol. Cancer Res. 2019, 17, 929–936. [Google Scholar] [CrossRef]
- Hoskin, V.; Szeto, A.; Ghaffari, A.; Greer, P.A.; Cote, G.P.; Elliott, B.E. Ezrin regulates focal adhesion and invadopodia dynamics by altering calpain activity to promote breast cancer cell invasion. Mol. Biol. Cell 2015, 26, 3464–3479. [Google Scholar] [CrossRef]
- McBeath, R.; Pirone, D.M.; Nelson, C.M.; Bhadriraju, K.; Chen, C.S. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev. Cell 2004, 6, 483–495. [Google Scholar] [CrossRef]
- Sun, X.; Li, K.; Hase, M.; Zha, R.; Feng, Y.; Li, B.Y.; Yokota, H. Suppression of breast cancer-associated bone loss with osteoblast proteomes via Hsp90ab1/moesin-mediated inhibition of TGFbeta/FN1/CD44 signaling. Theranostics 2022, 12, 929–943. [Google Scholar] [CrossRef]
- Zhu, X.; Morales, F.C.; Agarwal, N.K.; Dogruluk, T.; Gagea, M.; Georgescu, M.M. Moesin is a glioma progression marker that induces proliferation and Wnt/beta-catenin pathway activation via interaction with CD44. Cancer Res. 2013, 73, 1142–1155. [Google Scholar] [CrossRef] [PubMed]
- Hong, A.W.; Meng, Z.; Plouffe, S.W.; Lin, Z.; Zhang, M.; Guan, K.L. Critical roles of phosphoinositides and NF2 in Hippo pathway regulation. Genes Dev. 2020, 34, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Tian, Q.; Gao, H.; Zhou, Y.; Zhu, L.; Yang, J.; Wang, B.; Liu, P.; Yang, J. RICH1 inhibits breast cancer stem cell traits through activating kinases cascade of Hippo signaling by competing with Merlin for binding to Amot-p80. Cell Death Dis. 2022, 13, 71. [Google Scholar] [CrossRef] [PubMed]
- Cordenonsi, M.; Zanconato, F.; Azzolin, L.; Forcato, M.; Rosato, A.; Frasson, C.; Inui, M.; Montagner, M.; Parenti, A.R.; Poletti, A.; et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 2011, 147, 759–772. [Google Scholar] [CrossRef]
- Marino, N.; Marshall, J.C.; Collins, J.W.; Zhou, M.; Qian, Y.; Veenstra, T.; Steeg, P.S. Nm23-h1 binds to gelsolin and inactivates its actin-severing capacity to promote tumor cell motility and metastasis. Cancer Res. 2013, 73, 5949–5962. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Wang, P.W.; Shieh, D.B.; Chiu, K.Y.; Liou, Y.M. Involvement of gelsolin in TGF-beta 1 induced epithelial to mesenchymal transition in breast cancer cells. J. Biomed. Sci. 2015, 22, 90. [Google Scholar] [CrossRef]
- Liao, P.H.; Hsu, H.H.; Chen, T.S.; Chen, M.C.; Day, C.H.; Tu, C.C.; Lin, Y.M.; Tsai, F.J.; Kuo, W.W.; Huang, C.Y. Phosphorylation of cofilin-1 by ERK confers HDAC inhibitor resistance in hepatocellular carcinoma cells via decreased ROS-mediated mitochondria injury. Oncogene 2017, 36, 1978–1990. [Google Scholar] [CrossRef]
- Lu, L.I.; Fu, N.I.; Luo, X.U.; Li, X.Y.; Li, X.P. Overexpression of cofilin 1 in prostate cancer and the corresponding clinical implications. Oncol. Lett. 2015, 9, 2757–2761. [Google Scholar] [CrossRef]
- Ozeki, M.; Aini, W.; Miyagawa-Hayashino, A.; Tamaki, K. Prevention of Cell Growth by Suppression of Villin Expression in Lithocholic Acid-Stimulated HepG2 Cells. J. Histochem. Cytochem. 2019, 67, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Qiao, X.T.; Gumucio, D.L. Current molecular markers for gastric progenitor cells and gastric cancer stem cells. J. Gastroenterol. 2011, 46, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.X.; Liu, B.; Wang, Y.F.; Zhang, R.S.; Yu, B.; Lu, Z.F.; Shi, Q.L.; Zhou, X.J. Pulmonary enteric adenocarcinoma: A study of the clinicopathologic and molecular status of nine cases. Int. J. Clin. Exp. Pathol. 2014, 7, 1266–1274. [Google Scholar] [PubMed]
- Zheng, H.; Yang, Y.; Hong, Y.G.; Wang, M.C.; Yuan, S.X.; Wang, Z.G.; Bi, F.R.; Hao, L.Q.; Yan, H.L.; Zhou, W.P. Tropomodulin 3 modulates EGFR-PI3K-AKT signaling to drive hepatocellular carcinoma metastasis. Mol. Carcinog. 2019, 58, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Ito-Kureha, T.; Koshikawa, N.; Yamamoto, M.; Semba, K.; Yamaguchi, N.; Yamamoto, T.; Seiki, M.; Inoue, J. Tropomodulin 1 expression driven by NF-kappaB enhances breast cancer growth. Cancer Res. 2015, 75, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Wang, Y.; Zhang, H.; Piao, J.; Muthusamy, S.; Wang, L.; Deng, Y.; Zhang, W.; Kuang, R.; Billadeau, D.D.; et al. Vasodilator-stimulated phosphoprotein promotes liver metastasis of gastrointestinal cancer by activating a beta1-integrin-FAK-YAP1/TAZ signaling pathway. NPJ Precis. Oncol. 2018, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Dai, G.; Cai, Y.; Gong, Q.; Wu, W.; Gao, M.; Fei, Z. Vasodilator-stimulated phosphoprotein (VASP), a novel target of miR-4455, promotes gastric cancer cell proliferation, migration, and invasion, through activating the PI3K/AKT signaling pathway. Cancer Cell. Int. 2018, 18, 97. [Google Scholar] [CrossRef]
- Dybdal-Hargreaves, N.F.; Risinger, A.L.; Mooberry, S.L. Regulation of E-cadherin localization by microtubule targeting agents: Rapid promotion of cortical E-cadherin through p130Cas/Src inhibition by eribulin. Oncotarget 2018, 9, 5545–5561. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Whipple, R.A.; Matrone, M.A.; Cho, E.H.; Balzer, E.M.; Vitolo, M.I.; Yoon, J.R.; Ioffe, O.B.; Tuttle, K.C.; Yang, J.; Martin, S.S. Epithelial-to-mesenchymal transition promotes tubulin detyrosination and microtentacles that enhance endothelial engagement. Cancer Res. 2010, 70, 8127–8137. [Google Scholar] [CrossRef]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef]
- Bonneau, C.; Gurard-Levin, Z.A.; Andre, F.; Pusztai, L.; Rouzier, R. Predictive and Prognostic Value of the TauProtein in Breast Cancer. Anticancer Res. 2015, 35, 5179–5184. [Google Scholar] [PubMed]
- Correa-Saez, A.; Jimenez-Izquierdo, R.; Garrido-Rodriguez, M.; Morrugares, R.; Munoz, E.; Calzado, M.A. Updating dual-specificity tyrosine-phosphorylation-regulated kinase 2 (DYRK2): Molecular basis, functions and role in diseases. Cell Mol. Life Sci. 2020, 77, 4747–4763. [Google Scholar] [CrossRef] [PubMed]
- Kuo, T.C.; Li, L.W.; Pan, S.H.; Fang, J.M.; Liu, J.H.; Cheng, T.J.; Wang, C.J.; Hung, P.F.; Chen, H.Y.; Hong, T.M.; et al. Purine-Type Compounds Induce Microtubule Fragmentation and Lung Cancer Cell Death through Interaction with Katanin. J. Med. Chem. 2016, 59, 8521–8534. [Google Scholar] [CrossRef]
- Wang, L.; Tantai, J.; Zhu, X. Katanin P60: A potential biomarker for lymph node metastasis and prognosis for non-small cell lung cancer. World J. Surg. Oncol. 2020, 18, 157. [Google Scholar] [CrossRef]
- Zhao, T.; He, Q.; Liu, Z.; Ding, X.; Zhou, X.; Wang, A. Angiotensin II type 2 receptor-interacting protein 3a suppresses proliferation, migration and invasion in tongue squamous cell carcinoma via the extracellular signal-regulated kinase-Snai2 pathway. Oncol. Lett. 2016, 11, 340–344. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, B.; Zhang, G.; Cui, S.; Du, G. Upregulation of KIF11 in TP53 Mutant Glioma Promotes Tumor Stemness and Drug Resistance. Cell Mol. Neurobiol. 2022, 42, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Jiang, P.; Du, W.; Wu, Z.; Li, C.; Qiao, M.; Yang, X.; Wu, M. Siva1 suppresses epithelial-mesenchymal transition and metastasis of tumor cells by inhibiting stathmin and stabilizing microtubules. Proc. Natl. Acad. Sci. USA 2011, 108, 12851–12856. [Google Scholar] [CrossRef]
- Christofori, G. New signals from the invasive front. Nature 2006, 441, 444–450. [Google Scholar] [CrossRef]
- Pongrakhananon, V.; Wattanathamsan, O.; Takeichi, M.; Chetprayoon, P.; Chanvorachote, P. Loss of CAMSAP3 promotes EMT via the modification of microtubule-Akt machinery. J. Cell Sci. 2018, 131, jcs216168. [Google Scholar] [CrossRef]
- Harada, T.; Swift, J.; Irianto, J.; Shin, J.W.; Spinler, K.R.; Athirasala, A.; Diegmiller, R.; Dingal, P.C.; Ivanovska, I.L.; Discher, D.E. Nuclear lamin stiffness is a barrier to 3D migration, but softness can limit survival. J. Cell Biol. 2014, 204, 669–682. [Google Scholar] [CrossRef]
- Thomas, D.; Thiagarajan, P.S.; Rai, V.; Reizes, O.; Lathia, J.; Egelhoff, T. Increased cancer stem cell invasion is mediated by myosin IIB and nuclear translocation. Oncotarget 2016, 7, 47586–47592. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.G.; Yenepalli, A.; Denais, C.M.; Rape, A.; Beach, J.R.; Wang, Y.L.; Schiemann, W.P.; Baskaran, H.; Lammerding, J.; Egelhoff, T.T. Non-muscle myosin IIB is critical for nuclear translocation during 3D invasion. J. Cell Biol. 2015, 210, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Derycke, L.; Stove, C.; Vercoutter-Edouart, A.S.; De Wever, O.; Dolle, L.; Colpaert, N.; Depypere, H.; Michalski, J.C.; Bracke, M. The role of non-muscle myosin IIA in aggregation and invasion of human MCF-7 breast cancer cells. Int. J. Dev. Biol. 2011, 55, 835–840. [Google Scholar] [CrossRef]
- Beach, J.R.; Hussey, G.S.; Miller, T.E.; Chaudhury, A.; Patel, P.; Monslow, J.; Zheng, Q.; Keri, R.A.; Reizes, O.; Bresnick, A.R.; et al. Myosin II isoform switching mediates invasiveness after TGF-beta-induced epithelial-mesenchymal transition. Proc. Natl. Acad. Sci. USA 2011, 108, 17991–17996. [Google Scholar] [CrossRef]
- Wu, S.; Du, Y.; Beckford, J.; Alachkar, H. Upregulation of the EMT marker vimentin is associated with poor clinical outcome in acute myeloid leukemia. J. Transl. Med. 2018, 16, 170. [Google Scholar] [CrossRef] [PubMed]
- Vuoriluoto, K.; Haugen, H.; Kiviluoto, S.; Mpindi, J.P.; Nevo, J.; Gjerdrum, C.; Tiron, C.; Lorens, J.B.; Ivaska, J. Vimentin regulates EMT induction by Slug and oncogenic H-Ras and migration by governing Axl expression in breast cancer. Oncogene 2011, 30, 1436–1448. [Google Scholar] [CrossRef]
- Usman, S.; Waseem, N.H.; Nguyen, T.K.N.; Mohsin, S.; Jamal, A.; Teh, M.T.; Waseem, A. Vimentin Is at the Heart of Epithelial Mesenchymal Transition (EMT) Mediated Metastasis. Cancers 2021, 13, 4985. [Google Scholar] [CrossRef]
- Neradil, J.; Veselska, R. Nestin as a marker of cancer stem cells. Cancer Sci. 2015, 106, 803–811. [Google Scholar] [CrossRef]
- Matsuda, Y.; Hagio, M.; Ishiwata, T. Nestin: A novel angiogenesis marker and possible target for tumor angiogenesis. World J. Gastroenterol. 2013, 19, 42–48. [Google Scholar] [CrossRef]
- Zhao, Z.; Lu, P.; Zhang, H.; Xu, H.; Gao, N.; Li, M.; Liu, C. Nestin positively regulates the Wnt/beta-catenin pathway and the proliferation, survival and invasiveness of breast cancer stem cells. Breast Cancer Res. 2014, 16, 408. [Google Scholar] [CrossRef]
- Tsai, F.J.; Lai, M.T.; Cheng, J.; Chao, S.C.; Korla, P.K.; Chen, H.J.; Lin, C.M.; Tsai, M.H.; Hua, C.H.; Jan, C.I.; et al. Novel K6-K14 keratin fusion enhances cancer stemness and aggressiveness in oral squamous cell carcinoma. Oncogene 2019, 38, 5113–5126. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Jang, T.H.; Tung, S.L.; Yen, T.C.; Chan, S.H.; Wang, L.H. A novel miR-365-3p/EHF/keratin 16 axis promotes oral squamous cell carcinoma metastasis, cancer stemness and drug resistance via enhancing beta5-integrin/c-met signaling pathway. J. Exp. Clin. Cancer Res. 2019, 38, 89. [Google Scholar] [CrossRef] [PubMed]
- Jang, T.H.; Huang, W.C.; Tung, S.L.; Lin, S.C.; Chen, P.M.; Cho, C.Y.; Yang, Y.Y.; Yen, T.C.; Lo, G.H.; Chuang, S.E.; et al. MicroRNA-485-5p targets keratin 17 to regulate oral cancer stemness and chemoresistance via the integrin/FAK/Src/ERK/beta-catenin pathway. J. Biomed. Sci. 2022, 29, 42. [Google Scholar] [CrossRef]
- Kim, S.Y. Cancer Energy Metabolism: Shutting Power off Cancer Factory. Biomol. Ther. 2018, 26, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Juvekar, A.; Lyssiotis, C.A.; Lien, E.C.; Albeck, J.G.; Oh, D.; Varma, G.; Hung, Y.P.; Ullas, S.; Lauring, J.; et al. Phosphoinositide 3-Kinase Regulates Glycolysis through Mobilization of Aldolase from the Actin Cytoskeleton. Cell 2016, 164, 433–446. [Google Scholar] [CrossRef]
- Shankar, J.; Nabi, I.R. Actin cytoskeleton regulation of epithelial mesenchymal transition in metastatic cancer cells. PLoS ONE 2015, 10, e0119954. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, J.; Yu, M.; Xie, Y.; Huang, Y.; Wolff, D.W.; Abel, P.W.; Tu, Y. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 2013, 32, 4814–4824. [Google Scholar] [CrossRef]
- Taulet, N.; Delorme-Walker, V.D.; DerMardirossian, C. Reactive oxygen species regulate protrusion efficiency by controlling actin dynamics. PLoS ONE 2012, 7, e41342. [Google Scholar] [CrossRef]
- Bonello, T.T.; Stehn, J.R.; Gunning, P.W. New approaches to targeting the actin cytoskeleton for chemotherapy. Future Med. Chem. 2009, 1, 1311–1331. [Google Scholar] [CrossRef]
- Guerriero, C.J.; Weisz, O.A. N-WASP inhibitor wiskostatin nonselectively perturbs membrane transport by decreasing cellular ATP levels. Am. J. Physiol. Cell Physiol. 2007, 292, C1562–C1566. [Google Scholar] [CrossRef]
- Hetrick, B.; Han, M.S.; Helgeson, L.A.; Nolen, B.J. Small molecules CK-666 and CK-869 inhibit actin-related protein 2/3 complex by blocking an activating conformational change. Chem. Biol. 2013, 20, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.R.; Mitchison, T.J. Small molecules, big impact: A history of chemical inhibitors and the cytoskeleton. Chem. Biol. 2002, 9, 1275–1285. [Google Scholar] [CrossRef]
- Kim, H.C.; Jo, Y.J.; Kim, N.H.; Namgoong, S. Small molecule inhibitor of formin homology 2 domains (SMIFH2) reveals the roles of the formin family of proteins in spindle assembly and asymmetric division in mouse oocytes. PLoS ONE 2015, 10, e0123438. [Google Scholar] [CrossRef]
- Ziske, M.A.; Pettee, K.M.; Khaing, M.; Rubinic, K.; Eisenmann, K.M. SMIFH2-mediated mDia formin functional inhibition potentiates chemotherapeutic targeting of human ovarian cancer spheroids. Biochem. Biophys. Res. Commun. 2016, 472, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Orgaz, J.L.; Herraiz, C.; Sanz-Moreno, V. Rho GTPases modulate malignant transformation of tumor cells. Small GTPases 2014, 5, e29019. [Google Scholar] [CrossRef]
- Biber, G.; Ben-Shmuel, A.; Sabag, B.; Barda-Saad, M. Actin regulators in cancer progression and metastases: From structure and function to cytoskeletal dynamics. Int. Rev. Cell Mol. Biol. 2020, 356, 131–196. [Google Scholar] [CrossRef]
- Arias-Romero, L.E.; Chernoff, J. Targeting Cdc42 in cancer. Expert Opin. Ther. Targets 2013, 17, 1263–1273. [Google Scholar] [CrossRef]
- Guo, Y.; Kenney, S.R.; Muller, C.Y.; Adams, S.; Rutledge, T.; Romero, E.; Murray-Krezan, C.; Prekeris, R.; Sklar, L.A.; Hudson, L.G.; et al. R-Ketorolac Targets Cdc42 and Rac1 and Alters Ovarian Cancer Cell Behaviors Critical for Invasion and Metastasis. Mol. Cancer Ther. 2015, 14, 2215–2227. [Google Scholar] [CrossRef]
- Humphries-Bickley, T.; Castillo-Pichardo, L.; Hernandez-O’Farrill, E.; Borrero-Garcia, L.D.; Forestier-Roman, I.; Gerena, Y.; Blanco, M.; Rivera-Robles, M.J.; Rodriguez-Medina, J.R.; Cubano, L.A.; et al. Characterization of a Dual Rac/Cdc42 Inhibitor MBQ-167 in Metastatic Cancer. Mol. Cancer Ther. 2017, 16, 805–818. [Google Scholar] [CrossRef]
- Maldonado, M.D.M.; Dharmawardhane, S. Targeting Rac and Cdc42 GTPases in Cancer. Cancer Res. 2018, 78, 3101–3111. [Google Scholar] [CrossRef]
- Schofield, A.V.; Bernard, O. Rho-associated coiled-coil kinase (ROCK) signaling and disease. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 301–316. [Google Scholar] [CrossRef] [PubMed]
- Shahbazi, R.; Baradaran, B.; Khordadmehr, M.; Safaei, S.; Baghbanzadeh, A.; Jigari, F.; Ezzati, H. Targeting ROCK signaling in health, malignant and non-malignant diseases. Immunol. Lett. 2020, 219, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Kim, H.M. ROCK inhibition activates MCF-7 cells. PLoS ONE 2014, 9, e88489. [Google Scholar] [CrossRef] [PubMed]
- Lock, F.E.; Ryan, K.R.; Poulter, N.S.; Parsons, M.; Hotchin, N.A. Differential regulation of adhesion complex turnover by ROCK1 and ROCK2. PLoS ONE 2012, 7, e31423. [Google Scholar] [CrossRef]
- Wei, L.; Surma, M.; Shi, S.; Lambert-Cheatham, N.; Shi, J. Novel Insights into the Roles of Rho Kinase in Cancer. Arch. Immunol. Ther. Exp. 2016, 64, 259–278. [Google Scholar] [CrossRef]
- Pimm, M.L.; Hotaling, J.; Henty-Ridilla, J.L. Profilin choreographs actin and microtubules in cells and cancer. Int. Rev. Cell Mol. Biol. 2020, 355, 155–204. [Google Scholar] [CrossRef]
- Gau, D.; Lewis, T.; McDermott, L.; Wipf, P.; Koes, D.; Roy, P. Structure-based virtual screening identifies a small-molecule inhibitor of the profilin 1-actin interaction. J. Biol. Chem. 2018, 293, 2606–2616. [Google Scholar] [CrossRef]
- Huang, D.; Wang, S.; Wang, A.; Chen, X.; Zhang, H. Thymosin beta 4 silencing suppresses proliferation and invasion of non-small cell lung cancer cells by repressing Notch1 activation. Acta Biochim. Biophys. Sin. 2016, 48, 788–794. [Google Scholar] [CrossRef]
- Ristic, B.; Kopel, J.; Sherazi, S.A.A.; Gupta, S.; Sachdeva, S.; Bansal, P.; Ali, A.; Perisetti, A.; Goyal, H. Emerging Role of Fascin-1 in the Pathogenesis, Diagnosis, and Treatment of the Gastrointestinal Cancers. Cancers 2021, 13, 2536. [Google Scholar] [CrossRef]
- Montoro-Garcia, S.; Alburquerque-Gonzalez, B.; Bernabe-Garcia, A.; Bernabe-Garcia, M.; Rodrigues, P.C.; den-Haan, H.; Luque, I.; Nicolas, F.J.; Perez-Sanchez, H.; Cayuela, M.L.; et al. Novel anti-invasive properties of a Fascin1 inhibitor on colorectal cancer cells. J. Mol. Med. 2020, 98, 383–394. [Google Scholar] [CrossRef]
- Mahmoud, A.; Elkhalifa, D.; Alali, F.; Al Moustafa, A.E.; Khalil, A. Novel Polymethoxylated Chalcones as Potential Compounds Against KRAS-Mutant Colorectal Cancers. Curr. Pharm. Des. 2020, 26, 1622–1633. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Dey, R.; Wang, Y.; Jakoncic, J.; Kurinov, I.; Huang, X.Y. Structural Insights into the Induced-fit Inhibition of Fascin by a Small-Molecule Inhibitor. J. Mol. Biol. 2018, 430, 1324–1335. [Google Scholar] [CrossRef] [PubMed]
- Chung, V.; Jhaveri, K.L.; Hoff, D.D.V.; Huang, X.-Y.; Garmey, E.G.; Zhang, J.; Tsai, F.Y.-C. Phase 1A clinical trial of the first-in-class fascin inhibitor NP-G2-044 evaluating safety and anti-tumor activity in patients with advanced and metastatic solid tumors. J. Clin. Oncol. 2021, 39, 2548. [Google Scholar] [CrossRef]
- Alburquerque-Gonzalez, B.; Bernabe-Garcia, A.; Bernabe-Garcia, M.; Ruiz-Sanz, J.; Lopez-Calderon, F.F.; Gonnelli, L.; Banci, L.; Pena-Garcia, J.; Luque, I.; Nicolas, F.J.; et al. The FDA-Approved Antiviral Raltegravir Inhibits Fascin1-Dependent Invasion of Colorectal Tumor Cells In Vitro and In Vivo. Cancers 2021, 13, 861. [Google Scholar] [CrossRef]
- Schenk, M.; Aykut, B.; Teske, C.; Giese, N.A.; Weitz, J.; Welsch, T. Salinomycin inhibits growth of pancreatic cancer and cancer cell migration by disruption of actin stress fiber integrity. Cancer Lett. 2015, 358, 161–169. [Google Scholar] [CrossRef]
- Alburquerque-Gonzalez, B.; Bernabe-Garcia, M.; Montoro-Garcia, S.; Bernabe-Garcia, A.; Rodrigues, P.C.; Ruiz Sanz, J.; Lopez-Calderon, F.F.; Luque, I.; Nicolas, F.J.; Cayuela, M.L.; et al. New role of the antidepressant imipramine as a Fascin1 inhibitor in colorectal cancer cells. Exp. Mol. Med. 2020, 52, 281–292. [Google Scholar] [CrossRef]
- Bulut, G.; Hong, S.H.; Chen, K.; Beauchamp, E.M.; Rahim, S.; Kosturko, G.W.; Glasgow, E.; Dakshanamurthy, S.; Lee, H.S.; Daar, I.; et al. Small molecule inhibitors of ezrin inhibit the invasive phenotype of osteosarcoma cells. Oncogene 2012, 31, 269–281. [Google Scholar] [CrossRef]
- Celik, H.; Bulut, G.; Han, J.; Graham, G.T.; Minas, T.Z.; Conn, E.J.; Hong, S.H.; Pauly, G.T.; Hayran, M.; Li, X.; et al. Ezrin Inhibition Up-regulates Stress Response Gene Expression. J. Biol. Chem. 2016, 291, 13257–13270. [Google Scholar] [CrossRef]
- Jeong, J.; Choi, J.; Kim, W.; Dann, P.; Takyar, F.; Gefter, J.V.; Friedman, P.A.; Wysolmerski, J.J. Inhibition of ezrin causes PKCalpha-mediated internalization of erbb2/HER2 tyrosine kinase in breast cancer cells. J. Biol. Chem. 2019, 294, 887–901. [Google Scholar] [CrossRef]
- Gavert, N.; Ben-Shmuel, A.; Lemmon, V.; Brabletz, T.; Ben-Ze’ev, A. Nuclear factor-kappaB signaling and ezrin are essential for L1-mediated metastasis of colon cancer cells. J. Cell Sci. 2010, 123, 2135–2143. [Google Scholar] [CrossRef]
- Lugini, L.; Matarrese, P.; Tinari, A.; Lozupone, F.; Federici, C.; Iessi, E.; Gentile, M.; Luciani, F.; Parmiani, G.; Rivoltini, L.; et al. Cannibalism of live lymphocytes by human metastatic but not primary melanoma cells. Cancer Res. 2006, 66, 3629–3638. [Google Scholar] [CrossRef] [PubMed]
- Shang, X.; Wang, Y.; Zhao, Q.; Wu, K.; Li, X.; Ji, X.; He, R.; Zhang, W. siRNAs target sites selection of ezrin and the influence of RNA interference on ezrin expression and biological characters of osteosarcoma cells. Mol. Cell Biochem. 2012, 364, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Pignochino, Y.; Grignani, G.; Cavalloni, G.; Motta, M.; Tapparo, M.; Bruno, S.; Bottos, A.; Gammaitoni, L.; Migliardi, G.; Camussi, G.; et al. Sorafenib blocks tumour growth, angiogenesis and metastatic potential in preclinical models of osteosarcoma through a mechanism potentially involving the inhibition of ERK1/2, MCL-1 and ezrin pathways. Mol. Cancer 2009, 8, 118. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Hou, R.; Gao, S.; Song, D.; Feng, Y. Baicalein Inhibits Proliferation Activity of Human Colorectal Cancer Cells HCT116 Through Downregulation of Ezrin. Cell Physiol. Biochem. 2018, 49, 2035–2046. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, W.; Zhou, Y.B.; Xiang, Y.X.; Wang, L.S.; Hu, W.K.; Wang, W.J. Baicalein inhibits osteosarcoma cell proliferation and invasion through the miR183/Ezrin pathway. Mol. Med. Rep. 2018, 18, 1104–1112. [Google Scholar] [CrossRef]
- Mukhtar, E.; Adhami, V.M.; Mukhtar, H. Targeting microtubules by natural agents for cancer therapy. Mol. Cancer Ther. 2014, 13, 275–284. [Google Scholar] [CrossRef]
- Weaver, B.A. How Taxol/paclitaxel kills cancer cells. Mol. Biol. Cell 2014, 25, 2677–2681. [Google Scholar] [CrossRef]
- Abu Samaan, T.M.; Samec, M.; Liskova, A.; Kubatka, P.; Busselberg, D. Paclitaxel’s Mechanistic and Clinical Effects on Breast Cancer. Biomolecules 2019, 9, 789. [Google Scholar] [CrossRef]
- Sharma, P.; Alsharif, S.; Fallatah, A.; Chung, B.M. Intermediate Filaments as Effectors of Cancer Development and Metastasis: A Focus on Keratins, Vimentin, and Nestin. Cells 2019, 8, 497. [Google Scholar] [CrossRef]
- Lee, I.C.; Choi, B.Y. Withaferin-A--A Natural Anticancer Agent with Pleitropic Mechanisms of Action. Int. J. Mol. Sci. 2016, 17, 290. [Google Scholar] [CrossRef]
- Nagalingam, A.; Kuppusamy, P.; Singh, S.V.; Sharma, D.; Saxena, N.K. Mechanistic elucidation of the antitumor properties of withaferin a in breast cancer. Cancer Res. 2014, 74, 2617–2629. [Google Scholar] [CrossRef] [PubMed]
- Bhat, K.M.; Setaluri, V. Microtubule-associated proteins as targets in cancer chemotherapy. Clin. Cancer Res. 2007, 13, 2849–2854. [Google Scholar] [CrossRef] [PubMed]
- Smoter, M.; Bodnar, L.; Duchnowska, R.; Stec, R.; Grala, B.; Szczylik, C. The role of Tau protein in resistance to paclitaxel. Cancer Chemother. Pharmacol. 2011, 68, 553–557. [Google Scholar] [CrossRef]
- Kavallaris, M. Microtubules and resistance to tubulin-binding agents. Nat. Rev. Cancer 2010, 10, 194–204. [Google Scholar] [CrossRef]
- Sakowicz, R.; Finer, J.T.; Beraud, C.; Crompton, A.; Lewis, E.; Fritsch, A.; Lee, Y.; Mak, J.; Moody, R.; Turincio, R.; et al. Antitumor activity of a kinesin inhibitor. Cancer Res. 2004, 64, 3276–3280. [Google Scholar] [CrossRef] [PubMed]
- Gomez, H.L.; Philco, M.; Pimentel, P.; Kiyan, M.; Monsalvo, M.L.; Conlan, M.G.; Saikali, K.G.; Chen, M.M.; Seroogy, J.J.; Wolff, A.A.; et al. Phase I dose-escalation and pharmacokinetic study of ispinesib, a kinesin spindle protein inhibitor, administered on days 1 and 15 of a 28-day schedule in patients with no prior treatment for advanced breast cancer. Anticancer Drugs 2012, 23, 335–341. [Google Scholar] [CrossRef]
- Holen, K.D.; Belani, C.P.; Wilding, G.; Ramalingam, S.; Volkman, J.L.; Ramanathan, R.K.; Vasist, L.S.; Bowen, C.J.; Hodge, J.P.; Dar, M.M.; et al. A first in human study of SB-743921, a kinesin spindle protein inhibitor, to determine pharmacokinetics, biologic effects and establish a recommended phase II dose. Cancer Chemother. Pharmacol. 2011, 67, 447–454. [Google Scholar] [CrossRef]
- Lucanus, A.J.; Yip, G.W. Kinesin superfamily: Roles in breast cancer, patient prognosis and therapeutics. Oncogene 2018, 37, 833–838. [Google Scholar] [CrossRef]
- Belletti, B.; Baldassarre, G. Stathmin: A protein with many tasks. New biomarker and potential target in cancer. Expert Opin. Ther. Targets 2011, 15, 1249–1266. [Google Scholar] [CrossRef]
- Miceli, C.; Tejada, A.; Castaneda, A.; Mistry, S.J. Cell cycle inhibition therapy that targets stathmin in in vitro and in vivo models of breast cancer. Cancer Gene Ther. 2013, 20, 298–307. [Google Scholar] [CrossRef][Green Version]
- Long, M.; Yin, G.; Liu, L.; Lin, F.; Wang, X.; Ren, J.; Wei, J.; Dong, K.; Zhang, H. Adenovirus-mediated Aurora A shRNA driven by stathmin promoter suppressed tumor growth and enhanced paclitaxel chemotherapy sensitivity in human breast carcinoma cells. Cancer Gene Ther. 2012, 19, 271–281. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, H.; Chomyn, A.; Chan, D.C. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 2005, 280, 26185–26192. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.C.B.; Campos, J.C.; Qvit, N.; Qi, X.; Bozi, L.H.M.; Bechara, L.R.G.; Lima, V.M.; Queliconi, B.B.; Disatnik, M.H.; Dourado, P.M.M.; et al. A selective inhibitor of mitofusin 1-betaIIPKC association improves heart failure outcome in rats. Nat. Commun. 2019, 10, 329. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, E.; Cioffi, M.; Sancho, P.; Sanchez-Ripoll, Y.; Trabulo, S.M.; Dorado, J.; Balic, A.; Hidalgo, M.; Heeschen, C. Metformin targets the metabolic achilles heel of human pancreatic cancer stem cells. PLoS ONE 2013, 8, e76518. [Google Scholar] [CrossRef]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Choi, J.; Lee, J.H.; Koh, I.; Shim, J.K.; Park, J.; Jeon, J.Y.; Yun, M.; Kim, S.H.; Yook, J.I.; Kim, E.H.; et al. Inhibiting stemness and invasive properties of glioblastoma tumorsphere by combined treatment with temozolomide and a newly designed biguanide (HL156A). Oncotarget 2016, 7, 65643–65659. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Class | Proteins | Mechanism | Effect |
|---|---|---|---|
| Actin | Activation of the downstream Rho/ROCK pathway via YAP/TAZ | Facilitating the survival of CSCs | |
| Monomer-binding proteins | Profilin | Direct regulation of stem cell-associated transcription factors | Maintaining the stemness of CSCs |
| Thymosin β4 | Activation of the BMP pathway, followed by JNK activation via the TAB1 and TAK1 complexes | Maintaining the stemness of CSCs | |
| Twinfilin | Enhancing the activity of the MKL1 and actin cytoskeleton dynamics | Facilitating the survival of CSCs | |
| Cross-linking and bundling proteins | Fascin | Activation of β-catenin protein signaling via FAK | Promoting CSC function |
| Filaminin | Interacts with Rho GTPases that activate cell migration and Ras GTPases that inhibit cell migration | Promoting CSC function | |
| Spectrin | Inhibition of CSCs by β-catenin-induced differentiation | Inhibiting the properties of CSCs | |
| α actinin | Acts through the Akt/GSK-3β/β-catenin axis | Maintaining the stemness of CSCs | |
| Anchoring proteins | Ezrin | Regulation of actin polymerization by ROCK inhibition | Maintaining the properties of CSCs |
| Moesin | Enhancement of positive feedback on the Wnt/β-catenin signaling pathway by increasing the expression of CD44 | Promoting CSC function | |
| Merlin | Adjusting the YAP/TAZ pathway via the merlin/NF2/YAP/TAZ axis | Promoting CSC function | |
| Capping and severing proteins | Gelsolin | Direct regulation of stem cell-associated transcription factors | Maintaining the properties of CSCs |
| Cofilin | Acts by promoting EMT expression | Maintaining the properties of CSCs | |
| Stabilizing proteins | Tropomodulin | Increased expression of MMP-13 and NF-κB and the activation of the PI3K–AKT signaling pathway | Promoting CSC function |
| Signaling proteins | ENA/VASP | ECM-mediated β1-integrin-FAK–YAP/TAZ signaling pathway | Maintaining the properties of CSCs |
| Tubulin | Regulating EMT and contributing to the formation of lamellar filopodia | Promoting CSC function | |
| Microtubule lattice-binding proteins | Tau | Activating the MAPK pathway by binding to PI3K | Maintaining the properties of CSCs |
| katanin | Activation of JNK by cutting cell microtubules into short segments | Promoting CSC function | |
| ATIP3 | Inhibition through ATIP3/ERK1/2-Snai2 signaling | Inhibiting the properties of CSCs | |
| Microtubule motor proteins | Kinesins | Promoting the expression of stem transcription factors (NANOG and OCT4) | Maintaining the properties of CSCs |
| Multiple site microtubule-binding proteins | STMN1 | Activates Rho by promoting microtubule depolymerization | Promoting CSC function |
| Microtubule minus-end-binding proteins | CAMSAPs | Inhibition of Akt activity through microtubule regulation | Inhibiting the properties of CSCs |
| Myosin | Involved in promoting the EMT and enhancing the nuclear translocation of CSC | Maintaining the properties of CSCs | |
| Intermediate filaments | Vimentin | Regulation of EMT-related genes, including Twist, Snail, ZEB1/2 and Slug | Maintaining the properties of CSCs |
| Nestin | Upregulation of VEGF, cancer stem cell markers, and proteins that activate Wnt/β-catenin to initiate proliferation | Promoting CSC function | |
| Keratins | Interacting with the β5-integrin/c-Met signaling pathway | Promoting CSC function | |
| Binding to β4-integrin/FAK or Src or β-catenin |
| Categories | Drug Name | Mechanism | Clinical Trial | NCT Registry Number/Ref. |
|---|---|---|---|---|
| Actin | SMIFH2 | Inhibiting actin nucleation and elongation | Experimental | [254] |
| Toradol | GTPase inhibition | Active, not recruiting | NCT02470299 | |
| MBQ-167 | Rac/Cdc42 inhibitor | Experimental | [256] | |
| Profilin | C1 and C2 | Preventing profilin from interacting with actin monomers | Experimental | [267] |
| Tβ4 | Tβ4 inhibitors | Silencing the Tβ4 gene | Experimental | [172,268] |
| Fascin | NP-G2-044 | Inhibiting fascin-1-directed actin remodeling | Completed | NCT03199586 |
| Compounds 3 and 14 | Downregulating fascin-1 | Experimental | [271] | |
| Raltegravir | Inhibitor of human immunodeficiency virus 1 integrase | Experimental | [274] | |
| Salinomycin | Fascin-1 inhibition | Experimental | [275] | |
| Imipramine | Fascin-1 inhibition | Early Phase 1 | NCT03122444 | |
| II | NCT04863950 | |||
| Ezrin | NSC305787 | Inhibiting the phosphorylation of ezrin | Experimental | [278] |
| NSC668394 | Experimental | [279] | ||
| Cytochalasin B | Inhibition of actin assembly | Experimental | [281] | |
| LY294002 | PI3K inhibitor | Experimental | [197] | |
| MK2206 | AKT inhibitor | |||
| BAY43-9006 | Multi-kinase inhibitor | Experimental | [283] | |
| Baicalin | Inhibitor of ezrin | Experimental | [284] | |
| Celastrol | Impairing the phosphorylation of ezrin | Experimental | [285] | |
| Microtubules | Paclitaxel | Stabilizing microtubules | Clinical medication | [287] |
| Taxanes | Inhibitors of microtubule dynamics | Clinical medication | [12] | |
| Vinca alkaloids | ||||
| Vimentin | Withaferin-A | Inducing vimentin cleavage | Experimental | [290] |
| Kinesin | Purine compound 5a | Regulating katanin’s cut-off activities | Experimental | [223] |
| Ispinesib | A kinesin spindle protein inhibitor | Completed | NCT00089973 | |
| SB-743921 | NCT00136513 | |||
| Stathmin | Anti-stathmin adenovirus | Cell cycle inhibition | Experimental | [300] |
| Aurora A shRNA | Inhibitor of stathmin | Experimental | [301] | |
| Mitochondria | βIIPKC | Inhibitor of mitochondrial fusion proteins | Experimental | [303] |
| Metformin | Inhibitors of OXPHOS | Experimental | [304] | |
| Benzformin | Experimental | [305] | ||
| IM156 | Metformin derivative | Experimental | [306] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Wang, D.; Ge, H.; Güngör, C.; Gong, X.; Chen, Y. Cytoskeletal and Cytoskeleton-Associated Proteins: Key Regulators of Cancer Stem Cell Properties. Pharmaceuticals 2022, 15, 1369. https://doi.org/10.3390/ph15111369
Li Y, Wang D, Ge H, Güngör C, Gong X, Chen Y. Cytoskeletal and Cytoskeleton-Associated Proteins: Key Regulators of Cancer Stem Cell Properties. Pharmaceuticals. 2022; 15(11):1369. https://doi.org/10.3390/ph15111369
Chicago/Turabian StyleLi, Yuqiang, Dan Wang, Heming Ge, Cenap Güngör, Xuejun Gong, and Yongheng Chen. 2022. "Cytoskeletal and Cytoskeleton-Associated Proteins: Key Regulators of Cancer Stem Cell Properties" Pharmaceuticals 15, no. 11: 1369. https://doi.org/10.3390/ph15111369
APA StyleLi, Y., Wang, D., Ge, H., Güngör, C., Gong, X., & Chen, Y. (2022). Cytoskeletal and Cytoskeleton-Associated Proteins: Key Regulators of Cancer Stem Cell Properties. Pharmaceuticals, 15(11), 1369. https://doi.org/10.3390/ph15111369