1. Introduction
The vascular endothelium (VE) is characterized by an endothelial cells monolayer, covering the blood vessel’s lumen, which acts through paracrine, endocrine, and autocrine effects. VE modulates the vascular tone and regulates the expression of adhesion molecules (CAMs), interfering in the coagulation cascade and blood flow. Those intrinsic properties are correlated with the mediators produced by VE, which include nitric oxide (NO). NO has an important role in the control of vascular tone, promoting vascular relaxation and vasodilation, and maintaining vascular hemostasis [
1,
2]. In addition, it acts by modulating pathways, pro-inflammatory cytokines (IL-1β, IL-6, and IL-8), and platelet-activating factor (PAF). These modulations occur through strictly controlled interactions between endothelial cells and other cellular components, such as blood cells, the immune system, and proteins present in the blood [
3].
The imbalances in the NO levels are prone to induce endothelial dysfunction (ED). Such a deleterious event is responsible for creating an environment in which pro-inflammatory and pro-coagulant cytokines are released, expression of adhesion molecules on the cell surface increases (i.e., E-selectin, ICAM-1, and VCAM-1), and vasoconstriction increases, thus, favoring the formation of clots [
2].
ED could also be induced by the high expression of oxidases, especially xanthine oxidase and NADPH oxidase complex [
4], both enzymes that participate in the tissue ischemia–reperfusion cascade. The activation of oxidases can contribute to increasing the reactive oxygen species (ROS) levels, aggravating vascular endothelial damage and hampering the activity of enzymes with antioxidant effects, such as the superoxide dismutase enzyme (SOD) [
5]. Moreover, it is described that such imbalances are prone to increase levels of vasoconstrictor mediators, such as Angiotensin II (Ang II) and Endothelin-1 (ET-1). Due to the effect of such vasoconstrictors, a more pronounced reduction in the levels of NO and antioxidants is found, aggravating the ED [
6,
7].
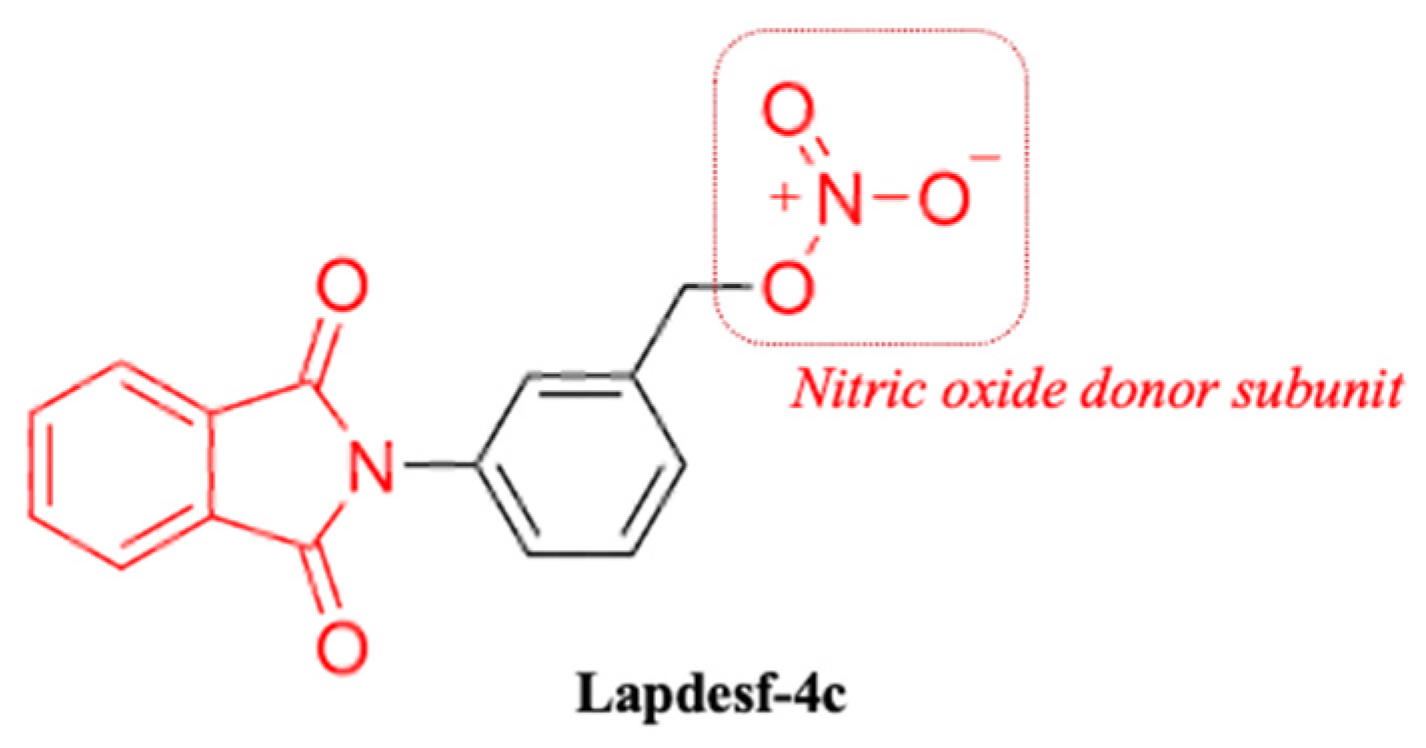
Compound 3-(1,3-dioxoisoindolin-2-yl) benzyl nitrate (
Lapdesf-4c) (
Figure 1) is described as the fetal hemoglobin-inducing agent that acts as NO donor, exhibiting pleiotropic effects as an anti-inflammatory and analgesic. These effects were associated, in parts, with the inhibition of pro-inflammatory cytokines, such as tumor necrosis factor-alpha (TNF-α) and IL-1 β [
8]. In vitro and in vivo experiments showed that
Lapdesf-4c did not exhibit mutagenic and genotoxic effects [
9].
Lapdesf-4c also reversed the priapism in sickle transgenic murine models displaying low NO bioavailability after 3-week treatment by inducing phosphodiesterase-5 (PDE-5) expression and reducing protein expression of ROS markers [
8,
10]. Although such activities suggested a protective profile of
Lapdesf-4c in the ED, there is no investigation of those vascular effects. Therefore, this work aims to evaluate the vasodilator potential of the compound and the endothelium participation in this response, in addition to verifying whether the compound is effective in promoting vasoprotection and if the antioxidant mechanism can reduce the ROS from the release of Ang II.
3. Discussion
ED is characterized by low levels of NO, increased expression of adhesion molecules (i.e., VCAM-1, ICAM-1), endothelin-1, Ang II, and pro-inflammatory cytokines (i.e., TNF-α, IL-1β, and IL-6). These deleterious environmental factors are prone to induce hypertension, clot formation, and vaso-occlusion [
11,
12,
13]. It has been described that NO donors can attenuate hypoxia, decrease cell adhesion, and release pro-inflammatory cytokines. Moreover, NO donors stimulate soluble guanylate cyclase (sGC) and decrease leukocyte adhesion in the vascular lumen [
14].
Strategies aiming to maintain appropriate levels of NO are important as therapeutic interventions to decrease the worsening of ED disease [
12,
14] Considering that
Lapdesf-4c acts as NO donor and is efficient to promote endothelium-independent vasodilation, it should be considered as a promising agent to prevent ED.
An interesting study to characterize the vasodilation in ED in humans through endothelium-dependent or independent mechanisms compared treatments after an intra-arterial infusion of NPS (a NO donor),
L-NAME (non-selective eNOS blocker), and methacholine (ACh analog). Brachial blood flow was monitored to assess the degree of arterial vasodilation [
15]. It was found that ED patients obtained a better response than healthy patients for NPS. Furthermore, the inhibition of eNOS by
L-NAME induced an inferior response in ED patients for methacholine, compared to healthy patients. Thus, these data demonstrated a reduced participation of endogenous NO in endothelium-dependent vasodilation. ED patients demonstrated lower levels of endogenous NO and a good response to exogenous NO donors [
15].
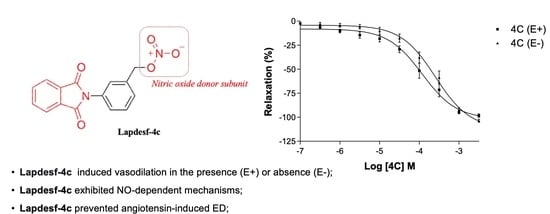
In this work, the NO donor
Lapdesf-4c was evaluated using the aorta artery of rats to study its potential vasodilator effect in the presence (E+) or absence (E−) of the endothelium. The results show that the percentage of vasodilation for
Lapdesf-4c in the presence (E+) and absence (E−) of the endothelium were similar, not showing any differences between them, suggesting that
Lapdesf-4c promotes endothelium-independent vasorelaxation (
Figure 2A). NPS, a well-established NO donor used in clinics, was used as a control in our experiments. Both
Lapdesf-4c and NPS act as sGC stimulators. However, pEC50 e Emax values for NPS are superior to those values found for
Lapdesf-4c (
Figure 3).
To study the mechanisms by which
Lapdesf-4c acts, vasodilation studies were carried out in the presence of hydroxocobalamin (NO scavenger), ODQ (sGC inhibitor), and
L-NAME (eNOS inhibitor).
Figure 4 shows that
L-NAME did not affect the maximum percentage of vasodilation, although it reduced the potency of the drug, according to its pEC50 values. The attenuated effect is due to the influence of endothelium on vascular tonus, which acted independently of the vasodilator mediator. Nitric oxide induces its synthesis, through the activation of GCs and accumulation of cGMP in endothelial cells [
16]. Our data corroborated the study of vasodilation in the presence and absence of endothelium and demonstrated that
Lapdesf-4c was effective to promote vasodilation in the absence of endothelium. However, ODQ and hydroxocobalamin were effective to inhibit vasodilation induced by
Lapdesf-4c, suggesting the involvement of NO pathway in our observations (
Figure 4).
For ODQ, it was reported that some NO donors can produce a vasodilator effect even in the presence of sGC inhibitors, such as linsidomine. However, other NO donors are unable to produce such an effect, due to the reflex blockage that exists to cGMP. The GCs blockage was already described for NPS [
17]. In our results, we conclude that
Lapdesf-4c promotes vasodilation independently of the endothelium; however, it is necessary to activate tissue through the cGMP pathway to promote vasodilation.
Vasculopathy in ED has been correlated with the low bioavailability of NO and increased levels of vasoconstrictors, such as Ang II and ET-1 [
6,
18]. Studies demonstrated that the interaction of Ang II with AT1 receptors in endothelial cells and vascular smooth muscle induces AT1 to activate the production of bET-1, a precursor to ET-1, which induces vasoconstriction and vascular/myocardial hypertrophies. Therefore, high levels of ET-1 are considered a cardiovascular risk for cardiovascular diseases [
19]. Furthermore, Ang II is correlated with an overstimulation of the sympathetic nervous system (SNS). Thus, ROS and ET-1 released by Ang II can stimulate the SNS, increasing its constricting response in the vasculature and developing an increase in sympathetic activity, which in turn is associated with the worsening of ED [
20]. Assuming that ED patients have high levels of Ang II, ROS, ET-1, and chronic activation of the SNS, it is reasonable that such patients are more prone to develop ED [
7,
20,
21]. In this context, compounds acting to protect against ED are useful to prevent cardiovascular damage. Previous work conducted by Sanchez and colleagues (2007) demonstrated that the incubation of aortic rings with Ang II (1 µM) induces ED in the vessels [
13]. Based on the work of Sanchez, the present study obtained similar data to those of Sanchez. After incubation of the rings with Ang II, a decrease in the response of the concentration–effect curve of Ach was observed, characterizing the ED. However, the results show that the previous incubation with
Lapdesf-4c promoted endothelial protection by comparasion of the Emax values for both groups, suggesting the vasoprotective effect of
Lapdesf-4c (
p < 0.0001) (
Figure 5).
We evaluated the protective effect of
Lapdesf-4c to prevent ED. For this, based on the concentration–effect curve, a low concentration of
Lapdesf-4c was used in order to avoid interfering with the Ach response, as demonstrated by the DAF-2DA experiment (
Figure 6). Indirect measurement of intracellular NO levels showed no differences for increased intracellular NO levels at the concentrations tested.
Previous reports have described that treatment with S-nitrous acetyl DL-penicillamine (SNAP), another NO donor, was able to cause gene suppression of AT1-type receptors. In addition to vasodilation properties, NO donors downregulate AT1 levels, which could be useful to prevent ED [
22]. Another work demonstrated that exposure of endothelial cells to different NO donors increased the gene expression of the Ang II receptor (AT2), which has opposite effects to AT1 [
22]. High levels of Ang II increase the number of reactive oxygen species through the activation of the NADPH oxidase complex [
6]. Ang II binding to the AT1 receptor occurs through the activation of G protein, promoting gp91phox activation. This intracellular mediator is regulated by the p47phox subunit, which undergoes phosphorylation and activates the Nox-1 subunit of the NADPH oxidase complex. Thus, it affects the catalytic activities and releasing of ROS, especially O
2−, which reacts with NO to form NOOO
−. High levels of NOOO
− induce vessel inflammation, release pro-inflammatory cytokines, and induce the expression of adhesion molecules. Altogether, these events are prone to cause more endothelial damage [
18,
20]. ROS are potent secondary messengers that mediate signaling in pathways, whose increased levels favor vessel inflammation and change the oxidative state of the iron present in the heme groups. The interactions between Ang II signaling and ROS lead to changes in the structural and functional characteristics of the vasculature [
23].
Silva and colleagues (2016) demonstrated that
Lapdesf-4c reduces the expression of gp91phox, responsible for increasing the NADPH oxidase complex [
10]. Based on these previous results and the effect of the compound in promoting endothelial protection, the present study decided to verify if, at the concentration used (1 μM), there was a decrease in O
2−. According to the results (
Figure 7), it was found that
Lapdesf-4c was able to promote the reduction of O
2− levels. The lucigenin experiment was also used to observe whether
Lapdesf-4c was able to reduce the levels of O
2−, at concentrations at which it did not release NO. Our results corroborate the previous observation from Silva and colleagues, since
Lapdesf-4c reduced the levels of O
2− in HUVEC cells, exhibiting cardiovascular protection effects.
Lapdesf-4c reduced the percentage of fluorescence levels compared to the Ang II group (
p < 0.001), suggesting that this compound exhibits an antioxidant effect, since in both experiments, we used concentrations that do not release NO. Silva et al. (2016) demonstrated in their previous studies on the compound that it does not present cytotoxicity (300 μM), which is in accordance with our results [
8,
9].
Lapdesf-4c is not cytotoxic for HUVEC cells up to high concentrations (200 μM).
4. Materials and Methods
Male Wistar rats, weighing 280–300 g, were housed at the Federal University of São Carlos (UFSCar) according to institutional guidelines for good laboratory practices using animals. Rats were maintained in temperature-controlled facilities on a 12 h light–dark cycle with ad libitum food (standard rat chow) and water access. All the procedures were previously approved by the Animal Care and Use Committee of the UFSCar (Protocol number CEUA nº 1295101219).
4.1. Vascular Reactivity Studies
The rats were euthanized by decapitation, the thoracic aortas were isolated, and n = 10 were used, with each aortic ring coming from the aorta of a different rat. Aortic rings were cut in 4 mm length, placed in bath chambers for isolated organs containing Krebs solution (5 mL) at 37 °C, and continuously bubbled with 95% O2 and 5% CO2, pH 7.4, in an isometric myograph (Mulvany-Halpern-model 610 DMT-USA, Marietta, GA, USA). The data were recorded by a PowerLab8/SP data acquisition system (AD Instruments Pty Ltd., Colorado Springs, CO, USA).
The aortic rings were submitted to a tension of 1.5 g, which was readjusted every 15 min throughout a 60 min equilibration period before the addition of the tested drug. Endothelial integrity (E+) was assessed by the degree of relaxation induced by 1 μM acetylcholine, after contraction of the aortic ring by phenylephrine (0.1 μM). The ring was discarded if relaxation with acetylcholine was lower than 80%. After the endothelial integrity test, aortic rings were pre-contracted with phenylephrine (0.1 μM), and then concentration–effect curves for Lapdesf-4c were obtained. The curves were also obtained in the absence of endothelium (E−), which was mechanically removed by rolling the lumen of the vessel on a thin wire. Endothelial integrity was assessed by relaxation induced by 1 μM of acetylcholine after contraction of the aortic ring by phenylephrine (0.1 μM). Sodium nitroprusside was used as control (NPS) (0.01 µM to 100 µM). The maximum relaxant effect (EMax) and the potency values (pEC50) were obtained. Each experiment was performed on rings prepared from different rats.
4.2. Mechanisms Pathways
Aortic rings with endothelium (E+) were treated for 30 min using different inhibitors, as described: (NG-mono-methyl-L-arginine ester) (L-NAME) at 5 μM, as a non-specific NOS inhibitor; ((1H)-(1,2,4)-oxadiazol(4,3-a)-quinoxaline-1-ona)) (ODQ) at 10 μM, as a soluble guanylate cyclase (sGC) inhibitor; hydroxocobalamin at 5 μmol/L, as NO scavenger. After incubation, aortic rings were pre-contracted using phenylephrine (0.1 μmol/L), and concentration–effect curves for Lapdesf-4c were obtained.
4.3. Prevention of Endothelial Dysfunction
Aortic rings were placed in 6 wells containing a Krebs solution (5 mL), antibiotic–antimycotic mixture (penicillin (50 µL) and amphotericin B (2.5 µL)) in a cell culture incubator in the absence or presence of the treatment with
Lapdesf-4c (1 μM) for one hour and after with Angiotensin II (Ang II) (2 μM) for six hours. Then, aorta rings were immediately used for contractile tension, and the values were recorded as described in
Section 2.2, providing the acetylcholine (ACh) curve. The Emax and the pEC50 were obtained and analyzed. Each experiment was performed on rings prepared from different rats.
4.4. Quantification of NO Levels
HUVEC cells (between the 15th and 20th passage) were placed in 96-well plates at the concentration of 10⁴ cells per well. The plate was incubated for 24 h in a humidified incubator containing 5% CO
2, at 37 °C. After 24 h, the treatment with the Lapdesf-4c (1 μM) was carried out, and the plates were incubated for 30 min in a humidified incubator. After the treatment, the Lapdesf-4c (1 μM) was removed, and the plates were gently washed with Phosphate Buffer Saline (PBS). The detection of intracellular NO was performed by incubation with a selective fluorescent probe 4,5-diaminofluorescein (DAF-2DA at 10 μM) for 30 min, to react with dinitrogen trioxide (N
2O
3) (an oxidation product of NO) and produce the fluorescent compound DAF-2DA. The reading was carried out with a SpectraMax GeminiXS fluorometer (Molecular Devices) at 435 nm excitation and 538 nm emission wavelength pair [
24].
4.5. Measurement of Superoxide Anion Production
HUVEC cells were seeded in 96-well plates at 104 cells per well and maintained at 37 °C in a humidified incubator containing 5% CO2 for 24 h. Cells were treated with Lapdesf-4c (1 μM) and lucigenin for 30 min, followed by treatment with Ang II 0.1 μmol/L for 30 min. The increase in fluorescence intensity was monitored using a fluorescence microplate reader (SpectraMaxGeminiXS, Molecular Devices) at 510 nm and 595 excitation wavelength pair.
4.6. Cellular Culture and Cell Viability Assay
Immortalized human umbilical endothelial cells (HUVEC) were grown in a DMEM medium (Inlab) supplemented with 10% of fetal calf serum, antibiotics, and antimycotics. Cultures were maintained at 37 ± 2 °C in a 5% CO2 atmosphere. The cells in confluence were trypsinized, centrifuged at 1200 rpm for 5 min, and plated in 96-well plates.
HUVEC cells were plated at a concentration of 5 × 104 cells per well and maintained at 37 °C in a humidified incubator containing 5% CO2 per 24 h. Cells were treated with Lapdesf-4c (200 μM) and triton 1%. Then, the viability of the cells was measured by using a colorimetric assay with MTT ([3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide). After 24 h of incubation, the medium was displaced, and 200 μL of MTT (5 mg/mL) was incubated for 4 h to form formazan crystals. Then, this solution was replaced by 100 μL of dimethyl sulfoxide (DMSO 100%) in each well and was incubated for 5 min, and the absorbance was read at 550 nm.
4.7. Statistical Analysis
Statistical analysis of the results was performed using GraphPad Prism version 8.0. Statistical significance was tested by one-way ANOVA (post hoc test: Newman–Keuls). Data are expressed as mean ± DP. In each set of experiments, n indicates the number of animals used in the study. Values of p < 0.05 were considered significant.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







