
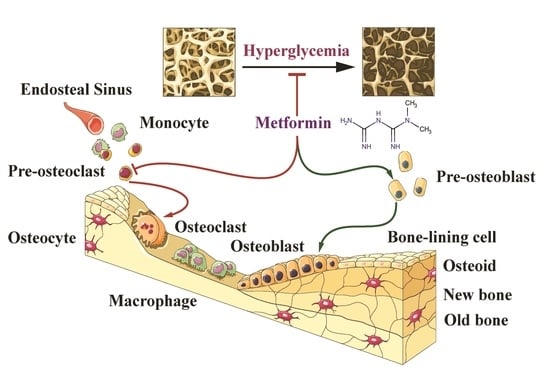
The Potential Therapeutic Role of Metformin in Diabetic and Non-Diabetic Bone Impairment



Abstract
{kind=link}
{kind=link}
{kind=link}
1. Introduction
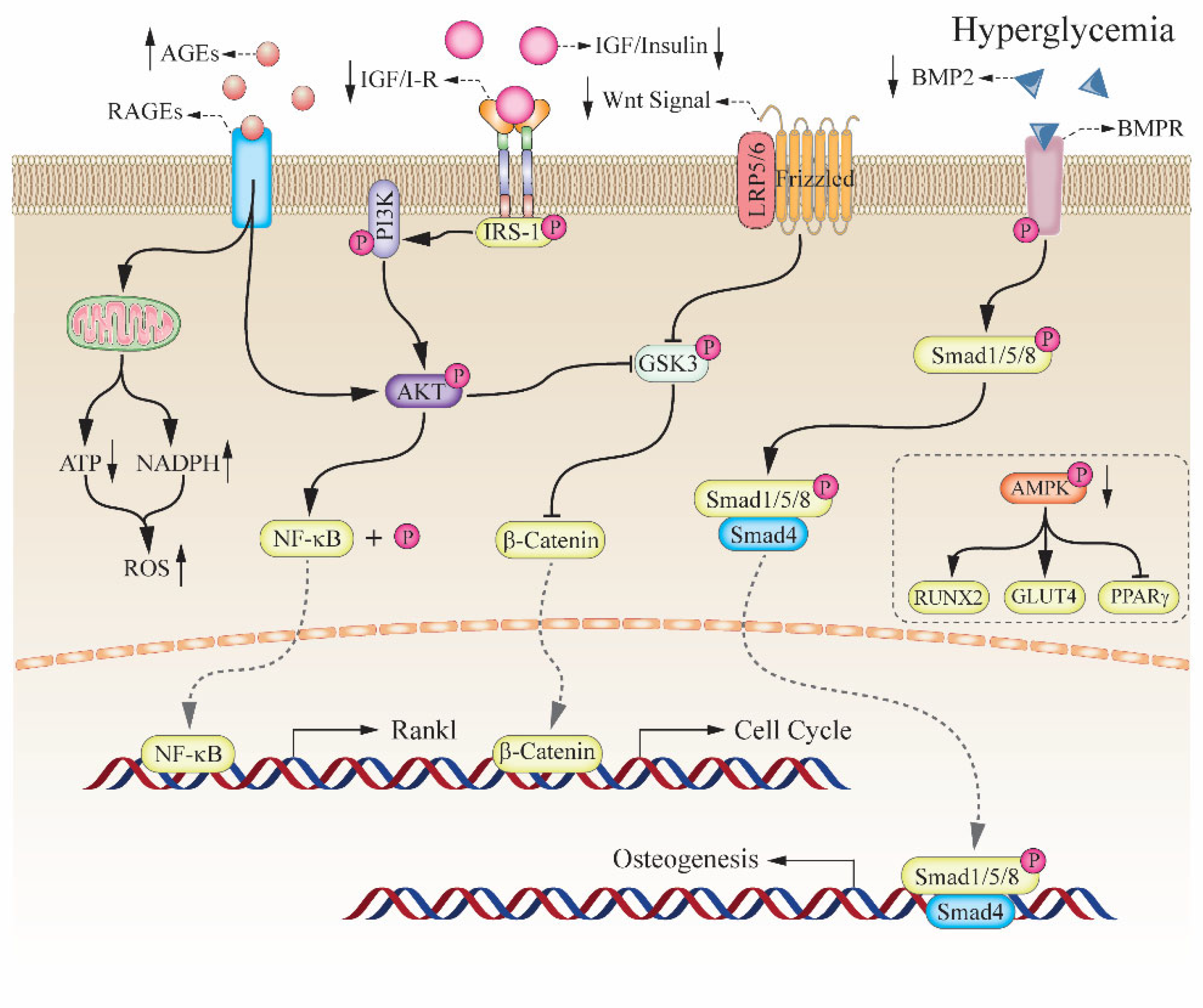
2. The Relationship between Hyperglycemia and Bone Impairment
2.1. Diabetic Induced the High Risk of Bone Fragility
2.2. Key Role of AMPK Complex in Bone Metabolism
2.3. Insulin and Insulin-like Growth Factor-1 in Bone Formation
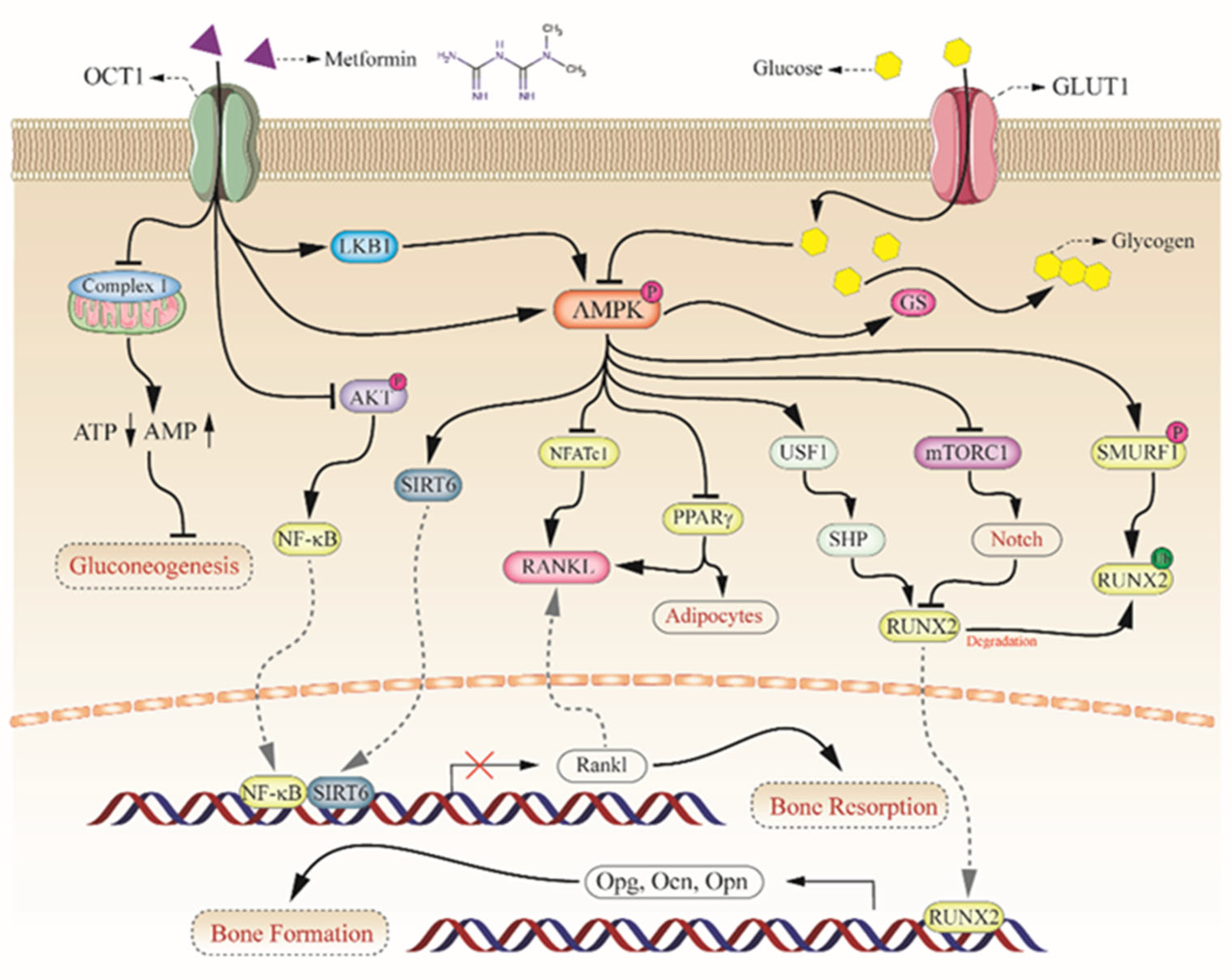
3. The Potential Mechanism of Metformin on Diabetic Bone Improvement
3.1. Activation of AMPKa and RUNX2 in Bone Formation
3.2. Upregulation of OPG/RANKL in Bone Resorption
3.3. Sirtuins in Bone Metabolism

4. Effects of Metformin on Cells In Vitro
4.1. Effects on Stem Cells
4.2. Acts on Other Bone Cells
5. Research on Bone Defect Animal Models
5.1. Promote Alveolar Bone Repair
5.2. Enhance Tendon-Bone Interface Healing
5.3. Single Use of Metformin in Bone Repair
5.4. Combinational Use of Metformin in Bone Repair
6. Investigations on Clinical Settings
7. Discussion and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huang, W.; Castelino, R.L.; Peterson, G.M. Metformin usage in type 2 diabetes mellitus: Are safety guidelines adhered to? Intern. Med. J. 2014, 44, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Shafiei-Irannejad, V.; Samadi, N.; Salehi, R.; Yousefi, B.; Zarghami, N. New insights into antidiabetic drugs: Possible applications in cancer treatment. Chem. Biol. Drug Des. 2017, 90, 1056–1066. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, H.M. AMPK and Exercise: Glucose Uptake and Insulin Sensitivity. Diabetes Metab. J. 2013, 37, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Chopra, I.; Li, H.F.; Wang, H.; Webster, K.A. Phosphorylation of the insulin receptor by AMP-activated protein kinase (AMPK) promotes ligand-independent activation of the insulin signalling pathway in rodent muscle. Diabetologia 2012, 55, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Bahne, E.; Sun, E.W.L.; Young, R.L.; Hansen, M.; Sonne, D.P.; Hansen, J.S.; Rohde, U.; Liou, A.P.; Jackson, M.L.; de Fontgalland, D.; et al. Metformin-induced glucagon-like peptide-1 secretion contributes to the actions of metformin in type 2 diabetes. JCI Insight 2018, 3, e93936. [Google Scholar] [CrossRef] [PubMed]
- Nash, R.J.; Kato, A.; Yu, C.Y.; Fleet, G.W.J. Iminosugars as therapeutic agents: Recent advances and promising trends. Future Med. Chem. 2011, 3, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.F.; Shimadate, Y.; Kato, A.; Li, Y.-X.; Jia, Y.-M.; Fleet, G.W.J.; Yu, C.-Y. Synthesis and glycosidase inhibition of N-substituted derivatives of DIM. Org. Biomol. Chem. 2020, 18, 999–1011. [Google Scholar] [CrossRef]
- Chennaiah, A.; Dahiya, A.; Dubbu, S.; Vankar, Y.D. A Stereoselective Synthesis of an Imino Glycal: Application in the Synthesis of (−)-1-Epi -Adenophorine and a Homoiminosugar. Eur. J. Org. Chem. 2018, 2018, 6574–6581. [Google Scholar] [CrossRef]
- Chennaiah, A.; Bhowmick, S.; Vankar, Y.D. Conversion of glycals into vicinal-1,2-diazides and 1,2-(or 2,1)-azidoacetates using hypervalent iodine reagents and Me3SiN3. Application in the synthesis of N-glycopeptides, pseudo-trisaccharides and an iminosugar. RSC Adv. 2017, 7, 41755–41762. [Google Scholar] [CrossRef]
- Rajasekaran, P.; Ande, C.; Vankar, Y.D. Synthesis of (5,6 & 6,6)-oxa-oxa annulated sugars as glycosidase inhibitors from 2-formyl galactal using iodocyclization as a key step. ARKIVOC 2022, 2022, 5–23. [Google Scholar]
- Vavanikunnel, J.; Charlier, S.; Becker, C.; Schneider, C.; Jick, S.S.; Meier, C.R.; Meier, C. Association Between Glycemic Control and Risk of Fracture in Diabetic Patients: A Nested Case-Control Study. J. Clin. Endocrinol. Metab. 2019, 104, 1645–1654. [Google Scholar] [CrossRef] [PubMed]
- Davie, G.S.; Pal, K.; Orton, E.; Tyrrell, E.G.; Petersen, I. Incident Type 2 Diabetes and Risk of Fracture: A Comparative Cohort Analysis Using U.K. Primary Care Records. Diabetes Care 2021, 44, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Chen, H.; Li, J.; Li, T.; Zheng, B.; Zheng, Y.; Jin, H.; He, Y.; Gu, Q.; Xu, X. Sirtuin 1-mediated cellular metabolic memory of high glucose via the LKB1/AMPK/ROS pathway and therapeutic effects of metformin. Diabetes 2012, 61, 217–228. [Google Scholar] [CrossRef]
- Drzewoski, J.; Hanefeld, M. The Current and Potential Therapeutic Use of Metformin-The Good Old Drug. Pharmaceuticals 2022, 14, 122. [Google Scholar] [CrossRef]
- Zhu, Z.N.; Jiang, Y.F.; Ding, T. Risk of fracture with thiazolidinediones: An updated meta-analysis of randomized clinical trials. Bone 2014, 68, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Molinuevo, M.S.; Schurman, L.; McCarthy, A.D.; Cortizo, A.M.; Tolosa, M.J.; Gangoiti, M.V.; Arnol, V.; Sedlinsky, C. Effect of metformin on bone marrow progenitor cell differentiation: In vivo and in vitro studies. J. Bone Min. Res. 2010, 25, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Lecka-Czernik, B. Diabetes, bone and glucose-lowering agents: Basic biology. Diabetologia 2017, 60, 1163–1169. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Shimazu, J.; Makinistoglu, M.P.; Maurizi, A.; Kajimura, D.; Zong, H.; Takarada, T.; Lezaki, T.; Pessin, J.E.; Hinoi, E.; et al. Glucose Uptake and Runx2 Synergize to Orchestrate Osteoblast Differentiation and Bone Formation. Cell 2015, 161, 1576–1591. [Google Scholar] [CrossRef]
- Paschou, S.A.; Dede, A.D.; Anagnostis, P.G.; Vryonidou, A.; Morganstein, D.; Goulis, D.G. Type 2 Diabetes and Osteoporosis: A Guide to Optimal Management. J. Clin. Endocrinol. Metab. 2017, 102, 3621–3634. [Google Scholar] [CrossRef]
- Farr, J.N.; Drake, M.T.; Amin, S.; Melton, L.J., 3rd; McCready, L.K.; Khosla, S. In vivo assessment of bone quality in postmenopausal women with type 2 diabetes. J. Bone Min. Res. 2014, 29, 787–795. [Google Scholar] [CrossRef]
- Katayama, Y.; Akatsu, T.; Yamamoto, M.; Kugai, N.; Nagata, N. Role of nonenzymatic glycosylation of type I collagen in diabetic osteopenia. J. Bone Min. Res. 1996, 11, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, C.; Zhou, Y.; Chen, W.; Luo, G.; Zhang, Z.; Wang, H.; Zhang, Y.; Xu, D.; Sheng, P. Advanced glycation end products biphasically modulate bone resorption in osteoclast-like cells. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E355–E366. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Guo, Q.; Xiao, Y.; Guo, Q.; Huang, Y.; Li, C.; Luo, X. Endocrine role of bone in the regulation of energy metabolism. Bone Res. 2021, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Lecka-Czernik, B.; Moerman, E.J.; Grant, D.F.; Lehmann, J.M.; Manolagas, S.C.; Jilka, R.L. Divergent effects of selective peroxisome proliferator-activated receptor-gamma 2 ligands on adipocyte versus osteoblast differentiation. Endocrinology 2002, 143, 2376–2384. [Google Scholar] [CrossRef] [PubMed]
- Manolagas, S.C. From estrogen-centric to aging and oxidative stress: A revised perspective of the pathogenesis of osteoporosis. Endocr. Rev. 2010, 31, 266–300. [Google Scholar] [CrossRef]
- Hamann, C.; Kirschner, S.; Gunther, K.P.; Hofbauer, L.C. Bone, sweet bone--osteoporotic fractures in diabetes mellitus. Nat. Rev. Endocrinol. 2012, 8, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, R.; Totsuka, Y.; Hamano, K.; Ajima, M.; Miura, M.; Hirota, Y.; Hata, K.; Fukumoto, S.; Matsumoto, T. Metabolic improvement of poorly controlled noninsulin-dependent diabetes mellitus decreases bone turnover. J. Clin. Endocrinol. Metab. 1997, 82, 2915–2920. [Google Scholar] [CrossRef]
- McCarthy, A.D.; Cortizo, A.M.; Sedlinsky, C. Metformin revisited: Does this regulator of AMP-activated protein kinase secondarily affect bone metabolism and prevent diabetic osteopathy. World J. Diabetes 2016, 7, 122–133. [Google Scholar] [CrossRef]
- Carling, D. AMPK signalling in health and disease. Curr. Opin. Cell Bio.l 2017, 45, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMP-activated/SNF1 protein kinases: Conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 2007, 8, 774–785. [Google Scholar] [CrossRef]
- Liu, Y.; Park, J.M.; Chang, K.H.; Huh, H.J.; Lee, K.; Lee, M.Y. AMP-Activated Protein Kinase Mediates the Antiplatelet Effects of the Thiazolidinediones Rosiglitazone and Pioglitazone. Mol. Pharmacol. 2016, 89, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Reznick, R.M.; Shulman, G.I. The role of AMP-activated protein kinase in mitochondrial biogenesis. J. Physiol. 2006, 574 Pt 1, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Bolster, D.R.; Crozier, S.J.; Kimball, S.R.; Jefferson, L.S. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J. Biol. Chem. 2002, 277, 23977–23980. [Google Scholar] [CrossRef]
- Wang, Y.G.; Qu, X.H.; Yang, Y.; Han, X.G.; Wang, L.; Qiao, H.; Fan, Q.M.; Tang, T.T.; Dai, K.R. AMPK promotes osteogenesis and inhibits adipogenesis through AMPK-Gfi1-OPN axis. Cell. Signal. 2016, 28, 1270–1282. [Google Scholar] [CrossRef]
- Takada, I.; Suzawa, M.; Matsumoto, K.; Kato, S. Suppression of PPAR transactivation switches cell fate of bone marrow stem cells from adipocytes into osteoblasts. Ann. N. Y. Acad. Sci. 2007, 1116, 182–195. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Rohatgi, N.; Chen, T.H.; Schilling, J.; Abu-Amer, Y.; Teitelbaum, S.L. PPAR-gamma regulates pharmacological but not physiological or pathological osteoclast formation. Nat. Med. 2016, 22, 1203–1205. [Google Scholar] [CrossRef] [PubMed]
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [PubMed]
- Cirmanova, V.; Zofkova, I.; Kasalicky, P.; Lanska, V.; Bayer, M.; Starka, L.; Kanceva, R. Hormonal and bone parameters in pubertal girls. Physiol. Res. 2017, 66 (Suppl. 3), S419–S424. [Google Scholar] [CrossRef]
- Fowlkes, J.L.; Bunn, R.C.; Thrailkill, K.M. Contributions of the Insulin/Insulin-Like Growth Factor-1 Axis to Diabetic Osteopathy. J. Diabetes Metab. 2011, 1, S1–S003. [Google Scholar] [PubMed]
- Shah, V.N.; Carpenter, R.D.; Ferguson, V.L.; Schwartz, A.V. Bone health in type 1 diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 2018, 25, 231–236. [Google Scholar] [CrossRef]
- Kanazawa, I.; Yamaguchi, T.; Sugimoto, T. Serum insulin-like growth factor-I is a marker for assessing the severity of vertebral fractures in postmenopausal women with type 2 diabetes mellitus. Osteoporos. Int. 2011, 22, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, I.; Yamaguchi, T.; Yano, S.; Yamauchi, M.; Yamamoto, M.; Sugimoto, T. Adiponectin and AMP kinase activator stimulate proliferation, differentiation, and mineralization of osteoblastic MC3T3-E1 cells. BMC Cell Biol. 2007, 8, 51. [Google Scholar] [CrossRef]
- Cao, W.; Li, J.; Yang, K.; Cao, D. An overview of autophagy: Mechanism, regulation and research progress. Bull. Cancer 2021, 108, 304–322. [Google Scholar] [CrossRef] [PubMed]
- Marsh, T.; Tolani, B.; Debnath, J. The pleiotropic functions of autophagy in metastasis. J. Cell Sci. 2021, 134, jcs247056. [Google Scholar] [CrossRef]
- Li, J.X.; Yan, Q.; Liu, N.; Zheng, W.J.; Hu, M.; Yu, Z.M.; Zhou, Y.D.; Wang, X.W.; Liang, F.X.; Chen, R. The Prognostic Value of Autophagy-Related Markers Bclin-1 and LC-3 in Colorectal Cancers: A Systematic Review and Meta-analysis. Evid. Based Complement. Altern. Med. 2020, 2020, 8475840. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Yee, S.W.; Seiser, E.L.; van Leeuwen, N.; Tavendale, R.; Bennett, A.J.; Groves, C.J.; Coleman, R.L.; van der Heijden, A.A.; Beulens, J.W.; et al. Variation in the glucose transporter gene SLC2A2 is associated with glycemic response to metformin. Nat. Genet. 2016, 48, 1055–1059. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, T.; Pafundi, P.C.; Galiero, R.; Gjeloshi, K.; Masini, F.; Acierno, C.; Di Martino, A.; Albanese, G.; Alfano, M.; Rinaldi, L.; et al. Metformin: A Potential Therapeutic Tool for Rheumatologists. Pharmaceuticals 2020, 13, 234. [Google Scholar] [CrossRef]
- Huang, B.; Wang, Y.; Wang, W.; Chen, J.; Lai, P.; Liu, Z.; Yan, B.; Xu, S.; Zhang, Z.; Zeng, C.; et al. mTORC1 Prevents Preosteoblast Differentiation through the Notch Signaling Pathway. PLoS Genet. 2015, 11, e1005426. [Google Scholar]
- Canalis, E.; Parker, K.; Feng, J.Q.; Zanotti, S. Osteoblast lineage-specific effects of notch activation in the skeleton. Endocrinology 2013, 154, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Engin, F.; Yao, Z.; Yang, T.; Zhou, G.; Bertin, T.; Jiang, M.M.; Chen, Y.; Wang, L.; Zheng, H.; Sutton, R.E.; et al. Dimorphic effects of Notch signaling in bone homeostasis. Nat. Med. 2008, 14, 299–305. [Google Scholar]
- Hilton, M.J.; Tu, X.; Wu, X.; Bai, S.; Zhao, H.; Kobayashi, T.; Kronenberg, H.M.; Teitelbaum, S.L.; Ross, F.P.; Kopan, R.; et al. Notch signaling maintains bone marrow mesenchymal progenitors by suppressing osteoblast differentiation. Nat. Med. 2008, 14, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Meng, Y.; Kwiatkowski, D.J.; Chen, X.; Peng, H.; Sun, Q.; Zha, X.; Wang, F.; Wang, Y.; Jing, Y.; et al. Mammalian target of rapamycin regulates murine and human cell differentiation through STAT3/p63/Jagged/Notch cascade. J. Clin. Investig. 2010, 120, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Ding, D.; Wang, L.; Yan, J.; Ma, L.; Jin, Q. Metformin attenuates osteoclast-mediated abnormal subchondral bone remodeling and alleviates osteoarthritis via AMPK/NF-kappaB/ERK signaling pathway. PLoS ONE 2021, 16, e0261127. [Google Scholar] [CrossRef] [PubMed]
- Jiating, L.; Buyun, J.; Yinchang, Z. Role of Metformin on Osteoblast Differentiation in Type 2 Diabetes. BioMed Res. Int. 2019, 2019, 9203934. [Google Scholar] [CrossRef]
- Batandier, C.; Guigas, B.; Detaille, D.; El-Mir, M.Y.; Fontaine, E.; Rigoulet, M.; Leverve, X.M. The ROS production induced by a reverse-electron flux at respiratory-chain complex 1 is hampered by metformin. J. Bioenergy Biomembr. 2006, 38, 33–42. [Google Scholar] [CrossRef]
- Kuang, Y.; Hu, B.; Feng, G.; Xiang, M.; Deng, Y.; Tan, M.; Li, J.; Song, J. Metformin prevents against oxidative stress-induced senescence in human periodontal ligament cells. Biogerontology 2020, 21, 13–27. [Google Scholar] [CrossRef]
- Wang, L.X.; Wang, G.Y.; Su, N.; Ma, J.; Li, Y.K. Effects of different doses of metformin on bone mineral density and bone metabolism in elderly male patients with type 2 diabetes mellitus. World J. Clin. Cases 2020, 8, 4010–4016. [Google Scholar] [CrossRef]
- Kanazawa, I.; Takeno, A.; Tanaka, K.I.; Notsu, M.; Sugimoto, T. Osteoblast AMP-activated protein kinase regulates glucose metabolism and bone mass in adult mice. Biochem. Biophys. Res. Commun. 2018, 503, 1955–1961. [Google Scholar] [CrossRef]
- Takarada, T.; Hinoi, E.; Nakazato, R.; Ochi, H.; Xu, C.; Tsuchikane, A.; Takeda, S.; Karsenty, G.; Abe, T.; Kiyonari, H.; et al. An analysis of skeletal development in osteoblast-specific and chondrocyte-specific runt-related transcription factor-2 (Runx2) knockout mice. J. Bone Min. Res. 2013, 28, 2064–2069. [Google Scholar] [CrossRef]
- Jang, W.G.; Kim, E.J.; Bae, I.H.; Lee, K.N.; Kim, Y.D.; Kim, D.K.; Kim, S.H.; Lee, C.H.; Franceschi, R.T.; Choi, H.S.; et al. Metformin induces osteoblast differentiation via orphan nuclear receptor SHP-mediated transactivation of Runx2. Bone 2011, 48, 885–893. [Google Scholar] [CrossRef]
- Colombo, J.S.; Balani, D.; Sloan, A.J.; Crean, S.J.; Okazaki, J.; Waddington, R.J. Delayed osteoblast differentiation and altered inflammatory response around implants placed in incisor sockets of type 2 diabetic rats. Clin Oral Implants Res 2011, 22, 578–586. [Google Scholar] [CrossRef]
- Hashiguchi, C.; Kawamoto, S.; Kasai, T.; Nishi, Y.; Nagaoka, E. Influence of an antidiabetic drug on biomechanical and histological parameters around implants in type 2 diabetic rats. Implant Dent. 2014, 23, 264–269. [Google Scholar] [CrossRef]
- Serrao, C.R.; Bastos, M.F.; Cruz, D.F.; de Souza Malta, F.; Vallim, P.C.; Duarte, P.M. Role of Metformin in Reversing the Negative Impact of Hyperglycemia on Bone Healing Around Implants Inserted in Type 2 Diabetic Rats. Int. J. Oral Maxillofac. Implants 2017, 32, 547–554. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, C.; Hu, Y.; Peng, B. Protective effect of metformin on periapical lesions in rats by decreasing the ratio of receptor activator of nuclear factor kappa B ligand/osteoprotegerin. J. Endod. 2012, 38, 943–947. [Google Scholar] [CrossRef]
- Mai, Q.G.; Zhang, Z.M.; Xu, S.; Lu, M.; Zhou, R.P.; Zhao, L.; Jia, C.H.; Wen, Z.H.; Jin, D.D.; Bai, X.C. Metformin stimulates osteoprotegerin and reduces RANKL expression in osteoblasts and ovariectomized rats. J. Cell. Biochem. 2011, 112, 2902–2909. [Google Scholar] [CrossRef] [PubMed]
- Mu, W.; Wang, Z.; Ma, C.; Jiang, Y.; Zhang, N.; Hu, K.; Li, L.; Wang, Z. Metformin promotes the proliferation and differentiation of murine preosteoblast by regulating the expression of sirt6 and oct4. Pharmacol. Res. 2018, 129, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Pei, L.; Zhou, S.; Tao, L.; Zhu, Y. Metformin attenuates H2O2-induced osteoblast apoptosis by regulating SIRT3 via the PI3K/AKT pathway. Exp. Ther. Med. 2021, 22, 1316. [Google Scholar] [CrossRef]
- Ren, C.; Hao, X.; Wang, L.; Hu, Y.; Meng, L.; Zheng, S.; Ren, F.; Bu, W.; Wang, H.; Li, D.; et al. Metformin Carbon Dots for Promoting Periodontal Bone Regeneration via Activation of ERK/AMPK Pathway. Adv. Healthc. Mater. 2021, 10, e2100196. [Google Scholar] [CrossRef]
- Zhu, J.; Ye, H.; Deng, D.; Li, J.; Wu, Y. Electrospun metformin-loaded polycaprolactone/chitosan nanofibrous membranes as promoting guided bone regeneration membranes: Preparation and characterization of fibers, drug release, and osteogenic activity in vitro. J. Biomater. Appl. 2020, 34, 1282–1293. [Google Scholar] [CrossRef]
- Shi, Q.; Zhang, T.; Chen, Y.; Xu, Y.; Deng, Z.; Xu, D. Local Administration of Metformin Improves Bone Microarchitecture and Biomechanical Properties During Ruptured Canine Achilles Tendon-Calcaneus Interface Healing. Am. J. Sports Med. 2022, 50, 2145–2154. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, Z.L.; Hu, X.T.; Wang, X.T.; Chen, A.M. Metformin promotes differentiation of human bone marrow derived mesenchymal stem cells into osteoblast via GSK3beta inhibition. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 7962–7968. [Google Scholar] [PubMed]
- Lin, H.; Shi, F.; Jiang, S.; Wang, Y.; Zou, J.; Ying, Y.; Huang, D.; Luo, L.; Yan, X.; Luo, Z. Metformin attenuates trauma-induced heterotopic ossification via inhibition of Bone Morphogenetic Protein signalling. J. Cell. Mol. Med. 2020, 24, 14491–14501. [Google Scholar] [CrossRef]
- Yagci, B.S.; Odabas, S.; Aksoy, E.A. Development of metformin chain extended polyurethane elastomers as bone regenerative films. Eur. J. Pharm. Sci. 2019, 131, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Kang, M.A.; Moon, Y.J.; Jang, K.Y.; Kim, J.R. Metformin coordinates osteoblast/osteoclast differentiation associated with ischemic osteonecrosis. Aging 2020, 12, 4727–4741. [Google Scholar] [CrossRef] [PubMed]
- Gamez, B.; Morris, E.V.; Olechnowicz, S.W.Z.; Webb, S.; Edwards, J.R.; Sowman, A.; Turner, C.J.; Edwards, C.M. The antidiabetic drug metformin acts on the bone microenvironment to promote myeloma cell adhesion to preosteoblasts and increase myeloma tumour burden in vivo. Transl. Oncol. 2022, 15, 101301. [Google Scholar] [CrossRef]
- Smieszek, A.; Tomaszewski, K.A.; Kornicka, K.; Marycz, K. Metformin Promotes Osteogenic Differentiation of Adipose-Derived Stromal Cells and Exerts Pro-Osteogenic Effect Stimulating Bone Regeneration. J. Clin. Med. 2018, 7, 482. [Google Scholar] [CrossRef]
- Fang, C.H.; Sun, C.K.; Lin, Y.W.; Hung, M.C.; Lin, H.Y.; Li, C.H.; Lin, I.P.; Chang, H.C.; Sun, J.S.; Chang, J.Z. Metformin-Incorporated Gelatin/Nano-Hydroxyapatite Scaffolds Promotes Bone Regeneration in Critical Size Rat Alveolar Bone Defect Model. Int. J. Mol. Sci. 2022, 23, 558. [Google Scholar] [CrossRef]
- Khajuria, D.K.; Patil, O.N.; Karasik, D.; Razdan, R. Development and evaluation of novel biodegradable chitosan based metformin intrapocket dental film for the management of periodontitis and alveolar bone loss in a rat model. Arch. Oral Biol. 2018, 85, 120–129. [Google Scholar] [CrossRef]
- Xu, W.; Tan, W.; Li, C.; Wu, K.; Zeng, X.; Xiao, L. Metformin-loaded beta-TCP/CTS/SBA-15 composite scaffolds promote alveolar bone regeneration in a rat model of periodontitis. J. Mater. Sci. Mater. Med. 2021, 32, 145. [Google Scholar] [CrossRef]
- Zhao, Z.; Liu, J.; Schneider, A.; Gao, X.; Ren, K.; Weir, M.D.; Zhang, N.; Zhang, K.; Zhang, L.; Bai, Y.; et al. Human periodontal ligament stem cell seeding on calcium phosphate cement scaffold delivering metformin for bone tissue engineering. J. Dent. 2019, 91, 103220. [Google Scholar] [CrossRef]
- Yildirim, T.T.; Dundar, S.; Bozoglan, A.; Karaman, T.; Kahraman, O.E.; Ozcan, E.C. The effects of metformin on the bone filling ration around of TiAl6Va4 implants in non diabetic rats. J. Oral Biol. Craniofac. Res. 2020, 10, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Shahrezaee, M.; Salehi, M.; Keshtkari, S.; Oryan, A.; Kamali, A.; Shekarchi, B. In vitro and in vivo investigation of PLA/PCL scaffold coated with metformin-loaded gelatin nanocarriers in regeneration of critical-sized bone defects. Nanomedicine 2018, 14, 2061–2073. [Google Scholar] [CrossRef] [PubMed]
- Fan, K.J.; Wu, J.; Wang, Q.S.; Xu, B.X.; Zhao, F.T.; Wang, T.Y. Metformin inhibits inflammation and bone destruction in collagen-induced arthritis in rats. Ann. Transl. Med. 2020, 8, 1565. [Google Scholar] [CrossRef]
- Neven, E.; Vervaet, B.; Brand, K.; Gottwald-Hostalek, U.; Opdebeeck, B.; De Mare, A.; Verhulst, A.; Lalau, J.D.; Kamel, S.; De Broe, M.E.; et al. Metformin prevents the development of severe chronic kidney disease and its associated mineral and bone disorder. Kidney Int. 2018, 94, 102–113. [Google Scholar] [CrossRef]
- Liu, Q.; Xu, X.; Yang, Z.; Liu, Y.; Wu, X.; Huang, Z.; Liu, J.; Huang, Z.; Kong, G.; Ding, J.; et al. Metformin Alleviates the Bone Loss Induced by Ketogenic Diet: An In Vivo Study in Mice. Calcif. Tissue Int. 2019, 104, 59–69. [Google Scholar] [CrossRef]
- Yan, Z.; Zhu, S.; Tian, X.; Ye, Z.; Zhai, D.; Zhu, Z.; Wei, D.; Zhu, Q.; Lu, Z.; Cao, X. Metformin protects bone mass in ultra-high-molecular-weight polyethylene particle-induced osteolysis by regulating osteocyte secretion. J. Bone Min. Metab. 2019, 37, 399–410. [Google Scholar] [CrossRef]
- Loh, D.K.W.; Kadirvelu, A.; Pamidi, N. Effects of Metformin on Bone Mineral Density and Adiposity-Associated Pathways in Animal Models with Type 2 Diabetes Mellitus: A Systematic Review. J. Clin. Med. 2022, 11, 4193. [Google Scholar] [CrossRef]
- Tan, W.; Gao, C.; Feng, P.; Liu, Q.; Liu, C.; Wang, Z.; Deng, Y.; Shuai, C. Dual-functional scaffolds of poly(L-lactic acid)/nanohydroxyapatite encapsulated with metformin: Simultaneous enhancement of bone repair and bone tumor inhibition. Mater. Sci. Eng. C Mater. Biol. Appl. 2021, 120, 111592. [Google Scholar] [CrossRef]
- Sun, C.K.; Weng, P.W.; Chang, J.Z.; Lin, Y.W.; Tsuang, F.Y.; Lin, F.H.; Tsai, T.H.; Sun, J.S. Metformin-Incorporated Gelatin/Hydroxyapatite Nanofiber Scaffold for Bone Regeneration. Tissue Eng. Part A 2022, 28, 1–12. [Google Scholar] [CrossRef]
- Zhou, Q.; Guan, Z.; Liu, S.; Xuan, Y.; Han, G.; Chen, H.; Jin, X.; Tao, K.; Guan, Z. The effects of metformin and alendronate in attenuating bone loss and improving glucose metabolism in diabetes mellitus mice. Aging 2022, 14, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Liu, Q.; He, H.; Jiang, L.; Lee, K.O.; Li, D.; Ma, J. Metformin treatment is associated with an increase in bone mineral density in type 2 diabetes mellitus patients in China: A retrospective single center study. Diabetes Metab. 2022, 48, 101350. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, X.; Kang, S.; Duan, W. Correlation of Serum IGF-1R, VEGF, and ET Levels with Bone Mineral Density in Type 2 Diabetic Mellitus Patients Treated with Metformin Plus alpha-Glucosidase Inhibitors. Evid. Based Complement. Altern. Med. 2022, 2022, 7707875. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.H. Metformin and primary bone cancer risk in Taiwanese patients with type 2 diabetes mellitus. Bone 2021, 151, 116037. [Google Scholar] [CrossRef] [PubMed]
- Vogel, F.; Braun, L.; Rubinstein, G.; Zopp, S.; Osswald, A.; Schilbach, K.; Schmidmaier, R.; Bidlingmaier, M.; Reincke, M. Metformin and Bone Metabolism in Endogenous Glucocorticoid Excess: An Exploratory Study. Front. Endocrinol. 2021, 12, 765067. [Google Scholar] [CrossRef]
- Bolen, S.; Feldman, L.; Vassy, J.; Wilson, L.; Yeh, H.C.; Marinopoulos, S.; Wiley, C.; Selvin, E.; Wilson, R.; Bass, E.B. Systematic review: Comparative effectiveness and safety of oral medications for type 2 diabetes mellitus. Ann. Intern. Med. 2007, 147, 386–399. [Google Scholar] [CrossRef]
- Khurana, R.; Malik, I.S. Metformin: Safety in cardiac patients. Postgrad. Med. J. 2010, 86, 371–373. [Google Scholar] [CrossRef]
- Heaf, J. Metformin in chronic kidney disease: Time for a rethink. Perit. Dial. Int. 2014, 34, 353–357. [Google Scholar] [CrossRef]
- Nathan, D.M.; Buse, J.B.; Davidson, M.B.; Ferrannini, E.; Holman, R.R.; Sherwin, R.; Zinman, B.; American Diabetes Association; European Association for the Study of Diabetes. Medical management of hyperglycaemia in type 2 diabetes mellitus: A consensus algorithm for the initiation and adjustment of therapy: A consensus statement from the American Diabetes Association and the European Association for the Study of Diabetes. Diabetologia 2009, 52, 17–30. [Google Scholar] [CrossRef]
- Stang, M.; Wysowski, D.K.; Butler-Jones, D. Incidence of lactic acidosis in metformin users. Diabetes Care 1999, 22, 925–927. [Google Scholar] [CrossRef]
- Salpeter, S.R.; Greyber, E.; Pasternak, G.A.; and Salpeter, E.E. Risk of fatal and nonfatal lactic acidosis with metformin use in type 2 diabetes mellitus: Systematic review and meta-analysis. Arch. Intern. Med. 2003, 163, 2594–2602. [Google Scholar] [CrossRef] [PubMed]
- Teale, K.F.; Devine, A.; Stewart, H.; and Harper, N.J. The management of metformin overdose. Anaesthesia 1998, 53, 698–701. [Google Scholar] [CrossRef]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef]
- Rena, G.; Pearson, E.R.; Sakamoto, K. Molecular mechanism of action of metformin: Old or new insights? Diabetologia 2013, 56, 1898–1906. [Google Scholar] [CrossRef] [PubMed]
- Misra, P.; Chakrabarti, R. The role of AMP kinase in diabetes. Indian J. Med. Res. 2007, 125, 389–398. [Google Scholar] [PubMed]
- Valsecchi, F.; Ramos-Espiritu, L.S.; Buck, J.; Levin, L.R.; Manfredi, G. cAMP and mitochondria. Physiology 2013, 28, 199–209. [Google Scholar] [CrossRef]
- Hunter, R.W.; Hughey, C.C.; Lantier, L.; Sundelin, E.I.; Peggie, M.; Zeqiraj, E.; Sicheri, F.; Jessen, N.; Wasserman, D.H.; Sakamoto, K. Metformin reduces liver glucose production by inhibition of fructose-1-6-bisphosphatase. Nat. Med. 2018, 24, 1395–1406. [Google Scholar] [CrossRef]
- Malta, F.S.; Garcia, R.P.; Azarias, J.S.; Ribeiro, G.; Miranda, T.S.; Shibli, J.A.; Bastos, M.F. Impact of hyperglycemia and treatment with metformin on ligature-induced bone loss, bone repair and expression of bone metabolism transcription factors. PLoS ONE 2020, 15, e0237660. [Google Scholar] [CrossRef]
- Rakel, A.; Sheehy, O.; Rahme, E.; LeLorier, J. Osteoporosis among patients with type 1 and type 2 diabetes. Diabetes Metab. 2008, 34, 193–205. [Google Scholar] [CrossRef]
- Cortizo, A.M.; Lettieri, M.G.; Barrio, D.A.; Mercer, N.; Etcheverry, S.B.; McCarthy, A.D. Advanced glycation end-products (AGEs) induce concerted changes in the osteoblastic expression of their receptor RAGE and in the activation of extracellular signal-regulated kinases (ERK). Mol. Cell. Biochem. 2003, 250, 1–10. [Google Scholar] [CrossRef]
- Schurman, L.; McCarthy, A.D.; Sedlinsky, C.; Gangoiti, M.V.; Arnol, V.; Bruzzone, L.; Cortizo, A.M. Metformin reverts deleterious effects of advanced glycation end-products (AGEs) on osteoblastic cells. Exp. Clin. Endocrinol. Diabetes 2008, 116, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Bilha, S.C.; Leustean, L.; Preda, C.; Branisteanu, D.D.; Mihalache, L.; Ungureanu, M.C. Bone mineral density predictors in long-standing type 1 and type 2 diabetes mellitus. BMC Endocr. Disord. 2021, 21, 156. [Google Scholar] [CrossRef]
- Yan, X.L.; Wang, Y.Y.; Yu, Z.F.; Tian, M.M.; Li, H. Peroxisome proliferator-activated receptor-gamma activation attenuates diabetic cardiomyopathy via regulation of the TGF-beta/ERK pathway and epithelial-to-mesenchymal transition. Life Sci. 2018, 213, 269–278. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kim, Y.S.; Lee, S.Y.; Kim, G.H.; Kim, B.J.; Lee, S.H.; Lee, K.U.; Kim, G.S.; Kim, S.W.; Koh, J.M. AMP kinase acts as a negative regulator of RANKL in the differentiation of osteoclasts. Bone 2010, 47, 926–937. [Google Scholar] [CrossRef]
- Oliaro-Bosso, S.; Calcio Gaudino, E.; Mantegna, S.; Giraudo, E.; Meda, C.; Viola, F.; Cravotto, G. Regulation of HMGCoA reductase activity by policosanol and octacosadienol, a new synthetic analogue of octacosanol. Lipids 2009, 44, 907–916. [Google Scholar] [CrossRef]
- Montagnani, A.; Gonnelli, S. Antidiabetic therapy effects on bone metabolism and fracture risk. Diabetes Obes. Metab. 2013, 15, 784–791. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mu, W.; Liang, G.; Feng, Y.; Jiang, Y.; Qu, F. The Potential Therapeutic Role of Metformin in Diabetic and Non-Diabetic Bone Impairment. Pharmaceuticals 2022, 15, 1274. https://doi.org/10.3390/ph15101274
Mu W, Liang G, Feng Y, Jiang Y, Qu F. The Potential Therapeutic Role of Metformin in Diabetic and Non-Diabetic Bone Impairment. Pharmaceuticals. 2022; 15(10):1274. https://doi.org/10.3390/ph15101274
Chicago/Turabian StyleMu, Wei, Guoqiang Liang, Yue Feng, Yunyun Jiang, and Falin Qu. 2022. "The Potential Therapeutic Role of Metformin in Diabetic and Non-Diabetic Bone Impairment" Pharmaceuticals 15, no. 10: 1274. https://doi.org/10.3390/ph15101274
APA StyleMu, W., Liang, G., Feng, Y., Jiang, Y., & Qu, F. (2022). The Potential Therapeutic Role of Metformin in Diabetic and Non-Diabetic Bone Impairment. Pharmaceuticals, 15(10), 1274. https://doi.org/10.3390/ph15101274