
Cu(II)-Catalysed Hydrocarboxylation of Imines Utilizing CO2 to Synthesize α-Unsaturated Aminocarboxylic Acids


Abstract
1. Introduction
2. Results and Discussion
2.1. Photocatalyst Characterization
2.1.1. UV-Vis and Band Gap
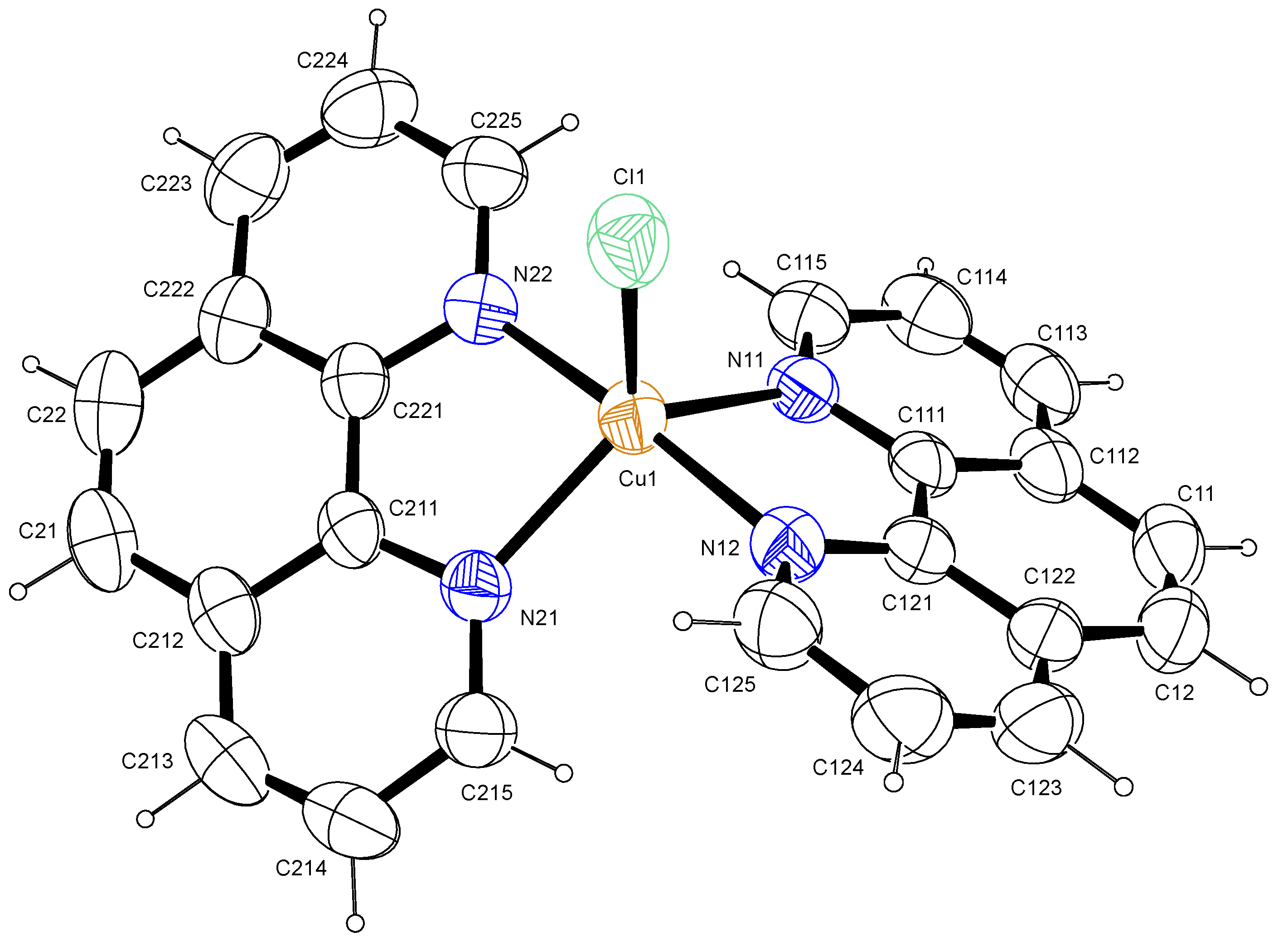
2.1.2. Crystal Structure: [Cu(phen)2Cl]
2.2. Schiff Bases and Corresponding Amino Acid (Hydrocarboxylation)
2.2.1. Schiff Bases
2.2.2. Hydrocarboxylation Reaction
2.2.3. Mechanistic Pathway of Cu(II) Photocatalyst
2.2.4. FT-IR
2.3. Theoretical Studies
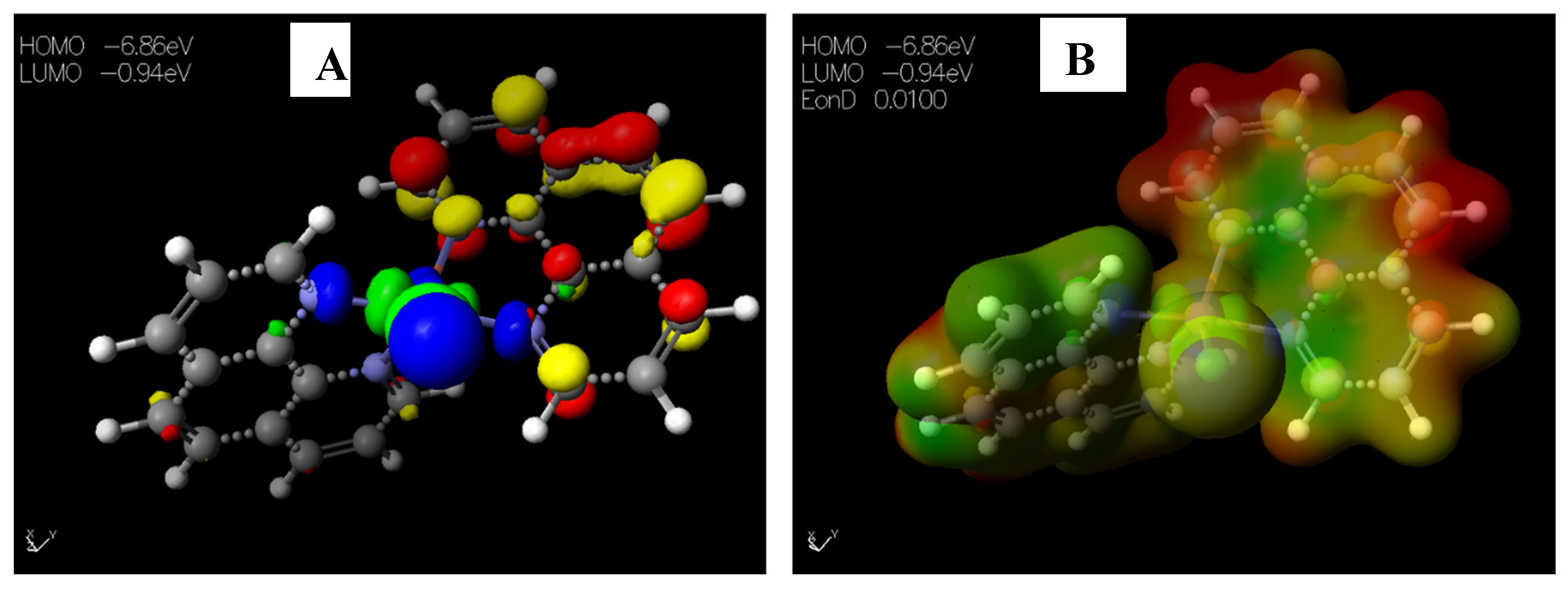
2.3.1. Chemical Descriptors of [Cu(phen)2Cl]
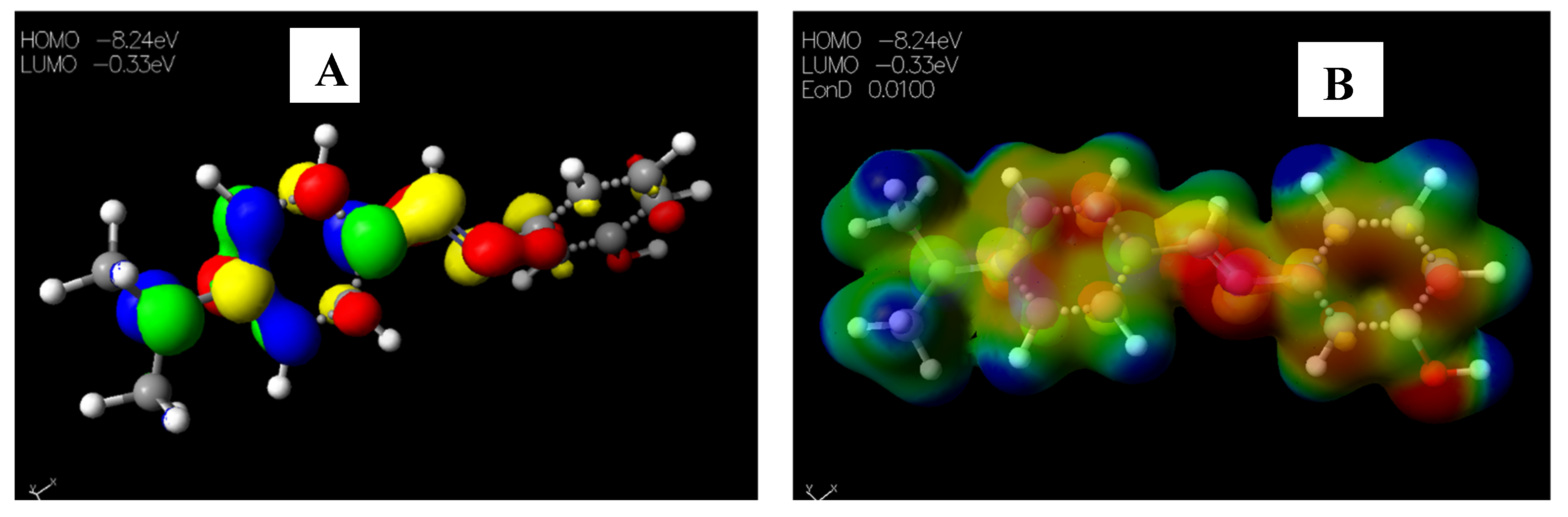
2.3.2. Chemical Descriptors of Schiff Bases and Envisaged Unnatural α-amino Acids
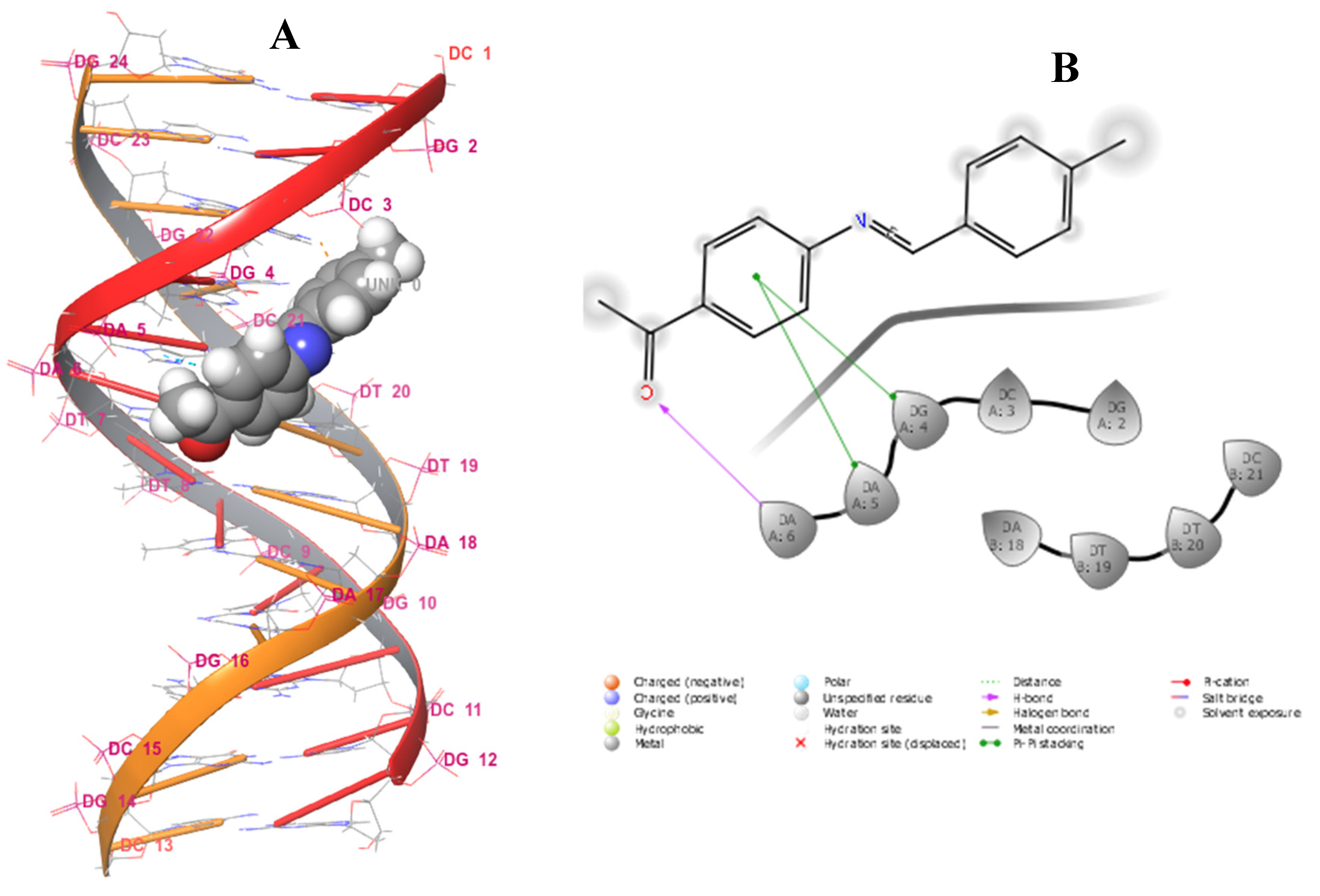
2.3.3. Docking Studies
2.3.4. ADMET Properties
3. Materials and Methods
3.1. General Chemistry Methods
3.2. Photocatalyst Synthesis
Copper (II) Complex
3.3. Schiff Bases
3.3.1. Synthesis of (E)-1-(4-((4-methylbenzylidene)amino)phenyl)ethanone (1)
3.3.2. Synthesis of (E)-3-((4-(dimethylamino)benzylidene)amino)phenol (2)
3.3.3. Synthesis of (E)-4-((4-hydroxybenzylidene)amino)-1,5-dimethyl-2-phenyl-1H-pyrazol-3(2H)-one (3)
3.3.4. Synthesis of (E)-1,5-dimethyl-4-((4-methylbenzylidene)amino)-2-phenyl-1H-pyrazol-3(2H)-one (4)
3.4. Hydrocarboxylation Reaction
3.5. Theoretical Studies
3.5.1. DFT Calculations
3.5.2. Docking Calculations
3.5.3. ADMET Determination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- He, L.-N.; Wang, J.-Q.; Wang, J.-L. Carbon dioxide chemistry: Examples and challenges in chemical utilization of carbon dioxide. Pure Appl. Chem. 2009, 81, 2069–2080. [Google Scholar] [CrossRef]
- Tamaki, Y.; Koike, K.; Ishitani, O. Highly efficient, selective, and durable photocatalytic system for CO2 reduction to formic acid. Chem. Sci. 2015, 6, 7213–7221. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Gong, X.; Ma, M.; Wang, R.; Walsh, P.J. Visible light-promoted CO2 fixation with imines to synthesize diaryl α-amino acids. Nat. Commun. 2018, 9, 4936. [Google Scholar] [CrossRef]
- Hossain, A.; Bhattacharyya, A.; Reiser, O. Copper’s rapid ascent in visible-light photoredox catalysis. Science 2019, 364, eaav9713. [Google Scholar] [CrossRef] [PubMed]
- Mulat, M.; Pandita, A.; Khan, F. Medicinal plant compounds for combating the multi-drug resistant pathogenic bacteria: A review. Curr. Pharm. Biotechnol. 2019, 20, 183–196. [Google Scholar] [CrossRef]
- Ashraf, M.A.; Mahmood, K.; Wajid, A.; Maah, M.J.; Yusoff, I. Synthesis, characterization and biological activity of Schiff bases. IPCBEE 2011, 10, 185. [Google Scholar]
- Perdih, A.; Dolenc, M.S. Recent advances in the synthesis of unnatural α-amino acids. Curr. Org. Chem. 2007, 11, 801–832. [Google Scholar] [CrossRef]
- Kalgutkar, A.S.; Daniel, J.S. Carboxylic Acids and their Bioisosteres. Impact of the Building Blocks of Medical Chemistry on ADMET; RSC: London, UK, 2010; pp. 99–167. [Google Scholar]
- Davis, E.; Mott, N. Conduction in non-crystalline systems V. Conductivity, optical absorption and photoconductivity in amorphous semiconductors. Philos. Mag. 1970, 22, 0903–0922. [Google Scholar] [CrossRef]
- Fouad, R.; Shaaban, I.A.; Ali, T.E.; Assiri, M.A.; Shenouda, S. Co (ii), Ni (ii), Cu (ii) and Cd (ii)-thiocarbonohydrazone complexes: Spectroscopic, DFT, thermal, and electrical conductivity studies. RSC Adv. 2021, 11, 37726–37743. [Google Scholar] [CrossRef]
- Abou-Melha, K. Spectral, modeling and anticancer activity studies on the newly synthesized N-allyl-2-(2, 4-dinitrophenyl) hydrazine-1-carbothioamide and some bivalent metal complexes. J. Mol. Struct. 2021, 1223, 128949. [Google Scholar] [CrossRef]
- Hosny, N.M.; Hassan, N.Y.; Mahmoud, H.M.; Abdel-Rhman, M.H. Spectral, optical and cytotoxicity studies on 2-isonicotinoyl-N-phenylhydrazine-1-carboxamide (H3L) and some of its metal complexes. J. Mol. Struct. 2018, 1156, 602–611. [Google Scholar] [CrossRef]
- Lu, L.; Qin, S.; Yang, P.; Zhu, M. Chlorobis (1, 10-phenanthroline) copper (II) chloride methanol solvate 4.5-hydrate. Acta Crystallogr. Sect. E Struct. Rep. Online 2004, 60, m574–m576. [Google Scholar] [CrossRef]
- Onawumi, O.; Adekunle, F.; Ibrahim, A.; Rajasekharan, M.; Odunola, O. Synthesis, Characterization, and Crystal Structures of [Cu(phen)2Cl]Cl. 6.5 H2O and [Cu(phen)2Br]Br. Synth. React. Inorg. Met. -Org. Nano-Met. Chem. 2010, 40, 78–83. [Google Scholar]
- Louis, B.; Detoni, C.; Carvalho, N.; Duarte, C.; Antunes, O. Cu (II) bipyridine and phenantroline complexes: Tailor-made catalysts for the selective oxidation of tetralin. Appl. Catal. A Gen. 2009, 360, 218–225. [Google Scholar] [CrossRef]
- Vafazadeh, R.; Namazian, M.; Chavoshiyan, M.; Willis, A.C.; Carr, P.D. Synthesis, X-ray structural characterization, and DFT calculations of binuclear mixed-ligand copper (II) complexes containing diamine, acetate and methacrylate ligands. Acta Chim. Slov. 2017, 64, 613–620. [Google Scholar] [CrossRef][Green Version]
- Vafazadeh, R.; Khaledi, B.; Willis, A.C.; Namazian, M. Synthesis, crystal structure and DFT analysis of a new trinuclear complex of copper. Polyhedron 2011, 30, 1815–1819. [Google Scholar] [CrossRef]
- Alpaslan, G.; Macit, M. Crystal structure, spectroscopic characterization and density functional studies of (E)-1-((3-methoxyphenylimino) methyl) naphthalen-2-ol. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 121, 372–380. [Google Scholar] [CrossRef]
- Xavier, A.; Srividhya, N. Synthesis and study of Schiff base ligands. IOSR J. Appl. Chem. 2014, 7, 6–15. [Google Scholar] [CrossRef]
- Sinha, D.; Tiwari, A.K.; Singh, S.; Shukla, G.; Mishra, P.; Chandra, H.; Mishra, A.K. Synthesis, characterization and biological activity of Schiff base analogues of indole-3-carboxaldehyde. Eur. J. Med. Chem. 2008, 43, 160–165. [Google Scholar] [CrossRef]
- Pour, N.T.; Khalighi, A.; Yousefi, M.; Amani, V. One-Dimensional Barium Coordination Polymer With 2, 2′-Bipyridine-5, 5′-Dicarboxylate Ligand: Synthesis, Spectroscopic Characterization, Thermal Analyses, and Crystal Structure. Synth. React. Inorg. Met.-Org. Nano-Met. Chem. 2015, 45, 1427–1433. [Google Scholar] [CrossRef]
- Delbari, A.S.; Shahvelayati, A.S.; Jodaian, V.; Amani, V. Mononuclear and dinuclear indium (III) complexes containing methoxy and hydroxy-bridge groups, nitrate anion and 4, 4′-dimethyl-2, 2′-bipyridine ligand: Synthesis, characterization, crystal structure determination, luminescent properties, and thermal analyses. J. Iran. Chem. Soc. 2015, 12, 223–232. [Google Scholar]
- Ogunlaja, A.S.; Hosten, E.; Tshentu, Z.R. Dispersion of asphaltenes in petroleum with ionic liquids: Evaluation of molecular interactions in the binary mixture. Ind. Eng. Chem. Res. 2014, 53, 18390–18401. [Google Scholar] [CrossRef]
- Olalekan, T.E.; Ogunlaja, A.S.; VanBrecht, B.; Watkins, G.M. Spectroscopic, structural and theoretical studies of copper (II) complexes of tridentate NOS Schiff bases. J. Mol. Struct. 2016, 1122, 72–79. [Google Scholar] [CrossRef]
- Rathi, P.C.; Ludlow, R.F.; Verdonk, M.L. Practical high-quality electrostatic potential surfaces for drug discovery using a graph-convolutional deep neural network. J. Med. Chem. 2019, 63, 8778–8790. [Google Scholar] [CrossRef]
- Altürk, S.; Tamer, Ö.; Avcı, D.; Atalay, Y. Synthesis, spectroscopic characterization, second and third-order nonlinear optical properties, and DFT calculations of a novel Mn (II) complex. J. Organomet. Chem. 2015, 797, 110–119. [Google Scholar] [CrossRef]
- Kargar, H.; Behjatmanesh-Ardakani, R.; Torabi, V.; Kashani, M.; Chavoshpour-Natanzi, Z.; Kazemi, Z.; Mirkhani, V.; Sahraei, A.; Tahir, M.N.; Ashfaq, M. Synthesis, characterization, crystal structures, DFT, TD-DFT, molecular docking and DNA binding studies of novel copper (II) and zinc (II) complexes bearing halogenated bidentate N, O-donor Schiff base ligands. Polyhedron 2021, 195, 114988. [Google Scholar] [CrossRef]
- Antypenko, L.M.; Kovalenko, S.I.; Los’, T.S.; Rebec’, O.L. Synthesis and Characterization of Novel N-(Phenyl, Benzyl, Hetaryl)-2-([1,2,4] Triazolo [1, 5-c] Quinazolin-2-ylthio) Acetamides by Spectral Data, Antimicrobial Activity, Molecular Docking and QSAR Studies. J. Heterocycl. Chem. 2017, 54, 1267–1278. [Google Scholar] [CrossRef]
- Sharma, P.C.; Jain, A.; Jain, S.; Pahwa, R.; Yar, M.S. Ciprofloxacin: Review on developments in synthetic, analytical, and medicinal aspects. J. Enzym. Inhib. Med. Chem. 2010, 25, 577–589. [Google Scholar] [CrossRef]
- Casalvieri, K.A.; Matheson, C.J.; Backos, D.S.; Reigan, P. Molecular docking of substituted pteridinones and pyrimidines to the ATP-binding site of the N-terminal domain of RSK2 and associated MM/GBSA and molecular field datasets. Data Brief 2020, 29, 105347. [Google Scholar] [CrossRef]
- Alves, J.E.F.; de Oliveira, J.F.; de Lima Souza, T.R.C.; de Moura, R.O.; de Carvalho Junior, L.B.; de Lima, M.d.C.A.; de Almeida, S.M.V. Novel indole-thiazole and indole-thiazolidinone derivatives as DNA groove binders. Int. J. Biol. Macromol. 2021, 170, 622–635. [Google Scholar] [CrossRef]
- Cai, X.; Gray, P.J., Jr.; von Hoff, D.D. DNA minor groove binders: Back in the groove. Cancer Treat. Rev. 2009, 35, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Rescifina, A.; Zagni, C.; Varrica, M.G.; Pistarà, V.; Corsaro, A. Recent advances in small organic molecules as DNA intercalating agents: Synthesis, activity, and modeling. Eur. J. Med. Chem. 2014, 74, 95–115. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, S.M.V.; Ribeiro, A.G.; Silva, G.C.d.; Alves, J.E.F.; Beltrão, E.I.C.; de Oliveira, J.F.; Junior, L.B.d.; de Lima, M.d.C.A. DNA binding and Topoisomerase inhibition: How can these mechanisms be explored to design more specific anticancer agents? Biomed. Pharmacother. 2017, 96, 1538–1556. [Google Scholar] [CrossRef] [PubMed]
- Froehlich, E.; Mandeville, J.; Weinert, C.; Kreplak, L.; Tajmir-Riahi, H. Bundling and aggregation of DNA by cationic dendrimers. Biomacromolecules 2011, 12, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Aleksić, M.; Kapetanović, V. An overview of the optical and electrochemical methods for detection of DNA-drug interactions. Acta Chim. Slov. 2014, 61, 555–573. [Google Scholar]
- Gupta, S.; Maurya, P.; Upadhyay, A.; Kushwaha, P.; Krishna, S.; Siddiqi, M.I.; Sashidhara, K.V.; Banerjee, D. Synthesis and bio-evaluation of indole-chalcone based benzopyrans as promising antiligase and antiproliferative agents. Eur. J. Med. Chem. 2018, 143, 1981–1996. [Google Scholar] [CrossRef]
- Mohanram, I.; Meshram, J. Synthesis and biological activities of 4-aminoantipyrine derivatives derived from betti-type reaction. Int. Sch. Res. Not. 2014, 2014, 639392. [Google Scholar] [CrossRef]
- Bruker, A. APEX2, SADABS and SAINT; Bruker, AXS: Madison, WI, USA, 2010. [Google Scholar]
- Sheldrick, G. XS. version 2013/1, Georg-August-Universität Göttingen, Germany, 2013; b) GM Sheldrick. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Gordon, A.T.; Abosede, O.O.; Ntsimango, S.; Hosten, E.C.; Myeza, N.; van Eyk, A.; Harmse, L.; Ogunlaja, A.S. Synthesis and anticancer evaluation of copper (II)-and manganese (II)-theophylline mixed ligand complexes. Polyhedron 2022, 214, 115649. [Google Scholar] [CrossRef]
- De, R.L.; Mukherjee, J.; Mandal, M.; Roy, L.; Bhowal, R.; Banerjee, I. The synthesis and structure of 1-[3-{(2-hydroxybenzylidene) amino} phenyl] ethanone. Indian J. Chem. Sect. B-Org. Chem. Incl. Med. Chem. 2009, 48, 595–598. [Google Scholar]
- Prokopenko, Y.S.; Perekhoda, L.O.; Georgiyants, A.V. Docking studies of biologically active substances from plant extracts with anticonvulsant activity. J. Appl. Pharm. Sci. 2019, 9, 066–072. [Google Scholar]
- Jędrzejczyk, M.; Janczak, J.; Huczyński, A. Molecular structure and spectroscopic studies of the product of acidic degradation of salinomycin and its potassium salt. J. Mol. Struct. 2022, 1263, 133129. [Google Scholar] [CrossRef]
- Olubode, S.O.; Bankole, M.O.; Akinnusi, P.A.; Adanlawo, O.S.; Ojubola, K.I.; Nwankwo, D.O.; Edjebah, O.E.; Adebesin, A.O.; Ayodele, A.O. Molecular modeling studies of natural inhibitors of androgen signaling in prostate cancer. Cancer Inform. 2022, 21, 11769351221118556. [Google Scholar] [CrossRef]
- Veeralakshmi, S.; Sabapathi, G.; Nehru, S.; Venuvanalingam, P.; Arunachalam, S. Surfactant–cobalt (III) complexes: The impact of hydrophobicity on interaction with HSA and DNA–insights from experimental and theoretical approach. Colloids Surf. B Biointerfaces 2017, 153, 85–94. [Google Scholar] [CrossRef]
- Palanivel, S.; Yli-Harja, O.; Kandhavelu, M. Molecular interaction study of novel indoline derivatives with EGFR-kinase domain using multiple computational analysis. J. Biomol. Struct. Dyn. 2022, 40, 7545–7554. [Google Scholar] [CrossRef]
- Pathania, S.; Singh, P.; Narang, R.; Rawal, R. Structure based designing of thiazolidinone-pyrimidine derivatives as ERK2 inhibitors: Synthesis and in vitro evaluation. SAR QSAR Environ. Res. 2021, 32, 793–816. [Google Scholar] [CrossRef]
- Bourne, C.R.; Wakeham, N.; Webb, N.; Nammalwar, B.; Bunce, R.A.; Berlin, K.D.; Barrow, W.W. The structure and competitive substrate inhibition of dihydrofolate reductase from Enterococcus faecalis reveal restrictions to cofactor docking. Biochemistry 2014, 53, 1228–1238. [Google Scholar] [CrossRef]
- Drew, H.R.; Wing, R.M.; Takano, T.; Broka, C.; Tanaka, S.; Itakura, K.; Dickerson, R.E. Structure of a B-DNA dodecamer: Conformation and dynamics. Proc. Natl. Acad. Sci. USA 1981, 78, 2179–2183. [Google Scholar] [CrossRef]
- Iakovenko, R.; Hlaváč, J. Visible light-mediated metal-free double bond deuteration of substituted phenylalkenes. Green Chem. 2021, 23, 440–446. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | [Cu(phen)2Cl] |
|---|---|
| Empirical formula | C24H16ClCuN4 |
| Formula weight | 459.40 |
| Estimated formula | C28H28Cl2CuN4O2 |
| Estimated moiety formula | C24H16ClCuN4, Cl, 2(C2H6O) |
| Estimated formula weight | 587.01 |
| Crystal colour | Green–purple |
| Crystal system | Monoclinic |
| Space group | C2/c (No.15) |
| Temperature (K) | 296 |
| a, b, c (Å) | 23.2998(6) 30.2646(8) 7.4844(2) |
| α, β, γ (⁰) | 90, 97.789(1), 90 |
| V (Å3) | 5229.0(2) |
| Z | 8 |
| F(000) | 1872 |
| ρcalc (g/cm3) | 1.167 |
| Radiation (Å) | Moka 0.71073 |
| Dataset | 30:31; −40:40; −9:9 |
| Theta Min–Max (De) | 1.3, 28.3 |
| Nref, Npar | 6494, 271 |
| Crystal Size (mm) | 0.06 × 0.34 × 0.54 |
| Min. and Max. Resd. Dens. (e/Ang^3) | −0.28, 0.32 |
| R, wR2, S | 0.0364, 0.1120, 1.01 |
| Bond Length Experimental (Å) | Bond angle Experimental (°) | ||
|---|---|---|---|
| Cu1-N11 | 2.1073(19) | N11-Cu1-N12 | 81.21(7) |
| Cu1-N12 | 1.9872(17) | N11-Cu1-N21 | 123.14(6) |
| Cu1-N21 | 2.0997(16) | N12-Cu1-N21 | 95.15(7) |
| Cu1-N22 | 1.9769(17) | N12-Cu1-N22 | 175.97(7) |
| Cu1-Cl1 | 2.3368(7) | Cl1-Cu1-N11 | 115.28(5) |
| Cl1-Cu1-N12 | 91.24(5) |
| Interactions | D—H (Å) | H···A (Å) | D···A (Å) | D—H···A (º) | Y―X…π (Å) | π···π (Å) |
|---|---|---|---|---|---|---|
| C21-H21…Cl1 i | 0.93 | 2.76 | 3.648(3) | 161 | ||
| Cu1-Cl1…Cg1 ii | 3.5854(10) | |||||
| Cg2…Cg3 iii | 0.93 | 2.58 | 3.498(7) | 171 | 3.7006(13) | |
| Cg4…Cg1 i | 3.6196(12) |
| Schiff Base Synthesis (Protocol 1) | Visible Light Hydrocarboxylation (Protocol 2) |
|---|---|
 |  |
 |  |
 |  |
 |  |
| Parameter | CAT | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|---|
| EHOMO (eV) | −6.86 | −9.04 | −8.25 | −8.55 | −8.51 | –9.29 | –8.47 | –9.02 | –8.81 |
| ELUMO (eV) | −0.94 | −1.12 | –0.20 | −0.42 | −0.30 | –0.77 | –0.16 | –0.82 | −0.66 |
| ΔEgap (eV) | 5.92 | 7.92 | 8.05 | 8.13 | 8.21 | 8.52 | 8.31 | 8.20 | 8.15 |
| I (eV) | 6.86 | 9.04 | 8.25 | 8.55 | 8.51 | 9.29 | 8.47 | 9.02 | 8.81 |
| A (eV) | 0.94 | 1.12 | 0.20 | 0.42 | 0.30 | 0.77 | 0.16 | 0.82 | 0.66 |
| μ (eV) | −3.90 | –5.08 | –4.23 | –4.49 | –4.41 | −5.03 | −4.31 | –4.92 | −4.74 |
| χ (eV) | 3.90 | 5.08 | 4.23 | 4.49 | 4.41 | 5.03 | 4.31 | 4.92 | 4.74 |
| ƞ (eV) | 2.96 | 3.96 | 4.03 | 4.07 | 4.11 | 4.26 | 4.16 | 4.10 | 4.08 |
| S (eV) | 0.34 | 0.25 | 0.25 | 0.25 | 0.24 | 0.24 | 0.24 | 0.24 | 0.25 |
| ω (eV) | 22.51 | 3.26 | 2.22 | 2.48 | 2.37 | 2.97 | 2.23 | 2.95 | 2.75 |
| G−Score | E−Model | Ligand Efficiency | ||||
|---|---|---|---|---|---|---|
| Receptor/DNA | 4M7U | 1BNA | 4M7U | 1BNA | 4M7U | 1BNA |
| 1 | −4.759 | −3.434 | −41.752 | −30.420 | −0.264 | −0.191 |
| 2 | −4.557 | −5.815 | −39.811 | −52.597 | −0.252 | −0.178 |
| 3 | −4.891 | −3.976 | −47.146 | −33.735 | −0.213 | −0.173 |
| 4 | −3.255 | −4.995 | −29.960 | −53.619 | −0.140 | −0.067 |
| 5 | −5.902 | −2.420 | −59.663 | −21.008 | −0.281 | −0.122 |
| 6 | −5.931 | −4.013 | −58.006 | −27.247 | −0.280 | −0.129 |
| 7 | −5.656 | −2.369 | −62.981 | −27.070 | −0.217 | −0.130 |
| 8 | −5.626 | −2.603 | −63.009 | −26.668 | −0.216 | −0.112 |
| Trimethoprim | −5.837 | −4.289 | −45.603 | −32.005 | −0.248 | −0.160 |
| Ciprofloxacin | −4.228 | −3.369 | −45.719 | −27.134 | −0.132 | −0.136 |
| Ligands | Enterococcus Faecalis 4M7U (kcal/mol) | |||||||
|---|---|---|---|---|---|---|---|---|
| ΔGBind | ΔGCoul | ΔGcov | ΔGHbond | ΔGPack | ΔGlipho | ΔGSolv_GB | ΔGVdW | |
| Schiff Bases | ||||||||
| 1 | −39.21 | −15.43 | 1.73 | −0.59 | 0.00 | −15.39 | 22.13 | −31.67 |
| 2 | −29.23 | −7.95 | 0.25 | −0.53 | 0.00 | −12.53 | 18.34 | −26.80 |
| 3 | −39.21 | −22.95 | 6.04 | −1.77 | −0.08 | −14.51 | 24.14 | −30.08 |
| 4 | −39.73 | −18.96 | 2.22 | −0.63 | −0.05 | −15.19 | 24.49 | −31.61 |
| α-unsaturated aminocarboxylic acids | ||||||||
| 5 | −37.87 | −16.63 | 3.41 | −3.06 | −0.12 | −11.16 | 22.98 | −33.29 |
| 6 | −37.64 | −23.74 | 1.67 | −2.04 | −0.19 | −12.64 | 31.33 | −32.02 |
| 7 | −41.45 | −29.35 | 10.04 | −2.27 | 0.00 | −14.79 | 29.71 | −34.79 |
| 8 | −39.27 | −23.82 | 10.13 | −1.67 | 0.00 | −15.51 | 29.57 | −37.97 |
| Control | ||||||||
| Trimethoprim | −36.94 | −34.03 | 2.39 | −3.18 | −0.97 | −8.45 | 30.82 | −23.51 |
| Ciprofloxacin | −31.72 | −40.54 | 0.42 | −2.22 | −1.27 | −9.99 | 50.27 | −28.39 |
| Enterococcus Faecalis (PDB id: 4M7U) with Interacting Residues | B-DNA Dodecamer (PDB id: 1BNA) with Interacting Nucleotides | ||||
|---|---|---|---|---|---|
| Entry | Hydrogen Bond (Å) | π-Interactions (Å) | Hydrogen Bond (Å) | π-π Stacking (Å) | |
| Schiff bases | |||||
| 1 | SER100 (1.86) | - | DA6 (2.27) | DA5 (5.23), DG4 (4.80) | |
| 2 | GLY18 (1.74) | GLY18 (2.30) π-alkyl | DA18 (2.37) | DC3 (5.21) | |
| 3 | ALA45 (2.69), SER65 (2.05), (1.80) | ARG44 (6.26) π-cation | DT19 (1.89), DC3 (1.97) | DG4 (5.00) | |
| 4 | VAL101 (2.73), GLY99 (2.68) | - | DG4 (2.75) | DG4 (5.14), DC3 (5.24) | |
| α-unsaturated aminocarboxylic acids | |||||
| 5 | VAL102 (2.04), ARG44 (2.60), SER65 (1.88), THR64 (2.07) | SER65 (2.38), (2.29) π-alkyl | DC3 (1.65) | - | |
| 6 | GLU105 (1.75), SER65(2.63) | - | DC3 (2.07), DC21 (2.74), DT19 (1.78) | - | |
| 7 | ALA45 (1.80), THR46 (2.60). GLY99 (2.60), THR126 (1.90) | ARG44 (6.52) π-cation | DC3(1.64), DC21 (2.74) | - | |
| 8 | ALA45 (1.83), THR46 (2.60) | DC3 (1.69), DC21 (2.33) | - | ||
| Control | |||||
| Trimethoprim | SER65 (2.04), ARG44 (1.93) | ARG44 (5.78) π-cation | DC3 (1.91), DG4 (1.92) | - | |
| Ciprofloxacin | ASP125 (1.80), VAL101 (2.11), THR46 (2.20) | - | DG4 (2.24), DA18 (2.55), DA17 (1.60) | DA18 (4.74) | |
| Entry | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | Trimethoprim | Ciprofloxacin |
|---|---|---|---|---|---|---|---|---|---|---|
| Mw | 237.30 | 240.31 | 307.35 | 305.38 | 283.33 | 286.33 | 353.38 | 351.41 | 290.32 | 331.34 |
| #stars | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| WPSA | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 31.43 |
| Volume (A3) | 889.07 | 879.93 | 1005.64 | 1051.19 | 969.90 | 950.591 | 1151.07 | 1182.23 | 930.09 | 1013.05 |
| QPpolrz (A3) | 29.70 | 28.86 | 34.57 | 36.62 | 31.86 | 30.32 | 40.02 | 41.58 | 27.50 | 34.40 |
| EA (eV) | 0.94 | 0.46 | 0.54 | 0.49 | 0.39 | −0.02 | 0.27 | 0.27 | −0.092 | 0.76 |
| QplogPoct | 11.42 | 11.97 | 10.71 | 14.86 | 16.12 | 17.09 | 23.09 | 21.15 | 17.52 | 17.79 |
| QplogPw | 5.52 | 6.60 | 10.16 | 7.81 | 10.52 | 11.43 | 15.87 | 13.31 | 12.11 | 9.94 |
| QplogPo/w | 3.59 | 3.55 | 2.79 | 3.82 | 2.98 | 2.69 | 0.25 | 1.23 | 0.91 | 0.280 |
| QplogS | −4.17 | −4.21 | −3.81 | −4.44 | −4.06 | −3.32 | −4.10 | −4.85 | −2.85 | −3.79 |
| QPPCaco (nm/s) | 2670.97 | 2535.63 | 1248.80 | 4244.01 | 117.72 | 88.16 | 10.26 | 36.48 | 2396.80 | 12.98 |
| #metab | 1 | 2 | 2 | 2 | 4 | 6 | 4 | 4 | 5 | 0 |
| %Human Oral Absor | 100 | 100 | 100 | 100 | 81.46 | 77.53 | 46.52 | 62.09 | 78.08 | 48.51 |
| PSA | 39.24 | 35.097 | 62.99 | 40.68 | 85.21 | 87.30 | 112.60 | 90.10 | 98.46 | 98.88 |
| Rule of 3 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 |
| Rule of 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gordon, A.T.; Hosten, E.C.; Ogunlaja, A.S. Cu(II)-Catalysed Hydrocarboxylation of Imines Utilizing CO2 to Synthesize α-Unsaturated Aminocarboxylic Acids. Pharmaceuticals 2022, 15, 1240. https://doi.org/10.3390/ph15101240
Gordon AT, Hosten EC, Ogunlaja AS. Cu(II)-Catalysed Hydrocarboxylation of Imines Utilizing CO2 to Synthesize α-Unsaturated Aminocarboxylic Acids. Pharmaceuticals. 2022; 15(10):1240. https://doi.org/10.3390/ph15101240
Chicago/Turabian StyleGordon, Allen T., Eric C. Hosten, and Adeniyi S. Ogunlaja. 2022. "Cu(II)-Catalysed Hydrocarboxylation of Imines Utilizing CO2 to Synthesize α-Unsaturated Aminocarboxylic Acids" Pharmaceuticals 15, no. 10: 1240. https://doi.org/10.3390/ph15101240
APA StyleGordon, A. T., Hosten, E. C., & Ogunlaja, A. S. (2022). Cu(II)-Catalysed Hydrocarboxylation of Imines Utilizing CO2 to Synthesize α-Unsaturated Aminocarboxylic Acids. Pharmaceuticals, 15(10), 1240. https://doi.org/10.3390/ph15101240