2.1. The Optimization Process of SIM-DOX-LCL
Usually, the implementation of the QbD approach starts by defining the QTPP and ends with establishing the control strategy [
18]. Setting the QTPP in the first steps of the development process helps the research group to focus on designing a product that meets both the patient needs and the international regulation [
21]. The QTPP generally contains features about the formulation, route of administration, release profile and other quality attributes such as morphology, crystallinity, water content, etc. [
18]. In view of this, the current study aimed to optimize a former developed liposomal formulation co-encapsulating SIM and DOX that has perspectives in cancer therapy [
14]. To accomplish this target, the long circulating liposomes loaded with SIM and DOX (SIM-DOX-LCL) were designed for intravenous administration to increase the bioavailability of both SIM and DOX due to their intensive hepatic metabolization [
22,
23]. Moreover, the choice of a liposomal formulation can reduce the intensity and the number of systemic side effects caused by the high doses of SIM and DOX needed in cancer therapy [
8,
24,
25].
According to a World Health Organization report, the first three most prevalent types of cancer in 2020 were breast, lung and colon and rectum cancer, accounting for more than three million deaths worldwide [
26]. This fact emphasizes the need to discover new drugs for cancer treatment, particularly for these three types of cancer. As a continuation of our previous study, which demonstrated that SIM can enhance the antiproliferative properties of DOX on C26 murine colon cancer cell line co-cultured with macrophages [
14], in the present study we intended to evaluate the same drug combination as free form and co-encapsulated in liposomes against two human cancerous cell lines, namely T47D-KBluc human mammary ductal carcinoma cell line and A549 human pulmonary cancer cell line. The benefits of associating statins with chemotherapeutic drugs have been demonstrated in both preclinical and clinical studies. The results have evidenced that an anticancer effect can be obtained in cancers such as lung cancer, breast cancer or prostate cancer [
27]. Moreover, the idea of repurposing SIM is in line with the latest trends of the European Medicine Agency, which started a pilot project aiming at drug repurposing to provide new therapeutic alternatives in conditions where the actual therapy is associated with numerous disadvantages, such as multi-organ side effects [
28]. On the other hand, a thorough understanding of cancer biology is essential for the selection of the appropriate drug combinations and administered doses, to achieve a synergistic effect in anticancer treatment [
29]. By assessing the same drug combination on two different human cancer cell lines using identical concentration levels, the differences in the antiproliferative responses will be determined by the differences in cellular biology, thus providing a better understanding of the influences that affect the anticancer activity of SIM and DOX combination.
Yet, a liposomal formulation can be considered suitable for in vivo administration only if the quality attributes of liposomes, i.e., size, PdI, zeta potential, are attaining the target values established by the scientific community. More precisely, the liposomes should be in the size range of 50 to 200 nm and monodispersed (PdI < 0.2) to guarantee the permeation and retention of the nanoparticles in the tumor, and to avoid their uptake by the macrophages [
30]. On the other hand, a zeta potential value lesser than −20 mV is preferred because it secures long-term stability and the developed nanoparticles are not noxious to health cells or blood components [
31]. In addition, we considered it essential that the drug release profile of both SIM and DOX be extended in order to avoid early drug release in the blood [
32], which can further lead to the occurrence of systemic side effects and the failure to ensure the right drug ratio at the tumor site.
The next step in the QbD approach is performing risk assessment, which has the role of identifying and quantifying the potential risk that endangers the achievement of the QTPP [
18]. According to the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) guideline, risk assessment “is typically performed early in the pharmaceutical development process and is repeated as more information becomes available and greater knowledge is obtained” [
18]. In the previous work [
14], risk identification via an Ishikawa diagram, and risk evaluation via Failure Mode and Effects Analysis (FMEA) were performed to identify those factors that might have a critical impact on the critical quality attributes (CQAs) of SIM-DOX-LCL formulation. Three formulation factors, namely the PL concentration, SIM and DOX concentration, and two process parameters, namely the pH of the ammonium sulfate (AS) solution and the incubation time of long circulating liposomes loaded with SIM (SIM-LCL) with DOX were formerly identified as potentially critical and were studied in a screening experimental design [
14].The obtained results were used in the current study as an outset for the optimization of SIM-DOX-LCL formulation. As the screening study has pointed out, PL, SIM and DOX concentrations, as well as the pH of the AS solution, presented the greatest influence on SIM-DOX-LCL CQAs [
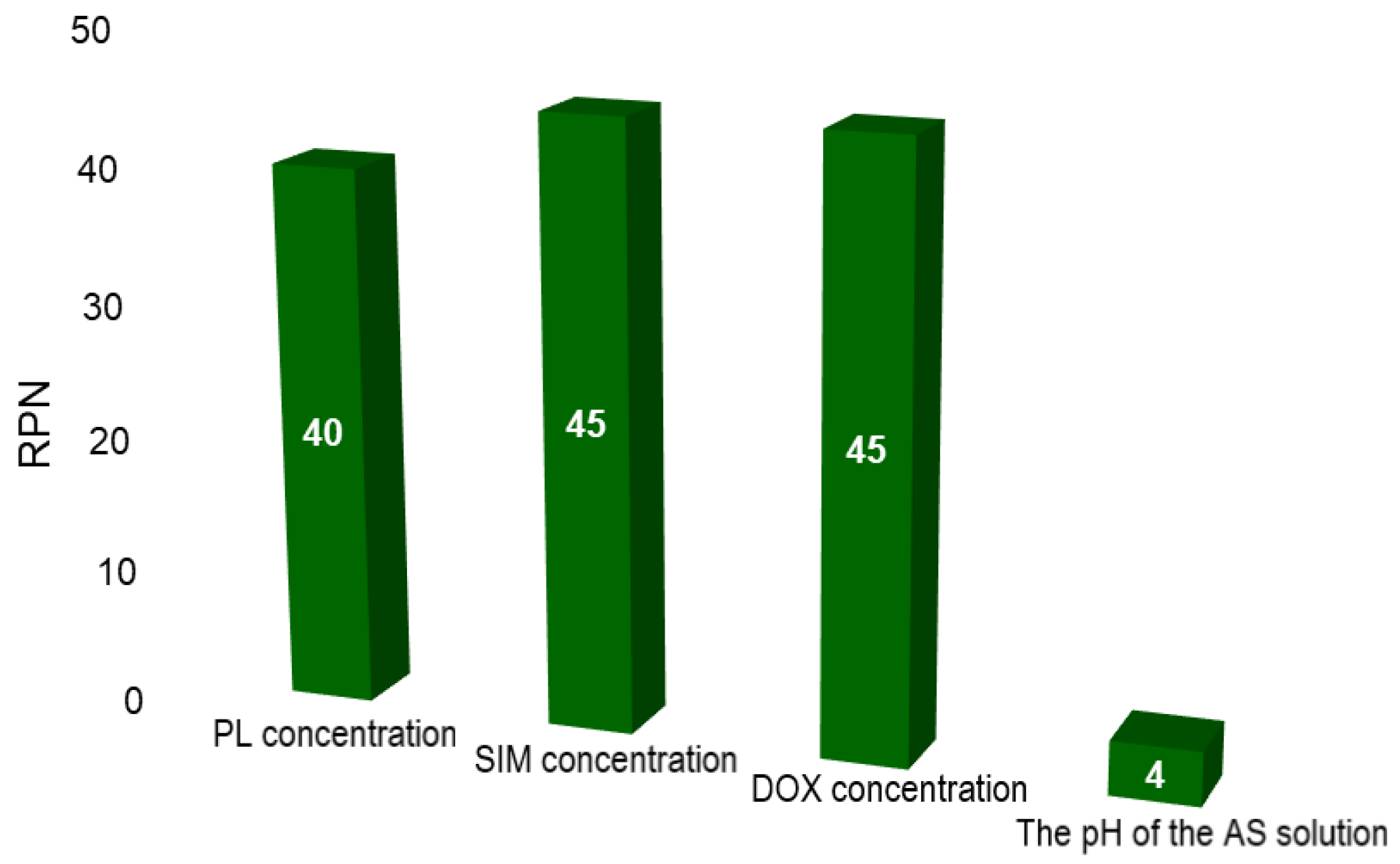
14]. Therefore, these factors were re-evaluated via FMEA and the scores are plotted in
Figure 1.
Compared to the previous results [
14], the risk priority number (RPN) for all factors decreased (
Figure 1). This was possible through the screening study, which provided an overview of the influences that the studied factors have on the CQAs of the SIM-DOX-LCL formulation, and contributed to the understanding of these effects. Nevertheless, the PL, SIM and DOX concentration still have the highest RPN. The criticality of these three factors arises from their major role in assuring the right ratio between SIM and DOX in the final formulation, as well as in guaranteeing a high encapsulation efficiency (EE%) [
14,
33]. The previous work demonstrated that the use of a low concentration of PL results in low EE% and entrapped drug concentration [
14] and therefore, higher levels of PL were proposed for the optimization DoE (between 44 and 77 mM). On the other hand, the pH of the AS solution became non-critical. The reason for this is that DOX can achieve a higher EE% when the pH of the AS solution is five [
14] and consequently, the central issue becomes the control method of the pH.
Considering these results, the concentrations of PL, SIM and DOX were studied as independent variables in an optimization DoE, while the entrapped drug concentration, EE%, liposomal size, PdI, and zeta potential were the dependent variables. The composition of the formulations and the experimental results of the DoE are presented in
Table 1.
The experimental results were fitted through multilinear regression model and the statistical parameters of the fitting model and Analysis of Variance (ANOVA) test are presented in
Table 2.
The statistical parameters of the summary of fit evidenced that the results fit well with the proposed model. However, the ANOVA statistical parameters showed that the formulation factors had a great impact on the responses, and there is no lack of fit for either of the responses (p > 0.1).
The equation that describes the influences of the formulation factors on the responses is:
where Y
n is the response in question, b
0 is the model constant, b
1, b
2 and b
3 are the linear coefficients, b
11, b
22 and b
33 are the quadratic coefficients, and b
12, b
13 and b
23 are the interaction coefficients of the three factors.
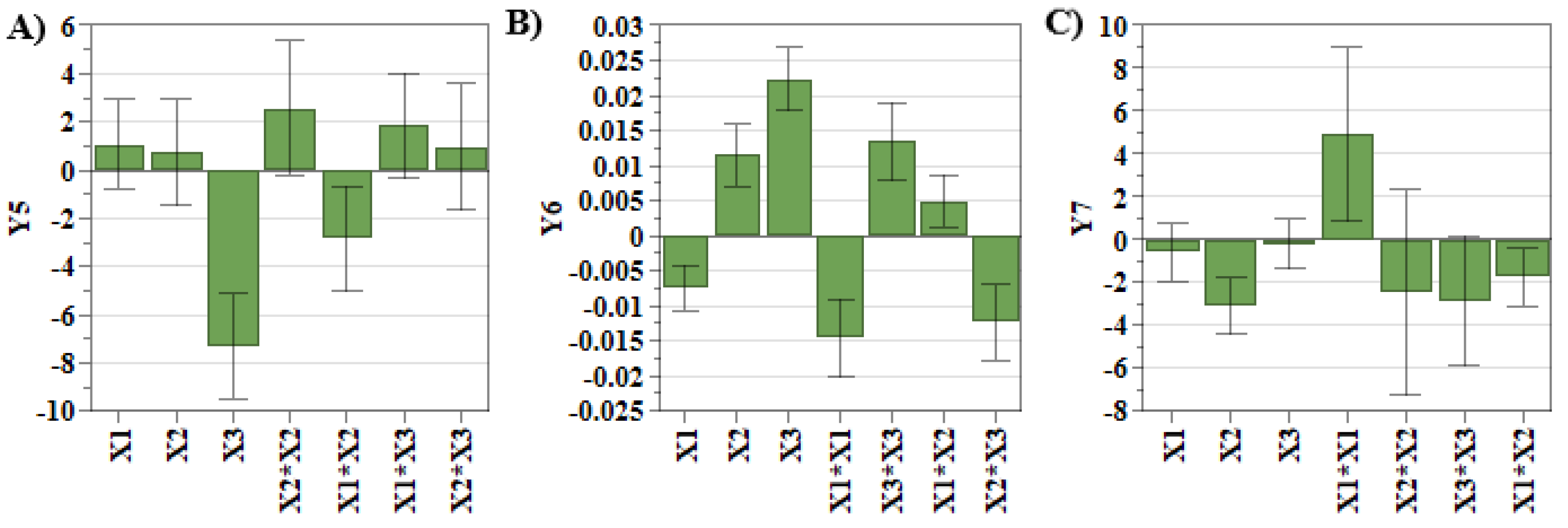
By looking at the experimental results (
Table 1), it can be observed that the liposomes dimension was in the size range of 105.5 nm and 169.4 nm, the PdI was between 0.02 and 0.08, and zeta potential values were below −20 mV. These results highlight that the referred quality attributes are within the established target ranges, and no additional optimization is required. However, the coefficients that show the manner in which each factor influenced the studied responses are presented in
Table 3 and
Figure 2.
Figure 2A,B evidence that SIM concentration had the greatest influence on the liposomal size and PdI. As shown in our previous work, SIM presents a stiffening effect on the lipid bilayer, causing ruptures in the liposomal membrane during the extrusion process. Due to this, the newly formed liposomes are small in size and polydisperse [
14,
34,
35]. Another factor that causes the increase in PdI values is DOX concentration (
Figure 2B). During the incubation step of SIM-LCL with DOX, DOX crosses the lipid bilayer due to a pH gradient and precipitates inside the liposomes as DOX sulfate. When high concentrations of DOX are used, the size and the shape of DOX sulfate crystals vary, influencing the shape of liposomes and the PdI values [
14,
36]. On the contrary, the use of high concentrations of PL results in the formation of a greater number of liposomes that can encapsulate DOX, and consequently, the dimensions of DOX sulfate crystals decrease [
14]. In addition to these, DOX concentration also has a significant impact on zeta potential response. More specifically, by increasing the concentration of DOX, the zeta potential values decrease (
Figure 2C). During the incubation step of SIM-LCL with DOX, DOX hydrochloride dissociates outside the liposomes in DOX, chloride ions and protons. DOX in a neutral form crosses the lipid bilayer, where it precipitates as DOX sulfate, and the resulting ammonium molecules from the dissociation of AS diffuse outside the liposomes [
37]. Considering these, we assume that the chloride ions resulted from the dissociation of DOX chloride exceed that of ammonia, and therefore, the zeta potential decreases with the increase in DOX concentration.
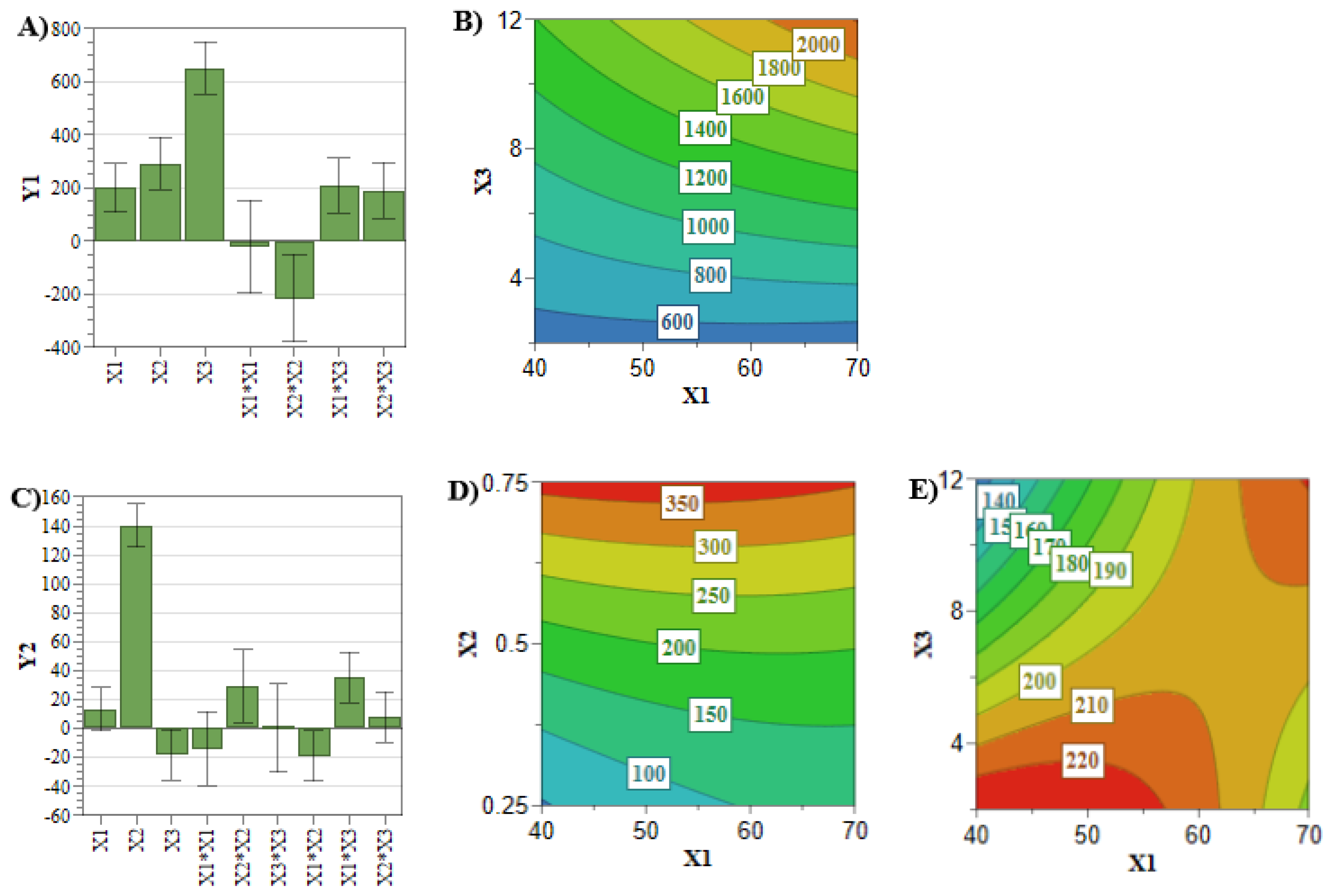
In comparison with the previously analyzed quality attributes, the entrapped drug concentration and the EE% responses did not attain the target values for all the experiments of the DoE. Therefore, an in-depth analysis of these two CQAs is mandatory for the optimization process. Achieving a high entrapped drug concentration and EE% is desirous in terms of costs and administered dose [
33]. These two responses are mainly influenced by the concentration of the PL and active pharmaceutical ingredient (API) (
Figure 3 and
Figure 4) [
33]. In this respect,
Figure 4A,B highlighted that by increasing the PL and SIM concentrations, the SIM EE% increases due to an increase in the number of formed liposomes that can encapsulate more API [
38]. On the other hand,
Figure 3A–D show that the API concentration had the greatest influence on the entrapped drug concentration of SIM and DOX, and these responses increased with the use of elevated concentrations of SIM or DOX.
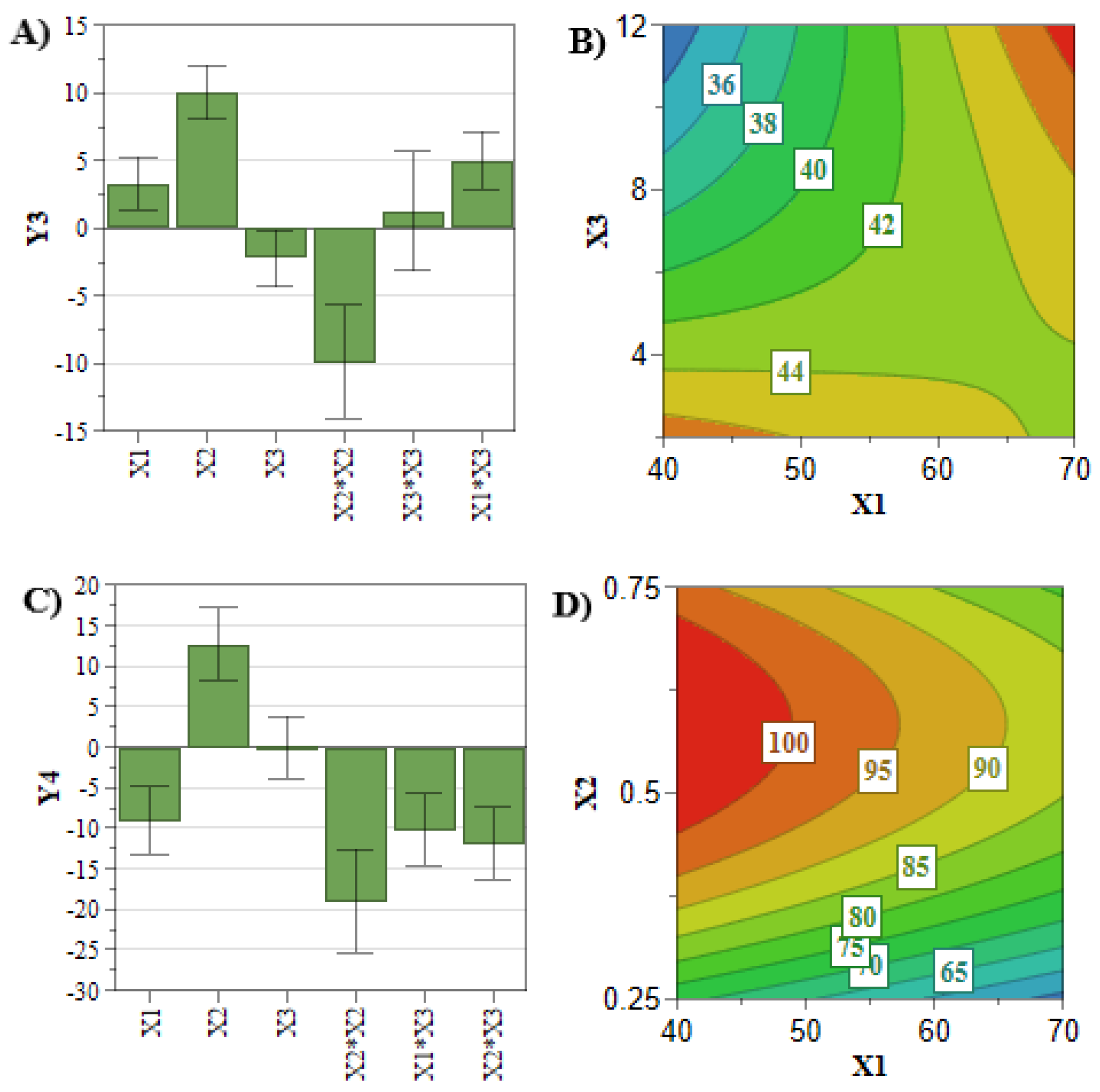
However, when co-encapsulating two APIs with different physiochemical properties, interferences in the encapsulation process of the APIs might occur [
39,
40]. In this view, the interaction coefficients (
Table 3) demonstrated linkage between SIM and PL concentrations for DOX entrapped concentration plotted in
Figure 3E. It can be noted that the highest DOX entrapped concentration was achieved at low levels of PL and SIM concentration, and inversely. This might be the consequence of the lipophilic character of SIM, which could prevent DOX from crossing the liposomal membrane during the incubation process, leading to a reduced entrapped concentration [
40]. At the same time, using high levels of SIM and PL hinders the extrusion process, leading to a reduction in SIM entrapped concentration and lipophilic character of liposomal membrane, thus promoting the encapsulation of DOX [
14].
These mathematical relationships established between input and output provide the scientific basis for defining the design space for SIM-DOX-LCL formulation [
18,
21]. The design space and the optimal formulation for SIM-DOX-LCL were generated using the optimization function of Modde 13 software after establishing the target ranges for each response.
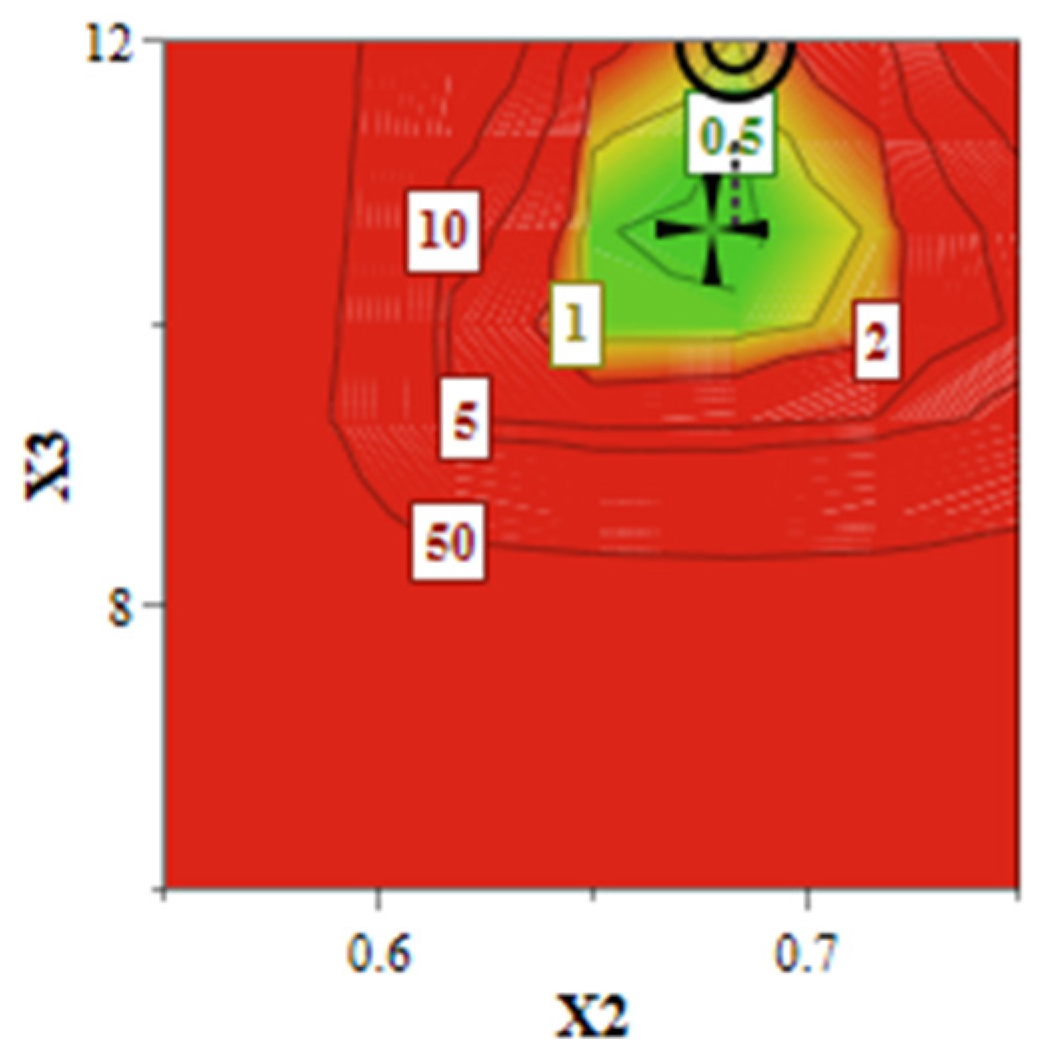
As illustrated in
Figure 5, the design space (green area) suggests that the achievement of a liposomal formulation that meets the QTPP is conditioned by the use of increased levels of SIM and DOX, attesting to the criticality of these two factors. The quantitative formulation of the optimized SIM-DOX-LCL is presented in
Table 4 along with the predicted values and the experimental results, showing that these two values are very close. In other words, it can be said that SIM-DOX-LCL optimal formulation complies with the QTPP, since the experimental results meet the target ranges.
2.3. Stability Study
Investigating the stability of liposomes is considered to be a critical issue in the pharmaceutical development process because the intended effect depends on maintaining the liposomes’ CQAs in the target ranges during storage [
30,
61,
62]. Previously, it was evidenced that the stability of liposomal formulations is dependent on factors such as the type of PL, cholesterol concentration, loading method of the API, temperature or pH [
61,
63,
64,
65]. The ICH guideline pointed out that “a 5% change in assay from its initial value” is considered significant, and additional tests are required [
66]. The stability of SIM-DOX-LCL optimal formulation was evaluated in terms of liposomal size, PdI, zeta potential and the percentage of drug retained over four weeks. The storage conditions were established by considering the specifications mentioned in the summary of product characteristics of Doxil
® and Caelyx [
67,
68], two liposomal products with DOX approved by the regulatory agencies.
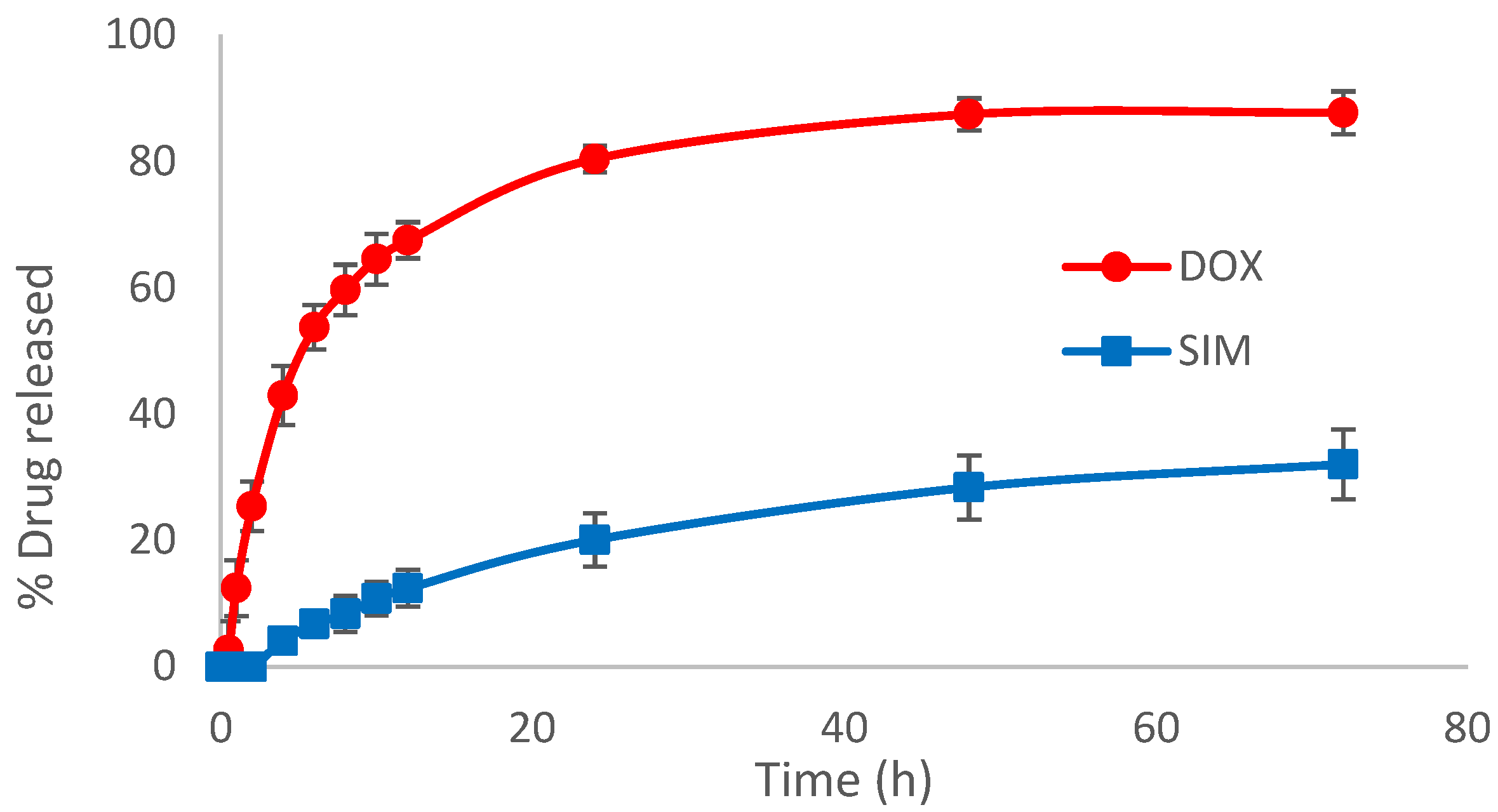
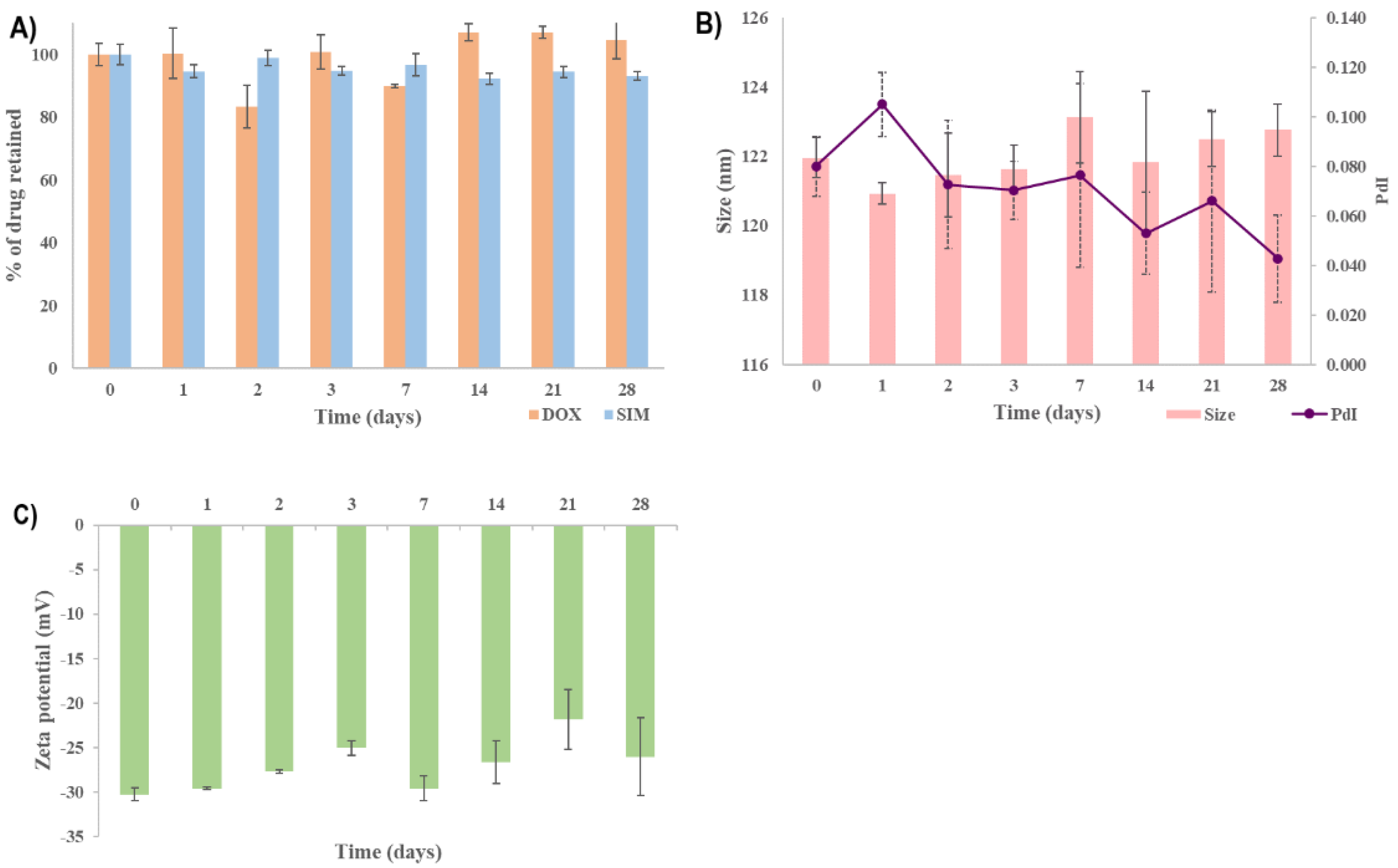
Under these experimental conditions, the SIM-DOX-LCL optimal formulation presented good stability over the tested period. The % of DOX retained (
Figure 9A) was constant for the entire duration of the study, while the % of SIM retained decreased to 93% after four weeks. Evidence has shown that the use of PL with a high chain length and T
m produces liposomes with increased stability, while the use of a low temperature (<T
m) to store the liposomal formulations minimizes lipid mobility, thus preventing drug leakage and liposomal coalescence [
63,
64]. Additionally, the encapsulation of DOX using the AS gradient determines the formation of DOX-sulfate precipitate with a very low permeability through the liposomal membrane, thereby ensuring efficient retention of the drug inside the liposomes for a long period of time [
32,
69]. On the other hand, the stability of SIM in aqueous mediums was evidenced to be highly affected by factors such as the composition of the aqueous medium, pH or temperature [
70,
71]. Because the molecular structure of SIM includes a lactone moiety, SIM is highly susceptible to degradation to an acidic form [
70]. Previous studies evidenced that the half-life of SIM in water at room temperature is 12.9 days [
72], and an acidic pH and a lower temperature can delay the conversion [
70]. Therefore, we assume that the low temperature used when storing the optimized SIM-DOX-LCL formulation prevented SIM degradation.
The other referenced CQAs, namely particle size, PdI and zeta potential, were preserved in the previously established target ranges, as can be seen in
Figure 9B,C. In light of this, it can be concluded that the increased stability of SIM-DOX-LCL optimal formulation is ascribed equally to the liposomal formulation, storage conditions and to the active loading method of DOX.
2.4. Cell Proliferation Assay
It is a fact that breast and lung cancer present the highest mortality among oncologic patients and, consequently, many studies are focusing on finding treatment options that can inhibit tumor progression [
6,
10,
26]. Usually, the therapeutic schemes for breast and lung cancer involve drug combinations that can target different intratumoral/intracellular pathways to inhibit tumor progression and avoid drug resistance [
6,
10]. The most recent results obtained from clinical trials have suggested that statins combined with chemotherapeutic agents have a negative influence on the progression of both lung and breast cancer [
6,
73]. The results obtained in our study also indicate that SIM and DOX present a synergistic effect on both T47D-Kbluc human mammary ductal carcinoma cell line and A549 human pulmonary cancer cell line when the two active substances are combined in a free form. In this case, the cell viability was less than 50% for all drug combinations, making it difficult to estimate the IC
50 (
Table 6).
The long circulating liposomes loaded with DOX (DOX-LCL) were observed to increase the IC
50 of DOX on both cell lines, possibly due to the prolonged drug release [
14,
74]. In the case of SIM-LCL formulation, a two-fold decrease in the IC
50 of SIM on T47D-KBluc cell line was noted. On the other hand, the results obtained on the A549 cell line highlighted that there is only a small difference in the IC
50 value of SIM-LCL and free SIM. It should be mentioned that empty liposomes (E-LCL) were assessed in conjunction with the treatments to exclude their influence on cell viability. The results highlighted that cell viability for E-LCL was 99% (data not presented), and all the reported results exclusively represent the influence of the treatments.
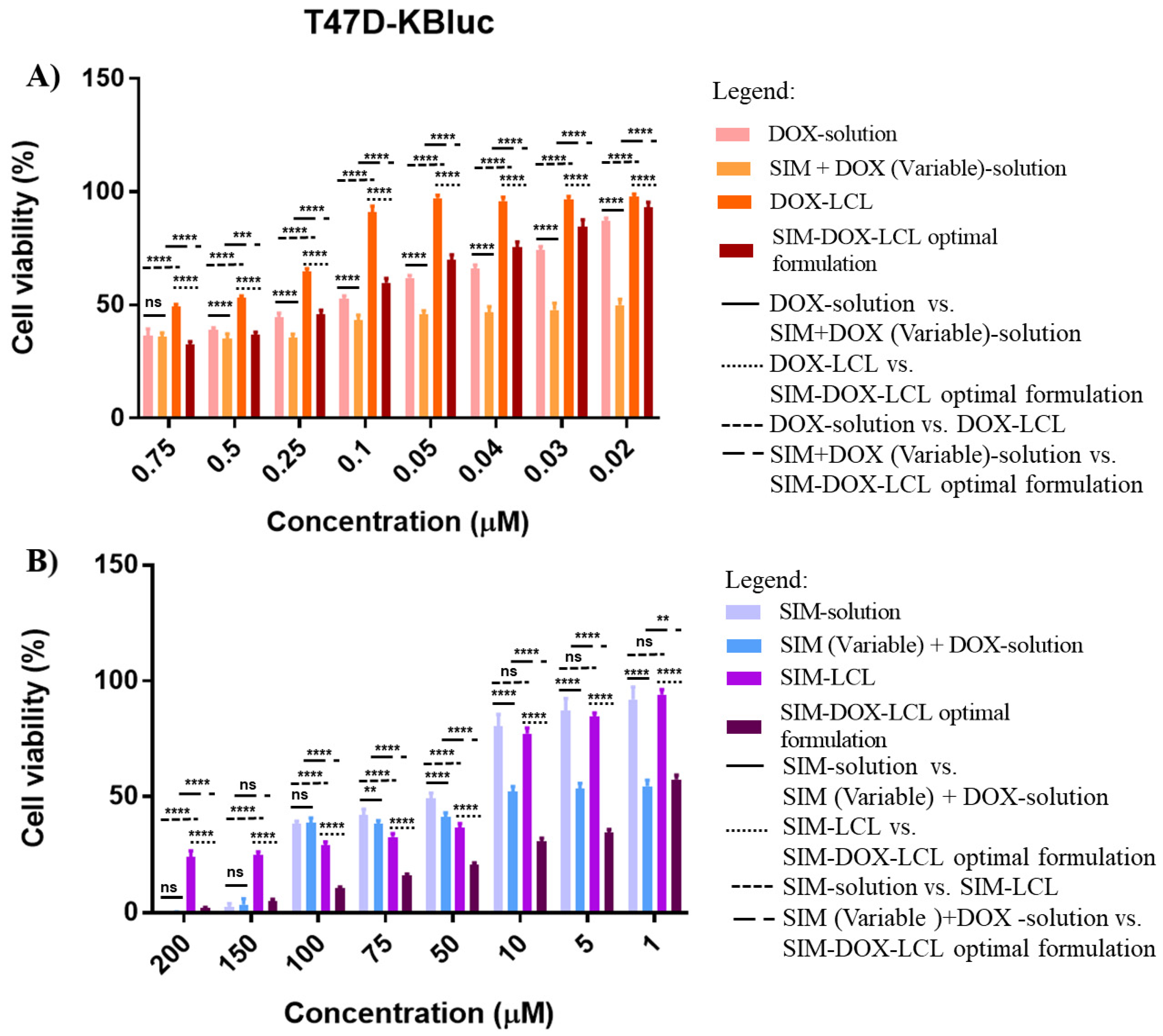
Apart from these results, SIM-DOX-LCL optimal formulation also documented increased resistance in the T47D-KBluc human mammary ductal carcinoma cell line when low concentrations of SIM (7.5–0.2 µM) and DOX (0.75–0.02 µM) were used (
Figure 10A), the DOX IC
50 value increasing 1.5 times for liposomes compared to free DOX. This result is in agreement with another study, which demonstrated that the T47D-KBluc cell line is more resistant to SIM therapy than other breast cancer cell lines, and higher concentrations are required to achieve an antiproliferative effect [
75]. Moreover, the same study evidenced that SIM, atorvastatin and rosuvastatin can increase the expression of HMGCoA reductase genes in some breast cancer cell lines, therefore inducing drug resistance [
75]. By comparing the results obtained with what was already reported, it can be observed that the use of higher concentrations of SIM (1–200 µM) and DOX (0.1–20 µM) in the liposomal formulation decrease the SIM IC
50 by 30 times compared to free SIM, and 16 times compared to SIM-LCL.
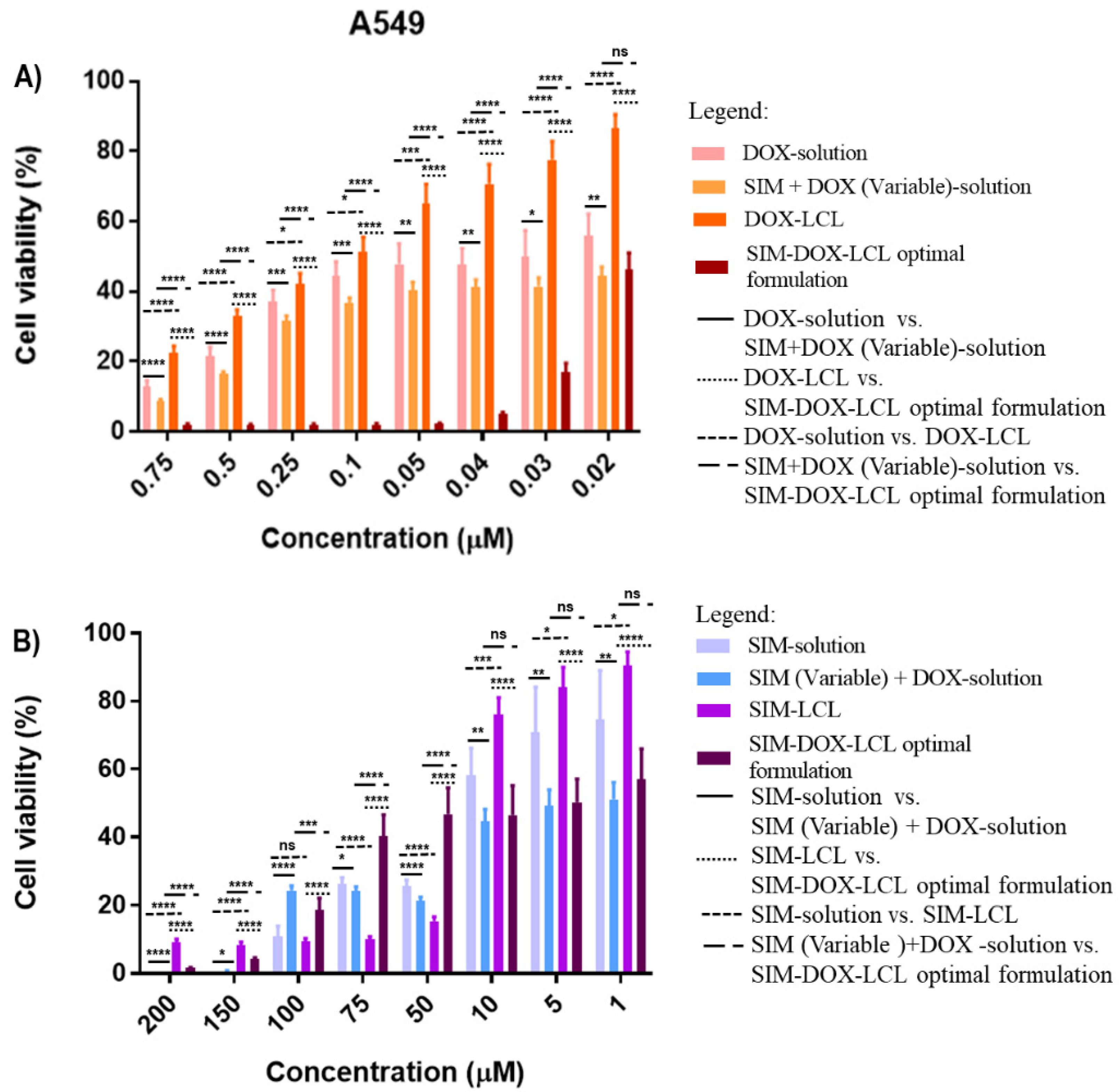
In contrast with the previous results, the SIM-DOX-LCL optimal formulation highlighted a higher level of activity against the A549 human pulmonary cancer cell line when smaller concentrations of SIM (7.5–0.2 µM) and DOX (0.75–0.02 µM) were used (
Figure 11A). The IC
50 value of DOX decreased 2.5 times compared to free DOX, and 6.8 times compared with DOX-LCL. In this respect, Aksoy and Ceylan demonstrated that statins have a different impact on A549 human pulmonary cancer cell line. More precisely, they reported that high concentrations of SIM determine an increase in cholesterol concentration in the cell membrane, with a direct impact on its fluidity and permeability [
76]. Bearing this in mind, we assume that higher concentrations of the SIM-DOX-LCL optimal formulation determined an increase in cholesterol content of cell membrane, which further limited the internalization of liposomes, thus inducing the resistance to treatment (
Figure 11B) and a 1.17 times increase in SIM IC
50 value compared to free SIM or SIM-LCL.
The results obtained in this study suggest that SIM enhances the anticancer properties of DOX in a free form and co-encapsulated in liposomes, achieving a synergistic effect on both studied human cancer cell lines. However, it must be taken into consideration that the antiproliferative potential of SIM-DOX-LCL is concentration dependent, and a thorough analysis should be performed before establishing the dosing regimens for the future in vivo studies. In addition to these, additional studies are required to establish the pharmacokinetic profile of SIM-DOX-LCL optimal formulation as a means to ensure the desired drug ratio at the tumor site.


 ,
, 




_Cherfan.png)













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}