
Combined In Silico and In Vitro Evidence Supporting an Aurora A Kinase Inhibitory Role of the Anti-Viral Drug Rilpivirine and an Anti-Proliferative Influence on Cancer Cells

 and
and
Abstract
1. Introduction
2. Results
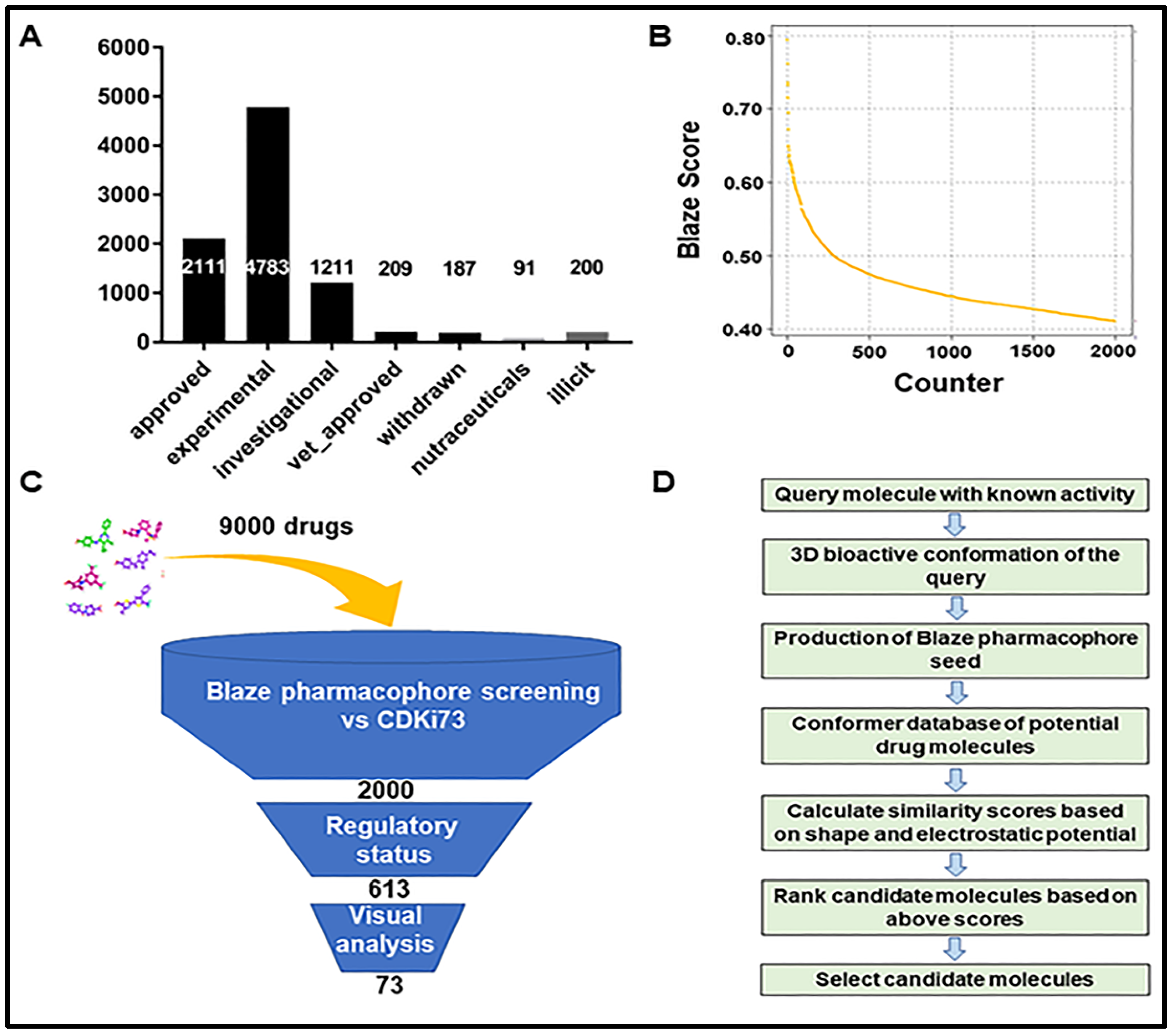
2.1. Identification of Repurposing Drug Candidates Utilising Virtual Screening
2.2. Assessment of Anti-Proliferative Activities of Drug Candidates
2.3. Drug Candidates Displaying Anti-Proliferative Activities Shared Similar Structural Features with the In Silico Ligand
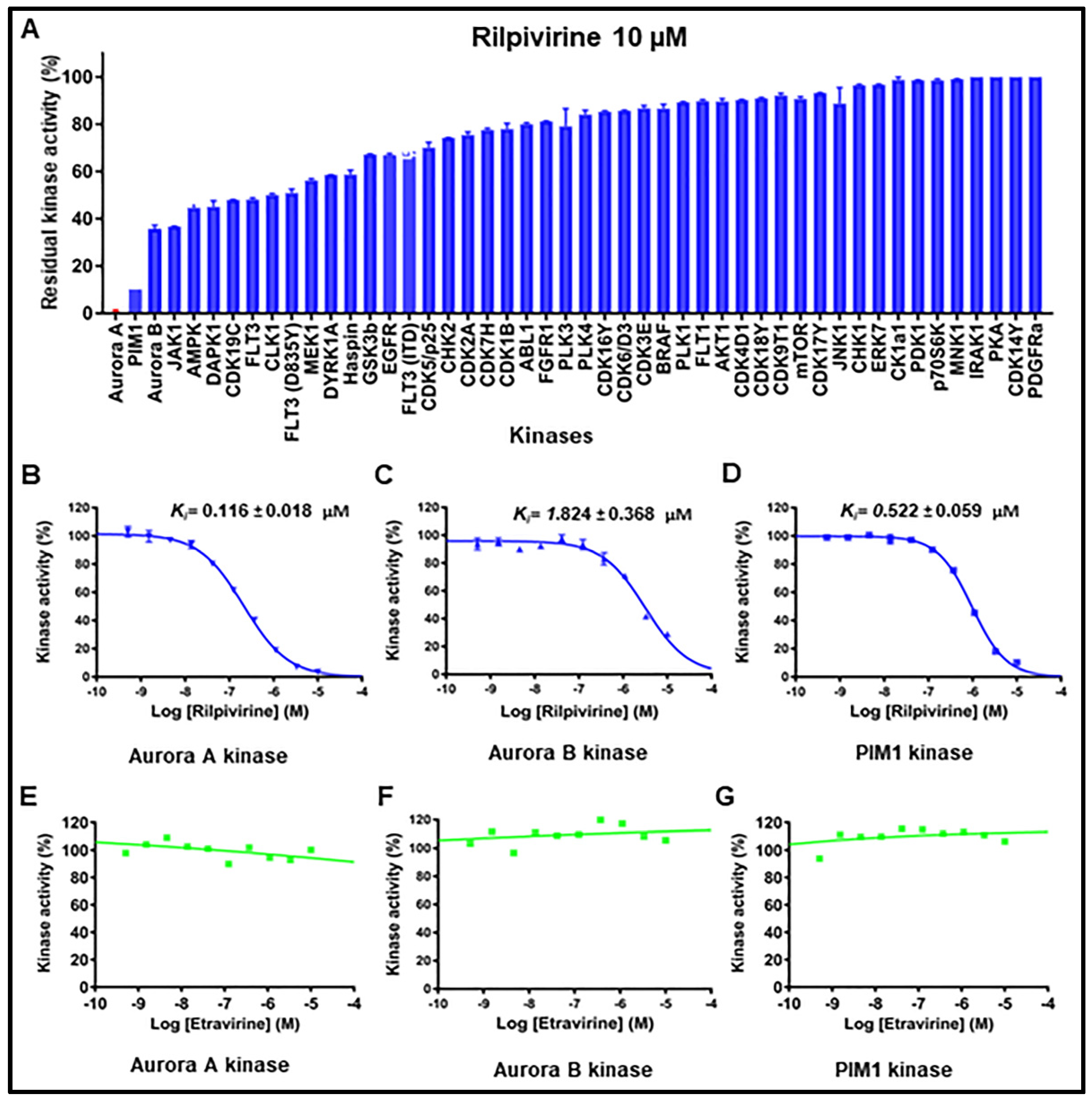
2.4. Kinase Inhibitory Profiles of Rilpivirine
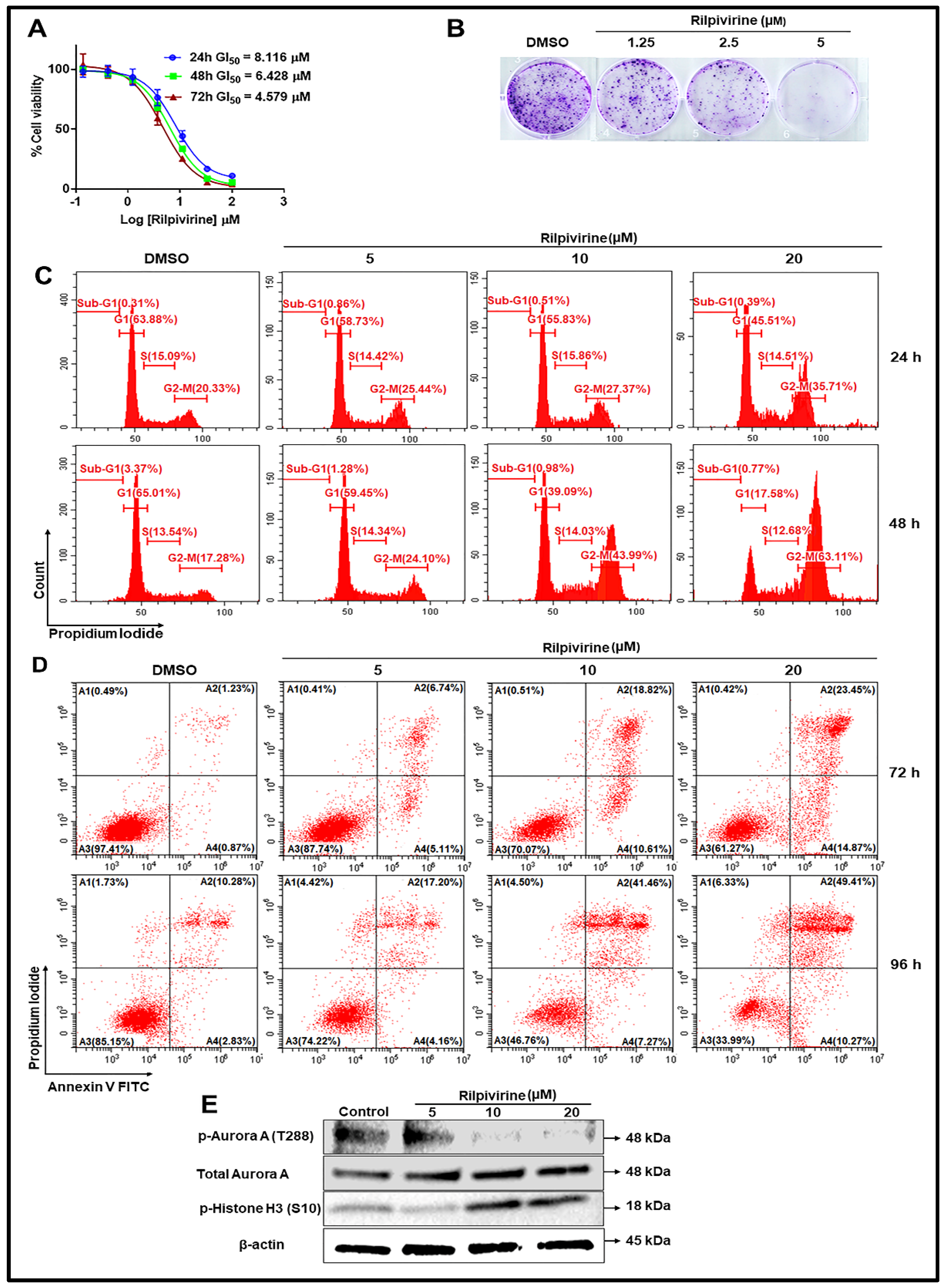
2.5. Cellular Mode of Action
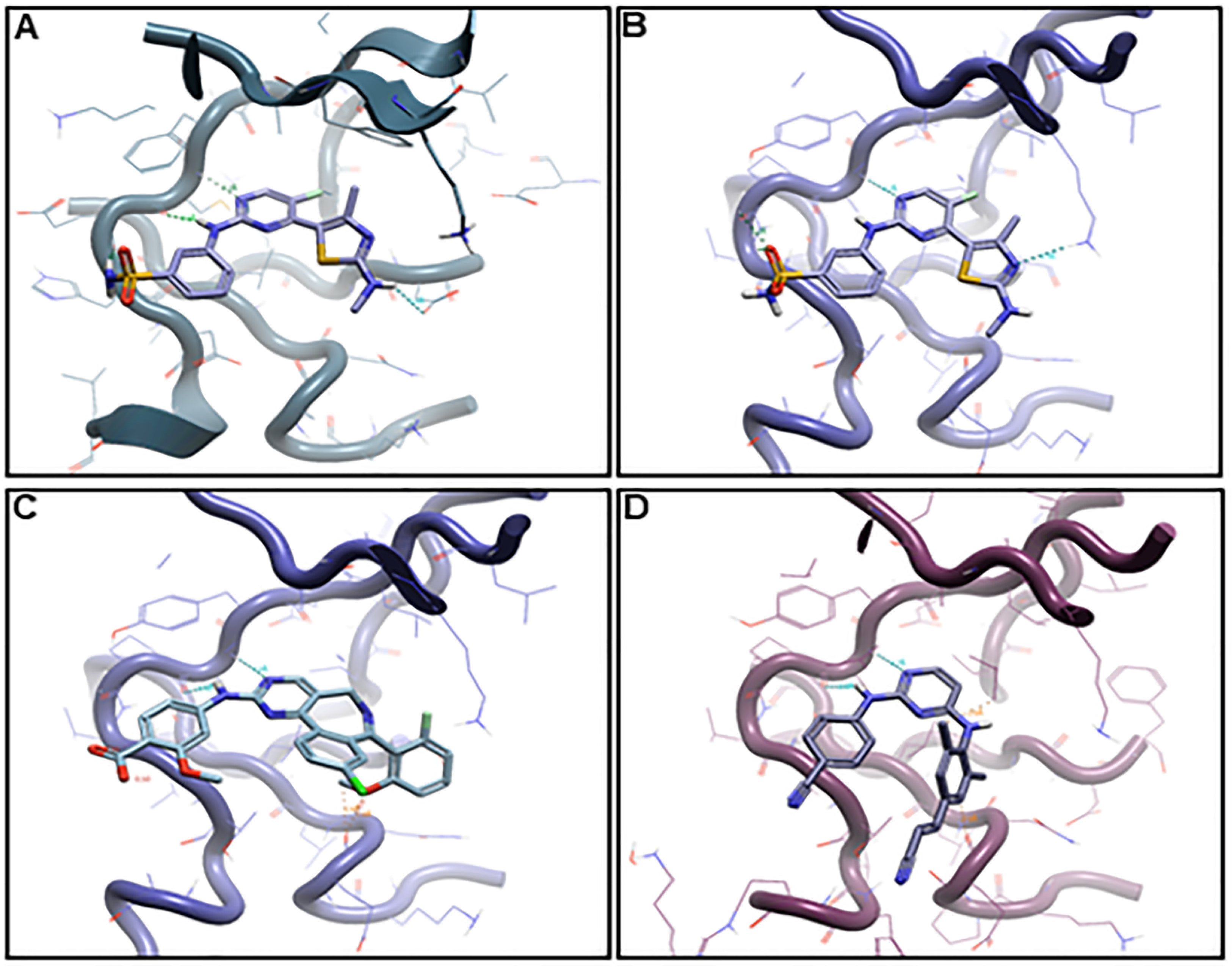
2.6. Binding Hypothesis Determination
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. Virtual Screening
4.4. Cell Viability Assays (MTT and Resazurin Assays)
4.5. Colony Formation Assay
4.6. Kinase Assays
4.7. Cell Cycle Analysis
4.8. Apoptosis Analysis
4.9. Western Blot Analysis
4.10. Molecular Modelling
4.11. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Islam, S.; Wang, S.; Bowden, N.; Martin, J.; Head, R. Repurposing existing therapeutics, its importance in oncology drug development: Kinases as a potential target. Br. J. Clin. Pharmacol. 2021, 88, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Bertolini, F.; Sukhatme, V.P.; Bouche, G. Drug repurposing in oncology--patient and health systems opportunities. Nat. Rev. Clin. Oncol. 2015, 12, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2018, 18, 41. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhou, L.; Xie, N.; Nice, E.C.; Zhang, T.; Cui, Y.; Huang, C. Overcoming cancer therapeutic bottleneck by drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 1–25. [Google Scholar] [CrossRef]
- Paolini, G.V.; Shapland, R.H.; van Hoorn, W.P.; Mason, J.S.; Hopkins, A.L. Global mapping of pharmacological space. Nat. Biotechnol. 2006, 24, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Glicksberg, B.S.; Li, L.; Cheng, W.-Y.; Shameer, K.; Hakenberg, J.; Castellanos, R.; Ma, M.; Shi, L.; Shah, H.; Dudley, J.T. An Integrative Pipeline for Multi-Modal Discovery of Disease Relationships; Pacific Symposium on Biocomputing Co-Chairs; World Scientific: Singapore, 2014; pp. 407–418. [Google Scholar]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef]
- Ferguson, F.M.; Gray, N.S. Kinase inhibitors: The road ahead. Nat. Rev. Drug Discov. 2018, 17, 353–377. [Google Scholar] [CrossRef]
- Dancey, J.; Sausville, E.A. Issues and progress with protein kinase inhibitors for cancer treatment. Nat. Rev. Drug Discov. 2003, 2, 296–313. [Google Scholar] [CrossRef]
- Bertolin, G.; Bulteau, A.-L.; Alves-Guerra, M.-C.; Burel, A.; Lavault, M.-T.; Gavard, O.; Le Bras, S.; Gagné, J.-P.; Poirier, G.G.; Le Borgne, R. Aurora kinase a localises to mitochondria to control organelle dynamics and energy production. Elife 2018, 7, e38111. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Lake, E.W.; Muretta, J.M.; Thompson, A.R.; Rasmussen, D.M.; Majumdar, A.; Faber, E.B.; Ruff, E.F.; Thomas, D.D.; Levinson, N.M. Quantitative conformational profiling of kinase inhibitors reveals origins of selectivity for aurora kinase activation states. Proc. Natl. Acad. Sci. USA 2018, 115, E11894–E11903. [Google Scholar] [CrossRef] [PubMed]
- Wainberg, M.A. Combination therapies, effectiveness, and adherence in patients with hiv infection: Clinical utility of a single tablet of emtricitabine, rilpivirine, and tenofovir. HIV/AIDS 2013, 5, 41. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yan, M.; Wang, C.; He, B.; Yang, M.; Tong, M.; Long, Z.; Liu, B.; Peng, F.; Xu, L.; Zhang, Y.; et al. Aurora-a kinase: A potent oncogene and target for cancer therapy. Med. Res. Rev. 2016, 36, 1036–1079. [Google Scholar] [CrossRef]
- Du, R.; Huang, C.; Liu, K.; Li, X.; Dong, Z. Targeting aurka in cancer: Molecular mechanisms and opportunities for cancer therapy. Mol. Cancer 2021, 20, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Carpinelli, P.; Moll, J. Aurora kinase inhibitors: Identification and preclinical validation of their biomarkers. Expert Opin. Ther. Targets 2008, 12, 69–80. [Google Scholar] [CrossRef]
- Gorgun, G.; Calabrese, E.; Hideshima, T.; Ecsedy, J.; Perrone, G.; Mani, M.; Ikeda, H.; Bianchi, G.; Hu, Y.; Cirstea, D.; et al. A novel aurora-a kinase inhibitor mln8237 induces cytotoxicity and cell-cycle arrest in multiple myeloma. Blood 2010, 115, 5202–5213. [Google Scholar] [CrossRef]
- Sells, T.B.; Chau, R.; Ecsedy, J.A.; Gershman, R.E.; Hoar, K.; Huck, J.; Janowick, D.A.; Kadambi, V.J.; LeRoy, P.J.; Stirling, M. Mln8054 and alisertib (mln8237): Discovery of selective oral aurora a inhibitors. ACS Med. Chem. Lett. 2015, 6, 630–634. [Google Scholar] [CrossRef]
- Shimomura, T.; Hasako, S.; Nakatsuru, Y.; Mita, T.; Ichikawa, K.; Kodera, T.; Sakai, T.; Nambu, T.; Miyamoto, M.; Takahashi, I. Mk-5108, a highly selective aurora-a kinase inhibitor, shows antitumor activity alone and in combination with docetaxel. Mol. Cancer Ther. 2010, 9, 157–166. [Google Scholar] [CrossRef]
- Knapp, S. New Opportunities for Kinase Drug Repurposing and Target Discovery; Nature Publishing Group: Berlin, Germany, 2018. [Google Scholar]
- Wang, L.; Chen, L.; Yu, M.; Xu, L.-H.; Cheng, B.; Lin, Y.-S.; Gu, Q.; He, X.-H.; Xu, J. Discovering new mtor inhibitors for cancer treatment through virtual screening methods and in vitro assays. Sci. Rep. 2016, 6, 18987. [Google Scholar] [CrossRef]
- Shi, X.-N.; Li, H.; Yao, H.; Liu, X.; Li, L.; Leung, K.-S.; Kung, H.-F.; Lu, D.; Wong, M.-H.; Lin, M.C.-M. In silico identification and in vitro and in vivo validation of anti-psychotic drug fluspirilene as a potential cdk2 inhibitor and a candidate anti-cancer drug. PLoS ONE 2015, 10, e0132072. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.; Foley, D.W.; Huang, S.; Abbas, A.Y.; Lam, F.; Gershkovich, P.; Bradshaw, T.D.; Pepper, C.; Fischer, P.M.; Wang, S. Structure-based design of highly selective 2, 4, 5-trisubstituted pyrimidine cdk9 inhibitors as anti-cancer agents. Eur. J. Med. Chem. 2021, 214, 113244. [Google Scholar] [CrossRef] [PubMed]
- Keeton, E.K.; McEachern, K.; Dillman, K.S.; Palakurthi, S.; Cao, Y.; Grondine, M.R.; Kaur, S.; Wang, S.; Chen, Y.; Wu, A. Azd1208, a potent and selective pan-pim kinase inhibitor, demonstrates efficacy in preclinical models of acute myeloid leukemia. Blood J. Am. Soc. Hematol. 2014, 123, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.-W.; Huang, C.-H.; Liu, L.-C.; Cheng, F.-J.; Wei, Y.-L.; Lin, Y.-M.; Wang, Y.-F.; Wei, C.-T.; Chen, Y.; Chen, Y.-J. Pim1 kinase inhibitors exert anti-cancer activity against her2-positive breast cancer cells through downregulation of her2. Front. Pharmacol. 2021, 12, 1637. [Google Scholar] [CrossRef]
- Brasó-Maristany, F.; Filosto, S.; Catchpole, S.; Marlow, R.; Quist, J.; Francesch-Domenech, E.; Plumb, D.A.; Zakka, L.; Gazinska, P.; Liccardi, G. Pim1 kinase regulates cell death, tumor growth and chemotherapy response in triple-negative breast cancer. Nat. Med. 2016, 22, 1303–1313. [Google Scholar] [CrossRef]
- Perna, A.; Lucariello, A.; Sellitto, C.; Agliata, I.; Carleo, M.A.; Sangiovanni, V.; Esposito, V.; Guerra, G.; Cobellis, L.; De Luca, A. Different cell cycle modulation in skov-3 ovarian cancer cell line by anti-hiv drugs. Oncol. Res. 2017, 25, 1617. [Google Scholar] [CrossRef]
- Hecht, M.; Erber, S.; Harrer, T.; Klinker, H.; Roth, T.; Parsch, H.; Fiebig, N.; Fietkau, R.; Distel, L.V. Efavirenz has the highest anti-proliferative effect of non-nucleoside reverse transcriptase inhibitors against pancreatic cancer cells. PLoS ONE 2015, 10, e0130277. [Google Scholar] [CrossRef]
- Mangiacasale, R.; Pittoggi, C.; Sciamanna, I.; Careddu, A.; Mattei, E.; Lorenzini, R.; Travaglini, L.; Landriscina, M.; Barone, C.; Nervi, C. Exposure of normal and transformed cells to nevirapine, a reverse transcriptase inhibitor, reduces cell growth and promotes differentiation. Oncogene 2003, 22, 2750–2761. [Google Scholar] [CrossRef]
- Sciamanna, I.; Landriscina, M.; Pittoggi, C.; Quirino, M.; Mearelli, C.; Beraldi, R.; Mattei, E.; Serafino, A.; Cassano, A.; Sinibaldi-Vallebona, P. Inhibition of endogenous reverse transcriptase antagonizes human tumor growth. Oncogene 2005, 24, 3923–3931. [Google Scholar] [CrossRef]
- Hecht, M.; Harrer, T.; Büttner, M.; Schwegler, M.; Erber, S.; Fietkau, R.; Distel, L.V. Cytotoxic effect of efavirenz is selective against cancer cells and associated with the cannabinoid system. AIDS 2013, 27, 2031–2040. [Google Scholar] [CrossRef]
- Apostolova, N.; Gomez-Sucerquia, L.J.; Moran, A.; Alvarez, A.; Blas-Garcia, A.; Esplugues, J. Enhanced oxidative stress and increased mitochondrial mass during efavirenz-induced apoptosis in human hepatic cells. Br. J. Pharmacol. 2010, 160, 2069–2084. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Perez de Castro, I. Aurora kinase a inhibitors: Promising agents in antitumoral therapy. Expert Opin. Ther. Targets 2014, 18, 1377–1393. [Google Scholar] [PubMed]
- Chakraborti, S.; Chakravarthi, P.; Srinivasan, N. A ligand-centric approach to identify potential drugs for repurposing: Case study with aurora kinase inhibitors. In Drug Repurposing in Cancer Therapy; Elsevier: Amsterdam, The Netherlands, 2020; pp. 15–54. [Google Scholar]
- Wang, S.; Griffiths, G.; Midgley, C.A.; Barnett, A.L.; Cooper, M.; Grabarek, J.; Ingram, L.; Jackson, W.; Kontopidis, G.; McClue, S.J. Discovery and characterization of 2-anilino-4-(thiazol-5-yl) pyrimidine transcriptional cdk inhibitors as anticancer agents. Chem. Biol. 2010, 17, 1111–1121. [Google Scholar] [CrossRef]
- Shao, H.; Shi, S.; Huang, S.; Hole, A.J.; Abbas, A.Y.; Baumli, S.; Liu, X.; Lam, F.; Foley, D.W.; Fischer, P.M. Substituted 4-(thiazol-5-yl)-2-(phenylamino) pyrimidines are highly active cdk9 inhibitors: Synthesis, x-ray crystal structures, structure–activity relationship, and anticancer activities. J. Med. Chem. 2013, 56, 640–659. [Google Scholar] [CrossRef] [PubMed]
- Cheeseright, T.J.; Mackey, M.D.; Melville, J.L.; Vinter, J.G. Fieldscreen: Virtual screening using molecular fields. Application to the dud data set. J. Chem. Inf. Modeling 2008, 48, 2108–2117. [Google Scholar] [CrossRef] [PubMed]
- Cheeseright, T.; Mackey, M.; Rose, S.; Vinter, A. Molecular field extrema as descriptors of biological activity: Definition and validation. J. Chem. Inf. Modeling 2006, 46, 665–676. [Google Scholar] [CrossRef]
- Grant, J.A.; Gallardo, M.A.; Pickup, B.T. A fast method of molecular shape comparison: A simple application of a gaussian description of molecular shape. J. Comput. Chem. 1996, 17, 1653–1666. [Google Scholar] [CrossRef]
- Wang, S.; Meades, C.; Wood, G.; Osnowski, A.; Anderson, S.; Yuill, R.; Thomas, M.; Mezna, M.; Jackson, W.; Midgley, C. 2-anilino-4-(thiazol-5-yl) pyrimidine cdk inhibitors: Synthesis, sar analysis, x-ray crystallography, and biological activity. J. Med. Chem. 2004, 47, 1662–1675. [Google Scholar] [CrossRef] [PubMed]
- Diab, S.; Abdelaziz, A.M.; Li, P.; Teo, T.; Basnet, S.K.; Noll, B.; Rahaman, M.H.; Lu, J.; Hou, J.; Yu, M. Dual inhibition of mnk2 and flt3 for potential treatment of acute myeloid leukaemia. Eur. J. Med. Chem. 2017, 139, 762–772. [Google Scholar] [CrossRef]
- Basnet, S.K.; Diab, S.; Schmid, R.; Yu, M.; Yang, Y.; Gillam, T.A.; Teo, T.; Li, P.; Peat, T.; Albrecht, H. Identification of a highly conserved allosteric binding site on mnk1 and mnk2. Mol. Pharmacol. 2015, 88, 935–948. [Google Scholar] [CrossRef]
- Yung-Chi, C.; Prusoff, W.H. Relationship between the inhibition constant (ki) and the concentration of inhibitor which causes 50 per cent inhibition (i50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
- Rahaman, M.H.; Yu, Y.; Zhong, L.; Adams, J.; Lam, F.; Li, P.; Noll, B.; Milne, R.; Peng, J.; Wang, S. Cdki-73: An orally bioavailable and highly efficacious cdk9 inhibitor against acute myeloid leukemia. Investig. New Drugs 2019, 37, 625–635. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Human Cancer Cell Lines | 72 h GI50 (µmol/L) ± SD | ||||
|---|---|---|---|---|---|
| Origin | Destination | Etravirine | Pimobendan | Rilpivirine | Revaprazan |
| Breast | MCF-7 | 5.174 ± 0.373 | 5.939 ± 0.085 | 5.928 ± 0.206 | 11.02 ± 0.675 |
| Colon | HCT-116 | 5.753 ± 0.520 | 5.450 ± 0.658 | 9.422 ± 0.812 | 17.60 ± 3.020 |
| HT-29 | 5.210 ± 0.634 | 6.121 ± 1.828 | 5.195 ± 0.469 | 11.42 ± 1.920 | |
| Ovarian | A2780 | 4.101 ± 1.266 | 4.514 ± 1.009 | 3.045 ± 0.823 | 6.301 ± 1.629 |
| Pancreatic | PANC-1 | 5.185 ± 0.256 | 7.195 ± 0.168 | 4.764 ± 0.340 | 14.04 ± 0.851 |
| Leukaemia | MV4-11 | 5.863 ± 1.121 | 3.788 ± 0.699 | 4.375 ± 0.516 | 7.926 ± 0.983 |
| MOLM-13 | 7.052 ± 1.142 | 4.053 ± 1.811 | 4.266 ± 0.644 | 7.828 ± 0.715 | |
| JURKAT | 7.785 ± 0.408 | 4.756 ± 0.224 | 4.281 ± 0.767 | 32.67 ± 4.428 | |
| HL-60 | 5.952 ± 0.473 | 5.254 ± 0.202 | 4.762 ± 1.619 | 38.06 ± 1.607 | |
| K-562 | 8.163 ± 0.602 | 7.033 ± 0.165 | 6.858 ± 0.745 | 25.37 ± 0.926 | |
| NB4 | 4.782 ± 0.341 | 4.062 ± 0.494 | 3.395 ± 1.720 | 42.35 ± 3.673 | |
| PL-21 | 4.465 ± 0.282 | 4.535 ± 0.226 | 3.737 ± 0.636 | 16.55 ± 1.232 | |
| U-937 | 4.794 ± 0.497 | 3.782 ± 0.138 | 3.628 ± 1.939 | 28.14 ± 2.887 | |
| THP-1 | 9.450 ± 0.730 | 3.118 ± 0.377 | 6.032 ± 0.394 | 18.15 ± 1.884 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Islam, S.; Teo, T.; Kumarasiri, M.; Slater, M.; Martin, J.H.; Wang, S.; Head, R. Combined In Silico and In Vitro Evidence Supporting an Aurora A Kinase Inhibitory Role of the Anti-Viral Drug Rilpivirine and an Anti-Proliferative Influence on Cancer Cells. Pharmaceuticals 2022, 15, 1186. https://doi.org/10.3390/ph15101186
Islam S, Teo T, Kumarasiri M, Slater M, Martin JH, Wang S, Head R. Combined In Silico and In Vitro Evidence Supporting an Aurora A Kinase Inhibitory Role of the Anti-Viral Drug Rilpivirine and an Anti-Proliferative Influence on Cancer Cells. Pharmaceuticals. 2022; 15(10):1186. https://doi.org/10.3390/ph15101186
Chicago/Turabian StyleIslam, Saiful, Theodosia Teo, Malika Kumarasiri, Martin Slater, Jennifer H. Martin, Shudong Wang, and Richard Head. 2022. "Combined In Silico and In Vitro Evidence Supporting an Aurora A Kinase Inhibitory Role of the Anti-Viral Drug Rilpivirine and an Anti-Proliferative Influence on Cancer Cells" Pharmaceuticals 15, no. 10: 1186. https://doi.org/10.3390/ph15101186
APA StyleIslam, S., Teo, T., Kumarasiri, M., Slater, M., Martin, J. H., Wang, S., & Head, R. (2022). Combined In Silico and In Vitro Evidence Supporting an Aurora A Kinase Inhibitory Role of the Anti-Viral Drug Rilpivirine and an Anti-Proliferative Influence on Cancer Cells. Pharmaceuticals, 15(10), 1186. https://doi.org/10.3390/ph15101186