
The First Dimeric Derivatives of the Glycopeptide Antibiotic Teicoplanin

 ,
,  , , , , and
, , , , and
Abstract
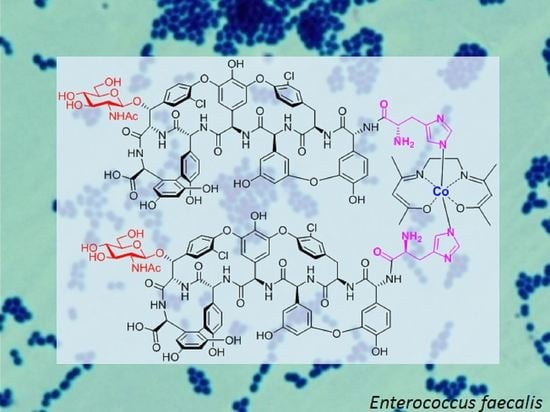
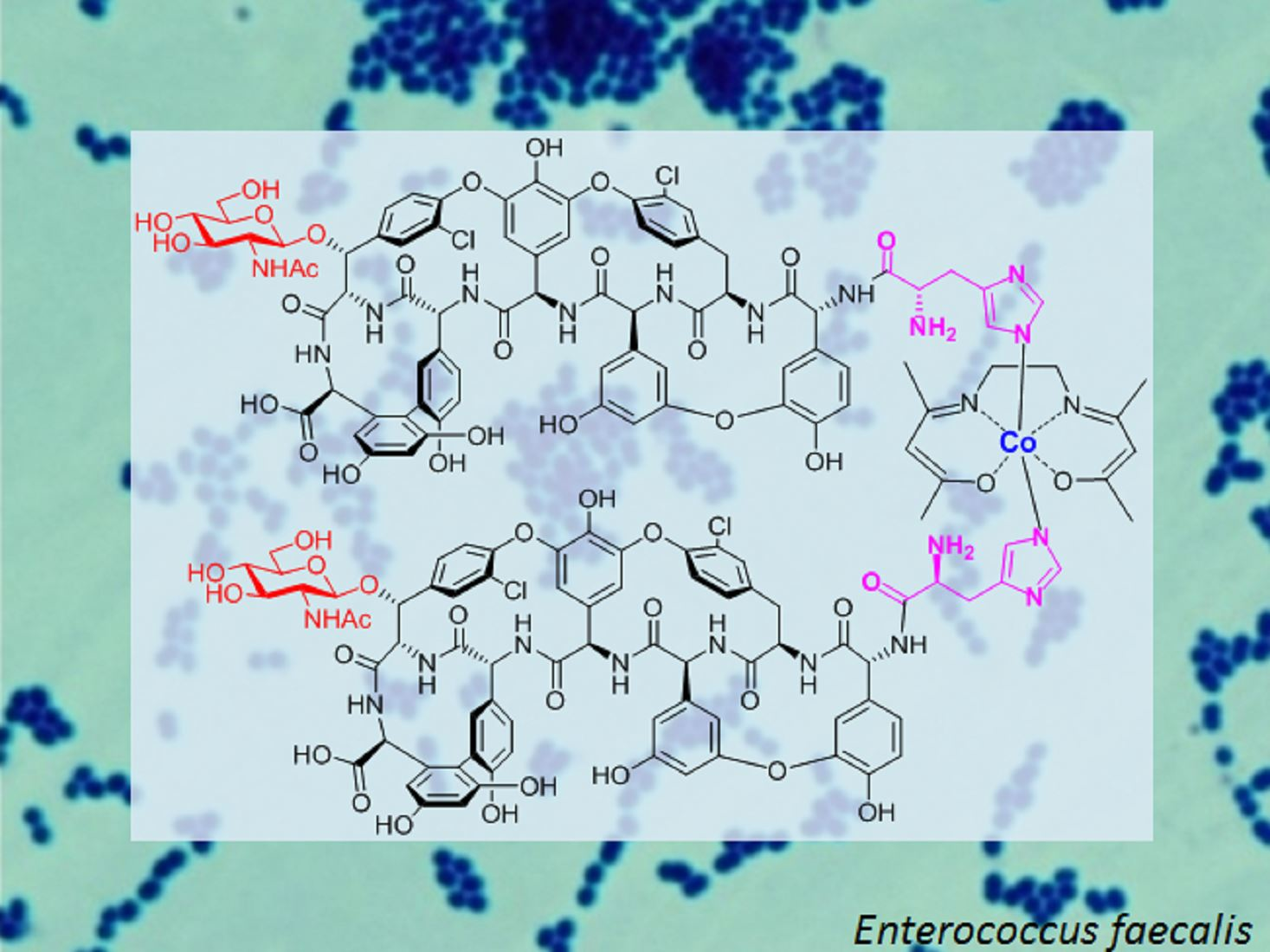
1. Introduction
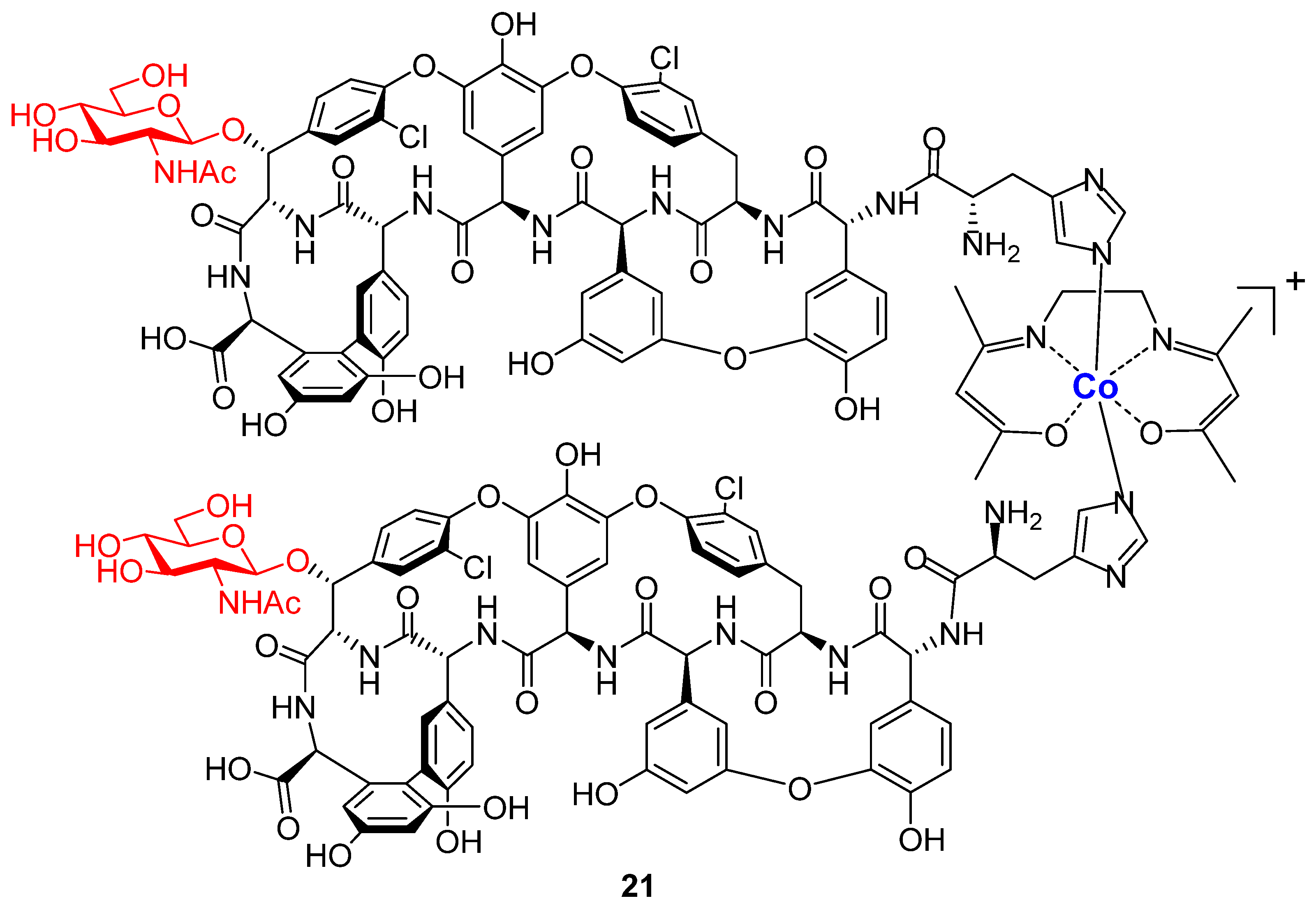
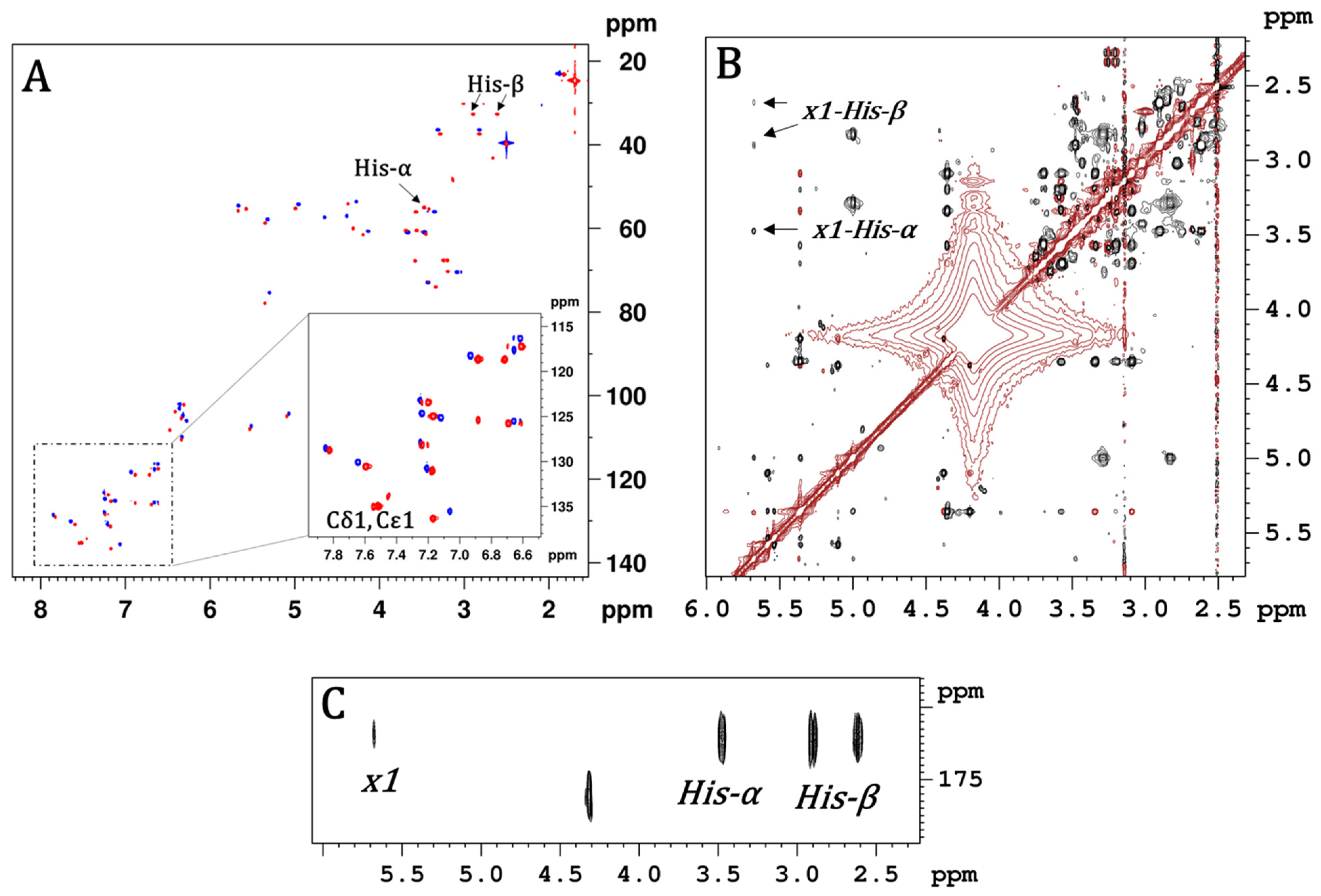
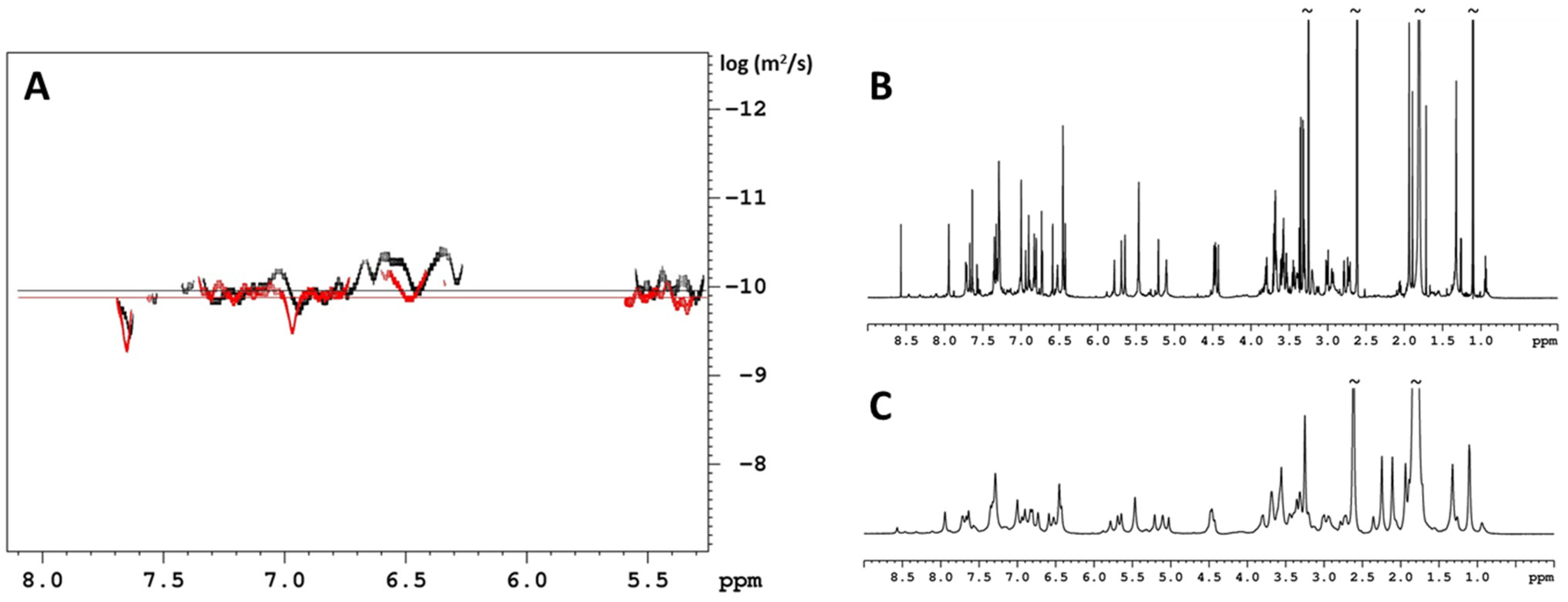
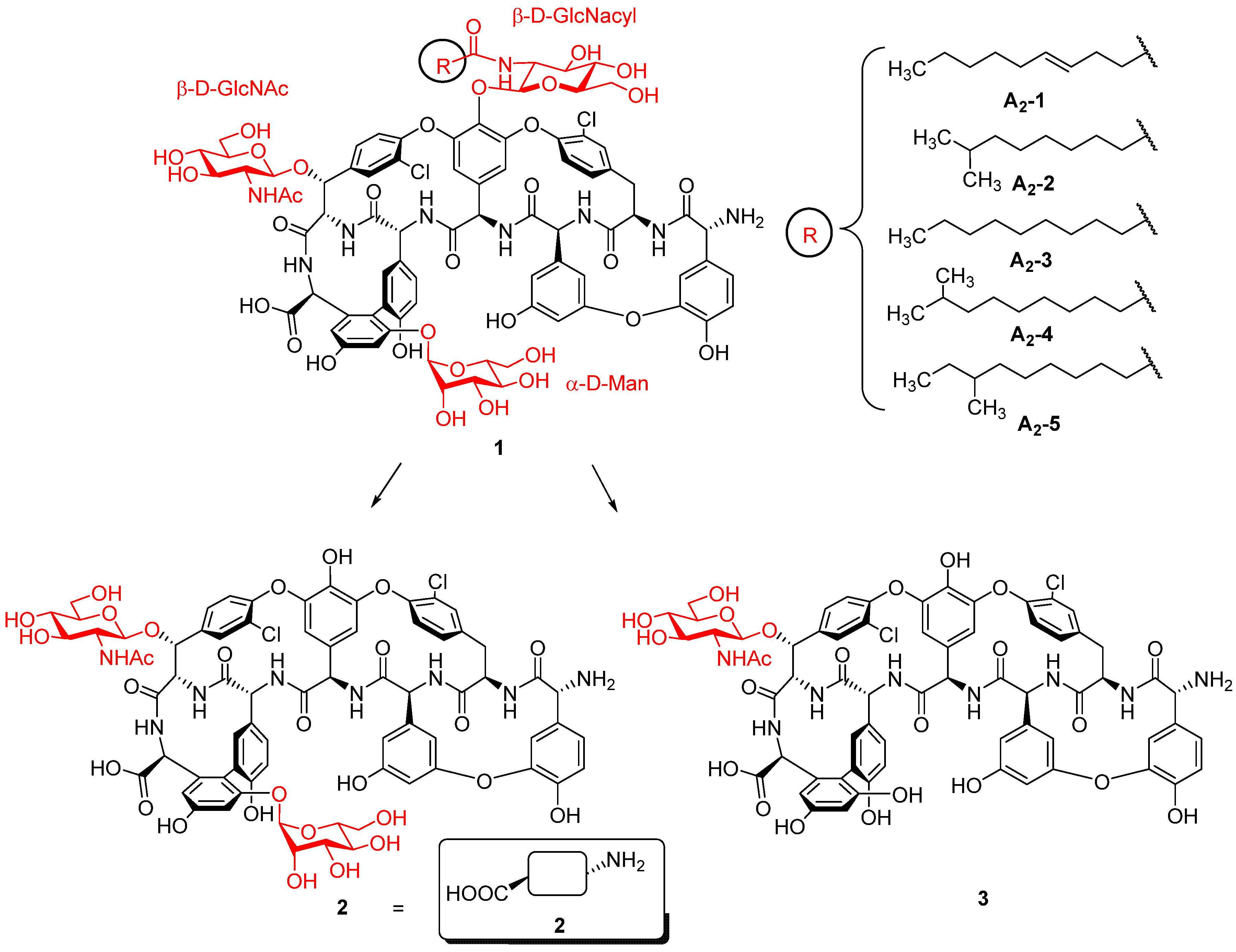
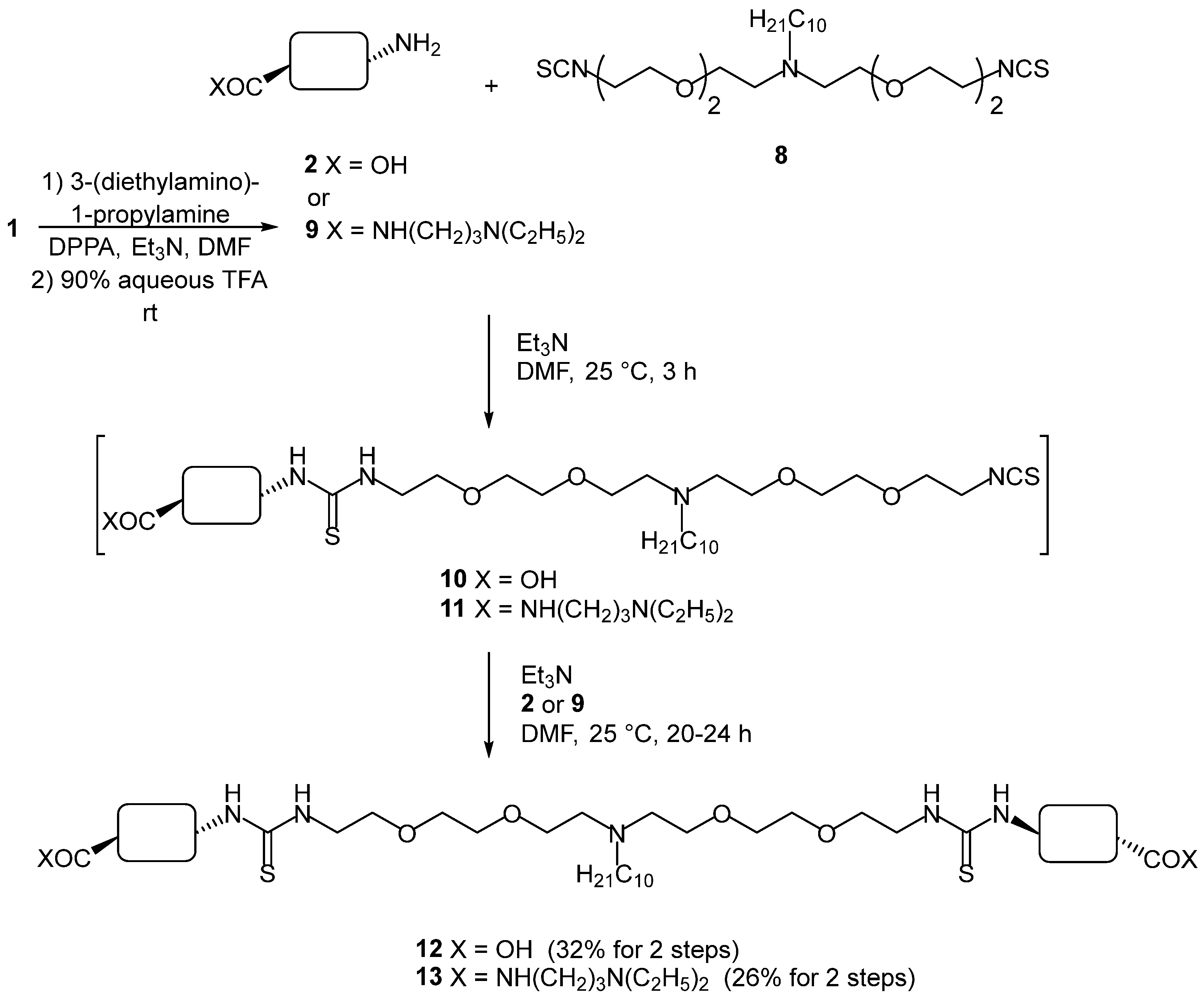
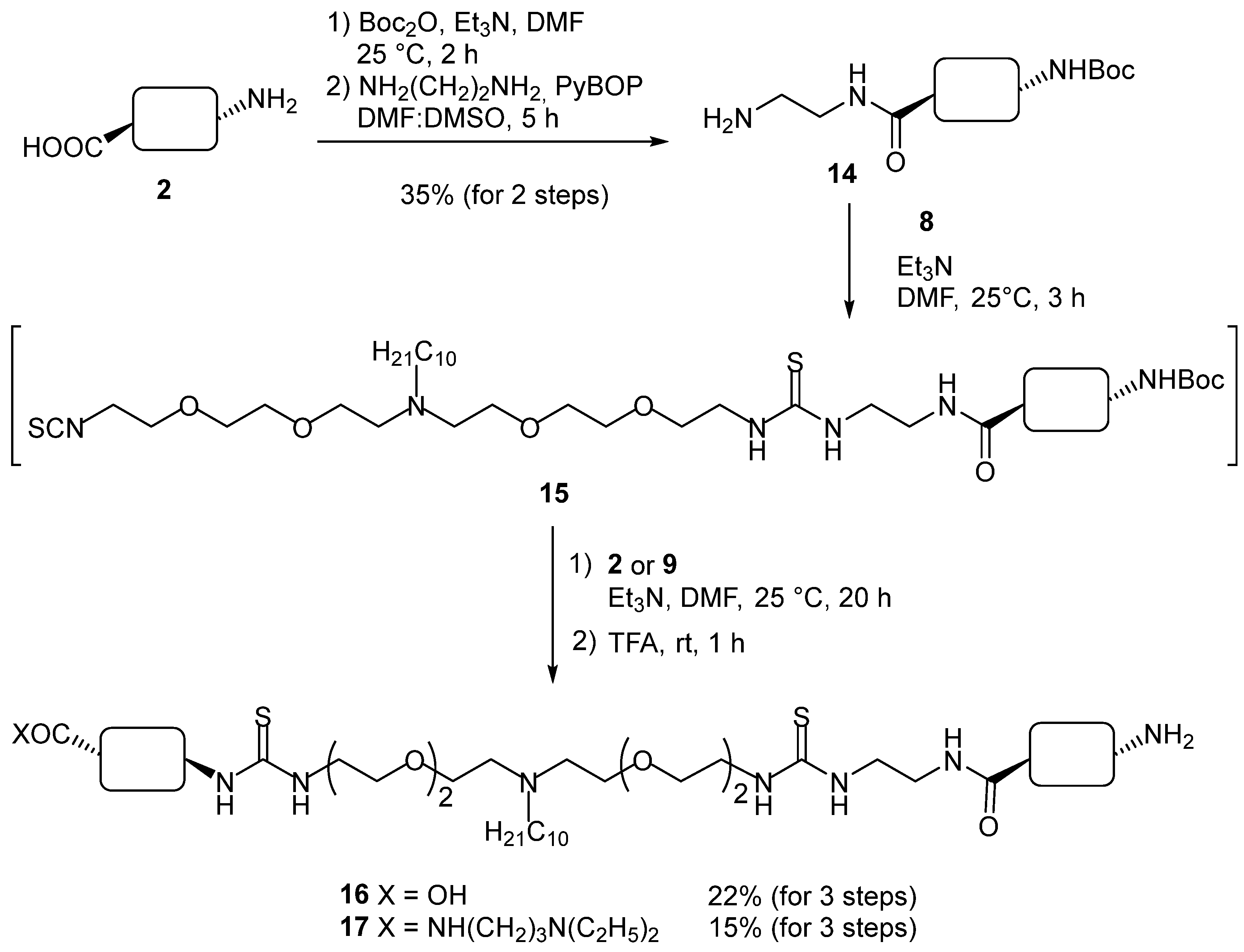
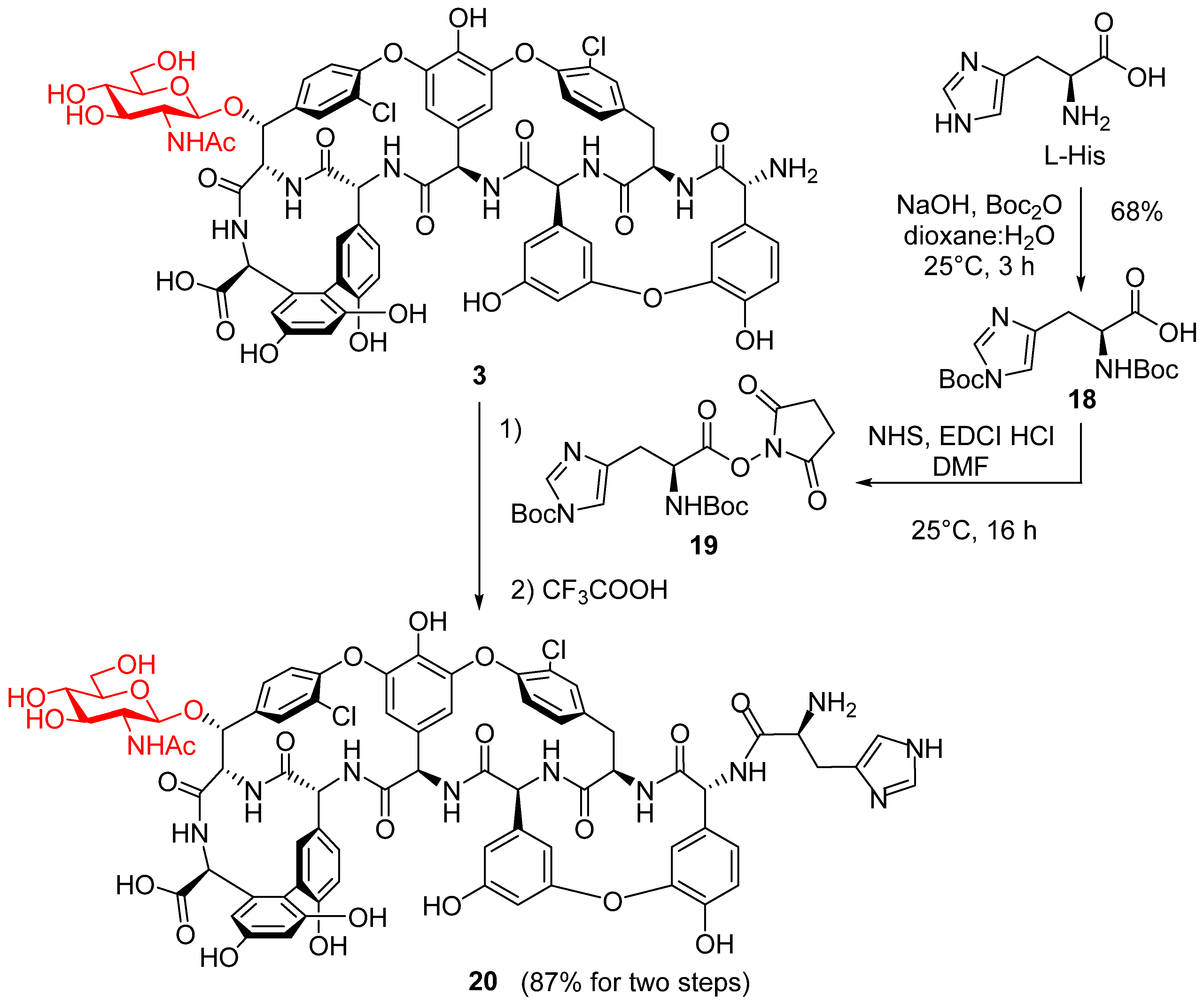
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Chemical Synthesis
3.2.1. 9-Decyl-3,6,12,15-Tetraoxa-9-Azaheptadecane-1,17-Diol (6)
3.2.2. N,N-Bis(2-(2-(2-Aminoethoxy)Ethoxy)Ethyl)Decan-1-Amine (7)
3.2.3. N,N-Bis(2-(2-(2-Isothiocyanatoethoxy)Ethoxy)Ethyl)Decan-1-Amine (8)
3.2.4. Compound 12
3.2.5. Compound 13
3.2.6. Compound 14
3.2.7. Compound 16
3.2.8. Compound 17
3.2.9. Compound 20
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nicolaou, K.C.; Boddy, C.N.C.; Brase, S.; Winssinger, N. Chemistry, biology, and medicine of the glycopeptide antibiotics. Angew. Chem. Int. Ed. 1999, 38, 2096–2152. [Google Scholar] [CrossRef]
- Biondi, S.; Chugunova, E.; Panunzio, M. From Natural Products to Drugs: Glyco- and Lipoglycopeptides, a New Generation of Potent Cell Wall Biosynthesis Inhibitors. Stud. Nat. Prod. Chem. 2016, 50, 249–297. [Google Scholar]
- Butler, M.S.; Hansford, K.A.; Blaskovich, M.A.T.; Halai, R.; Cooper, M.A. Glycopeptide antibiotics: Back to the future. J. Antibiot. 2014, 67, 631–644. [Google Scholar] [CrossRef]
- Olsufyeva, E.N.; Tevyashova, A.N. Synthesis, Properties, and Mechanism of Action of New Generation of Polycyclic Glycopeptide Antibiotics. Curr. Top. Med. Chem. 2017, 17, 2166–2198. [Google Scholar] [CrossRef]
- Blaskovich, M.A.T.; Hansford, K.A.; Butler, M.S.; Jia, Z.G.; Mark, A.E.; Cooper, M.A. Developments in Glycopeptide Antibiotics. Acs Infect. Dis. 2018, 4, 715–735. [Google Scholar] [CrossRef]
- Zeng, D.; Debabov, D.; Hartsell, T.L.; Cano, R.J.; Adams, S.; Schuyler, J.A.; McMillan, R.; Pace, J.L. Approved Glycopeptide Antibacterial Drugs: Mechanism of Action and Resistance. Cold Spring Harb. Perspect. Med. 2016, 6, a026989. [Google Scholar] [CrossRef]
- Waltho, J.P.; Williams, D.H. Aspects of Molecular Recognition—Solvent Exclusion and Dimerization of the Antibiotic Ristocetin When Bound to a Model Bacterial Cell-Wall Precursor. J. Am. Chem. Soc. 1989, 111, 2475–2480. [Google Scholar] [CrossRef]
- Batta, G.; Sztaricskai, F.; Kover, K.E.; Rudel, C.; Berdnikova, T.F. An NMR Study of Eremomycin and its Derivatives Full H-1 and C-13 Assignment, Motional Behavior, Dimerization and Complexation with Ac-D-ALA-D-ALA. J. Antibiot. 1991, 44, 1208–1221. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Allen, N.E.; Nicas, T.I. Mechanism of action of oritavancin and related glycopeptide antibiotics. FEMS Microbiol. Rev. 2003, 26, 511–532. [Google Scholar] [CrossRef] [PubMed]
- Loll, P.J.; Miller, R.; Weeks, C.M.; Axelsen, P.H. A ligand-mediated dimerization mode for vancomycin. Chem. Biol. 1998, 5, 293–298. [Google Scholar] [CrossRef]
- Izsépi, L.; Erdei, R.; Tevyashova, A.N.; Grammatikova, N.E.; Shchekotikhin, A.E.; Herczegh, P.; Batta, G. Cell Wall Analogue Peptides Control the Oligomeric States and Activity of the Glycopeptide Antibiotic Eremomycin. Solution NMR and Antimicrobial Studies. Pharmaceuticals 2021, 13, 83. [Google Scholar] [CrossRef]
- Cheng, M.; Ziora, Z.M.; Hansford, K.A.; Blaskovich, M.A.; Butler, M.S.; Cooper, M.A. Anti-cooperative ligand binding and dimerisation in the glycopeptide antibiotic dalbavancin. Org. Biomol. Chem. 2014, 12, 2568–2575. [Google Scholar] [CrossRef]
- Loll, P.J.; Derhovanessian, A.; Shapovalov, M.V.; Kaplan, J.; Yang, L.; Axelsen, P.H. Vancomycin Forms Ligand-Mediated Supramolecular Complexes. J. Mol. Biol. 2009, 385, 200–211. [Google Scholar] [CrossRef]
- Batta, G.; Cristofaro, M.F.; Sharman, G.J.; Williams, D.H. Demonstration of the difference in binding affinity between the two binding sites of the ristocetin A asymmetric dimer. Chem. Commun. 1996, 1, 101–103. [Google Scholar] [CrossRef]
- Williams, D.H.; Bardsley, B. The Vancomycin Group of Antibiotics and the Fight against Resistant Bacteria. Angew. Chem. Int. Ed. 1999, 38, 1172–1193. [Google Scholar] [CrossRef]
- Mackay, J.P.; Gerhard, U.; Beauregard, D.A.; Maplestone, R.A.; Williams, D.H. Dissection of the contributions toward dimerization of glycopeptide antibiotics. J. Am. Chem. Soc. 1994, 116, 4573–4580. [Google Scholar] [CrossRef]
- Shiozawa, H.; Chia, B.C.S.; Davies, N.L.; Zerella, R.; Williams, D.H. Cooperative binding interactions of glycopeptide antibiotics. J. Am. Chem. Soc. 2002, 124, 3914–3919. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, H.; Zerella, R.; Bardsley, B.; Tuck, K.L.; Williams, D.H. Noncovalent bond lengths and their cooperative shortening: Dimers of vancomycin group antibiotics in crystals and in solution. Helv. Chim. Acta 2003, 86, 1359–1370. [Google Scholar] [CrossRef]
- Sundram, V.N.; Griffin, J.H.; Nicas, T.I. Novel Vancomycin Dimers with Activity against Vancomycin-Resistant Enterococci. J. Am. Chem. Soc. 1996, 118, 13107–13108. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Hughes, R.; Cho, S.Y.; Winssinger, N.; Labischinski, H.; Endemann, R. Synthesis and biological evaluation of vancomycin dimers with potent activity against vancomycin-resistant bacteria: Target-accelerated combinatorial synthesis. Chem. 2001, 7, 3824–3843. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Hughes, R.; Cho, S.Y.; Winssinger, N.; Smethhurst, C.; Labischinski, H.; Endemann, R. Target-Accelerated Combinatorial Synthesis and Discovery of Highly Potent Antibiotics Effective Against Vancomycin-Resistant Bacteria. Angew. Chem. Int. Ed. Engl. 2000, 39, 3823–3828. [Google Scholar] [CrossRef]
- Rao, J.; Whitesides, G.M. Tight Binding of a Dimeric Derivative of Vancomycin with Dimeric l-Lys-d-Ala-d-Ala. J. Am. Chem. Soc. 1997, 119, 10286–10290. [Google Scholar] [CrossRef]
- Staroske, T.; Williams, D.H. Synthesis of covalent head-to-tail dimers of vancomycin. Tetrahedron Lett. 1998, 39, 4917–4920. [Google Scholar] [CrossRef]
- Adamczyk, M.; Moore, J.A.; Rege, S.D.; Yu, Z. Investigations into self-association of vancomycin covalent dimers using surface plasmon resonance technology. Bioorg. Med. Chem. Lett. 1999, 9, 2437–2440. [Google Scholar] [CrossRef]
- Griffin, J.H.; Linsell, M.S.; Nodwell, M.B.; Chen, Q.; Pace, J.L.; Quast, K.L.; Krause, K.M.; Farrington, L.; Wu, T.X.; Higgins, D.L.; et al. Multivalent drug design. Synthesis and in vitro analysis of an array of vancomycin dimers. J. Am. Chem. Soc. 2003, 125, 6517–6531. [Google Scholar] [CrossRef]
- Lu, J.; Yoshida, O.; Hayashi, S.; Arimoto, H. Synthesis of rigidly-linked vancomycin dimers and their in vivo efficacy against resistant bacteria. Chem. Commun. 2007, 2007, 251–253. [Google Scholar] [CrossRef] [PubMed]
- Yarlagadda, V.; Sarkar, P.; Manjunath, G.B.; Haldar, J. Lipophilic vancomycin aglycon dimer with high activity against vancomycin-resistant bacteria. Bioorg. Med. Chem. Lett. 2015, 25, 5477–5480. [Google Scholar] [CrossRef]
- Silverman, S.M.; Moses, J.E.; Sharpless, K.B. Reengineering Antibiotics to Combat Bacterial Resistance: Click Chemistry [1,2,3]-Triazole Vancomycin Dimers with Potent Activity against MRSA and VRE. Chem. Eur. J. 2017, 23, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.-W.; Xu, L.; Fu, W.; Lin, H.; Yu, J.-M.; Sun, X. Design, synthesis and biological activity of novel demethylvancomycin dimers against vancomycin-resistant enterococcus faecalis. Tetrahedron 2018, 74, 3527–3533. [Google Scholar] [CrossRef]
- Malabarba, A.; Strazzolini, P.; Depaoli, A.; Landi, M.; Berti, M.; Cavalleri, B. Teicoplanin, antibiotics from Actinoplanes teichomyceticus nov. sp. VI. Chemical degradation: Physico-chemical and biological properties of acid hydrolysis products. J. Antibiot. 1984, 37, 988–999. [Google Scholar] [CrossRef] [PubMed]
- Pintér, G.; Batta, G.; Kéki, S.; Mándi, A.; Komáromi, I.; Takács-Novák, K.; Sztaricskai, F.; Rőth, E.; Ostorházi, E.; Rozgonyi, F.; et al. Diazo transfer-click reaction route to new, lipophilic teicoplanin and ristocetin aglycon derivatives with high antibacterial and anti-influenza virus activity: An aggregation and receptor binding study. J. Med. Chem. 2009, 52, 6053–6061. [Google Scholar] [CrossRef]
- Szűcs, Z.; Bereczki, I.; Rőth, E.; Milánkovits, M.; Ostorházi, E.; Batta, G.; Nagy, L.; Dombrádi, Z.; Borbás, A.; Herczegh, P. N-terminal guanidine derivatives of teicoplanin antibiotics strongly active against glycopeptide resistant Enterococcus faecium. J. Antibiot. 2020, 73, 603–614. [Google Scholar] [CrossRef]
- Szűcs, Z.; Ostorházi, E.; Kicsák, M.; Nagy, L.; Borbás, A.; Herczegh, P. New semisynthetic teicoplanin derivatives have comparable in vitro activity to that of oritavancin against clinical isolates of VRE. J. Antibiot. 2019, 72, 524–534. [Google Scholar] [CrossRef]
- Munch, H.; Hansen, J.S.; Pittelkow, M.; Christensen, J.B.; Boas, U. A new efficient synthesis of isothiocyanates from amines using di-tert-butyl dicarbonate. Tetrahedron Lett. 2008, 49, 3117–3119. [Google Scholar] [CrossRef]
- Malabarba, A.; Trani, A.; Strazzolini, P.; Cietto, G.; Ferrari, P.; Tarzia, G.; Pallanza, R.; Berti, M. Synthesis and biological properties of N63-carboxamides of teicoplanin antibiotics. Structure-activity relationships. J. Med. Chem. 1989, 32, 2450–2460. [Google Scholar] [CrossRef] [PubMed]
- Heffern, M.C.; Yamamoto, N.; Holbrook, R.J.; Eckermann, A.L.; Meade, T.J. Cobalt derivatives as promising therapeutic agents. Curr. Opin. Chem. Biol. 2013, 17, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Matusziuk, L.M.; Holbrook, R.J.; Manus, L.M.; Heffern, M.C.; Ratner, M.A.; Meade, T.J. Rational design of [Co(acacen)L2]+inhibitors of protein function. Dalton Trans. 2013, 42, 4002–4012. [Google Scholar] [CrossRef] [PubMed]
- Costa, G.; Mestroni, G.; Tauzher, G.; Stefani, L. Organometallic derivatives of cobalt chelates of bis(acetylacetone) ethylendiamine. J. Organomet. Chem. 1966, 6, 181–187. [Google Scholar] [CrossRef]
- Huang, K.J.; Huang, Y.C.; Lin, Y.A. Synthesis of Histidine-Containing Oligopeptides via Histidine-Promoted Peptide Ligation. Chem. Asian J. 2018, 13, 400–403. [Google Scholar] [CrossRef]
- Szűcs, Z.; Bereczki, I.; Csávás, M.; Rőth, E.; Borbás, A.; Batta, G.; Ostorházi, E.; Szatmári, R.; Herczegh, P. Lipophilic teicoplanin pseudoaglycon derivatives are active against vancomycin- and teicoplanin-resistant enterococci. J. Antibiot. 2017, 70, 664–670. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial Strains. | Teico (1) | 2 | 3 | Co (acac) | 12 | 13 | 16 | 17 | 20 | 21 |
|---|---|---|---|---|---|---|---|---|---|---|
| MIC [g] (μg/mL) | ||||||||||
| Bacillus subtilis ATCC [a] 6633 | 0.5 | 2 | 2 | 256 | 64 | 4 | 4 | 8 | 2 | 4 |
| Staphylococcus aureus MSSA [b] ATCC 29213 | 0.5 | 4 | 2 | 256 | 64 | 8 | 16 | 64 | 4 | 4 |
| Staphylococcus aureus MRSA [c] ATCC 33591 | 0.5 | 2 | 1 | 256 | 256 | 4 | 8 | 16 | 4 | 4 |
| Staphylococcus epidermidis ATCC 35984 biofilm | 2 | 2 | 2 | 256 | 16 | 4 | 16 | 8 | 4 | 4 |
| Staphylococcus epidermidis mecA [d] | 16 | 4 | 1 | 256 | 64 | 4 | 64 | 16 | 2 | 2 |
| Enterococcus faecalis ATCC 29212 | 2 | 8 | 4 | 256 | 4 | 8 | 16 | 16 | 4 | 8 |
| Enterococcus faecalis 15376 VanA [e] | 256 | 4 | 256 | 256 | 16 | 4 | 8 | 8 | 4 | 8 |
| Enterococcus faecalis ATCC 51299 VanB [f] | 4 | 8 | 2 | 256 | 32 | 8 | 32 | 16 | 4 | 4 |
 | |||||
| 13C Shift (ppm) | 1H Shift (ppm) | Annotation | 13C Shift (ppm) | 1H Shift (ppm) | Annotation |
| 96.3 | 5.26 | M1 | 75.1 | 5.33 | z6 |
| 98.3 | 4.35 | G1 | 60.6 | 4.14 | x6 |
| 104.4 | 5.08 | 4f | 43.5 | 3.58 | La |
| 107.7 | 5.62 | 4b | 56.0 | 3.35 | G2 |
| 100.7 | 6.71 | 7d | 60.4 | 3.50 | M6 |
| 104.4 | 6.34 | 3f | 60.8 | 3.68 | G6 |
| 105.7 | 6.50 | 3d | 60.8 | 3.47 | G6 |
| 107.6 | 6.49 | 7f | 53.2 | 2.59 | Lf |
| 110.7 | 6.34 | 3b | 54.2 | 2.43 | D1 |
| 115.7 | 6.72 | 5e | 69.4 | 3.49 | Lb-d |
| 116.2 | 6.68 | 1b | 68.8 | 3.43 | Le |
| 117.7 | 6.94 | 1e | 69.6 | 3.26 | M3 |
| 123.1 | 7.26 | 6e | 70.4 | 3.23 | M2 |
| 123.2 | 7.05 | 1f | 70.3 | 3.08 | G4 |
| 125.6 | 6.73 | 5f | 72.7 | 3.44 | G3 |
| 124.5 | 7.26 | 2e | 73.4 | 3.49 | M5 |
| 128.0 | 7.25 | 6f | 76.6 | 3.05 | G5 |
| 129.8 | 7.60 | 2f | 65.7 | 3.48 | M4 |
| 128.4 | 7.81 | 6b | 36.3 | 3.29 | z2/z’2 |
| 130.7 | 7.22 | 2b | 36.4 | 2.81 | z2/z’2 |
| 135.1 | 7.02 | 5b | 22.9 | 1.88 | G-Me |
| 54.4 | 5.65 | x4 | 21.8 | 1.24 | D9 |
| 60.0 | 6.02 | x1 | 26.5 | 1.34 | D2 |
| 57.8 | 5.25 | x3 | 26.4 | 1.21 | D3 |
| 54.1 | 4.85 | x2 | 28.6 | 1.22 | D4–7 |
| 53.3 | 4.40 | x5 | 31.0 | 1.23 | D8 |
| 56.6 | 4.47 | x7 | 13.6 | 0.85 | D10 |
 | |||||
| 13C Shift (ppm) | 1H Shift (ppm) | Annotation | 13C Shift (ppm) | 1H Shift (ppm) | Annotation |
| 60.1 | 6.01 | x1 | 23.0 | 1.91 | G-Me |
| 54.1 | 4.86 | x2 | 26.2 | 1.64 | P2 |
| 57.9 | 5.27 | x3 | 49.8 | 2.46 | P3 |
| 54.3 | 5.69 | x4 | 37.2 | 3.19 | P1 |
| 53.4 | 4.40 | x5 | 54.4 | 2.43 | D1 |
| 56.7 | 4.47 | x7 | 26.5 | 1.34 | D2 |
| 60.8 | 4.16 | x6 | 26.4 | 1.22 | D3 |
| 74.9 | 5.42 | z6 | 28.7 | 1.25 | D4–7 |
| 61.2 | 3.73 | G6 | 31.0 | 1.23 | D8 |
| 61.2 | 3.42 | G6 | 21.7 | 1.26 | D9 |
| 96.4 | 5.25 | M1 | 45.9 | 2.51 | E1/E1′ |
| 98.3 | 4.34 | G1 | 135.2 | 7.12 | 5b |
| 104.3 | 5.11 | 4f | 125.6 | 6.74 | 5f |
| 107.4 | 5.62 | 4b | 100.6 | 6.72 | 7d |
| 55.9 | 3.29 | G2 | 107.6 | 6.41 | 7f |
| 76.5 | 3.05 | G5 | 110.4 | 6.34 | 3b |
| 60.5 | 3.52 | M6 | 104.1 | 6.33 | 3f |
| 70.4 | 3.19 | M2 | 105.5 | 6.50 | 3d |
| 70.6 | 2.99 | G4 | 116.0 | 6.73 | 5e |
| 69.7 | 3.22 | M3 | 123.3 | 7.02 | 1f |
| 72.5 | 3.49 | G3 | 116.3 | 6.67 | 1b |
| 73.5 | 3.48 | M5 | 118.0 | 6.92 | 1e |
| 65.8 | 3.50 | M4 | 123.0 | 7.29 | 6e |
| 69.3 | 3.51 | Lb-d | 128.5 | 7.82 | 6b |
| 68.9 | 3.43 | Le | 129.8 | 7.61 | 2f |
| 43.5 | 3.59 | La | 130.8 | 7.23 | 2b |
| 53.2 | 2.59 | Lf | 124.5 | 7.28 | 2e |
| 11.1 | 0.98 | E2/E2′ | 127.8 | 7.26 | 6f |
| 13.8 | 0.85 | D10 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bereczki, I.; Szűcs, Z.; Batta, G.; Nagy, T.M.; Ostorházi, E.; Kövér, K.E.; Borbás, A.; Herczegh, P. The First Dimeric Derivatives of the Glycopeptide Antibiotic Teicoplanin. Pharmaceuticals 2022, 15, 77. https://doi.org/10.3390/ph15010077
Bereczki I, Szűcs Z, Batta G, Nagy TM, Ostorházi E, Kövér KE, Borbás A, Herczegh P. The First Dimeric Derivatives of the Glycopeptide Antibiotic Teicoplanin. Pharmaceuticals. 2022; 15(1):77. https://doi.org/10.3390/ph15010077
Chicago/Turabian StyleBereczki, Ilona, Zsolt Szűcs, Gyula Batta, Tamás Milán Nagy, Eszter Ostorházi, Katalin E. Kövér, Anikó Borbás, and Pál Herczegh. 2022. "The First Dimeric Derivatives of the Glycopeptide Antibiotic Teicoplanin" Pharmaceuticals 15, no. 1: 77. https://doi.org/10.3390/ph15010077
APA StyleBereczki, I., Szűcs, Z., Batta, G., Nagy, T. M., Ostorházi, E., Kövér, K. E., Borbás, A., & Herczegh, P. (2022). The First Dimeric Derivatives of the Glycopeptide Antibiotic Teicoplanin. Pharmaceuticals, 15(1), 77. https://doi.org/10.3390/ph15010077