
Discovery of Cofactor Competitive Inhibitors against the Human Methyltransferase Fibrillarin

 , ,
, ,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
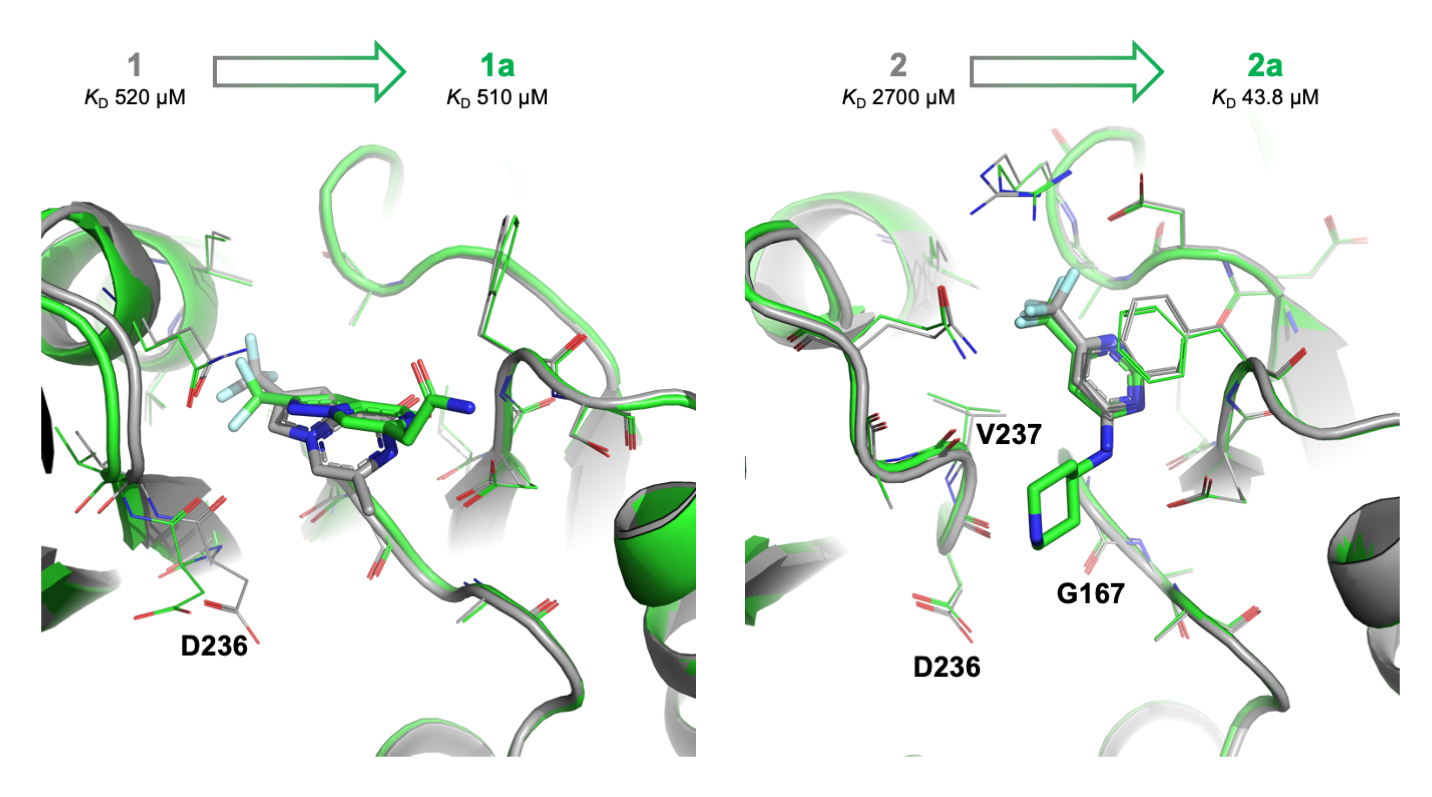
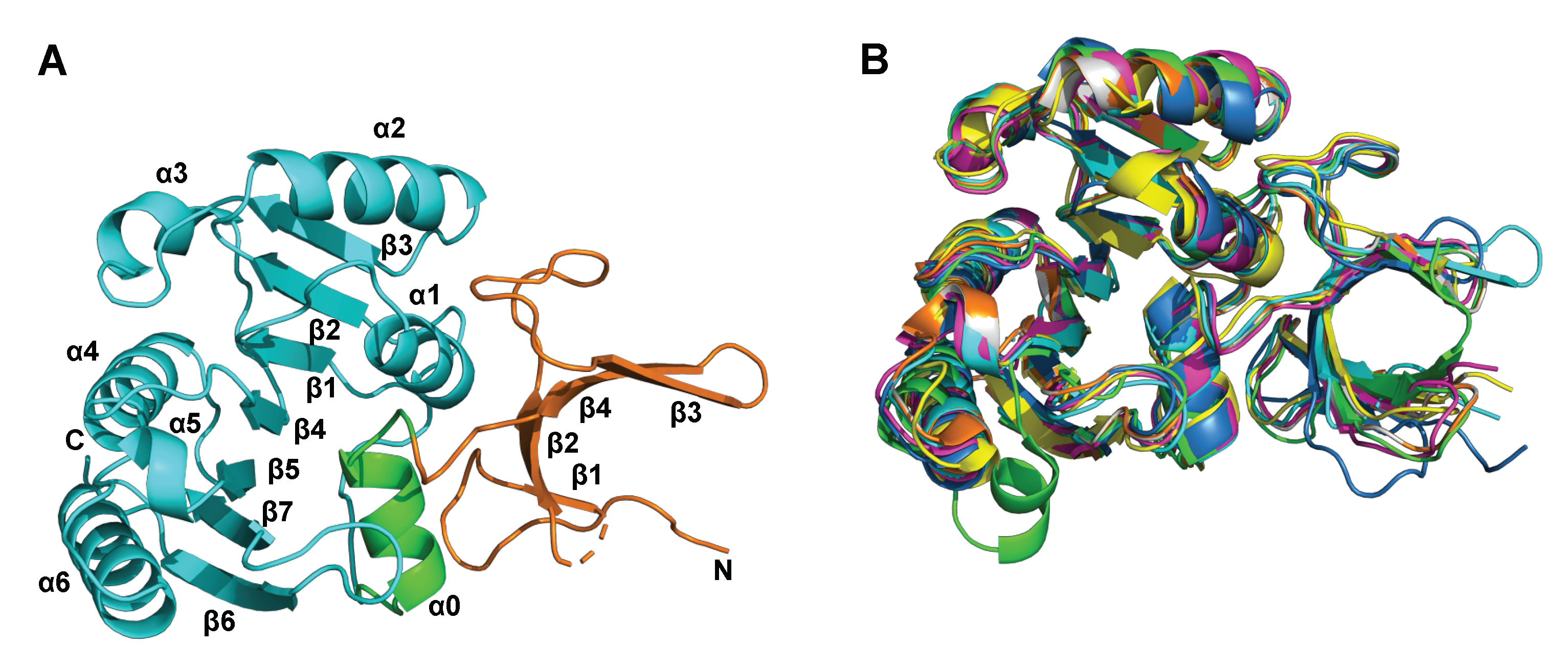
2.1. Apo and SAM-Bound hFBL Structures
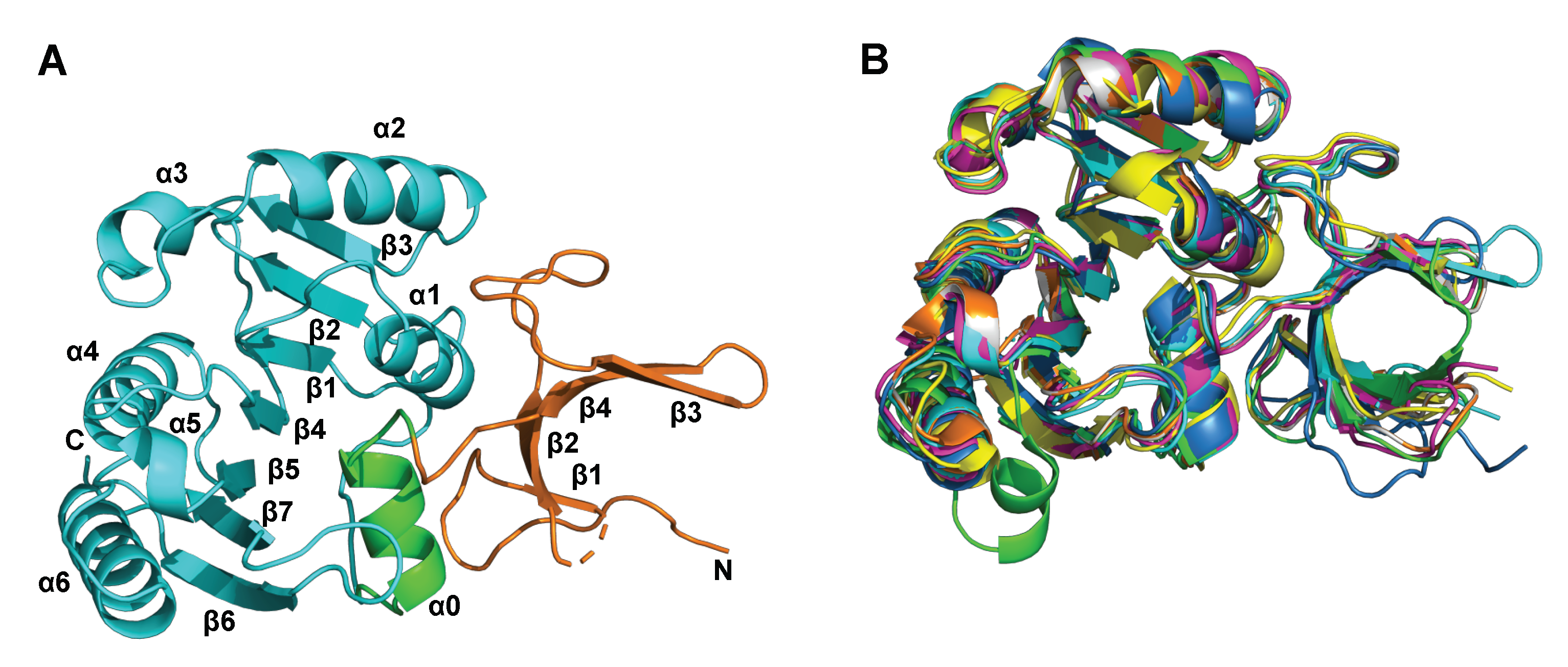
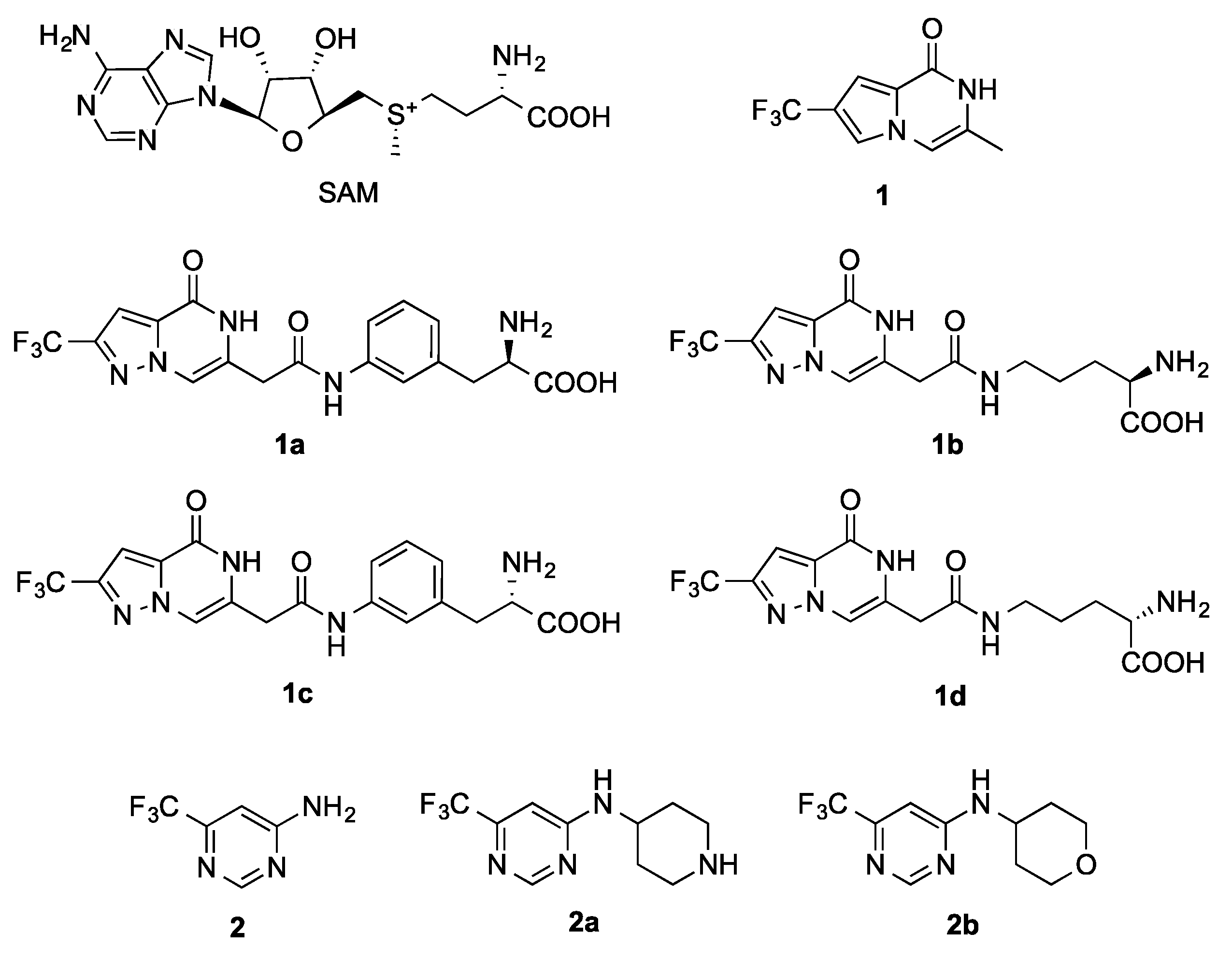
2.2. Characterisation of hFBL–SAM Interactions
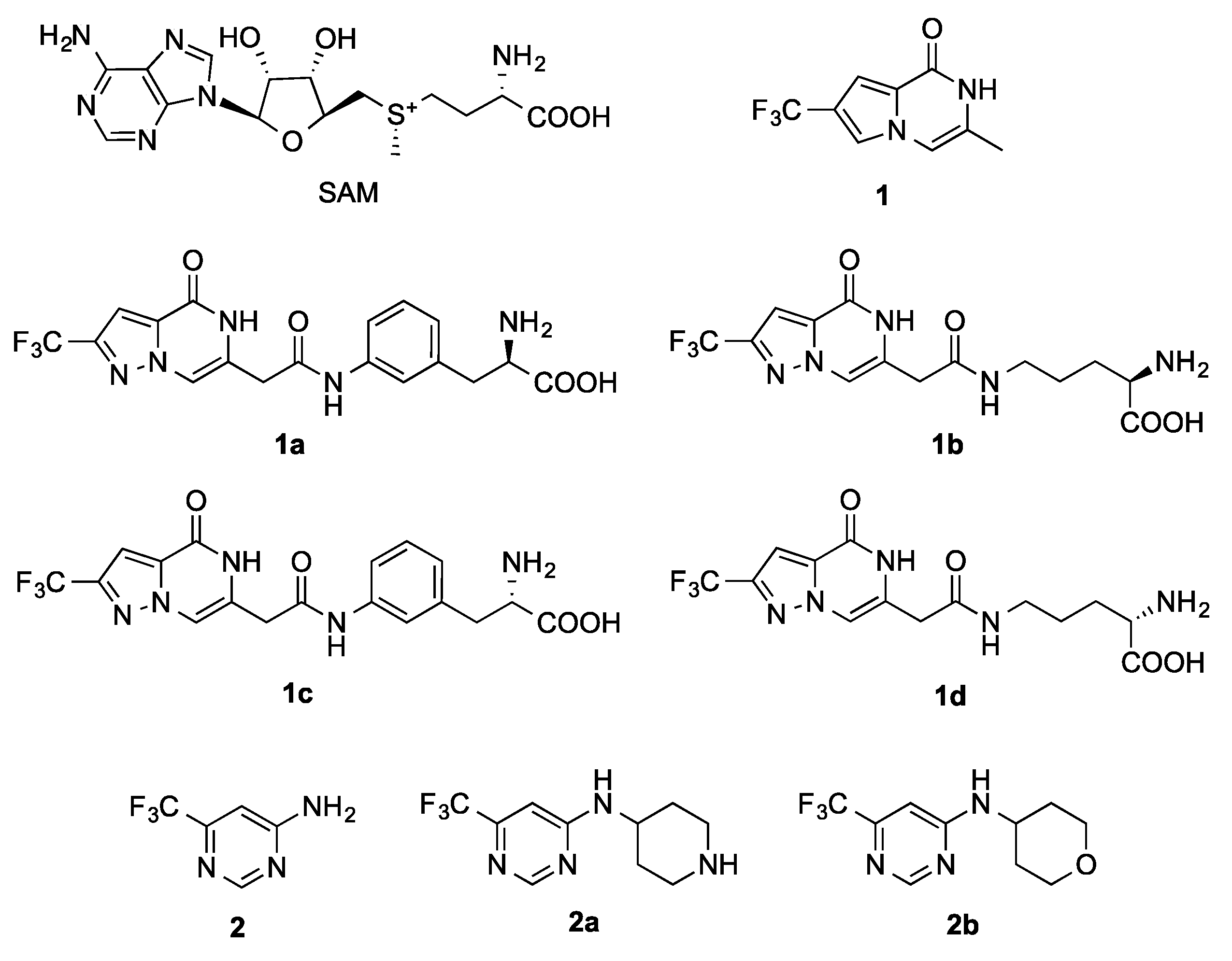
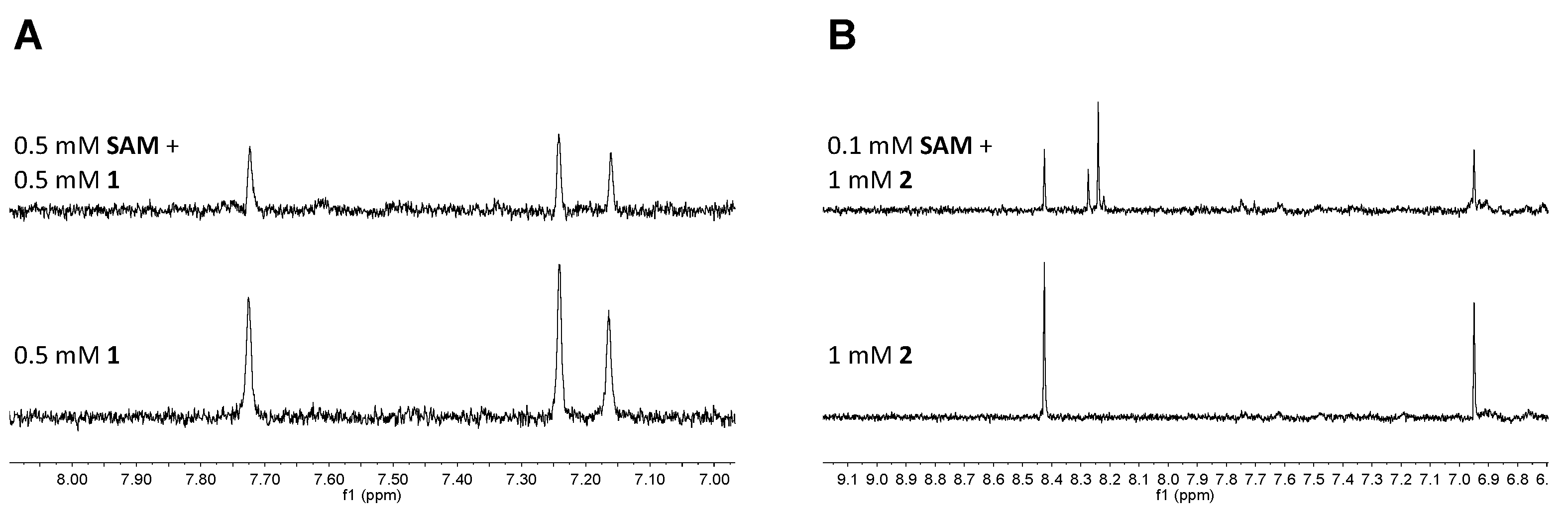
2.3. Fragment Screening
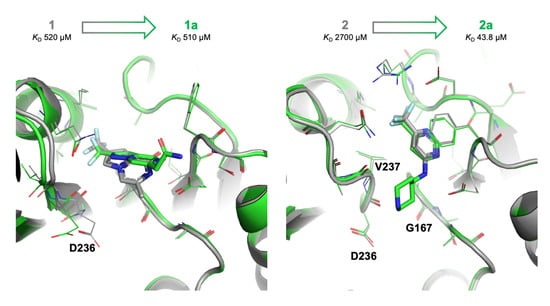
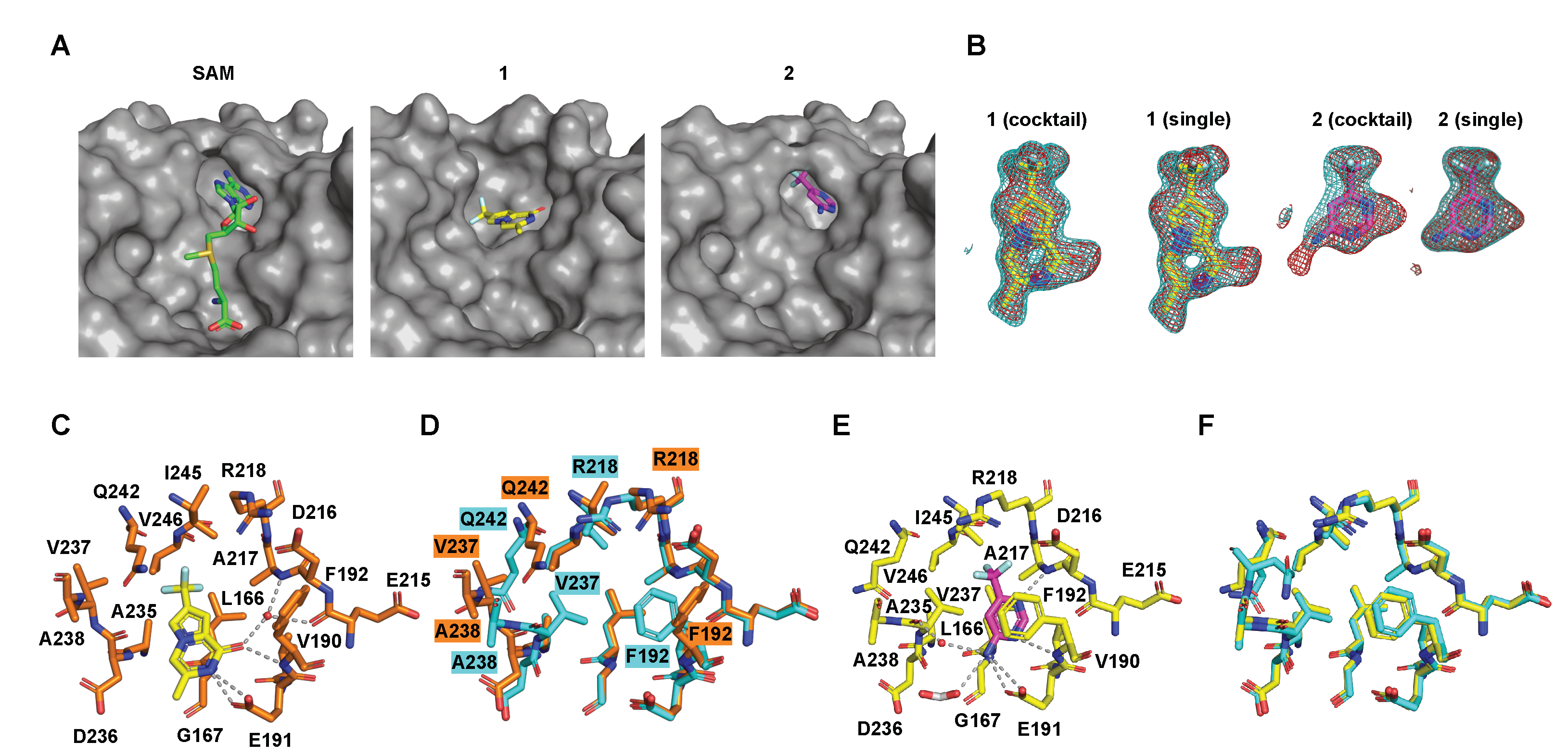
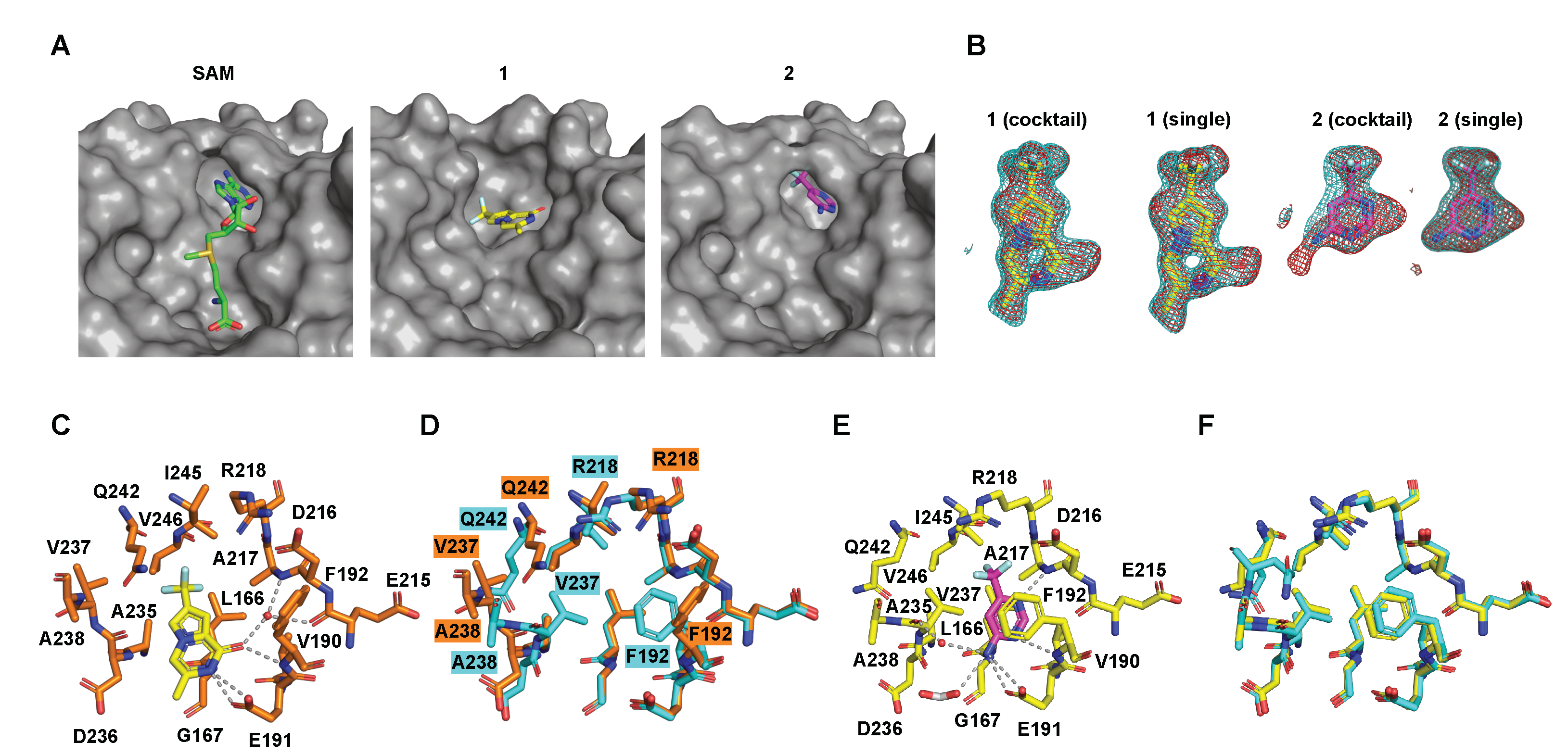
2.4. Details of the Binding Modes of Compounds 1 and 2
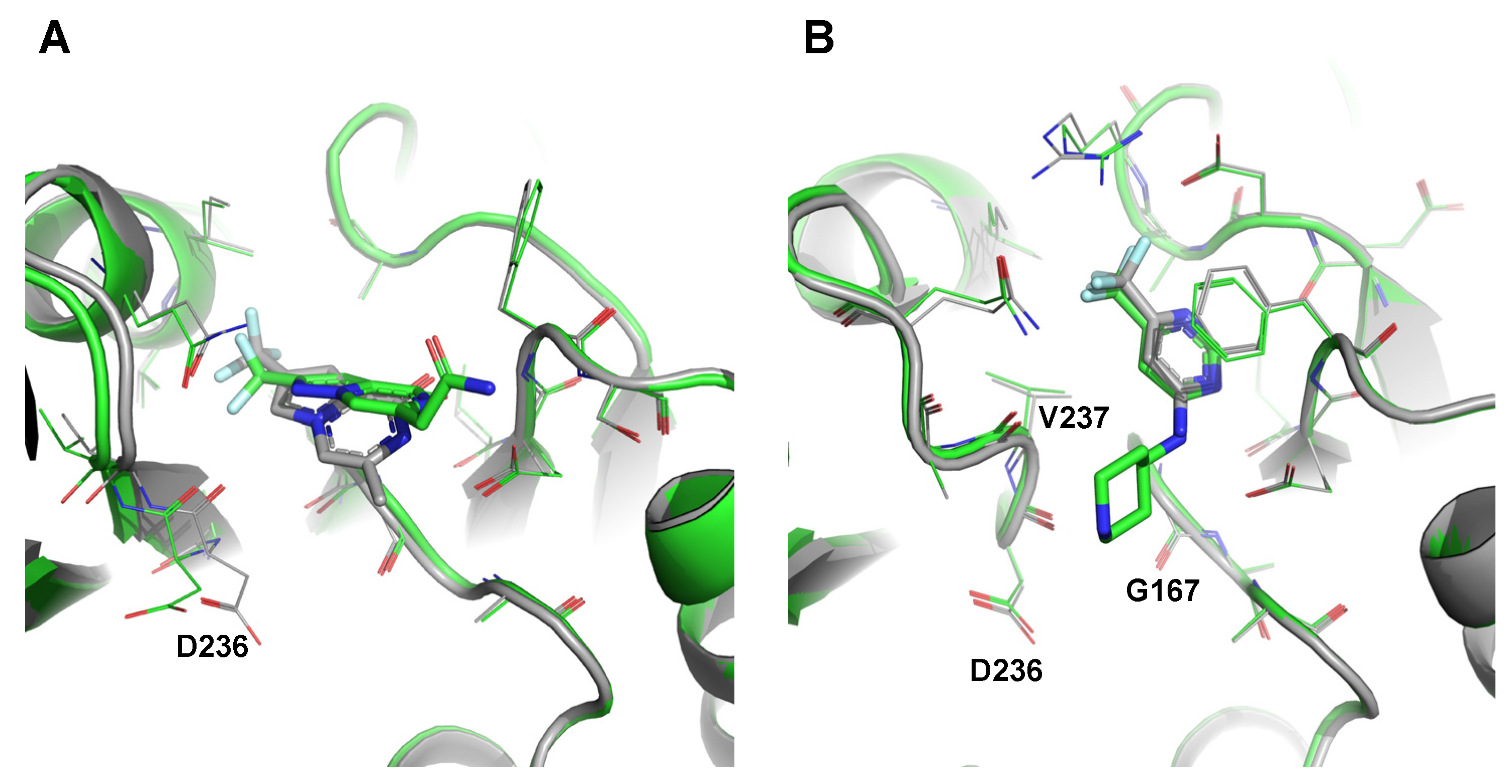
2.5. Preliminary Elaboration of Fragment Hits
3. Discussion
4. Materials and Methods
4.1. Protein Production and Purification
4.2. Fragment Library
4.3. Crystallisation
4.4. Crystallographic Fragment Screening
4.5. Preparation of hFBL-Compound Crystals
4.6. Data Collection and Processing, Structure Determination, and Refinement
4.7. Structural Analyses
4.8. Molecular Dynamics Simulations
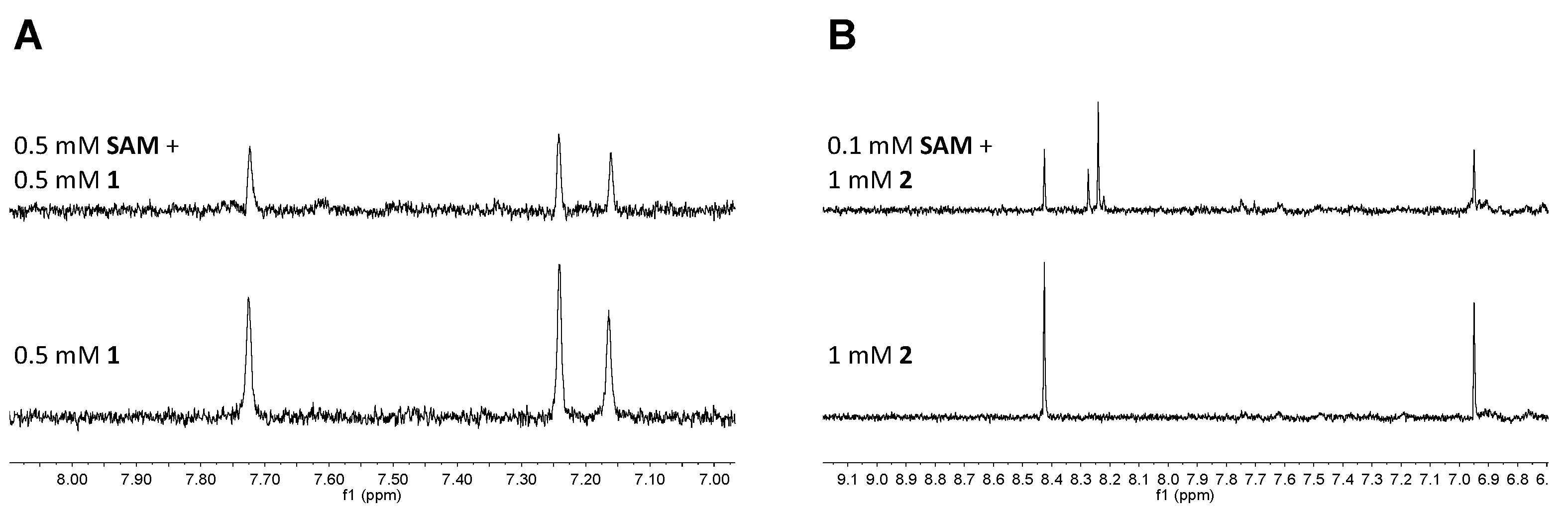
4.9. NMR Fragment Screening
4.10. Molecular Docking
4.11. STD NMR
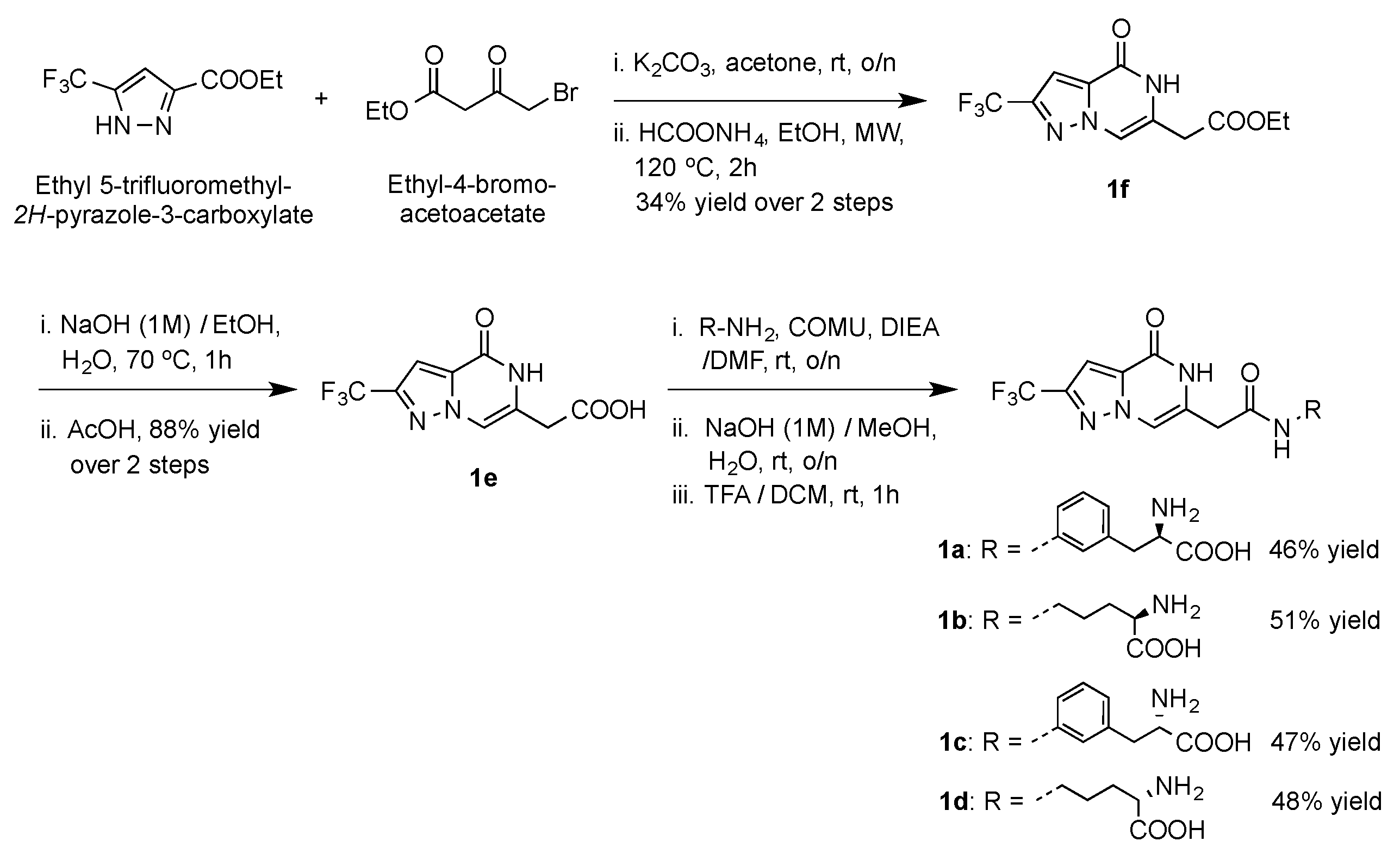
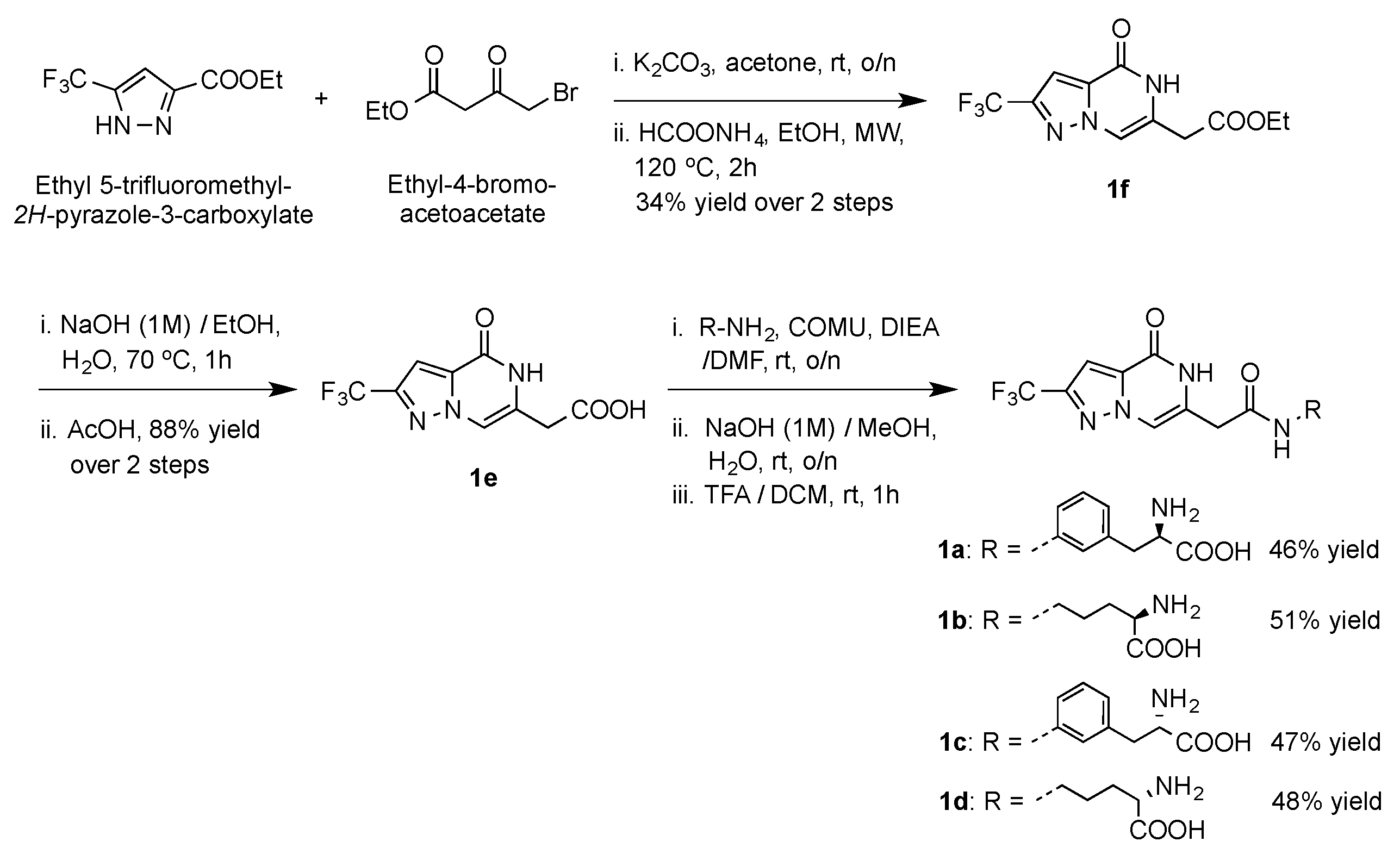
4.12. Chemical Synthesis
- Ethyl 2-(4-oxo-2-(trifluoromethyl)-4,5-dihydropyrazolo[1,5-a]pyrazin-6-yl)acetate (3), Ethyl 4-bromo-acetoacetate (335 μL, 2.4 mmol) was added to a solution of ethyl 5-trifluoromethyl-2H-pyrazole-3-carboxylate (500 mg, 2.4 mmol) in acetone (10 mL), followed by finely powdered dry K2CO3 (400 mg, 12.9 mmol). The reaction mixture was stirred at room temperature (rt) overnight (o/n) and was then concentrated under reduced pressure to remove the reaction solvent. The crude product was diluted with dichloromethane (DCM) (100 mL) and washed with water (50 mL). The organic layer was separated, washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was dissolved in absolute EtOH (5 mL) followed by the addition of ammonium formate (2.27 g, 36 mmol). The mixture was heated under microwave irradiation at 393 K for 2 h. After cooling to rt, the solvent was removed, the residue was diluted with H2O (20 mL), and was extracted with EtOAc (50 mL × 2). The combined organic extract was washed with brine, dried over Na2SO4, and concentrated under reduced pressure. Purification of the residue by silica gel chromatography using hexane:EtOAc (3:2) yielded pure 3 (235 mg, 34% yield) as a white powder. 1H NMR (400 MHz, DMSO-d6): δ 1.21 (t, J = 7.0 Hz, 3H), 3.64 (s, 2H), 4.13 (q, J = 7.1 Hz, 2H), 7.50 (s, 1H), 7.79 (s, 1H), 11.70 (s, 1H); 13C NMR (101 MHz, DMSO-d6): δ 14.52, 35.23, 61.33, 103.65 (d, J = 2.3 Hz), 109.77, 121.50 (q, J = 268.7 Hz), 126.45, 134.10, 141.71 (q, J = 38.0 Hz), 155.34, 169.28; LRMS [C11H10F3N3O3] (m/z): (+ve ion mode) 311.9 [M+Na]+.
- 2-(4-Oxo-2-(trifluoromethyl)-4,5-dihydropyrazolo[1,5-a]pyrazin-6-yl)acetic acid (4), Compound 3 (200 mg, 0.69 mmol) was suspended in EtOH/H2O (1:1, 10 mL) and NaOH (1M) was added to the suspension (under stirring) in a dropwise manner until pH = 14. The reaction mixture was heated at 243 K °C for 1 h, then allowed to cool down to rt. The reaction mixture was then acidified to pH = 4 using AcOH, concentrated under reduced pressure, and the residue was purified by silica gel chromatography using EtOAc:MeOH:H2O (9:1:0.5) to yield the pure acid 4 (160 mg, 88% yield) as a pale yellow powder. 1H NMR (400 MHz, DMSO-d6): δ 3.54 (s, 2H), 7.49 (s, 1H), 7.78 (s, 1H), 11.68 (s, 1H); 13C NMR (101 MHz, DMSO-d6): δ 35.45, 103.55 (d, J = 2.6 Hz), 109.60, 121.52 (q, J = 268.7 Hz), 127.08, 134.04, 141.62 (q, J = 38.1 Hz), 155.35, 170.80; LRMS [C9H6F3N3O3] (m/z): (+ve ion mode) 283.1 [M+Na]+.
- General method for the synthesis of1a–d, To a solution of the carboxylic acid 4 (50 mg, 0.19 mmol) in anhydrous DMF (2 mL) under argon the amine component (0.38 mmol) and COMU (160 mg, 0.38 mmol) were added followed by N,N-Diisopropylethylamine (DIEA) (132 μL, 0.76 mmol). The reaction mixture was stirred at rt o/n and was then concentrated under reduced pressure. The residue was triturated with H2O (10 mL) and extracted with EtOAc (50 mL). The organic layer was washed with brine, dried over Na2SO4, and concentrated under reduced pressure to yield the crude product. Purification by silica gel chromatography using the proper solvent system yielded the pure amide. The protected amide was suspended in a MeOH:H2O mixture (1:1, 2 mL), treated with NaOH (1M) to pH = 14, stirred at rt o/n, then the reaction was acidified with Amberlite@IR-120 (H+) resin (to pH = 5), filtered, and washed with MeOH (10 mL) and H2O (10 mL). The combined filtrate and washings were then concentrated under vacuum and the residue was dissolved in DCM (2 mL) and treated with TFA (300 μL, 3.8 mmol, 20 eq). After stirring for 1 h at rt, the solvent and excess TFA were removed by concentration and reduced pressure, and the residue was purified by silica gel chromatography using the suitable solvent system to yield the pure, deprotected amide 1a–d.
- (R)-2-Amino-3-(3-(2-(4-oxo-2-(trifluoromethyl)-4,5-dihydropyrazolo[1,5-a]pyrazin-6-yl)acetamido)phenyl)propanoic acid IE1775-42 (1a), The amide was formed by coupling with (R)-methyl 3-(3-aminophenyl)-2-((tert-butoxycarbonyl)amino)propanoate and the protected product was purified by silica gel chromatography using Hexane:EtOAc:MeOH (4:2:0.5). The final deprotected amide (1a) was obtained in 46% yield (over three steps) after purification by silica gel chromatography using EtOAc:MeOH:H2O (9:2:1) as a solvent. 1H NMR (400 MHz, D2O): δ 2.81 (dd, J = 13.5, 7.5 Hz, 1H), 2.99 (dd, J = 13.5, 5.5 Hz, 1H), 3.49 (dd, J = 7.5, 5.5 Hz, 1H), 3.66 (d, J = 7.0 Hz, 1H), 7.10 (d, J = 6.3 Hz, 2H), 7.27 (s, 1H), 7.32–7.41 (m, 2H), 7.78 (s, 1H); 13C NMR (101 MHz, D2O): δ 23.24, 40.64, 57.34, 98.63 (d, J = 2.8 Hz), 109.98, 120.47, 121.21 (d, J = 268.3 Hz), 123.01, 126.62, 129.15, 132.44, 136.65, 136.76, 139.33, 140.87 (q, J = 38.0 Hz), 163.73, 171.83, 182.23; LRMS [C18H16F3N5O4] (m/z): (-ve ion mode) 421.9 [M-H]−.
- (R)-2-Amino-5-(2-(4-oxo-2-(trifluoromethyl)-4,5-dihydropyrazolo[1,5-a]pyrazin-6-yl)acetamido)pentanoic acid IE1775-41 (1b), The amide was formed by coupling with (R)-methyl 5-amino-2-((tert-butoxycarbonyl)amino)pentanoate and the protected product was purified by silica gel chromatography using Hexane:EtOAc:MeOH (4:2:0.5). The final deprotected amide (1b) was obtained in 51% yield (over three steps) after purification by silica gel chromatography using EtOAc:MeOH:H2O (7:2:1) as a solvent. 1H NMR (400 MHz, D2O): δ 1.49–1.68 (m, 4H), 3.24 (ddd, J = 9.4, 7.9, 4.5 Hz, 3H), 3.49 (s, 2H), 7.10 (s, 1H), 7.72 (s, 1H); 13C NMR (101 MHz, DMSO-d6): δ 24.76, 31.75, 39.44, 41.09, 55.59, 98.73, 109.84, 121.19 (d, J = 268.2 Hz), 132.41, 136.58, 140.91 (q, J = 38.4 Hz), 163.54, 172.87, 182.73; LRMS [C14H16F3N5O4] (m/z): (+ve ion mode) 398.1 [M+Na]+.
- (S)-2-Amino-3-(3-(2-(4-oxo-2-(trifluoromethyl)-4,5-dihydropyrazolo[1,5-a]pyrazin-6-yl)acetamido)phenyl)propanoic acid IE1775-50 (1c), The amide was formed by coupling with (S)-methyl 3-(3-aminophenyl)-2-((tert-butoxycarbonyl)amino)-propanoate and the protected product was purified by silica gel chromatography using Hexane:EtOAc:MeOH (4:2:0.5). The final deprotected amide (1c) was obtained in 47% yield (over three steps) after purification by silica gel chromatography using EtOAc:MeOH:H2O (9:2:1) as a solvent. 1H NMR (400 MHz, D2O): δ 1.92 (s, 1H), 2.87 (dd, J = 13.8, 7.7 Hz, 1H), 3.05 (dd, J = 13.8, 5.4 Hz, 1H), 3.61 (dd, J = 7.7, 5.4 Hz, 1H), 7.10 (m, 1H), 7.15 (s, 1H), 7.28 (s, 1H), 7.32–7.40 (m, 2H), 7.74 (s, 1H); 13C NMR (101 MHz, D2O): δ 23.24, 39.53, 56.97, 99.68, 110.18, 121.44 (q, J = 269.7 Hz), 122.81, 125.05, 126.55, 129.29, 132.56, 134.16, 136.73, 138.51, 141.22 (q, J = 38.4 Hz), 162.27, 171.16, 180.08; LRMS [C18H16F3N5NaO4] (m/z): (+ve ion mode) 468.2 [M+Na]+.
- (S)-2-Amino-5-(2-(4-oxo-2-(trifluoromethyl)-4,5-dihydropyrazolo[1,5-a]pyrazin-6-yl)acetamido)pentanoic acid IE1775-48 (1d), The amide was formed by coupling with (S)-methyl 5-amino-2-((tert-butoxycarbonyl)amino)pentanoate and the protected product was purified by silica gel chromatography using Hexane:EtOAc:MeOH (4:2:0.5). The final deprotected amide (1d) was obtained in 48% yield (over three steps) after purification by silica gel chromatography using EtOAc:MeOH:H2O (7:2:1) as a solvent. 1H NMR (400 MHz, DMSO-d6): δ 1.51 (q, J = 7.0 Hz, 2H), 1.65 (d, J = 7.8 Hz, 2H), 3.06 (s, 3H), 3.20 (s, 4H), 6.94 (s, 1H), 7.53 (s, 1H), 8.37 (s, 1H); 13C NMR (101 MHz, DMSO-d6): δ 25.60, 29.44, 30.79, 31.75, 55.06, 98.89, 106.80, 122.31 (d, J = 268.3 Hz), 133.50, 136.66, 139.46 (d, J = 37.1 Hz), 161.82, 169.45, 177.64; LRMS [C14H16F3N5O4] (m/z): (-ve ion mode) 395.9 [M-H]−.
4.13. Isothermal Titration Calorimetry
4.14. Surface Plasmon Resonance
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rodriguez-Corona, U.; Sobol, M.; Rodriguez-Zapata, L.C.; Hozak, P.; Castano, E. Fibrillarin from Archaea to Human. Biol. Cell. 2015, 107, 159–174. [Google Scholar] [CrossRef]
- Ochs, R.L.; Lischwe, M.A.; Spohn, W.H.; Busch, H. Fibrillarin: A New Protein of the Nucleolus Identified by Autoimmune Sera. Biol. Cell. 1985, 54, 123–133. [Google Scholar] [CrossRef]
- Sobol, M.; Yildirim, S.; Philimonenko, V.V.; Marasek, P.; Castano, E.; Hozak, P. UBF Complexes with Phosphatidylinositol 4,5-Bisphosphate in Nucleolar Organizer Regions Regardless of Ongoing RNA Polymerase I Activity. Nucleus 2013, 4, 478–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feric, M.; Vaidya, N.; Harmon, T.S.; Mitrea, D.M.; Zhu, L.; Richardson, T.M.; Kriwacki, R.W.; Pappu, R.V.; Brangwynne, C.P. Coexisting Liquid Phases Underlie Nucleolar Subcompartments. Cell 2016, 165, 1686–1697. [Google Scholar] [CrossRef] [Green Version]
- Kozbial, P.Z.; Mushegian, A.R. Natural History of S-Adenosylmethionine-Binding Proteins. BMC Struct. Biol. 2005, 5, 19. [Google Scholar] [CrossRef] [Green Version]
- Omer, A.D.; Ziesche, S.; Ebhardt, H.; Dennis, P.P. In Vitro Reconstitution and Activity of a C/D Box Methylation Guide Ribonucleoprotein Complex. Proc. Natl. Acad. Sci. USA 2002, 99, 5289–5294. [Google Scholar] [CrossRef] [Green Version]
- Erales, J.; Marchand, V.; Panthu, B.; Gillot, S.; Belin, S.; Ghayad, S.E.; Garcia, M.; Laforets, F.; Marcel, V.; Baudin-Baillieu, A.; et al. Evidence for RRNA 2′-O-Methylation Plasticity: Control of Intrinsic Translational Capabilities of Human Ribosomes. Proc. Natl. Acad. Sci. USA 2017, 114, 12934–12939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tollervey, D.; Lehtonen, H.; Jansen, R.; Kern, H.; Hurt, E.C. Temperature-Sensitive Mutations Demonstrate Roles for Yeast Fibrillarin in Pre-RRNA Processing, Pre-RRNA Methylation, and Ribosome Assembly. Cell 1993, 72, 443–457. [Google Scholar] [CrossRef]
- Watkins, N.J.; Bohnsack, M.T. The Box C/D and H/ACA SnoRNPs: Key Players in the Modification, Processing and the Dynamic Folding of Ribosomal RNA. Wiley Interdiscip. Rev. RNA 2012, 3, 397–414. [Google Scholar] [CrossRef] [PubMed]
- Tessarz, P.; Santos-Rosa, H.; Robson, S.C.; Sylvestersen, K.B.; Nelson, C.J.; Nielsen, M.L.; Kouzarides, T. Glutamine Methylation in Histone H2A Is an RNA-Polymerase-I-Dedicated Modification. Nature 2014, 505, 564–568. [Google Scholar] [CrossRef] [Green Version]
- Koh, C.M.; Gurel, B.; Sutcliffe, S.; Aryee, M.J.; Schultz, D.; Iwata, T.; Uemura, M.; Zeller, K.I.; Anele, U.; Zheng, Q.; et al. Alterations in Nucleolar Structure and Gene Expression Programs in Prostatic Neoplasia Are Driven by the MYC Oncogene. Am. J. Pathol. 2011, 178, 1824–1834. [Google Scholar] [CrossRef]
- Marcel, V.; Ghayad, S.E.; Belin, S.; Therizols, G.; Morel, A.P.; Solano-Gonzalez, E.; Vendrell, J.A.; Hacot, S.; Mertani, H.C.; Albaret, M.A.; et al. P53 Acts as a Safeguard of Translational Control by Regulating Fibrillarin and RRNA Methylation in Cancer. Cancer Cell 2013, 24, 318–330. [Google Scholar] [CrossRef] [Green Version]
- Su, H.; Xu, T.; Ganapathy, S.; Shadfan, M.; Long, M.; Huang, T.H.; Thompson, I.; Yuan, Z.M. Elevated SnoRNA Biogenesis Is Essential in Breast Cancer. Oncogene 2014, 33, 1348–1358. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, G.; Li, Q.; Xie, F. Increased Fibrillarin Expression Is Associated with Tumor Progression and an Unfavorable Prognosis in Hepatocellular Carcinoma. Oncol. Lett. 2021, 21, 92. [Google Scholar] [CrossRef]
- El Hassouni, B.; Sarkisjan, D.; Vos, J.C.; Giovannetti, E.; Peters, G.J. Targeting the Ribosome Biogenesis Key Molecule Fibrillarin to Avoid Chemoresistance. Curr. Med. Chem. 2019, 26, 6020–6032. [Google Scholar] [CrossRef] [PubMed]
- Deffrasnes, C.; Marsh, G.A.; Foo, C.H.; Rootes, C.L.; Gould, C.M.; Grusovin, J.; Monaghan, P.; Lo, M.K.; Tompkins, S.M.; Adams, T.E.; et al. Genome-Wide SiRNA Screening at Biosafety Level 4 Reveals a Crucial Role for Fibrillarin in Henipavirus Infection. PLoS Pathog. 2016, 12, e1005478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luby, S.P.; Hossain, M.J.; Gurley, E.S.; Ahmed, B.N.; Banu, S.; Khan, S.U.; Homaira, N.; Rota, P.A.; Rollin, P.E.; Comer, J.A.; et al. Recurrent Zoonotic Transmission of Nipah Virus into Humans, Bangladesh, 2001–2007. Emerg. Infect. Dis. 2009, 15, 1229–1235. [Google Scholar] [CrossRef]
- Ferreira de Freitas, R.; Ivanochko, D.; Schapira, M. Methyltransferase Inhibitors: Competing with, or Exploiting the Bound Cofactor. Molecules 2019, 24, 4492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erlanson, D.A.; McDowell, R.S.; O’Brien, T. Fragment-Based Drug Discovery. J. Med. Chem. 2004, 47, 3463–3482. [Google Scholar] [CrossRef]
- Romasanta, A.K.S.; van der Sijde, P.; Hellsten, I.; Hubbard, R.E.; Keseru, G.M.; van Muijlwijk-Koezen, J.; de Esch, I.J.P. When Fragments Link: A Bibliometric Perspective on the Development of Fragment-Based Drug Discovery. Drug Discov. Today 2018, 23, 1596–1609. [Google Scholar] [CrossRef]
- Erlanson, D.A.; Fesik, S.W.; Hubbard, R.E.; Jahnke, W.; Jhoti, H. Twenty Years on: The Impact of Fragments on Drug Discovery. Nat. Rev. Drug Discov. 2016, 15, 605–619. [Google Scholar] [CrossRef]
- Congreve, M.; Carr, R.; Murray, C.; Jhoti, H. A ‘Rule of Three’ for Fragment-Based Lead Discovery? Drug Discov. Today 2003, 8, 876–877. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Hann, M.M.; Leach, A.R.; Harper, G. Molecular Complexity and Its Impact on the Probability of Finding Leads for Drug Discovery. J. Chem. Inf. Comput. Sci. 2001, 41, 856–864. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Groom, C.R.; Alex, A. Ligand Efficiency: A Useful Metric for Lead Selection. Drug Discov. Today 2004, 9, 430–431. [Google Scholar] [CrossRef]
- Murray, C.W.; Rees, D.C. The Rise of Fragment-Based Drug Discovery. Nat. Chem. 2009, 1, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Ferreira de Freitas, R.; Eram, M.S.; Szewczyk, M.M.; Steuber, H.; Smil, D.; Wu, H.; Li, F.; Senisterra, G.; Dong, A.; Brown, P.J.; et al. Discovery of a Potent Class I Protein Arginine Methyltransferase Fragment Inhibitor. J. Med. Chem. 2016, 59, 1176–1183. [Google Scholar] [CrossRef]
- Scheufler, C.; Möbitz, H.; Gaul, C.; Ragot, C.; Be, C.; Fernández, C.; Beyer, K.S.; Tiedt, R.; Stauffer, F. Optimization of a Fragment-Based Screening Hit toward Potent DOT1L Inhibitors Interacting in an Induced Binding Pocket. ACS Med. Chem. Lett. 2016, 7, 730–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benmansour, F.; Trist, I.; Coutard, B.; Decroly, E.; Querat, G.; Brancale, A.; Barral, K. Discovery of Novel Dengue Virus NS5 Methyltransferase Non-Nucleoside Inhibitors by Fragment-Based Drug Design. Eur. J. Med. Chem. 2017, 125, 865–880. [Google Scholar] [CrossRef]
- de Silva, U.; Zhou, Z.; Brown, B.A. Structure of Aeropyrum Pernix Fibrillarin in Complex with Natively Bound S-Adenosyl-L-Methionine at 1.7 A Resolution. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2012, 68, 854–859. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Lai, S.; Jia, R.; Xu, A.; Zhang, L.; Lu, J.; Ye, K. Structural Basis for Site-Specific Ribose Methylation by Box C/D RNA Protein Complexes. Nature 2011, 469, 559–563. [Google Scholar] [CrossRef]
- Aittaleb, M.; Visone, T.; Fenley, M.O.; Li, H. Structural and Thermodynamic Evidence for a Stabilizing Role of Nop5p in S-Adenosyl-L-Methionine Binding to Fibrillarin. J. Biol. Chem. 2004, 279, 41822–41829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Zhao, Y.; Li, H. The Multistructural Forms of Box C/D Ribonucleoprotein Particles. RNA 2018, 24, 1625–1633. [Google Scholar] [CrossRef] [Green Version]
- Schapira, M. Chemical Inhibition of Protein Methyltransferases. Cell Chem. Biol. 2016, 23, 1067–1076. [Google Scholar] [CrossRef] [Green Version]
- Brooun, A.; Gajiwala, K.S.; Deng, Y.-L.; Liu, W.; Bolaños, B.; Bingham, P.; He, Y.-A.; Diehl, W.; Grable, N.; Kung, P.-P.; et al. Polycomb Repressive Complex 2 Structure with Inhibitor Reveals a Mechanism of Activation and Drug Resistance. Nat. Commun. 2016, 7, 11384. [Google Scholar] [CrossRef] [PubMed]
- Basavapathruni, A.; Jin, L.; Daigle, S.R.; Majer, C.R.A.; Therkelsen, C.A.; Wigle, T.J.; Kuntz, K.W.; Chesworth, R.; Pollock, R.M.; Scott, M.P.; et al. Conformational Adaptation Drives Potent, Selective and Durable Inhibition of the Human Protein Methyltransferase DOT1L. Chem. Biol. Drug Des. 2012, 80, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Chory, E.J.; Wernimont, A.K.; Tempel, W.; Scopton, A.; Federation, A.; Marineau, J.J.; Qi, J.; Barsyte-Lovejoy, D.; Yi, J.; et al. Catalytic Site Remodelling of the DOT1L Methyltransferase by Selective Inhibitors. Nat. Commun. 2012, 3, 1288. [Google Scholar] [CrossRef] [PubMed]
- Badger, J. Crystallographic Fragment Screening. In Structure-Based Drug Discovery; Tari, L.W., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2012; Volume 841, pp. 161–177. ISBN 978-1-61779-519-0. [Google Scholar]
- Schiebel, J.; Radeva, N.; Köster, H.; Metz, A.; Krotzky, T.; Kuhnert, M.; Diederich, W.E.; Heine, A.; Neumann, L.; Atmanene, C.; et al. One Question, Multiple Answers: Biochemical and Biophysical Screening Methods Retrieve Deviating Fragment Hit Lists. ChemMedChem 2015, 10, 1511–1521. [Google Scholar] [CrossRef]
- Schiebel, J.; Radeva, N.; Krimmer, S.G.; Wang, X.; Stieler, M.; Ehrmann, F.R.; Fu, K.; Metz, A.; Huschmann, F.U.; Weiss, M.S.; et al. Six Biophysical Screening Methods Miss a Large Proportion of Crystallographically Discovered Fragment Hits: A Case Study. ACS Chem. Biol. 2016, 11, 1693–1701. [Google Scholar] [CrossRef]
- Stols, L.; Gu, M.; Dieckman, L.; Raffen, R.; Collart, F.R.; Donnelly, M.I. A New Vector for High-Throughput, Ligation-Independent Cloning Encoding a Tobacco Etch Virus Protease Cleavage Site. Protein Expr. Purif. 2002, 25, 8–15. [Google Scholar] [CrossRef] [Green Version]
- Studier, F.W. Protein Production by Auto-Induction in High-Density Shaking Cultures. Protein Expr. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; von Itzstein, M. How Size Matters: Diversity for Fragment Library Design. Molecules 2019, 24, 2838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 Suite and Current Developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCoy, A.J. Solving Structures of Protein Complexes by Molecular Replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr. 2007, 63, 32–41. [Google Scholar] [CrossRef] [Green Version]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards Automated Crystallographic Structure Refinement with Phenix.Refine. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 352–367. [Google Scholar] [CrossRef] [Green Version]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-Atom Structure Validation for Macromolecular Crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of Multiple Amber Force Fields and Development of Improved Protein Backbone Parameters. Proteins Struct. Funct. Bioinform. 2006, 65, 712–725. [Google Scholar] [CrossRef] [Green Version]
- Saez, D.A.; Vöhringer-Martinez, E. A Consistent S-Adenosylmethionine Force Field Improved by Dynamic Hirshfeld-I Atomic Charges for Biomolecular Simulation. J. Comput. Aided Mol. Des. 2015, 29, 951–961. [Google Scholar] [CrossRef]
- Páll, S.; Hess, B. A Flexible Algorithm for Calculating Pair Interactions on SIMD Architectures. Comput. Phys. Commun. 2013, 184, 2641–2650. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N⋅log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Hess, B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2007, 4, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Vulpetti, A.; Dalvit, C. Design and Generation of Highly Diverse Fluorinated Fragment Libraries and Their Efficient Screening with Improved 19F NMR Methodology. ChemMedChem 2013, 8, 2057–2069. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.; Meyer, B. Characterization of Ligand Binding by Saturation Transfer Difference NMR Spectroscopy. Angew. Chem. Int. Ed. 1999, 38, 1784–1788. [Google Scholar] [CrossRef]
- Piotto, M.; Saudek, V.; Sklenár, V. Gradient-Tailored Excitation for Single-Quantum NMR Spectroscopy of Aqueous Solutions. J. Biomol. NMR 1992, 2, 661–665. [Google Scholar] [CrossRef]
- Kuttan, A.; Nowshudin, S.; Rao, M.N.A. Ceric Ammonium Nitrate (CAN) Mediated Esterification of N-Boc Amino Acids Allows Either Retention or Removal of the N-Boc Group. Tetrahedron Lett. 2004, 45, 2663–2665. [Google Scholar] [CrossRef]
- Ylikangas, H.; Malmioja, K.; Peura, L.; Gynther, M.; Nwachukwu, E.O.; Leppänen, J.; Laine, K.; Rautio, J.; Lahtela-Kakkonen, M.; Huttunen, K.M.; et al. Quantitative Insight into the Design of Compounds Recognized by the L-Type Amino Acid Transporter 1 (LAT1). ChemMedChem 2014, 9, 2699–2707. [Google Scholar] [CrossRef] [PubMed]
- Evans, P. Scaling and Assessment of Data Quality. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 72–82. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, Y.; El-Deeb, I.M.; Masic, V.; Hartley-Tassell, L.; Maggioni, A.; Itzstein, M.v.; Ve, T. Discovery of Cofactor Competitive Inhibitors against the Human Methyltransferase Fibrillarin. Pharmaceuticals 2022, 15, 26. https://doi.org/10.3390/ph15010026
Shi Y, El-Deeb IM, Masic V, Hartley-Tassell L, Maggioni A, Itzstein Mv, Ve T. Discovery of Cofactor Competitive Inhibitors against the Human Methyltransferase Fibrillarin. Pharmaceuticals. 2022; 15(1):26. https://doi.org/10.3390/ph15010026
Chicago/Turabian StyleShi, Yun, Ibrahim M. El-Deeb, Veronika Masic, Lauren Hartley-Tassell, Andrea Maggioni, Mark von Itzstein, and Thomas Ve. 2022. "Discovery of Cofactor Competitive Inhibitors against the Human Methyltransferase Fibrillarin" Pharmaceuticals 15, no. 1: 26. https://doi.org/10.3390/ph15010026
APA StyleShi, Y., El-Deeb, I. M., Masic, V., Hartley-Tassell, L., Maggioni, A., Itzstein, M. v., & Ve, T. (2022). Discovery of Cofactor Competitive Inhibitors against the Human Methyltransferase Fibrillarin. Pharmaceuticals, 15(1), 26. https://doi.org/10.3390/ph15010026