
Manufacturing Bacteriophages (Part 1 of 2): Cell Line Development, Upstream, and Downstream Considerations

Abstract
1. Introduction
2. Cell Line Development (CLD)
2.1. Manufacturing Inclusion Criteria for Candidate Phages
2.2. Purity and Identity
2.3. Master Phage Bank Design
2.4. Storage
2.5. Phage Engineering Opportunities and Challenges in Cell Line Development
3. Upstream Processing (USP)
3.1. Phage Engineering Opportunities and Challenges in USP
3.2. Critical Process Parameters for USP Phage Production
3.2.1. The Effect of Temperature on Phage Production
3.2.2. The Effect of Media Composition on Phage Production
3.2.3. Multiplicity of Infection (MOI)
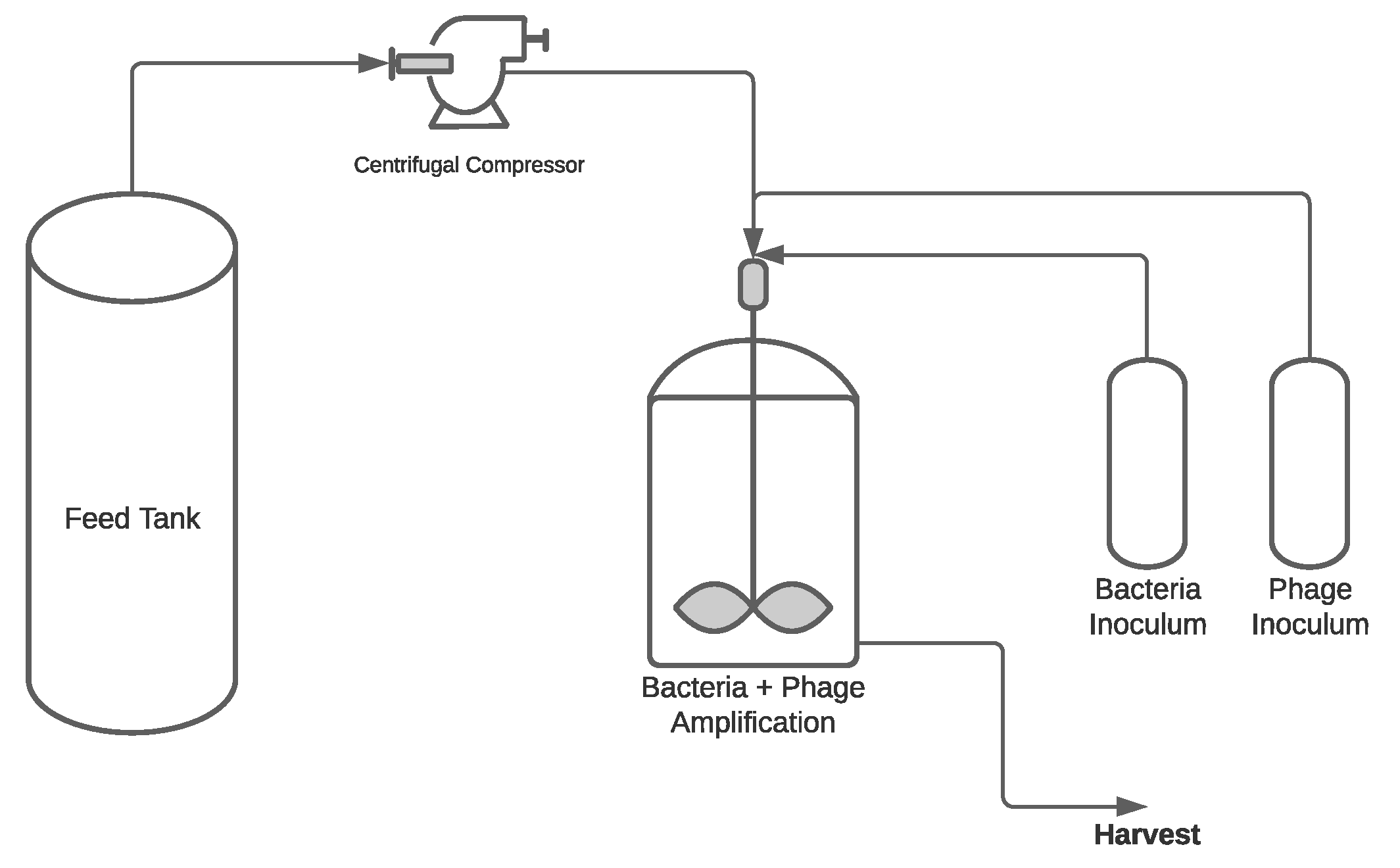
3.2.4. Production Modes for Phage Manufacturing
Batch Process
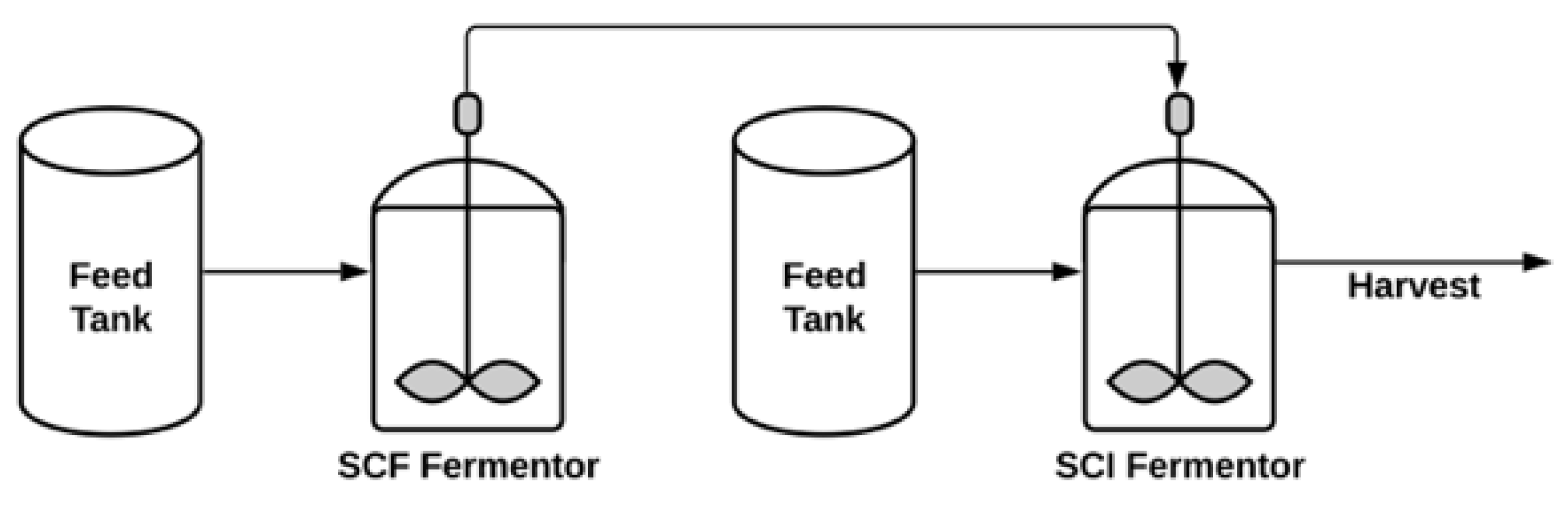
Semi-Continuous Process
Continuous Process
Single-Use vs. Stainless Steel
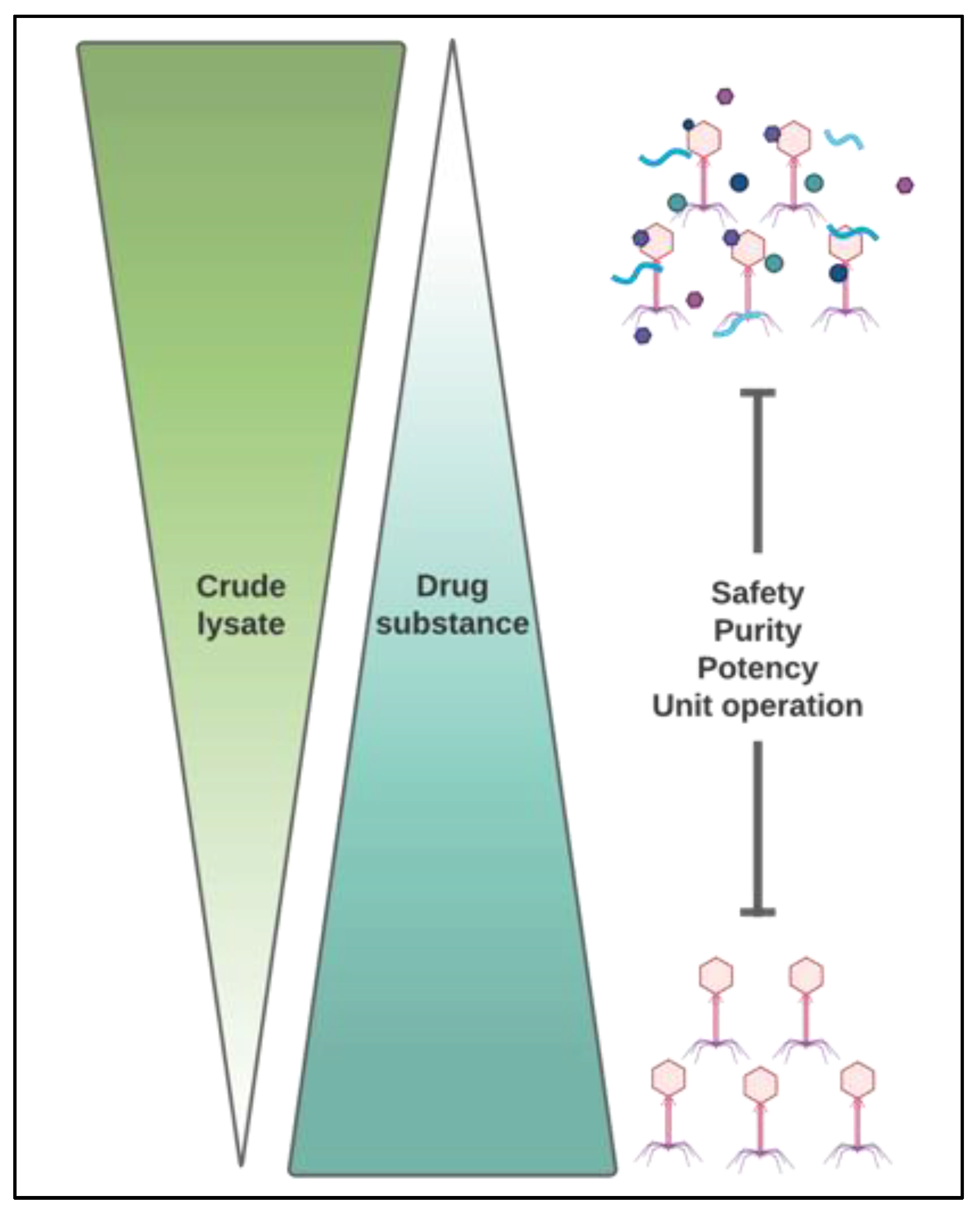
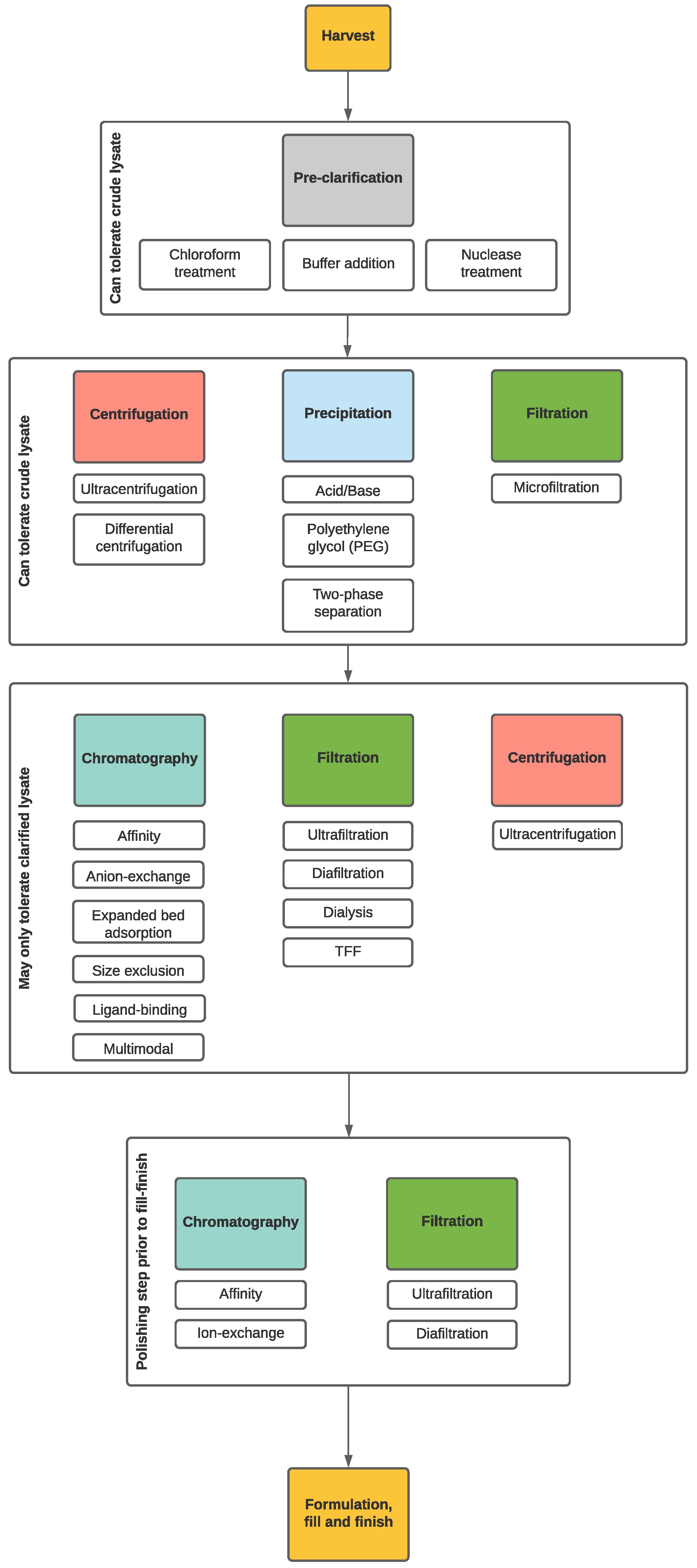
4. Downstream Processing (DSP)
4.1. Lysate Pre-Treatment
4.2. Crude Lysate Treatments
4.3. Clarified Lysate Treatments
4.3.1. Filtration
4.3.2. Chromatography
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schiff, L.J. Production, Characterization, and Testing of Banked Mammalian Cell Substrates Used to Produce Biological Products. In Vitro Cell. Dev. Biol. Anim. 2005, 41, 65–70. [Google Scholar] [CrossRef]
- Center for Biologics Evaluation and Research. Guidance for Industry: Characterization and Qualification of Cell Substrates and Other Biological Materials Used in the Production of Viral Vaccines for Infectious Disease Indications. 2010; Docket Number: FDA-2006-D-0223. Available online: http://www.fda.gov/downloads/biologicsbloodvaccines/Guidancecomplianceregulatoryinformation/guidances/vaccines/ucm202439.pdf (accessed on 14 September 2021).
- Carmen, J.; Burger, S.R.; McCaman, M.; Rowley, J.A. Developing assays to address identity, potency, purity and safety: Cell characterization in cell therapy process development. Regen. Med. 2012, 7, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Abedon, S.T. Phage therapy dosing: The problem(s) with multiplicity of infection (MOI). Bacteriophage 2016, 6, e1220348. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.-N. Lysis Timing and Bacteriophage Fitness. Genetics 2006, 172, 17–26. [Google Scholar] [CrossRef]
- Shao, Y.; Wang, I.-N. Bacteriophage Adsorption Rate and Optimal Lysis Time. Genetics 2008, 180, 471–482. [Google Scholar] [CrossRef]
- Nabergoj, D.; Kuzmić, N.; Drakslar, B.; Podgornik, A. Effect of dilution rate on productivity of continuous bacteriophage production in cellstat. Appl. Microbiol. Biotechnol. 2018, 102, 3649–3661. [Google Scholar] [CrossRef] [PubMed]
- Wiggins, B.A.; Alexander, M. Minimum bacterial density for bacteriophage replication: Implications for significance of bacteriophages in natural ecosystems. Appl. Environ. Microbiol. 1985, 49, 19–23. [Google Scholar] [CrossRef]
- Nabergoj, D.; Modic, P.; Podgornik, A. Effect of bacterial growth rate on bacteriophage population growth rate. MicrobiologyOpen 2018, 7, e00558. [Google Scholar] [CrossRef]
- Jérôme, G. Prophage in Phage Manufacturing: Is the Risk Overrated Compared to Other Therapies or Food? Antibiotics 2020, 9, 435. [Google Scholar] [CrossRef]
- Mavrich, T.N.; Casey, E.; Oliveira, J.; Bottacini, F.; James, K.; Franz, C.M.A.P.; Lugli, G.A.; Neve, H.; Ventura, M.; Hatfull, G.F.; et al. Characterization and induction of prophages in human gut-associated Bifidobacterium hosts. Sci. Rep. 2018, 8, 12772. [Google Scholar] [CrossRef]
- Pfeifer, E.; Michniewski, S.; Gätgens, C.; Münch, E.; Müller, F.; Polen, T.; Millard, A.; Blombach, B.; Frunzke, J. Generation of a Prophage-Free Variant of the Fast-Growing Bacterium Vibrio natriegens. Appl. Environ. Microbiol. 2019, 85, e00853-19. [Google Scholar] [CrossRef]
- Zaburlin, D.; Mercanti, D.J.; Quiberoni, A. A fast PCR-based method for the characterization of prophage profiles in strains of the Lactobacillus casei group. J. Virol. Methods 2017, 248, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Marcu, A.; Liang, Y.; Wishart, D.S. PHAST, PHASTER and PHASTEST: Tools for finding prophage in bacterial genomes. Brief. Bioinform. 2019, 20, 1560–1567. [Google Scholar] [CrossRef] [PubMed]
- Reis-Cunha, J.L.; Bartholomeu, D.C.; Manson, A.L.; Earl, A.M.; Cerqueira, G.C. ProphET, prophage estimation tool: A stand-alone prophage sequence prediction tool with self-updating reference database. PLoS ONE 2019, 14, e0223364. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, A.L.; Maués, D.; Lobato, A.; Franco, E.F.; Pinheiro, K.; Araújo, F.; Pantoja, Y.; Costa da Silva, A.L.; Morais, J.; Ramos, R.T.J. PhageWeb—Web Interface for Rapid Identification and Characterization of Prophages in Bacterial Genomes. Front. Genet. 2018, 9, 644. [Google Scholar] [CrossRef] [PubMed]
- Siow, W.T.; Koay, E.S.-C.; Lee, C.K.; Lee, H.K.; Ong, V.; Ngerng, W.J.; Lim, H.F.; Tan, A.; Tang, J.W.-T.; Phua, J. The Use of Polymerase Chain Reaction Amplification for the Detection of Viruses and Bacteria in Severe Community-Acquired Pneumonia. Respiration 2016, 92, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Abedon, S.T.; Culler, R.R. Optimizing bacteriophage plaque fecundity. J. Theor. Biol. 2007, 249, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Ács, N.; Gambino, M.; Brøndsted, L. Bacteriophage Enumeration and Detection Methods. Front. Microbiol. 2020, 11, 594868. [Google Scholar] [CrossRef]
- Anderson, B.; Rashid, M.H.; Carter, C.; Pasternack, G.; Rajanna, C.; Revazishvili, T.; Dean, T.; Senecal, A.; Sulakvelidze, A. Enumeration of bacteriophage particles: Comparative analysis of the traditional plaque assay and real-time QPCR- and NanoSight-based assays. Bacteriophage 2011, 1, 86–93. [Google Scholar] [CrossRef]
- Duyvejonck, H.; Merabishvili, M.; Pirnay, J.-P.; De Vos, D.; Verbeken, G.; Van Belleghem, J.; Gryp, T.; De Leenheer, J.; Van der Borght, K.; Van Simaey, L.; et al. Development of a qPCR platform for quantification of the five bacteriophages within bacteriophage cocktail 2 (BFC2). Sci. Rep. 2019, 9, 13893. [Google Scholar] [CrossRef] [PubMed]
- Refardt, D. Real-time quantitative PCR to discriminate and quantify lambdoid bacteriophages of Escherichia coli K-12. Bacteriophage 2012, 2, 98–104. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Łobocka, M.B.; Głowacka, A.; Golec, P. Methods for Bacteriophage Preservation. In Bacteriophage Therapy; Azeredo, J., Sillankorva, S., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2018; Volume 1693, pp. 219–230. ISBN 978-1-4939-7394-1. [Google Scholar]
- Clark, W.A. Comparison of Several Methods for Preserving Bacteriophages. Appl. Microbiol. 1962, 10, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Peters, T.L.; Bryan, D.W.; Hudson, L.K.; Denes, T.G. Characterization of a Novel Group of Listeria Phages That Target Serotype 4b Listeria monocytogenes. Viruses 2021, 13, 671. [Google Scholar] [CrossRef] [PubMed]
- Warren, J.C.; Hatch, M.T. Survival of T3 coliphage in varied extracellular environments. I. Viability of the coliphage during storage and in aerosols. Appl. Microbiol. 1969, 17, 256–261. [Google Scholar] [CrossRef]
- Ackermann, H.-W.; Tremblay, D.; Moineau, S. Long-Term Bacteriophage Preservation. 2004. Available online: https://www.researchgate.net/profile/Sylvain-Moineau/publication/285783875_Long-term_bacteriophage_preservation/links/56965e4b08ae34f3cf1dbb34/Long-term-bacteriophage-preservation.pdf (accessed on 14 September 2021).
- Clokie, M.R.J.; Kropinski, A.M. (Eds.) Bacteriophages: Methods and Protocols; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2009; ISBN 978-1-58829-682-5. [Google Scholar]
- Golec, P.; Dąbrowski, K.; Hejnowicz, M.S.; Gozdek, A.; Łoś, J.M.; Węgrzyn, G.; Łobocka, M.B.; Łoś, M. A reliable method for storage of tailed phages. J. Microbiol. Methods 2011, 84, 486–489. [Google Scholar] [CrossRef]
- Merabishvili, M.; Pirnay, J.-P.; Verbeken, G.; Chanishvili, N.; Tediashvili, M.; Lashkhi, N.; Glonti, T.; Krylov, V.; Mast, J.; van Parys, L.; et al. Quality-Controlled Small-Scale Production of a Well-Defined Bacteriophage Cocktail for Use in Human Clinical Trials. PLoS ONE 2009, 4, e4944-10. [Google Scholar] [CrossRef] [PubMed]
- Manohar, P.; Ramesh, N. Improved lyophilization conditions for long-term storage of bacteriophages. Sci. Rep. 2019, 9, 15242. [Google Scholar] [CrossRef]
- Kilcher, S.; Loessner, M.J. Engineering Bacteriophages as Versatile Biologics. Trends Microbiol. 2019, 27, 355–367. [Google Scholar] [CrossRef]
- Rostøl, J.T.; Marraffini, L. (Ph)ighting Phages: How Bacteria Resist Their Parasites. Cell Host Microbe 2019, 25, 184–194. [Google Scholar] [CrossRef]
- Rauch, B.J.; Silvis, M.R.; Hultquist, J.F.; Waters, C.S.; McGregor, M.J.; Krogan, N.J.; Bondy-Denomy, J. Inhibition of CRISPR-Cas9 with Bacteriophage Proteins. Cell 2017, 168, 150–158.e10. [Google Scholar] [CrossRef]
- Bhoobalan-Chitty, Y.; Johansen, T.B.; Di Cianni, N.; Peng, X. Inhibition of Type III CRISPR-Cas Immunity by an Archaeal Virus-Encoded Anti-CRISPR Protein. Cell 2019, 179, 448–458.e11. [Google Scholar] [CrossRef]
- Yehl, K. Engineering Phage Host-Range and Suppressing Bacterial Resistance through Phage Tail Fiber Mutagenesis. Cell 2019, 179, 459–469. [Google Scholar] [CrossRef]
- Namjoshi, S.; Dabbaghi, M.; Roberts, M.S.; Grice, J.E.; Mohammed, Y. Quality by Design: Development of the Quality Target Product Profile (QTPP) for Semisolid Topical Products. Pharmaceutics 2020, 12, 287. [Google Scholar] [CrossRef]
- Yu, L.X.; Amidon, G.; Khan, M.A.; Hoag, S.W.; Polli, J.; Raju, G.K.; Woodcock, J. Understanding Pharmaceutical Quality by Design. AAPS J. 2014, 16, 771–783. [Google Scholar] [CrossRef]
- Niazi, S.K. Biosimilars and Interchangeable Biologics: Tactical Elements; CRC Press: Boca Raton, FL, USA, 2016; ISBN 978-1-4987-4350-1. [Google Scholar]
- Weinbauer, M.G. Ecology of prokaryotic viruses. FEMS Microbiol. Rev. 2004, 28, 127–181. [Google Scholar] [CrossRef]
- Hyman, P.; Abedon, S.T. Practical Methods for Determining Phage Growth Parameters. In Bacteriophages: Methods and Protocols, Volume 1: Isolation, Characterization, and Interactions; Clokie, M.R.J., Kropinski, A.M., Eds.; Methods in Molecular BiologyTM; Humana Press: Totowa, NJ, USA, 2009; pp. 175–202. ISBN 978-1-60327-164-6. [Google Scholar]
- You, L.; Suthers, P.F.; Yin, J. Effects of Escherichia coli Physiology on Growth of Phage T7 In Vivo and In Silico. J. Bacteriol. 2002, 184, 1888–1894. [Google Scholar] [CrossRef] [PubMed]
- João, J.; Lampreia, J.; Prazeres, D.M.F.; Azevedo, A.M. Manufacturing of bacteriophages for therapeutic applications. Biotechnol. Adv. 2021, 49, 107758. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, R.; Chapman-McQuiston, E.; Wu, X.L. On Kinetics of Phage Adsorption. Biophys. J. 2007, 93, 303–315. [Google Scholar] [CrossRef]
- Koskella, B.; Brockhurst, M.A. Bacteria–phage coevolution as a driver of ecological and evolutionary processes in microbial communities. FEMS Microbiol. Rev. 2014, 38, 916–931. [Google Scholar] [CrossRef]
- Lenneman, B.R.; Fernbach, J.; Loessner, M.J.; Lu, T.K.; Kilcher, S. Enhancing phage therapy through synthetic biology and genome engineering. Curr. Opin. Biotechnol. 2021, 68, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Krom, R.J.; Bhargava, P.; Lobritz, M.A.; Collins, J.J. Engineered Phagemids for Nonlytic, Targeted Antibacterial Therapies. Nano Lett. 2015, 15, 4808–4813. [Google Scholar] [CrossRef]
- Tey, B.T.; Ooi, S.T.; Yong, K.C.; Ng, M.Y.T.; Ling, T.C.; Tan, W.S. Production of fusion m13 phage bearing the di-sulphide constrained peptide sequence (C-WSFFSNI-C) that interacts with hepatitis B core antigen. Afr. J. Biotechnol. 2009, 19, 268–273. [Google Scholar]
- Pollard, E.; Woodyatt, S. The Effect of Temperature on the Formation of T1 and T2r Bacteriophage. Biophys. J. 1964, 4, 367–385. [Google Scholar] [CrossRef]
- Grieco, S.-H.H.; Wong, A.Y.K.; Dunbar, W.S.; MacGillivray, R.T.A.; Curtis, S.B. Optimization of fermentation parameters in phage production using response surface methodology. J. Ind. Microbiol. Biotechnol. 2012, 39, 1515–1522. [Google Scholar] [CrossRef] [PubMed]
- Ali, J.; Rafiq, Q.; Ratcliffe, E. Improving phage titre through examining point of infection. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Shan, J.; Korbsrisate, S.; Withatanung, P.; Adler, N.L.; Clokie, M.R.J.; Galyov, E.E. Temperature dependent bacteriophages of a tropical bacterial pathogen. Front. Microbiol. 2014, 5, 599. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-W.; Dutta, V.; Elhanafi, D.; Lee, S.; Osborne, J.A.; Kathariou, S. A novel restriction-modification system is responsible for temperature-dependent phage resistance in Listeria monocytogenes ECII. Appl. Environ. Microbiol. 2012, 78, 1995–2004. [Google Scholar] [CrossRef]
- Tokman, J.I.; Kent, D.J.; Wiedmann, M.; Denes, T. Temperature Significantly Affects the Plaquing and Adsorption Efficiencies of Listeria Phages. Front. Microbiol. 2016, 7, 631. [Google Scholar] [CrossRef]
- Sochocka, M.; Tomczyk, T.; Sobczyński, M.; Szermer-Olearnik, B.; Boratyński, J. The kinetics of Escherichia coli B growth and bacteriophage T4 multiplication in SM-1 novel minimal culture medium. J. Gen. Appl. Microbiol. 2015, 61, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Mills, D.A.; Block, D.E. Development of Chemically Defined Media Supporting High-Cell-Density Growth of Lactococci, Enterococci, and Streptococci. Appl. Environ. Microbiol. 2009, 75, 1080–1087. [Google Scholar] [CrossRef]
- Mutti, M.; Corsini, L. Robust Approaches for the Production of Active Ingredient and Drug Product for Human Phage Therapy. Front. Microbiol. 2019, 10, 2289. [Google Scholar] [CrossRef]
- D’Humières, C.; Touchon, M.; Dion, S.; Cury, J.; Ghozlane, A.; Garcia-Garcera, M.; Bouchier, C.; Ma, L.; Denamur, E.; Rocha, E.P.C. A simple, reproducible and cost-effective procedure to analyse gut phageome: From phage isolation to bioinformatic approach. Sci. Rep. 2019, 9, 11331. [Google Scholar] [CrossRef]
- Kasman, L.M.; Kasman, A.; Westwater, C.; Dolan, J.; Schmidt, M.G.; Norris, J.S. Overcoming the Phage Replication Threshold: A Mathematical Model with Implications for Phage Therapy. J. Virol. 2002, 76, 5557–5564. [Google Scholar] [CrossRef]
- Ng, S.; Gisonni-Lex, L.; Azizi, A. New approaches for characterization of the genetic stability of vaccine cell lines. Hum. Vaccines Immunother. 2017, 13, 1669–1672. [Google Scholar] [CrossRef]
- Sanjuán, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral Mutation Rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef]
- Drake, J.W.; Charlesworth, B.; Charlesworth, D.; Crow, J.F. Rates of Spontaneous Mutation. Genetics 1998, 148, 1667–1686. [Google Scholar] [CrossRef] [PubMed]
- Kupczok, A.; Neve, H.; Huang, K.D.; Hoeppner, M.P.; Heller, K.J.; Franz, C.M.A.P.; Dagan, T. Rates of Mutation and Recombination in Siphoviridae Phage Genome Evolution over Three Decades. Mol. Biol. Evol. 2018, 35, 1147–1159. [Google Scholar] [CrossRef] [PubMed]
- Richards, K.; Malik, D.J. Microencapsulation of Bacteriophages Using Membrane Emulsification in Different pH-Triggered Controlled Release Formulations for Oral Administration. Pharmaceuticals 2021, 14, 424. [Google Scholar] [CrossRef]
- Jurač, K.; Nabergoj, D.; Podgornik, A. Bacteriophage production processes. Appl. Microbiol. Biotechnol. 2018, 103, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Warner, C.M.; Barker, N.; Lee, S.-W.; Perkins, E.J. M13 bacteriophage production for large-scale applications. Bioprocess Biosyst. Eng. 2014, 37, 2067–2072. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, F.; Shi, J.; Malik, D. High Throughput Manufacturing of Bacteriophages Using Continuous Stirred Tank Bioreactors Connected in Series to Ensure Optimum Host Bacteria Physiology for Phage Production. Viruses 2018, 10, 537. [Google Scholar] [CrossRef]
- Kim, S.G.; Kwon, J.; Giri, S.S.; Yun, S.; Kim, H.J.; Kim, S.W.; Kang, J.W.; Lee, S.B.; Jung, W.J.; Park, S.C. Strategy for mass production of lytic Staphylococcus aureus bacteriophage pSa-3: Contribution of multiplicity of infection and response surface methodology. Microb. Cell Factories 2021, 20, 56. [Google Scholar] [CrossRef] [PubMed]
- Sauvageau, D.; Cooper, D.G. Two-stage, self-cycling process for the production of bacteriophages. Microb. Cell Factories 2010, 9, 81. [Google Scholar] [CrossRef]
- Shukla, A.A.; Gottschalk, U. Single-use disposable technologies for biopharmaceutical manufacturing. Trends Biotechnol. 2013, 31, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Branston, S.; Stanley, E.; Keshavarz-Moore, E.; Ward, J. Precipitation of filamentous bacteriophages for their selective recovery in primary purification. Biotechnol. Prog. 2012, 28, 129–136. [Google Scholar] [CrossRef]
- Regulski, K.; Champion-Arnaud, P.; Gabard, J. Bacteriophage Manufacturing: From Early Twentieth-Century Processes to Current GMP. In Bacteriophages; Harper, D.R., Abedon, S.T., Burrowes, B.H., McConville, M.L., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 1–31. ISBN 978-3-319-40598-8. [Google Scholar]
- Bachrach, U.; Friedmann, A. Practical Procedures for the Purification of Bacterial Viruses. Appl. Microbiol. 1971, 22, 10. [Google Scholar] [CrossRef] [PubMed]
- Carroll-Portillo, A.; Coffman, C.N.; Varga, M.G.; Alcock, J.; Singh, S.B.; Lin, H.C. Standard Bacteriophage Purification Procedures Cause Loss in Numbers and Activity. Viruses 2021, 13, 328. [Google Scholar] [CrossRef]
- Wolf, M.W.; Reichl, U. Downstream processing of cell culture-derived virus particles. Expert Rev. Vaccines 2011, 10, 1451–1475. [Google Scholar] [CrossRef]
- Adriaenssens, E.M.; Lehman, S.M.; Vandersteegen, K.; Vandenheuvel, D.; Philippe, D.L.; Cornelissen, A.; Clokie, M.R.J.; García, A.J.; De Proft, M.; Maes, M.; et al. CIM® monolithic anion-exchange chromatography as a useful alternative to CsCl gradient purification of bacteriophage particles. Virology 2012, 434, 265–270. [Google Scholar] [CrossRef]
- Hyman, P. Phages for Phage Therapy: Isolation, Characterization, and Host Range Breadth. Pharmaceuticals 2019, 12, 35. [Google Scholar] [CrossRef]
- Fontes, L.V.Q.; Campos, G.S.; Beck, P.A.; Brandão, C.F.L.; Sardi, S.I. Precipitation of bovine rotavirus by polyethylen glycol (PEG) and its application to produce polyclonal and monoclonal antibodies. J. Virol. Methods 2005, 123, 147–153. [Google Scholar] [CrossRef]
- Kröber, T.; Knöchlein, A.; Eisold, K.; Kalbfuß-Zimmermann, B.; Reichl, U. DNA Depletion by Precipitation in the Purification of Cell Culture-Derived Influenza Vaccines. Chem. Eng. Technol. 2010, 33, 941–959. [Google Scholar] [CrossRef]
- Mullan, W. Isolation and Purification of Bacteriophages. Available online: https://www.dairyscience.info/index.php/isolation-and-purification-of-bacteriophages.html (accessed on 6 August 2021).
- Binabaji, E.; Ma, J.; Rao, S.; Zydney, A.L. Ultrafiltration of highly concentrated antibody solutions: Experiments and modeling for the effects of module and buffer conditions. Biotechnol. Prog. 2016, 32, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Wickramasinghe, S.R.; Bower, S.E.; Chen, Z.; Mukherjee, A.; Husson, S.M. Relating the pore size distribution of ultrafiltration membranes to dextran rejection. J. Membr. Sci. 2009, 340, 1–8. [Google Scholar] [CrossRef]
- Wickramasinghe, S.R.; Kalbfuß, B.; Zimmermann, A.; Thom, V.; Reichl, U. Tangential flow microfiltration and ultrafiltration for human influenza A virus concentration and purification. Biotechnol. Bioeng. 2005, 92, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Luong, T.; Salabarria, A.-C.; Edwards, R.A.; Roach, D.R. Standardized bacteriophage purification for personalized phage therapy. Nat. Protoc. 2020, 15, 2867–2890. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Munford, R. LPS stimulates IgM production in vivo without help from non-B cells. Innate Immun. 2016, 22, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Hietala, V.; Horsma-Heikkinen, J.; Carron, A.; Skurnik, M.; Kiljunen, S. The Removal of Endo- and Enterotoxins from Bacteriophage Preparations. Front. Microbiol. 2019, 10, 1674. [Google Scholar] [CrossRef]
- Kramberger, P.; Honour, R.C.; Herman, R.E.; Smrekar, F.; Peterka, M. Purification of the Staphylococcus aureus bacteriophages VDX-10 on methacrylate monoliths. J. Virol. Methods 2010, 166, 60–64. [Google Scholar] [CrossRef]
- Monjezi, R.; Tey, B.T.; Sieo, C.C.; Tan, W.S. Purification of bacteriophage M13 by anion exchange chromatography. J. Chromatogr. B 2010, 878, 1855–1859. [Google Scholar] [CrossRef]
- Smrekar, F.; Ciringer, M.; Jančar, J.; Raspor, P.; Štrancar, A.; Podgornik, A. Optimization of lytic phage manufacturing in bioreactor using monolithic supports. J. Sep. Sci. 2011, 34, 2152–2158. [Google Scholar] [CrossRef]
- Vandenheuvel, D.; Rombouts, S.; Adriaenssens, E.M. Purification of Bacteriophages Using Anion-Exchange Chromatography. In Bacteriophages; Clokie, M.R.J., Kropinski, A.M., Lavigne, R., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2018; Volume 1681, pp. 59–69. ISBN 978-1-4939-7341-5. [Google Scholar]
- Bourdin, G.; Schmitt, B.; Marvin Guy, L.; Germond, J.-E.; Zuber, S.; Michot, L.; Reuteler, G.; Brüssow, H. Amplification and Purification of T4-Like Escherichia coli Phages for Phage Therapy: From Laboratory to Pilot Scale. Appl. Environ. Microbiol. 2014, 80, 1469–1476. [Google Scholar] [CrossRef]
- Jiang, Q.; Song, C.; Nangreave, J.; Liu, X.; Lin, L.; Qiu, D.; Wang, Z.-G.; Zou, G.; Liang, X.; Yan, H.; et al. DNA Origami as a Carrier for Circumvention of Drug Resistance. J. Am. Chem. Soc. 2012, 134, 13396–13403. [Google Scholar] [CrossRef] [PubMed]
- Kattur Venkatachalam, A.; Szyporta, M.; Kiener, T.; Balraj, P.; Kwang, J. Concentration and purification of enterovirus 71 using a weak anion-exchange monolithic column. Virol. J. 2014, 11, 99. [Google Scholar] [CrossRef]
- Kramberger, P.; Urbas, L.; Štrancar, A. Downstream processing and chromatography based analytical methods for production of vaccines, gene therapy vectors, and bacteriophages. Hum. Vaccines Immunother. 2015, 11, 1010–1021. [Google Scholar] [CrossRef] [PubMed]
- Vandenheuvel, D.; Singh, A.; Vandersteegen, K.; Klumpp, J.; Lavigne, R.; Van den Mooter, G. Feasibility of spray drying bacteriophages into respirable powders to combat pulmonary bacterial infections. Eur. J. Pharm. Biopharm. 2013, 84, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Vandenheuvel, D.; Lavigne, R.; Brüssow, H. Bacteriophage Therapy: Advances in Formulation Strategies and Human Clinical Trials. Annu. Rev. Virol. 2015, 2, 599–618. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Operation Mode | Advantages | Disadvantages | References |
|---|---|---|---|
| Batch | High titers (from 5 × 1012 PFU/mL to 1 × 1016 PFU/mL) Cheapest to produce phage | Conditions change during the process Long preparation time Reduction in overall productivity Limited by the maximum equipment volume available Potential batch to batch variation | Jurač, Nabergoj and Podgornik, 2018 [65]; Warner et al., 2014 [66]; Sochocka et al., 2015; Nabergoj et al., 2018 [9] |
| Semi-Continuous | In processes where bacteria are grown separately from phages, phage-resistance is avoided. | Self-cycling system requires advanced and integrated control and monitoring | Mancuso, Shi and Malik, 2018 [67] |
| Continuous | Higher overall productivity Cost savings Consistent and higher quality product is obtained due to easier control Operational complexity is reduced | Laborious because the system is complex A totally continuous process may lead generation of phage-resistant strains if the required measures are not taken Requires close monitoring to sustain steady-state Expensive to implement the system | Mancuso. Shi and Malik, 2018 [67] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanir, T.; Orellana, M.; Escalante, A.; Moraes de Souza, C.; Koeris, M.S. Manufacturing Bacteriophages (Part 1 of 2): Cell Line Development, Upstream, and Downstream Considerations. Pharmaceuticals 2021, 14, 934. https://doi.org/10.3390/ph14090934
Tanir T, Orellana M, Escalante A, Moraes de Souza C, Koeris MS. Manufacturing Bacteriophages (Part 1 of 2): Cell Line Development, Upstream, and Downstream Considerations. Pharmaceuticals. 2021; 14(9):934. https://doi.org/10.3390/ph14090934
Chicago/Turabian StyleTanir, Tayfun, Marvin Orellana, Aster Escalante, Carolina Moraes de Souza, and Michael S. Koeris. 2021. "Manufacturing Bacteriophages (Part 1 of 2): Cell Line Development, Upstream, and Downstream Considerations" Pharmaceuticals 14, no. 9: 934. https://doi.org/10.3390/ph14090934
APA StyleTanir, T., Orellana, M., Escalante, A., Moraes de Souza, C., & Koeris, M. S. (2021). Manufacturing Bacteriophages (Part 1 of 2): Cell Line Development, Upstream, and Downstream Considerations. Pharmaceuticals, 14(9), 934. https://doi.org/10.3390/ph14090934