
In Silico-Based Design and In Vivo Evaluation of an Anthranilic Acid Derivative as a Multitarget Drug in a Diet-Induced Metabolic Syndrome Model


 ,
,  ,
,  and
and 
Abstract
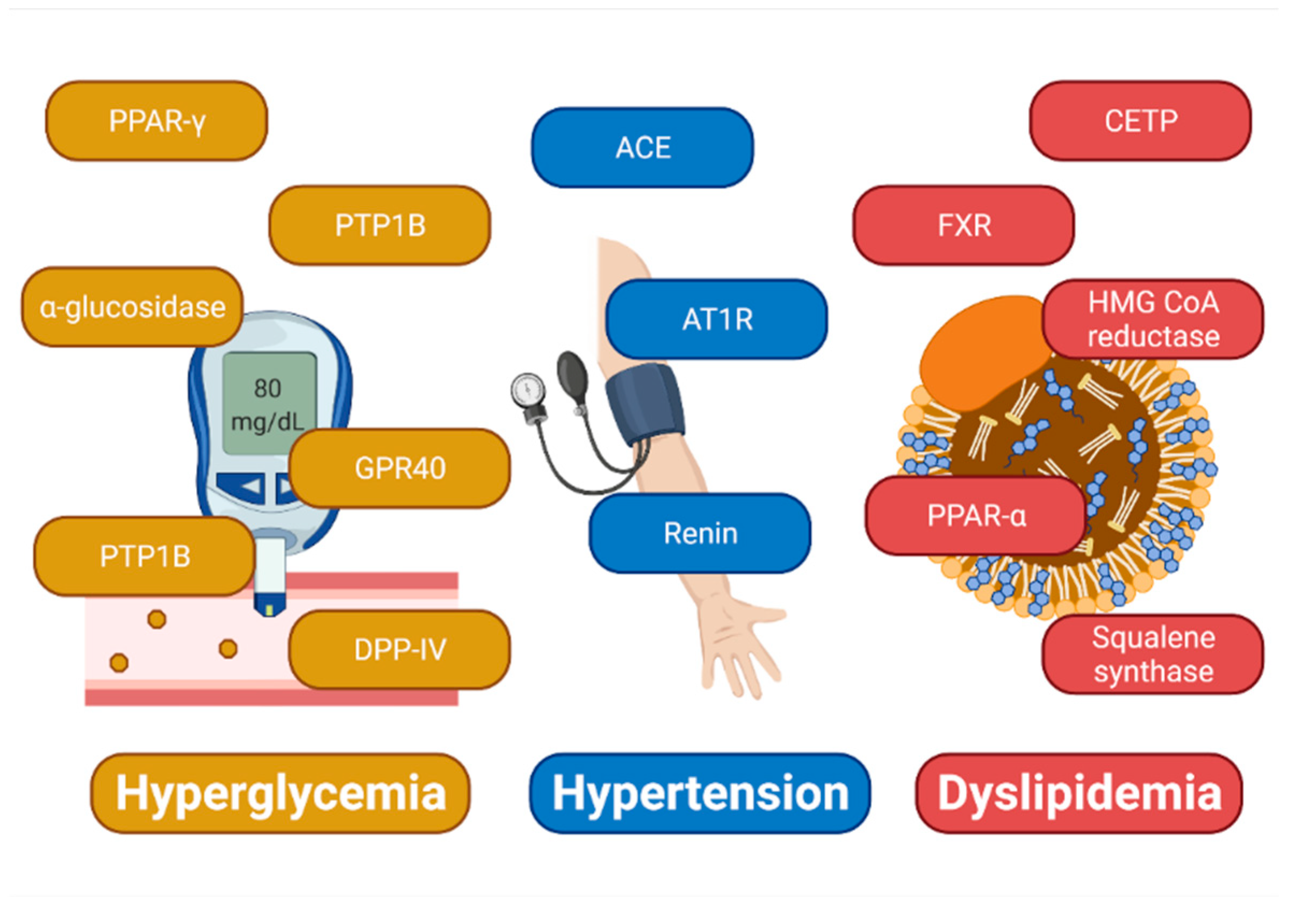
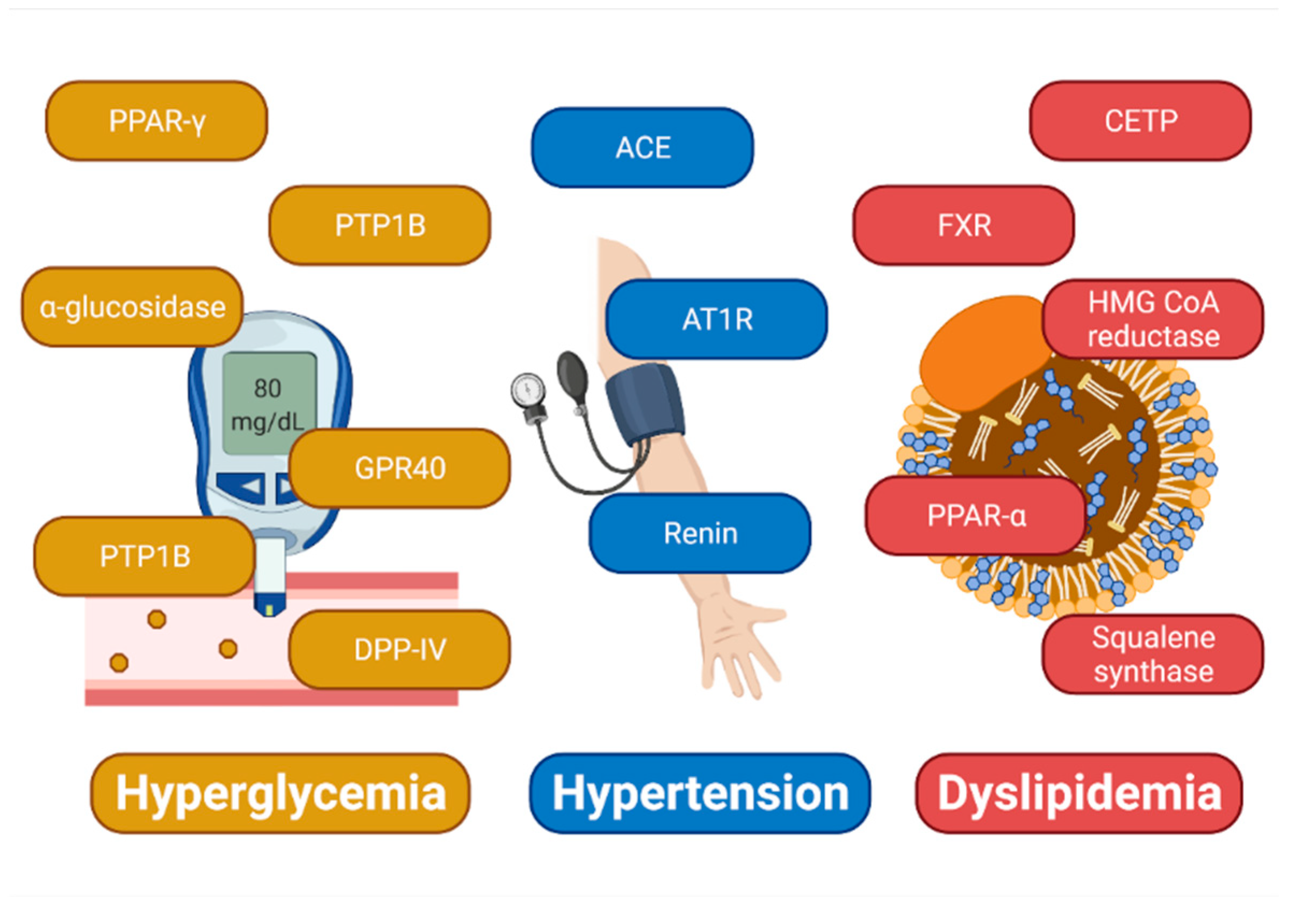
:1. Introduction
2. Results
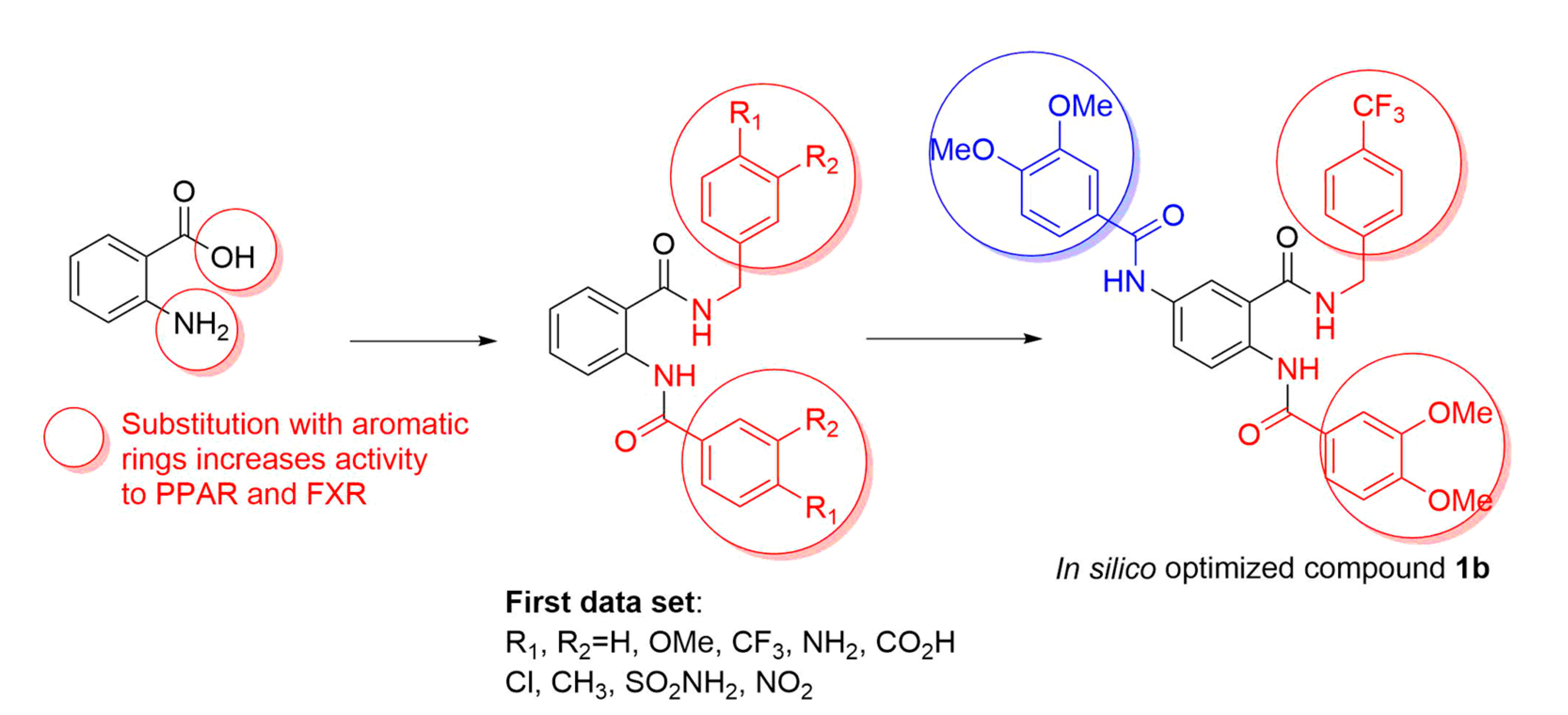
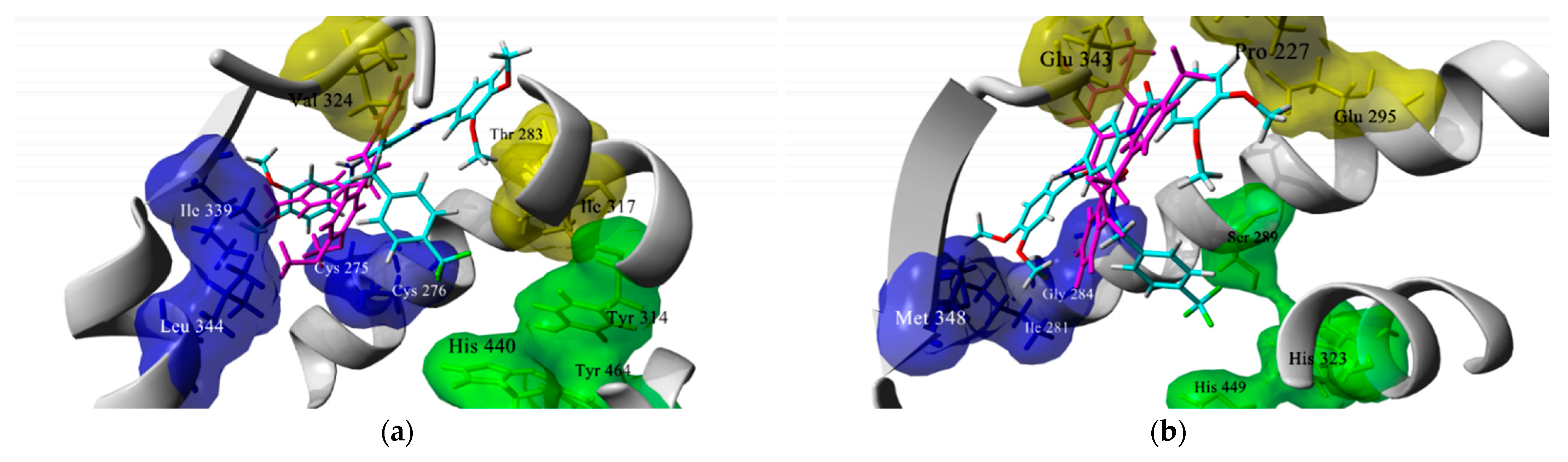
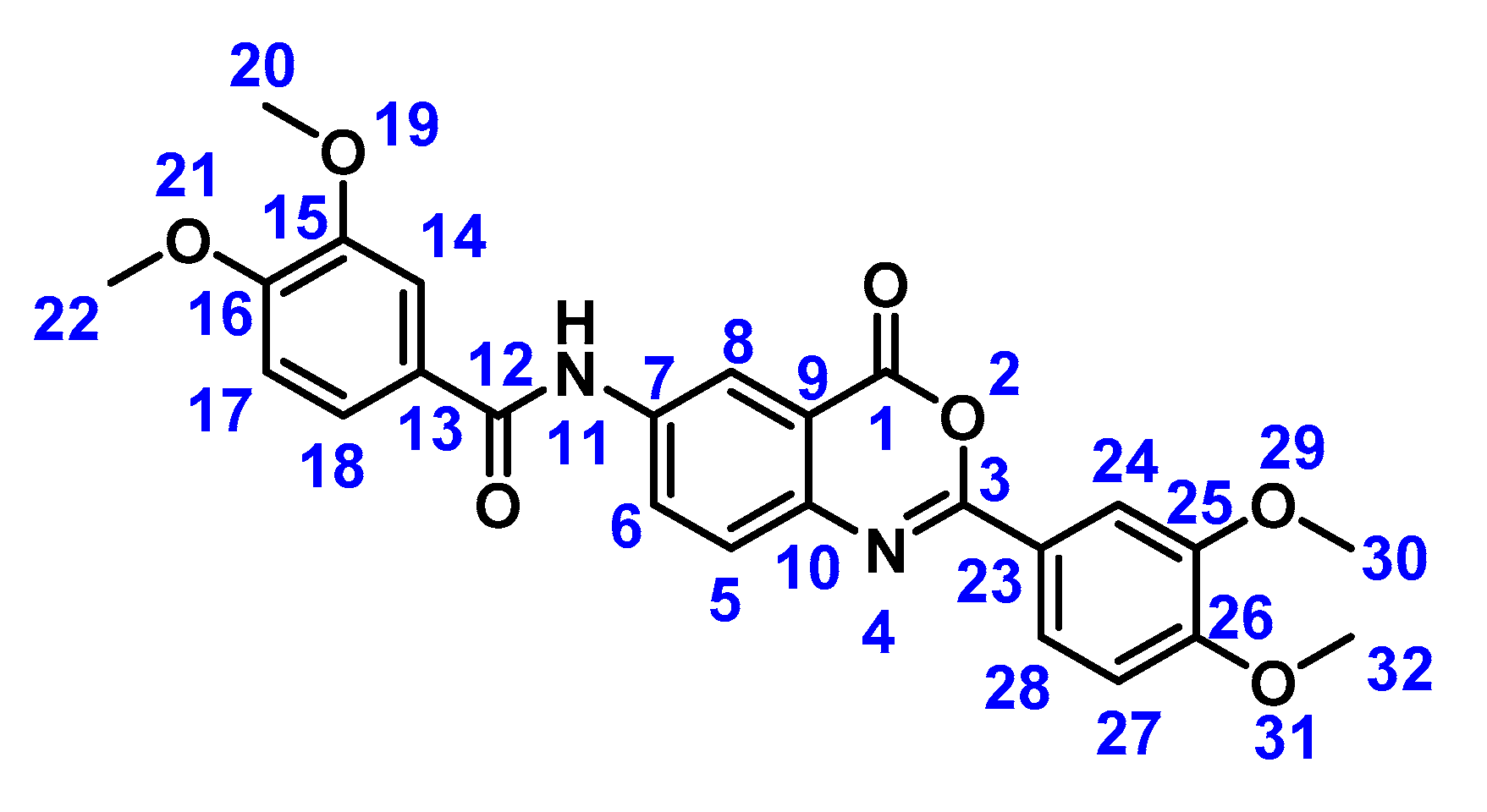
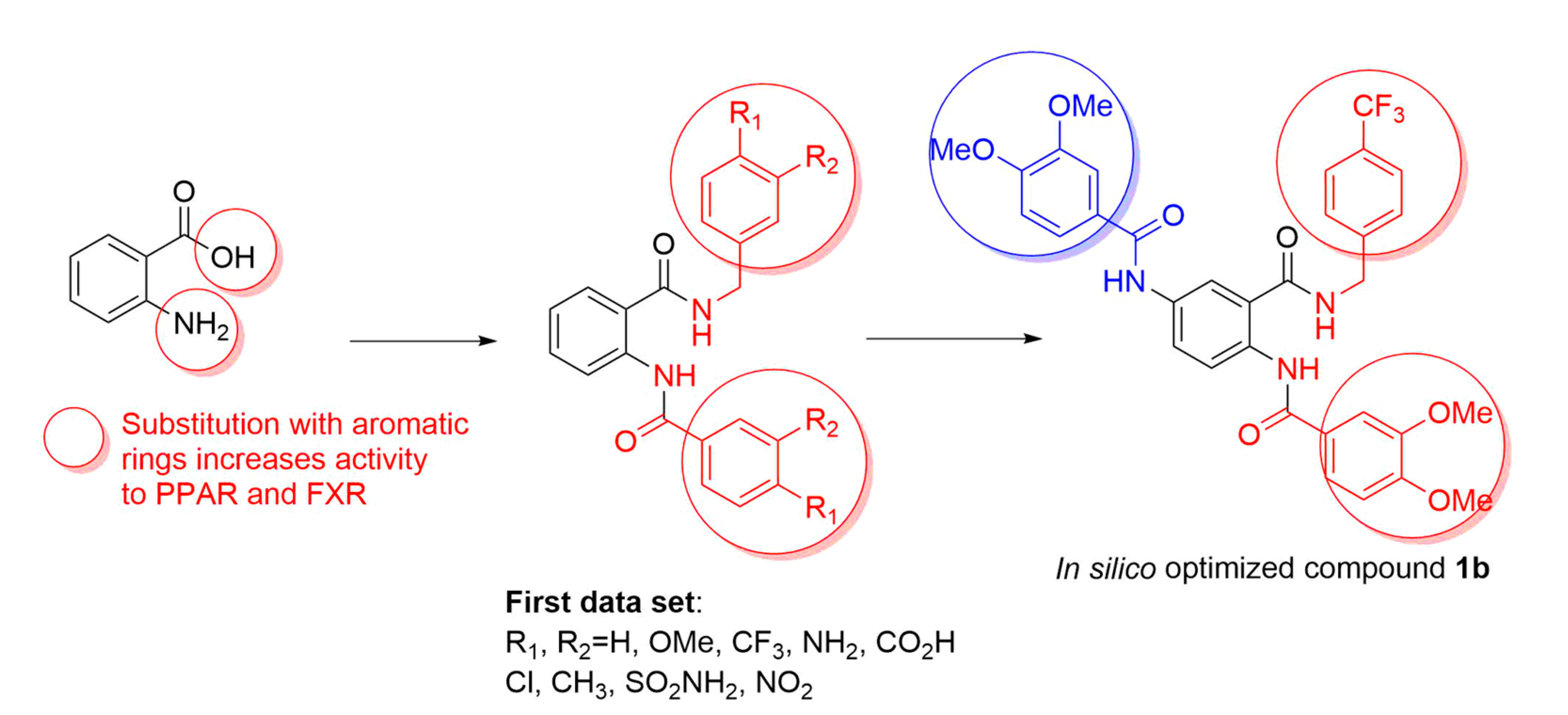
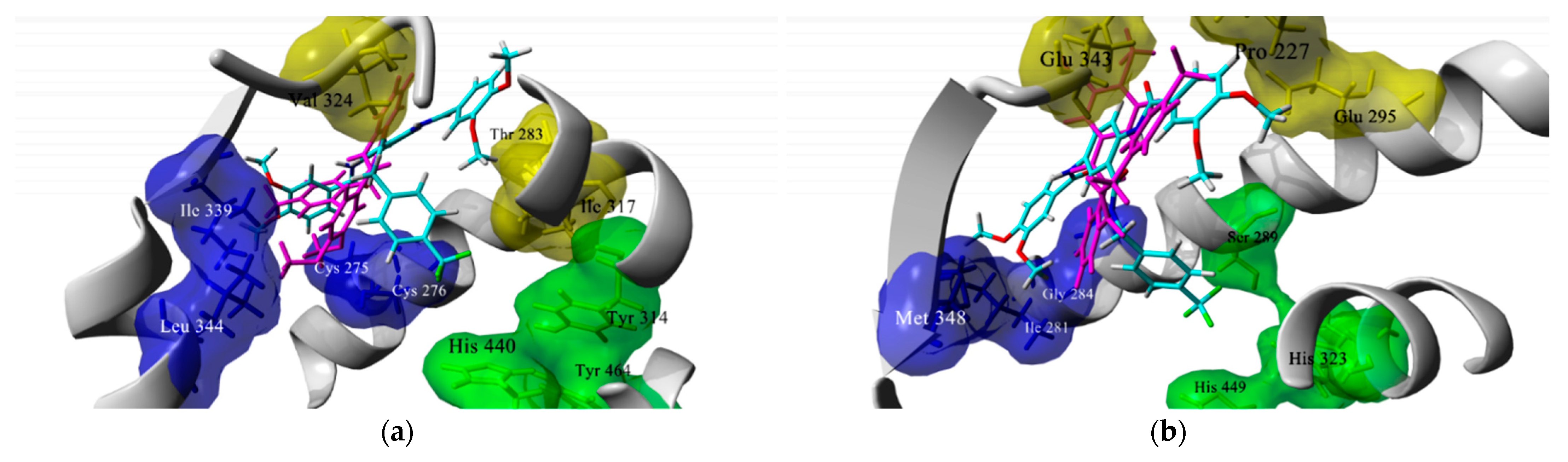
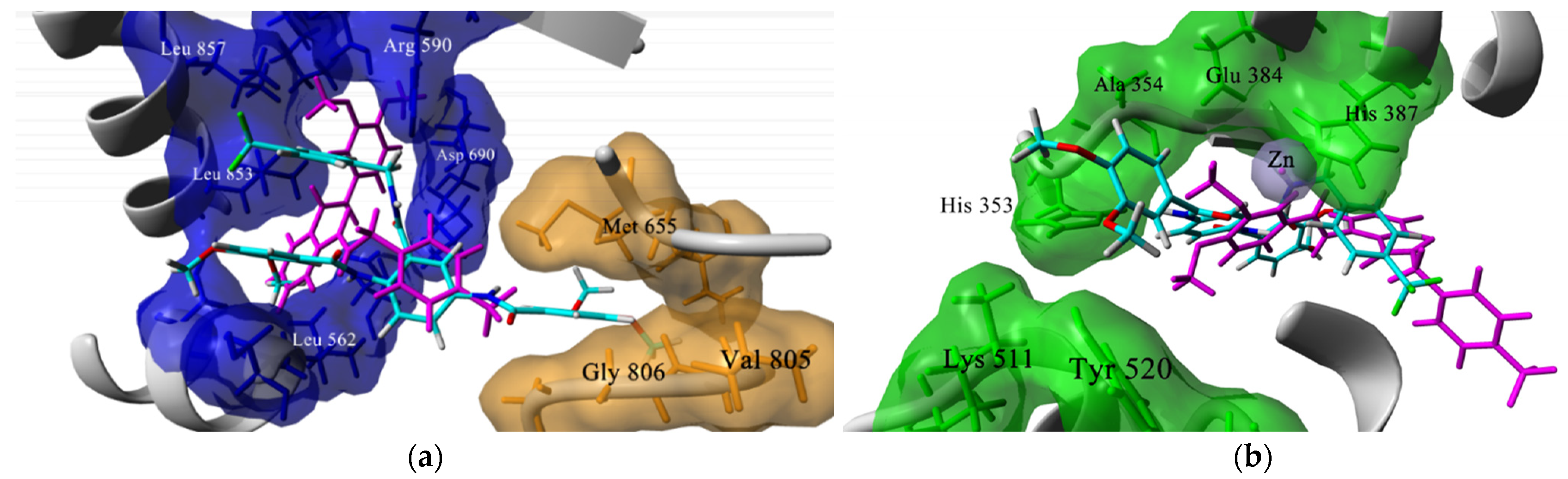
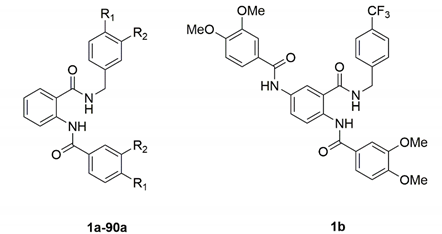
2.1. Docking-Based Design
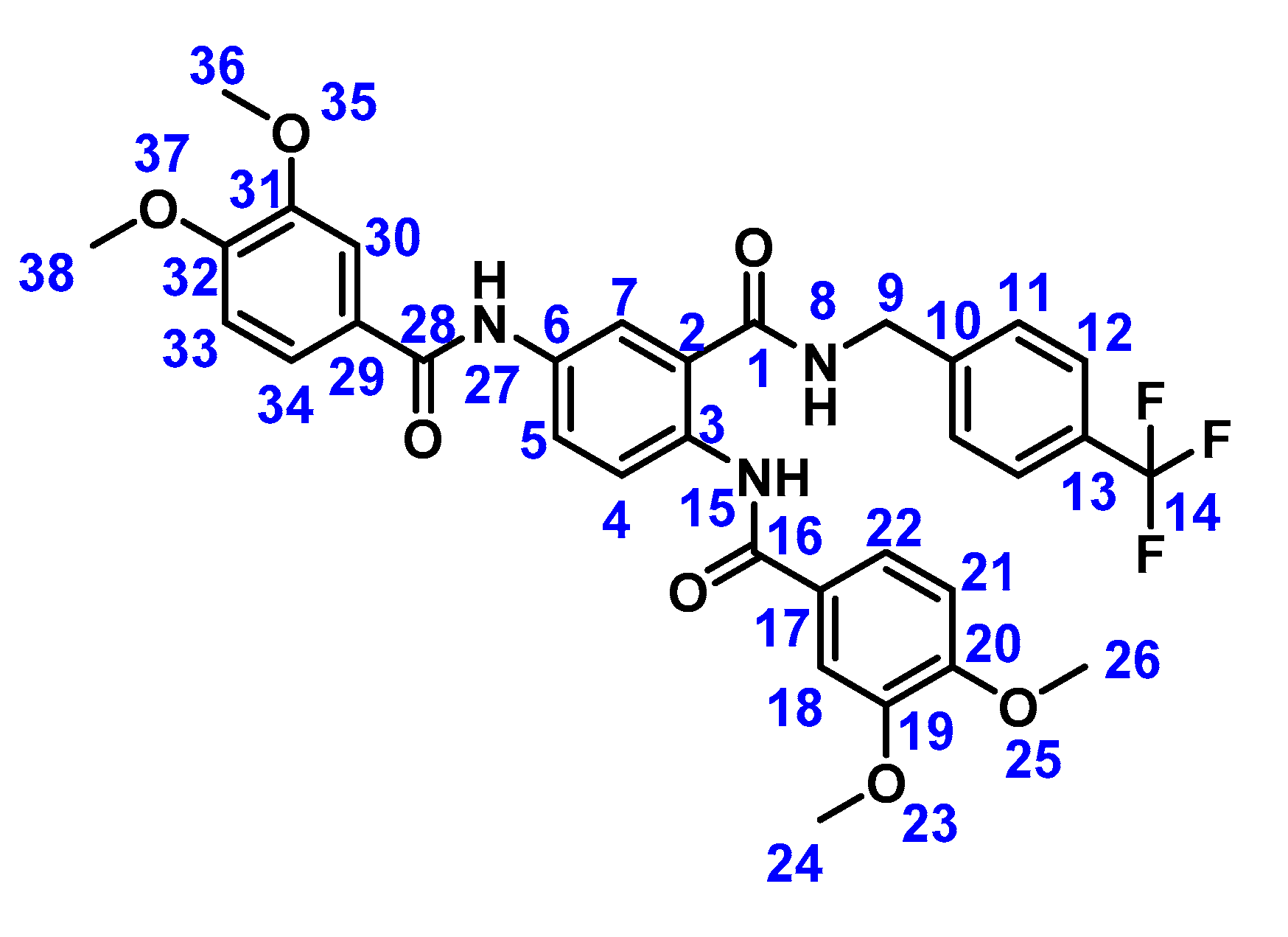
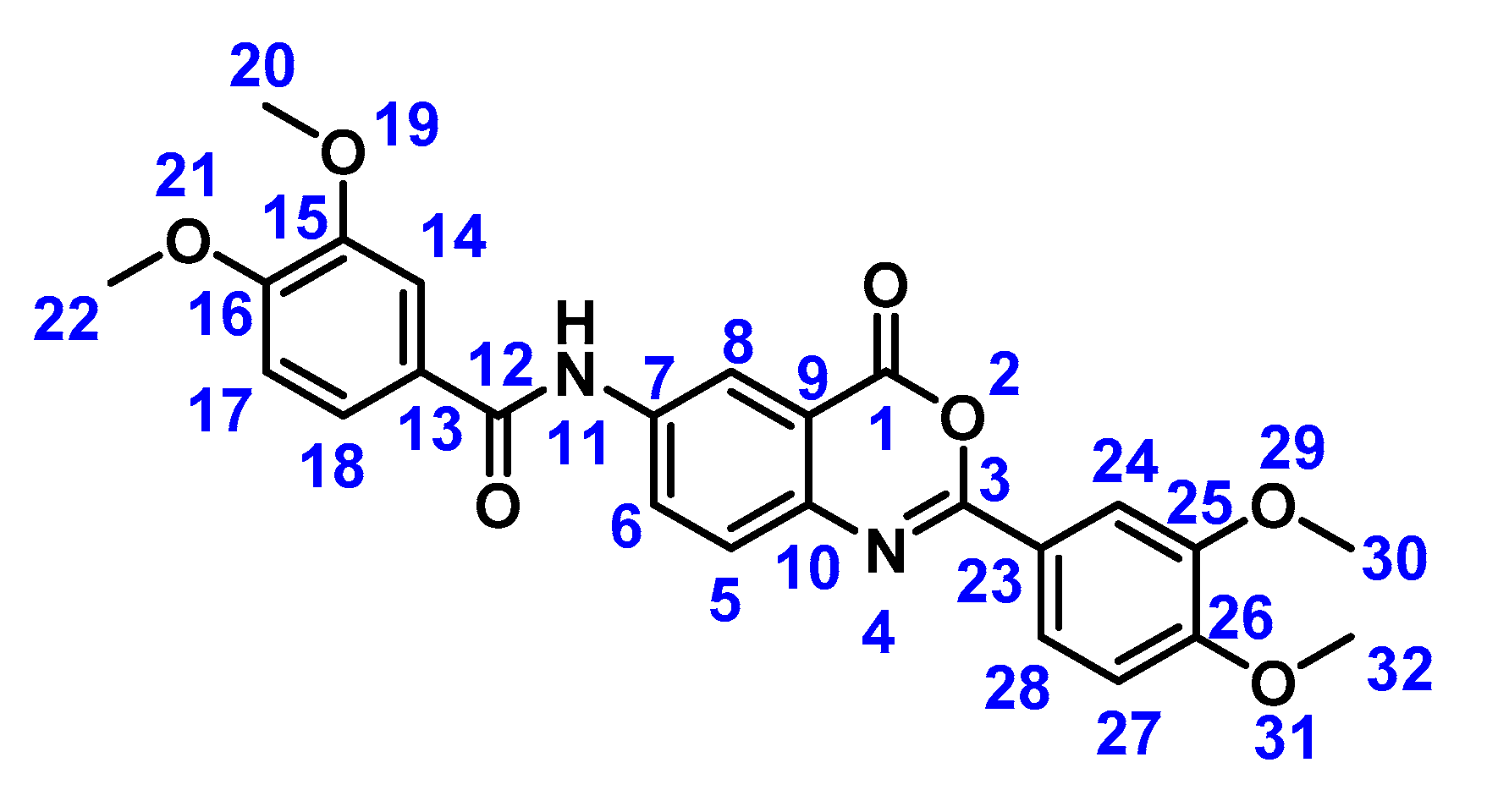
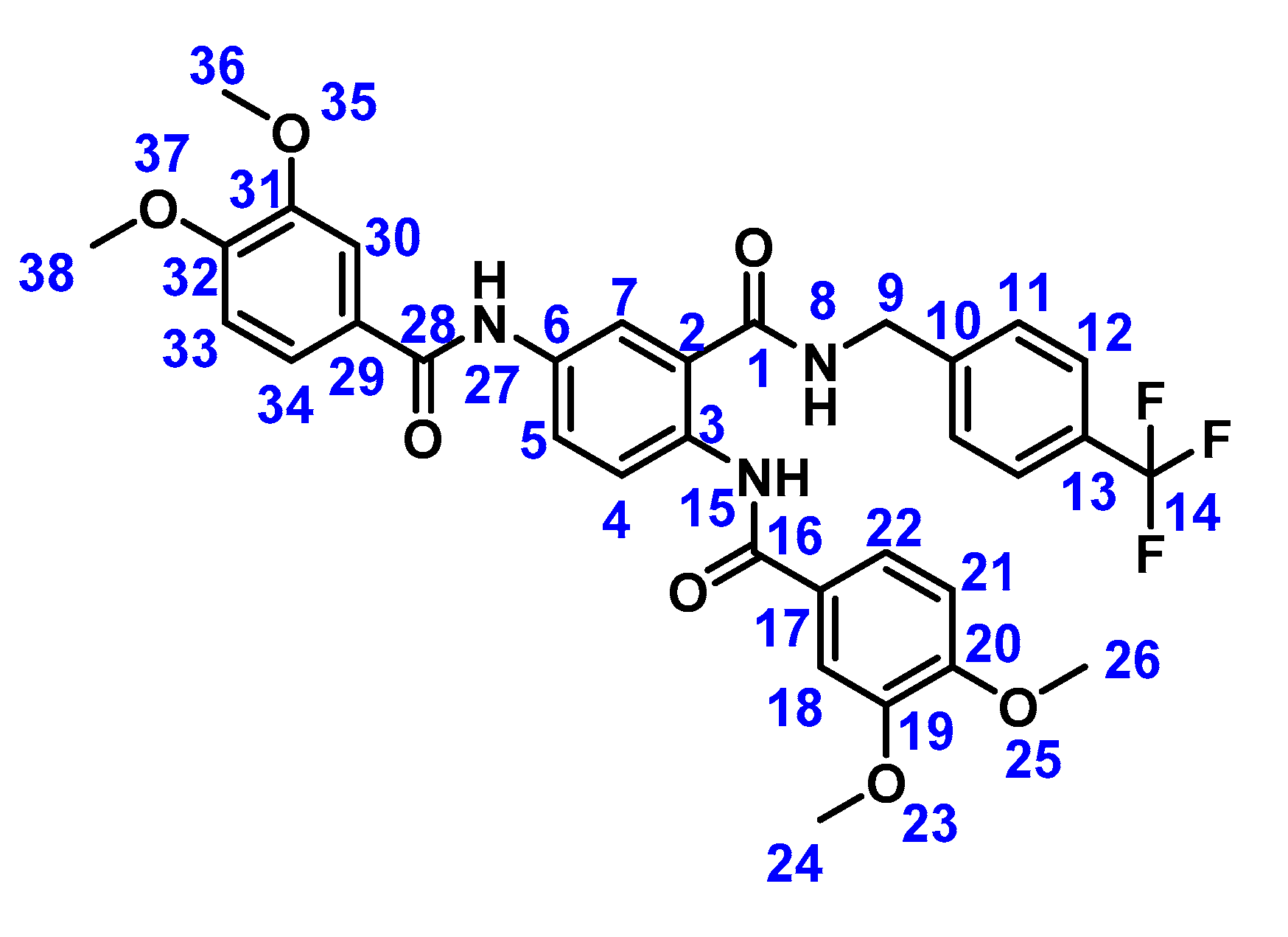
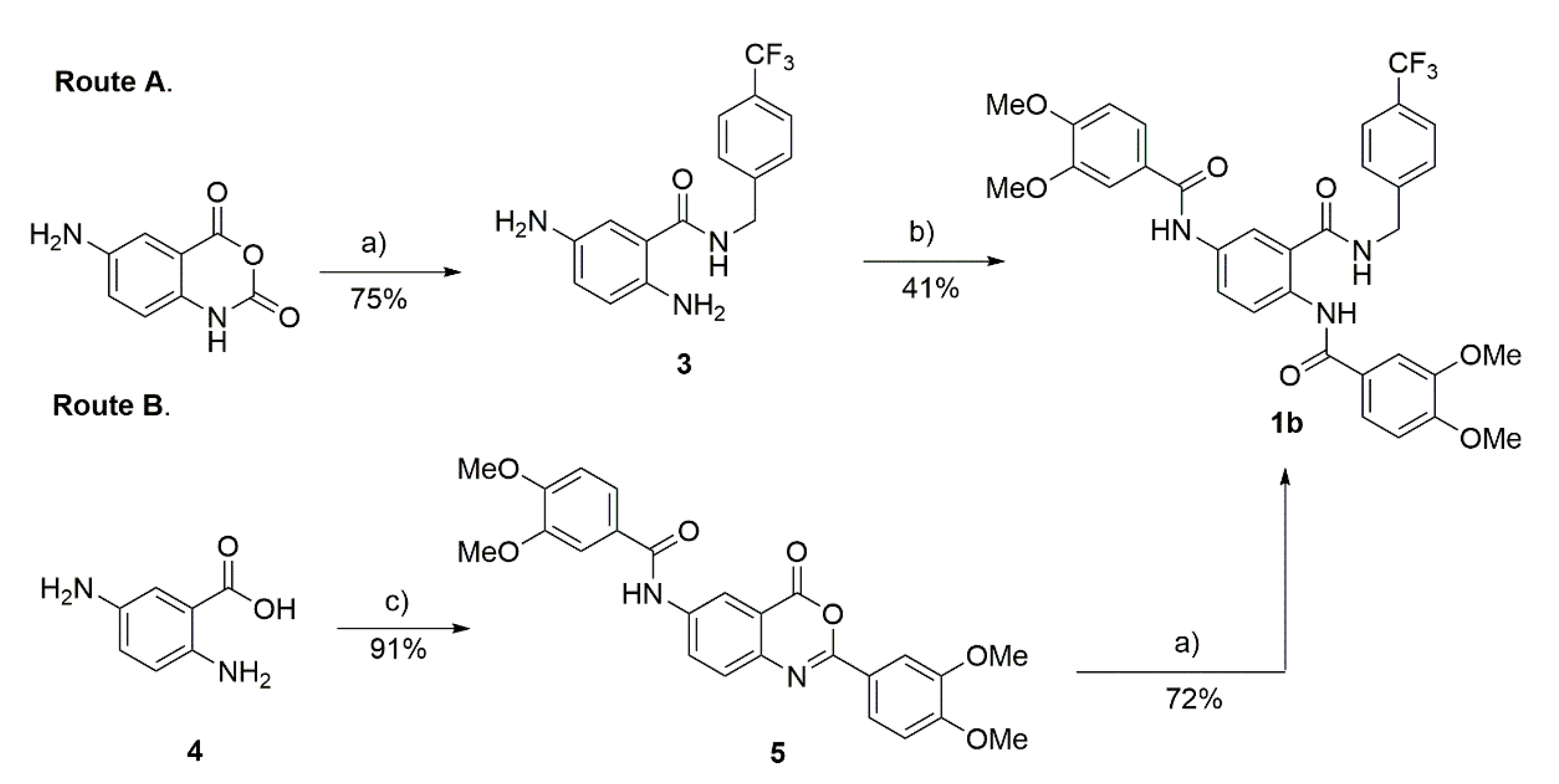
2.2. Preparation of Compound 1b
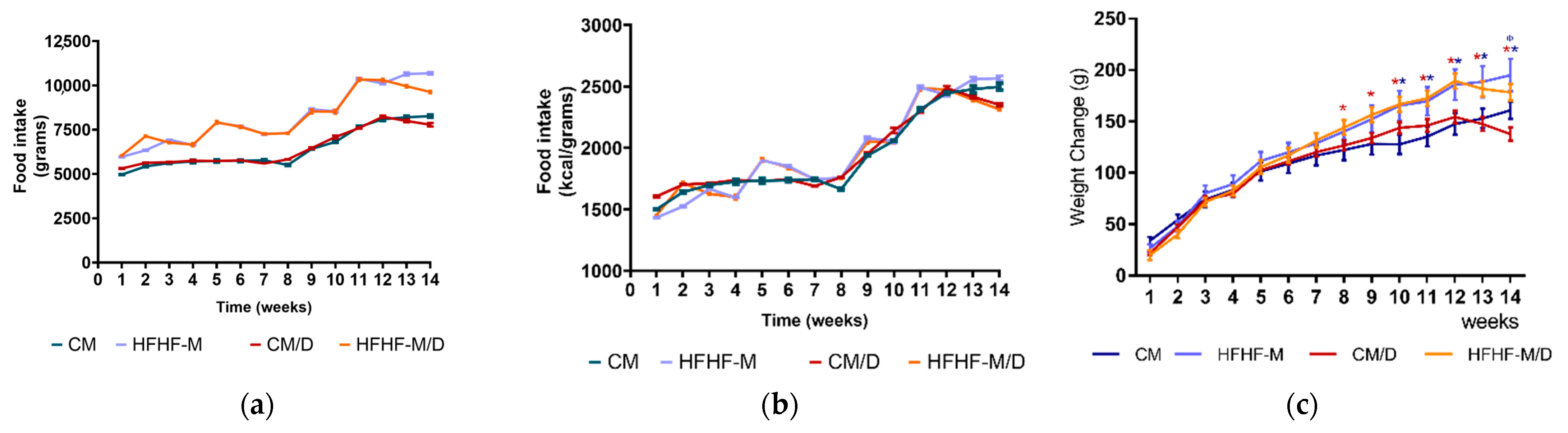
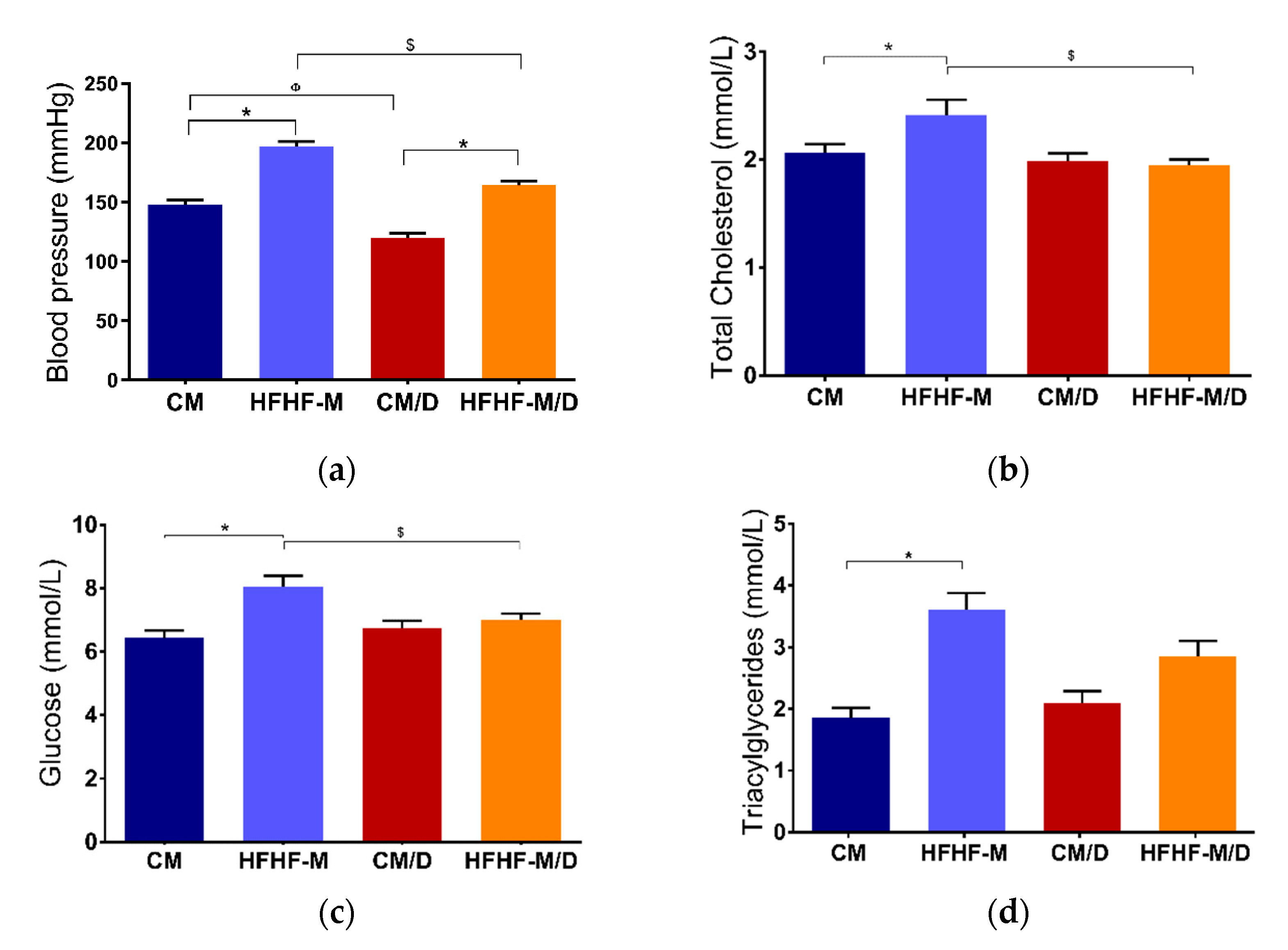
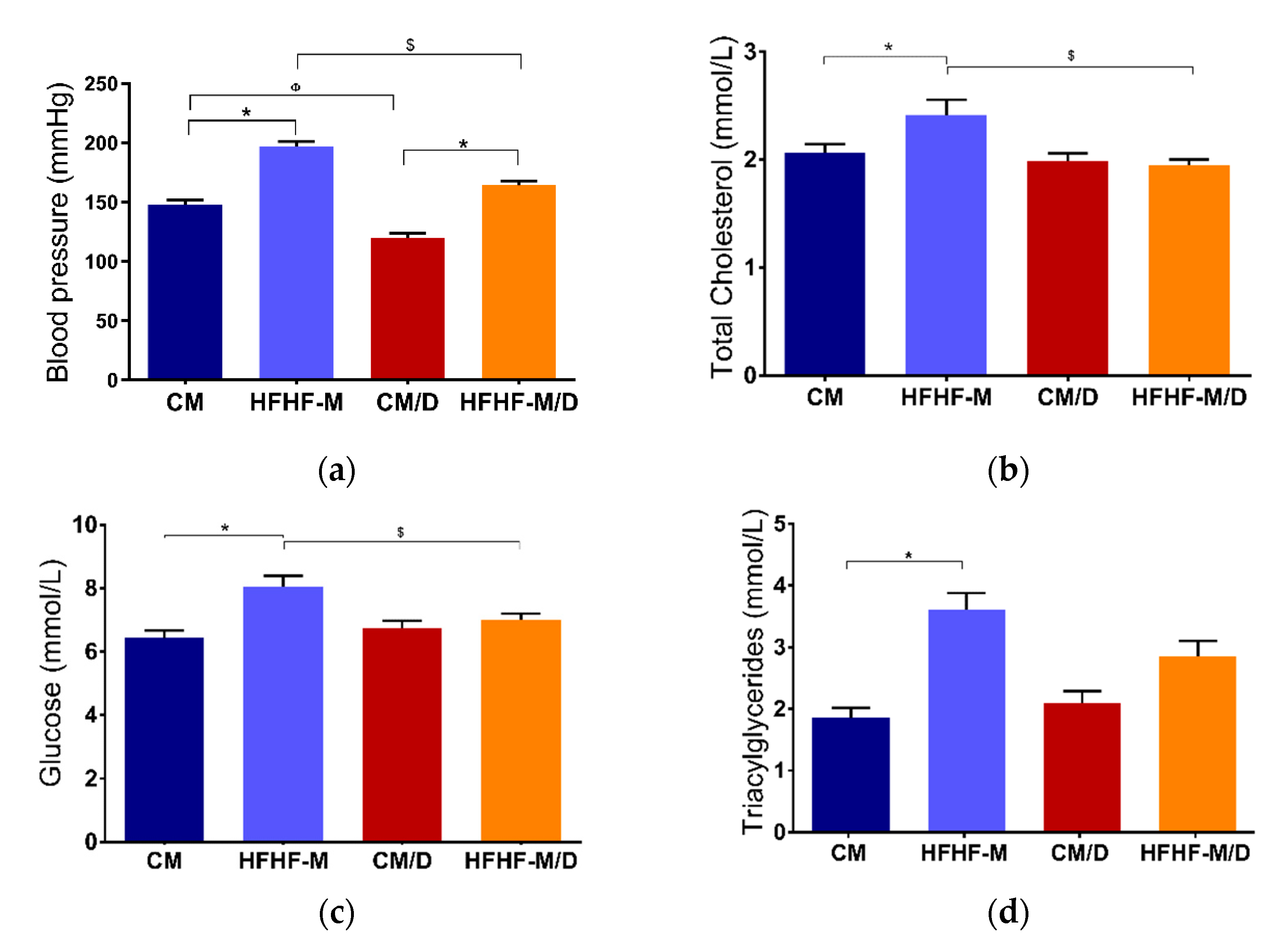
2.3. In Vivo Evaluation
3. Discussion
3.1. In Silico Studies
3.2. Preparation of Compound 1b
3.3. In Vivo Evaluation
4. Materials and Methods
4.1. In Silico Studies
4.1.1. Ligand Design
4.1.2. Docking Studies
4.2. Chemistry
4.2.1. 2,5-Diaminobenzoic Acid (4)
4.2.2. N-(2-(3,4-Dimethoxyphenyl)-4-oxo-4H-benzo[d][1,3]oxazin-6-yl)-3,4-DIMETHOXYBENZAMIDE (5)
4.2.3. N, N′-(2-((4-(Trifluoromethyl)benzyl)carbamoyl)-1,4-phenylene)bis(3,4-dimethoxybenzamide) (1b)
4.3. In Vivo Evaluation in Metabolic Syndrome
4.3.1. Animals
4.3.2. MetS Induction and Treatment with Compound 1b
4.3.3. In Vivo Acute Toxicity Determination
4.3.4. Triacylglycerides, Cholesterol and Glucose Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACAT | Acyl-coenzyme A cholesterol acyltransferase |
| ACE | Angiotensin-converting enzyme |
| ADME/Tox profile | Absorption, Distribution, Metabolism, Excretion and Toxicity profile |
| CETP | Cholesterylester transfer protein |
| DPP4 | Dipeptidyl peptidase 4 |
| FFAR–1 | Free fatty acid receptor -1 |
| FXR | Farnesoid X receptor |
| hERG | Human ether-à-go-go-related gene |
| HFHF diet | High fructose and high-fat diet |
| HMG–CoA reductase | Hydroxymethylglutaryl coenzyme A reductase |
| MetS | Metabolic syndrome |
| PPAR | Peroxisome proliferator–activated receptor |
| PTP1B | Protein tyrosine phosphatase 1B |
References
- Grundy, S.M. Metabolic syndrome update. Trends Cardiovasc. Med. 2016, 26, 364–373. [Google Scholar] [CrossRef]
- Alberti, K.; Eckel, R.; Grundy, S.; Zimmet, P.; Cleeman, J.; Donato, K.; Fruchart, J.; James, W.; Loria, C.; Smith, S.J. Harmonizing the Metabolic Syndrome: A Joint Interim Statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009, 120, 1640–1645. [Google Scholar]
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, P.M.; Tuomilehto, J.; Rydén, L. The metabolic syndrome—What is it and how should it be managed? Eur. J. Prev. Cardiol. 2019, 26, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Martínez, P.; Mikhailidis, D.P.; Athyros, V.G.; Bullo, M.; Couture, P.; Covas, M.I.; de Koning, L.; Delgado-Lista, J.; Díaz-López, A.; Drevon, C.A.; et al. Lifestyle recommendations for the prevention and management of metabolic syndrome: An international panel recommendation. Nutr. Rev. 2017, 75, 307–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Li, X.; Adams, H.; Kubena, K.; Guo, S. Etiology of Metabolic Syndrome and Dietary Intervention. Int. J. Mol. Sci. 2019, 20, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundy, S.M. Drug therapy of the metabolic syndrome: Minimizing the emerging crisis in polypharmacy. Nat. Rev. Drug Discov. 2006, 5, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Masnoon, N.; Shakib, S.; Kalisch-Ellett, L.; Caughey, G.E. What is polypharmacy? A systematic review of definitions. BMC Geriatr. 2017, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, L.E.; Spiers, G.; Kingston, A.; Todd, A.; Adamson, J.; Hanratty, B. Adverse Outcomes of Polypharmacy in Older People: Systematic Review of Reviews. J. Am. Med. Dir. Assoc. 2020, 21, 181–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proschak, E. Reconsidering the drug discovery pipeline for designed multitarget drugs. Drug Discov. Today 2013, 18, 1129–1130. [Google Scholar] [CrossRef]
- Prati, F.; Cavalli, A.; Bolognesi, M.L. Navigating the Chemical Space of Multitarget-Directed Ligands: From Hybrids to Fragments in Alzheimer’s Disease. Molecules 2016, 21, 466. [Google Scholar] [CrossRef] [Green Version]
- Ammazzalorso, A.; Maccallini, C.; Amoia, P.; Amoroso, R. Multitarget PPARγ agonists as innovative modulators of the metabolic syndrome. Eur. J. Med. Chem. 2019, 173, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Huang, B.; Zhang, Y. Recent advances in multitarget-directed ligands targeting G-protein-coupled receptors. Drug Discov. Today 2020, 25, 1682–1692. [Google Scholar] [CrossRef]
- Talevi, A. Multi-target pharmacology: Possibilities and limitations of the "skeleton key approach" from a medicinal chemist perspective. Front. Pharmacol. 2015, 6, 205. [Google Scholar] [CrossRef] [Green Version]
- Medina-Franco, J.L.; Giulianotti, M.A.; Welmaker, G.S.; Houghten, R.A. Shifting from the single to the multitarget paradigm in drug discovery. Drug Discov. Today 2013, 18, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Bolognesi, M.L.; Cavalli, A. Multitarget Drug Discovery and Polypharmacology. ChemMedChem 2016, 11, 1190–1192. [Google Scholar] [CrossRef] [Green Version]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multitarget drug discovery and design for complex diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Katselou, M.G.; Matralis, A.N.; Kourounakis, A.P. Multi-Target Drug Design Approaches for Multifactorial Diseases: From Neurodegenerative to Cardiovascular Applications. Curr. Med. Chem. 2014, 21, 2743–2787. [Google Scholar] [CrossRef] [PubMed]
- Alberca, L.N.; Talevi, A. The Efficiency of Multi-target Drugs: A Network Approach. Approaching Complex Dis. 2020, 3, 63–75. [Google Scholar] [CrossRef]
- Moller, D.E. Metabolic disease drug discovery—“Hitting the target” is easier said than done. Cell Metab. 2012, 15, 19–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakshmi, P.K.; Kumar, S.; Pawar, S.; Kuriakose, B.B.; Sudheesh, M.S.; Pawar, R.S. Targeting metabolic syndrome with phytochemicals: Focus on the role of molecular chaperones and hormesis in drug discovery. Pharmacol. Res. 2020, 159, 104925. [Google Scholar] [CrossRef]
- De Oliveira, A.A.; Davis, D.; Nunes, K.P. Pattern recognition receptors as potential therapeutic targets in metabolic syndrome: From bench to bedside. Diabetes Metab. Syndr. Clin. Res. Rev. 2019, 13, 1117–1122. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Kong, L.-D. High fructose diet-induced metabolic syndrome: Pathophysiological mechanism and treatment by traditional Chinese medicine. Pharmacol. Res. 2018, 130, 438–450. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Pei, J.; Lai, L. Computational Multitarget Drug Design. J. Chem. Inf. Model. 2017, 57, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Levin, J.I.; Chen, J.M.; Du, M.T.; Nelson, F.C.; Wehr, T.; DiJoseph, J.F.; Killar, L.M.; Skala, S.; Sung, A.; Sharr, M.A.; et al. The discovery of anthranilic acid-based MMP inhibitors. Part 3: Incorporation of basic amines. Bioorganic Med. Chem. Lett. 2001, 11, 2975–2978. [Google Scholar] [CrossRef]
- Shi, L.; Hu, R.; Wei, Y.; Liang, Y.; Yang, Z.; Ke, S. Anthranilic acid-based diamides derivatives incorporating aryl-isoxazoline pharmacophore as potential anticancer agents: Design, synthesis and biological evaluation. Eur. J. Med. Chem. 2012, 54, 549–556. [Google Scholar] [CrossRef]
- Ye, B.; Arnaiz, D.O.; Chou, Y.-L.; Griedel, B.D.; Karanjawala, R.; Lee, W.; Morrissey, M.M.; Sacchi, K.L.; Sakata, S.T.; Shaw, K.J.; et al. Thiophene-Anthranilamides as Highly Potent and Orally Available Factor Xa Inhibitors1. J. Med. Chem. 2007. [Google Scholar] [CrossRef]
- El-Azab, A.S.; Abdel-Aziz, A.A.M.; Bua, S.; Nocentini, A.; AlSaif, N.A.; Almehizia, A.A.; Alanazi, M.M.; Hefnawy, M.M.; Supuran, C.T. New anthranilic acid-incorporating N-benzenesulfonamidophthalimides as potent inhibitors of carbonic anhydrases I, II, IX, and XII: Synthesis, in vitro testing, and in silico assessment. Eur. J. Med. Chem. 2019, 181, 111573. [Google Scholar] [CrossRef]
- Han, S.H.; Suh, H.S.; Jo, H.; Oh, Y.; Mishra, N.K.; Han, S.; Kim, H.S.; Jung, Y.H.; Lee, B.M.; Kim, I.S. Synthesis and anti-inflammatory evaluation of N-sulfonyl anthranilic acids via Ir(III)-catalyzed C–H amidation of benzoic acids. Bioorganic Med. Chem. Lett. 2017, 27, 2129–2134. [Google Scholar] [CrossRef]
- Hartung, I.V.; Hammer, S.; Hitchcock, M.; Neuhaus, R.; Scholz, A.; Siemeister, G.; Bohlmann, R.; Hillig, R.C.; Pühler, F. Optimization of allosteric MEK inhibitors. Part 2: Taming the sulfamide group balances compound distribution properties. Bioorganic Med. Chem. Lett. 2016, 26, 186–193. [Google Scholar] [CrossRef]
- Merk, D.; Gabler, M.; Gomez, R.C.; Flesch, D.; Hanke, T.; Kaiser, A.; Lamers, C.; Werz, O.; Schneider, G.; Schubert-Zsilavecz, M. Anthranilic acid derivatives as novel ligands for farnesoid X receptor (FXR). Bioorganic Med. Chem. 2014, 22, 2447–2460. [Google Scholar] [CrossRef]
- Merk, D.; Lamers, C.; Weber, J.; Flesch, D.; Gabler, M.; Proschak, E.; Schubert-Zsilavecz, M. Anthranilic acid derivatives as nuclear receptor modulators—Development of novel PPAR selective and dual PPAR/FXR ligands. Bioorganic Med. Chem. 2015, 23, 499–514. [Google Scholar] [CrossRef]
- Heitel, P.; Faudone, G.; Helmstädter, M.; Schmidt, J.; Kaiser, A.; Tjaden, A.; Schröder, M.; Müller, S.; Schierle, S.; Pollinger, J.; et al. A triple farnesoid X receptor and peroxisome proliferator-activated receptor α/δ activator reverses hepatic fibrosis in diet-induced NASH in mice. Commun. Chem. 2020, 3, 1–16. [Google Scholar] [CrossRef]
- Gagnon, M.K.J.; Hausner, S.H.; Marik, J.; Abbey, C.K.; Marshall, J.F.; Sutcliffe, J.L. High-throughput in vivo screening of targeted molecular imaging agents. Proc. Natl. Acad. Sci. USA 2009, 106, 17904–17909. [Google Scholar] [CrossRef] [Green Version]
- Ben-Yakar, A. High-content and high-throughput in vivo drug screening platforms using microfluidics. Assay Drug Dev. Technol. 2019, 17, 8–13. [Google Scholar] [CrossRef] [Green Version]
- White, D.T.; Eroglu, A.U.; Wang, G.; Zhang, L.; Sengupta, S.; Ding, D.; Rajpurohit, S.K.; Walker, S.L.; Ji, H.; Qian, J.; et al. ARQiv-HTS, a versatile whole-organism screening platform enabling in vivo drug discovery at high-throughput rates. Nat. Protoc. 2016, 11, 2432–2453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, L.; Panchal, S.K. Rodent models for metabolic syndrome research. J. Biomed. Biotechnol. 2011, 2011, 351982. [Google Scholar] [CrossRef] [Green Version]
- Fellmann, L.; Nascimento, A.R.; Tibiriça, E.; Bousquet, P. Murine models for pharmacological studies of the metabolic syndrome. Pharmacol. Ther. 2013, 137, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.J.; Ellacott, K.L.J.; King, V.L.; Hasty, A.H. Mouse models of the metabolic syndrome. DMM Dis. Model. Mech. 2010, 3, 156–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, S.K.; Chin, K.Y.; Suhaimi, F.H.; Fairus, A.; Ima-Nirwana, S. Animal models of metabolic syndrome: A review. Nutr. Metab. 2016, 13, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinosa-García, C.; Fuentes-Venado, C.E.; Guerra-Araiza, C.; Segura-Uribe, J.; Chávez-Gutiérrez, E.; Farfán-García, E.D.; Estrada Cruz, N.A.; Pinto-Almazán, R. Sex differences in the performance of cognitive tasks in a murine model of metabolic syndrome. Eur. J. Neurosci. 2020, 52, 2724–2736. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Grundy, S.M. Metabolic syndrome: Therapeutic considerations. In Handbook of Experimental Pharmacology; Springer: Berlin, Germany, 2005; pp. 107–133. [Google Scholar]
- Kim, S.W.; Kang, H.J.; Jhon, M.; Kim, J.W.; Lee, J.Y.; Walker, A.J.; Agustini, B.; Kim, J.M.; Berk, M. Statins and inflammation: New therapeutic opportunities in psychiatry. Front. Psychiatry 2019, 10, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamagishi, S.; Takeuchi, M. Telmisartan is a promising cardiometabolic sartan due to its unique PPAR-γ-inducing property. Med. Hypotheses 2005, 64, 476–478. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Z.; Huang, S.T.; Wen, Y.W.; Chen, L.K.; Hsiao, F.Y. Combined Effects of Frailty and Polypharmacy on Health Outcomes in Older Adults: Frailty Outweighs Polypharmacy. J. Am. Med. Dir. Assoc. 2020, 22. [Google Scholar] [CrossRef]
- Kawasaki, M.; Kambe, A.; Yamamoto, Y.; Arulmozhiraja, S.; Ito, S.; Nakagawa, Y.; Tokiwa, H.; Nakano, S.; Shimano, H. Elucidation of Molecular Mechanism of a Selective PPARα Modulator, Pemafibrate, through Combinational Approaches of X-ray Crystallography, Thermodynamic Analysis, and First-Principle Calculations. Int. J. Mol. Sci. 2020, 21, 361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhajed, S.S.; Shinde, P.E.; Kshirsagar, S.J.; Sangshetti, J.N.; Gupta, P.P.; Parab, M.M.; Dasgupta, D. De-novo design and synthesis of conformationally restricted thiazolidine-2,4-dione analogues: Highly selective PPAR-γ agonist in search of anti-diabetic agent. Struct. Chem. 2020, 31, 1375–1385. [Google Scholar] [CrossRef]
- Garcia-Vallvé, S.; Guasch, L.; Tomas-Hernández, S.; Del Bas, J.M.; Ollendorff, V.; Arola, L.; Pujadas, G.; Mulero, M. Peroxisome Proliferator-Activated Receptor γ (PPARγ) and Ligand Choreography: Newcomers Take the Stage. J. Med. Chem. 2015, 58, 5381–5394. [Google Scholar] [CrossRef]
- Capelli, D.; Cerchia, C.; Montanari, R.; Loiodice, F.; Tortorella, P.; Laghezza, A.; Cervoni, L.; Pochetti, G.; Lavecchia, A. Structural basis for PPAR partial or full activation revealed by a novel ligand binding mode. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Abe, K.; Toma, T.; Nishikawa, M.; Ozawa, H.; Okuda, A.; Araki, T.; Oda, S.; Inoue, K.; Shibuya, K.; et al. Design and synthesis of highly potent and selective human peroxisome proliferator-activated receptor α agonists. Bioorganic Med. Chem. Lett. 2007, 17, 4689–4693. [Google Scholar] [CrossRef]
- Shvekhgeimer, M.G.A. Synthesis of heterocyclic compounds based on isatoic anhydrides (2H-3,1-benzoxazine-2,4-diones). (Review). Chem. Heterocycl. Compd. 2001, 37, 385–443. [Google Scholar] [CrossRef]
- Marasini, B.P.; Rahim, F.; Perveen, S.; Karim, A.; Mohammed Khan, K.; Atta-ur-Rahman; Choudhary, M.I. Synthesis, structure-activity relationships studies of benzoxazinone derivatives as α-chymotrypsin inhibitors. Bioorganic Chem. 2017, 70, 210–221. [Google Scholar] [CrossRef]
- Shariat, M.; Abdollahi, S. Synthesis of Benzoxazinone Derivatives: A New Route to 2 (N Phthaloylmethyl)-4H-3,1-benzoxazin-4-one. Molecules 2004, 9, 705–712. [Google Scholar] [CrossRef]
- Berthelsen, R.; Holm, R.; Jacobsen, J.; Kristensen, J.; Abrahamsson, B.; Müllertz, A. Kolliphor surfactants affect solubilization and bioavailability of fenofibrate. Studies of in vitro digestion and absorption in rats. Mol. Pharm. 2015, 12, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Tenenbaum, A.; Fisman, E.Z.; Motro, M. Metabolic syndrome and type 2 diabetes mellitus: Focus on peroxisome proliferator activated receptors (PPAR). Cardiovasc. Diabetol. 2003, 2, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Staels, B. PPAR Agonists and the Metabolic Syndrome. Therapies 2007, 62, 319–326. [Google Scholar] [CrossRef]
- Larsen, P.J.; Jensen, P.B.; Sørensen, R.V.; Larsen, L.K.; Vrang, N.; Wulff, E.M.; Wassermann, K. Differential Influences of Peroxisome Proliferator–Activated Receptorsγ and -α on Food Intake and Energy Homeostasis. Diabetes 2003, 52, 2249–2259. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Liu, H.; Jia, Z.; Guan, G.; Yang, T. Effects of Endogenous PPAR Agonist Nitro-Oleic Acid on Metabolic Syndrome in Obese Zucker Rats. PPAR Res. 2010, 2010. [Google Scholar] [CrossRef] [Green Version]
- Castrejón-Tellez, V.; Rodríguez-Pérez, J.M.; Pérez-Torres, I.; Pérez-Hernández, N.; Cruz-Lagunas, A.; Guarner-Lans, V.; Vargas-Alarcón, G.; Rubio-Ruiz, M.E. The Effect of Resveratrol and Quercetin Treatment on PPAR Mediated Uncoupling Protein (UCP-) 1, 2, and 3 Expression in Visceral White Adipose Tissue from Metabolic Syndrome Rats. Int. J. Mol. Sci. 2016, 17, 1069. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Gaetani, S.; Oveisi, F.; Lo Verme, J.; Serrano, A.; Rodríguez de Fonseca, F.; Rosengarth, A.; Luecke, H.; Di Giacomo, B.; Tarzia, G.; et al. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-α. Nature 2003, 425, 90–93. [Google Scholar] [CrossRef]
- Das, U.N. Essential fatty acids and their metabolites could function as endogenous HMG-CoA reductase and ACE enzyme inhibitors, anti-arrhythmic, antihypertensive, anti-atherosclerotic, anti-inflammatory, cytoprotective, and cardioprotective molecules. Lipids Health Dis. 2008, 7, 37. [Google Scholar] [CrossRef] [Green Version]
- Hetzel, J.; Balletshofer, B.; Rittig, K.; Walcher, D.; Kratzer, W.; Hombach, V.; Häring, H.-U.; Koenig, W.; Marx, N. Rapid Effects of Rosiglitazone Treatment on Endothelial Function and Inflammatory Biomarkers. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1804–1809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calkin, A.C.; Forbes, J.M.; Smith, C.M.; Lassila, M.; Cooper, M.E.; Jandeleit-Dahm, K.A.; Allen, T.J. Rosiglitazone Attenuates Atherosclerosis in a Model of Insulin Insufficiency Independent of Its Metabolic Effects. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1903–1909. [Google Scholar] [CrossRef] [Green Version]
- Ketsawatsomkron, P.; Pelham, C.J.; Groh, S.; Keen, H.L.; Faraci, F.M.; Sigmund, C.D. Does Peroxisome Proliferator-activated Receptor-γ (PPARγ) Protect from Hypertension Directly through Effects in the Vasculature? J. Biol. Chem. 2010, 285, 9311–9316. [Google Scholar] [CrossRef] [Green Version]
- De las Heras, N.; Valero-Muñoz, M.; Ballesteros, S.; Gómez-Hernández, A.; Martín-Fernández, B.; Blanco-Rivero, J.; Cachofeiro, V.; Benito, M.; Balfagón, G.; Lahera, V. Factors involved in rosuvastatin induction of insulin sensitization in rats fed a high fat diet. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 1107–1114. [Google Scholar] [CrossRef]
- Shiomi, Y.; Yamauchi, T.; Iwabu, M.; Okada-Iwabu, M.; Nakayama, R.; Orikawa, Y.; Yoshioka, Y.; Tanaka, K.; Ueki, K.; Kadowaki, T. A Novel Peroxisome Proliferator-activated Receptor (PPAR)α Agonist and PPARγ Antagonist, Z-551, Ameliorates High-fat Diet-induced Obesity and Metabolic Disorders in Mice. J. Biol. Chem. 2015, 290, 14567–14581. [Google Scholar] [CrossRef] [Green Version]
- Lorke, D. A new approach to practical acute toxicity testing. Arch. Toxicol. 1983, 54, 275–287. [Google Scholar] [CrossRef]
- Spartan 10 for Windows; Wavefunction Inc.: Irvine, CA, USA, 2010.
- Thomsen, R.; Christensen, M.H. MolDock: A new technique for high-accuracy molecular docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Guan, C.; Niu, Y.; Chen, S.-C.; Kang, Y.; Wu, J.-X.; Nishi, K.; Chang, C.C.Y.; Chang, T.-Y.; Luo, T.; Chen, L. Structural insights into the inhibition mechanism of human sterol O-acyltransferase 1 by a competitive inhibitor. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Howard, E.I.; Sanishvili, R.; Cachau, R.E.; Mitschler, A.; Chevrier, B.; Barth, P.; Lamour, V.; Van Zandt, M.; Sibley, E.; Bon, C.; et al. Ultrahigh resolution drug design I: Details of interactions in human aldose reductase–inhibitor complex at 0.66 Å. Proteins Struct. Funct. Bioinform. 2004, 55, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Sutton, J.M.; Clark, D.E.; Dunsdon, S.J.; Fenton, G.; Fillmore, A.; Harris, N.V.; Higgs, C.; Hurley, C.A.; Krintel, S.L.; MacKenzie, R.E.; et al. Novel heterocyclic DPP-4 inhibitors for the treatment of type 2 diabetes. Bioorganic Med. Chem. Lett. 2012, 22, 1464–1468. [Google Scholar] [CrossRef] [PubMed]
- Natesh, R.; Schwager, S.L.U.; Sturrock, E.D.; Acharya, K.R. Crystal structure of the human angiotensin-converting enzyme–lisinopril complex. Nature 2003, 421, 551–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Downes, M.; Verdecia, M.A.; Roecker, A.J.; Hughes, R.; Hogenesch, J.B.; Kast-Woelbern, H.R.; Bowman, M.E.; Ferrer, J.-L.; Anisfeld, A.M.; Edwards, P.A.; et al. A Chemical, Genetic, and Structural Analysis of the Nuclear Bile Acid Receptor FXR. Mol. Cell 2003, 11, 1079–1092. [Google Scholar] [CrossRef]
- Srivastava, A.; Yano, J.; Hirozane, Y.; Kefala, G.; Gruswitz, F.; Snell, G.; Lane, W.; Ivetac, A.; Aertgeerts, K.; Nguyen, J.; et al. High-resolution structure of the human GPR40 receptor bound to allosteric agonist TAK-875. Nature 2014, 513, 124–127. [Google Scholar] [CrossRef]
- Istvan, E.S.; Deisenhofer, J. Structural Mechanism for Statin Inhibition of HMG-CoA Reductase. Science 2001, 292, 1160–1164. [Google Scholar] [CrossRef] [Green Version]
- Cronet, P.; Petersen, J.F.W.; Folmer, R.; Blomberg, N.; Sjöblom, K.; Karlsson, U.; Lindstedt, E.L.; Bamberg, K. Structure of the PPARα and -γ ligand binding domain in complex with AZ 242; ligand selectivity and agonist activation in the PPAR family. Structure 2001, 9, 699–706. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Liu, G.; Xin, Z.; Serby, M.D.; Pei, Z.; Szczepankiewicz, B.G.; Hajduk, P.J.; Abad-Zapatero, C.; Hutchins, C.W.; Lubben, T.H.; et al. Isoxazole carboxylic acids as protein tyrosine phosphatase 1B (PTP1B) inhibitors. Bioorganic Med. Chem. Lett. 2004, 14, 5543–5546. [Google Scholar] [CrossRef]
- Loza-Mejía, M.A.; Salazar, J.R. Sterols and triterpenoids as potential anti-inflammatories: Molecular docking studies for binding to some enzymes involved in inflammatory pathways. J. Mol. Graph. Model. 2015, 62, 18–25. [Google Scholar] [CrossRef]
- Harris, R.K.; Becker, E.D.; de Menezes, S.M.C.; Goodfellow, R.; Granger, P. NMR nomenclature: Nuclear spin properties and conventions for chemical shifts. IUPAC Recommendations 2001. International Union of Pure and Applied Chemistry. Physical Chemistry Division. Commission on Molecular Structure and Spectroscopy. Magn. Reson. Chem. 2002, 40, 489–505. [Google Scholar] [CrossRef]
- Berger, S.; Braun, S. 200 and More NMR Experiments: A Practical Course; Wiley-VCH: Weinheim, Germany, 2004; 838p. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Ligand. | Benzylamine Substituents | Benzamide Substituents | PPAR-α | PPAR-γ | HMG-CoA Reductase | ACE |
| 48a | 4′-CF3 | 3′,4′-diOMe | −118.7 | −127.9 | −111.8 | −111.3 |
| 45a | 2′,4′-di-OMe | 3′,4′-diOMe | −119.0 | −124.2 | −113.3 | −97.3 |
| 44a | 3′,4′-diOMe | 3′,4′-diOMe | −125.0 | −119.5 | −112.5 | −90.9 |
| 28a | 4′-CF3 | 4′-NO2 | −113.8 | −116.7 | −99.0 | −119.7 |
| 88a | 4′-CF3 | 4′-SO2NH2 | −118.7 | −127.9 | −111.8 | −111.6 |
| 1b | -- | -- | −151.6 | −165.2 | −156.2 | −137.2 |
| Reference 2 | -- | -- | −113.8 | −116.7 | −164.5 | −123.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Álvarez, H.; Bravo-Jiménez, A.; Martínez-Arellanes, M.; Gamboa-Osorio, G.O.; Chávez-Gutiérrez, E.; González-Hernández, L.A.; Gallardo-Ignacio, K.; Quintana-Romero, O.J.; Ariza-Castolo, A.; Guerra-Araiza, C.; et al. In Silico-Based Design and In Vivo Evaluation of an Anthranilic Acid Derivative as a Multitarget Drug in a Diet-Induced Metabolic Syndrome Model. Pharmaceuticals 2021, 14, 914. https://doi.org/10.3390/ph14090914
González-Álvarez H, Bravo-Jiménez A, Martínez-Arellanes M, Gamboa-Osorio GO, Chávez-Gutiérrez E, González-Hernández LA, Gallardo-Ignacio K, Quintana-Romero OJ, Ariza-Castolo A, Guerra-Araiza C, et al. In Silico-Based Design and In Vivo Evaluation of an Anthranilic Acid Derivative as a Multitarget Drug in a Diet-Induced Metabolic Syndrome Model. Pharmaceuticals. 2021; 14(9):914. https://doi.org/10.3390/ph14090914
Chicago/Turabian StyleGonzález-Álvarez, Héctor, Astrid Bravo-Jiménez, Matilda Martínez-Arellanes, Gabriela Odette Gamboa-Osorio, Edwin Chávez-Gutiérrez, Lino A. González-Hernández, Karina Gallardo-Ignacio, Osvaldo J. Quintana-Romero, Armando Ariza-Castolo, Christian Guerra-Araiza, and et al. 2021. "In Silico-Based Design and In Vivo Evaluation of an Anthranilic Acid Derivative as a Multitarget Drug in a Diet-Induced Metabolic Syndrome Model" Pharmaceuticals 14, no. 9: 914. https://doi.org/10.3390/ph14090914
APA StyleGonzález-Álvarez, H., Bravo-Jiménez, A., Martínez-Arellanes, M., Gamboa-Osorio, G. O., Chávez-Gutiérrez, E., González-Hernández, L. A., Gallardo-Ignacio, K., Quintana-Romero, O. J., Ariza-Castolo, A., Guerra-Araiza, C., Martino-Roaro, L., Meneses-Ruiz, D. M., Pinto-Almazán, R., & Loza-Mejía, M. A. (2021). In Silico-Based Design and In Vivo Evaluation of an Anthranilic Acid Derivative as a Multitarget Drug in a Diet-Induced Metabolic Syndrome Model. Pharmaceuticals, 14(9), 914. https://doi.org/10.3390/ph14090914