
Transient Receptor Potential Ankyrin-1 (TRPA1) Block Protects against Loss of White Matter Function during Ischaemia in the Mouse Optic Nerve



Abstract

1. Introduction
2. Results
2.1. Oligodendrocytes Express Tonically Active TRPA1
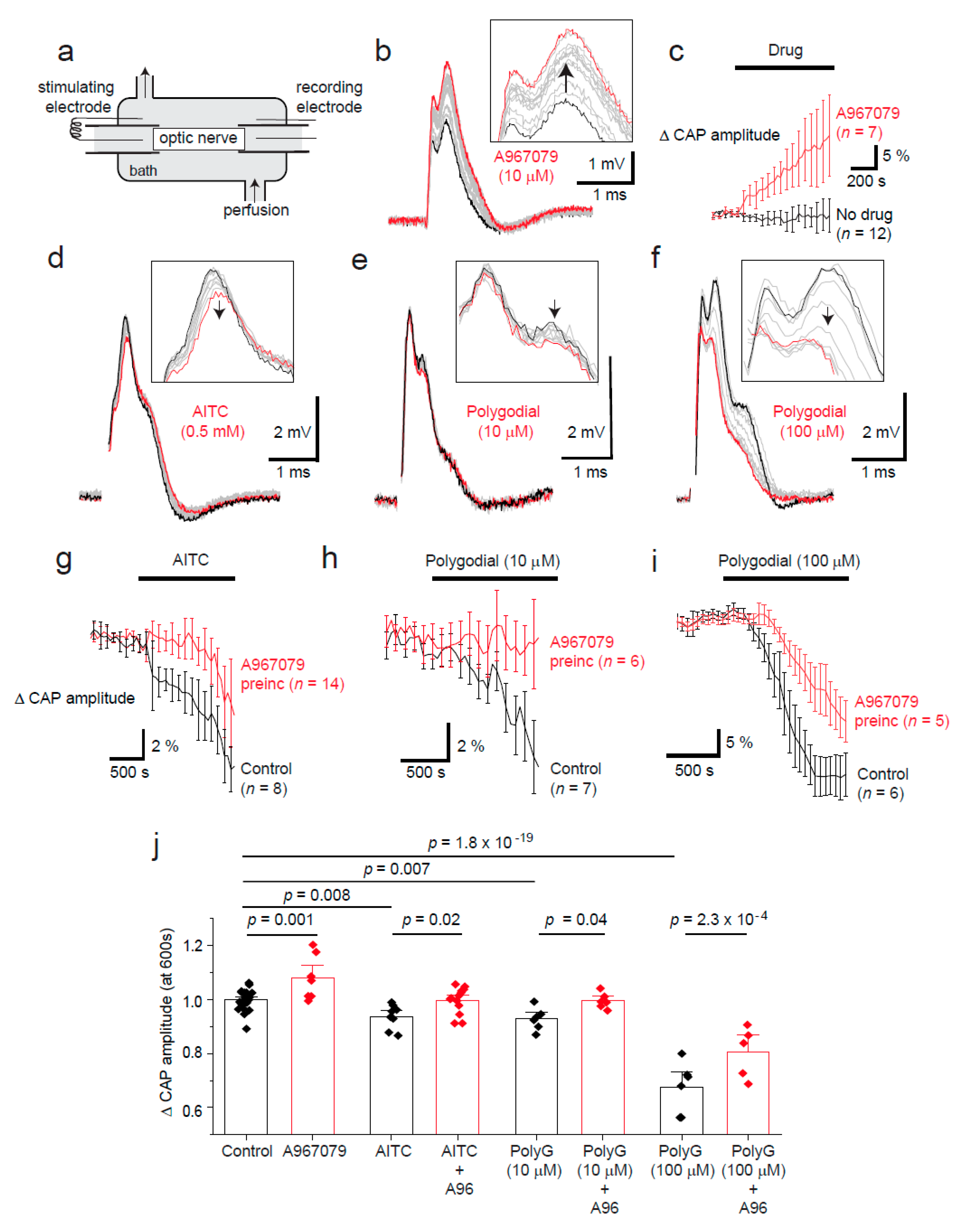
2.2. Tonic TRPA1 Activation Regulates the Compound Action Potential Amplitude in the Optic Nerve
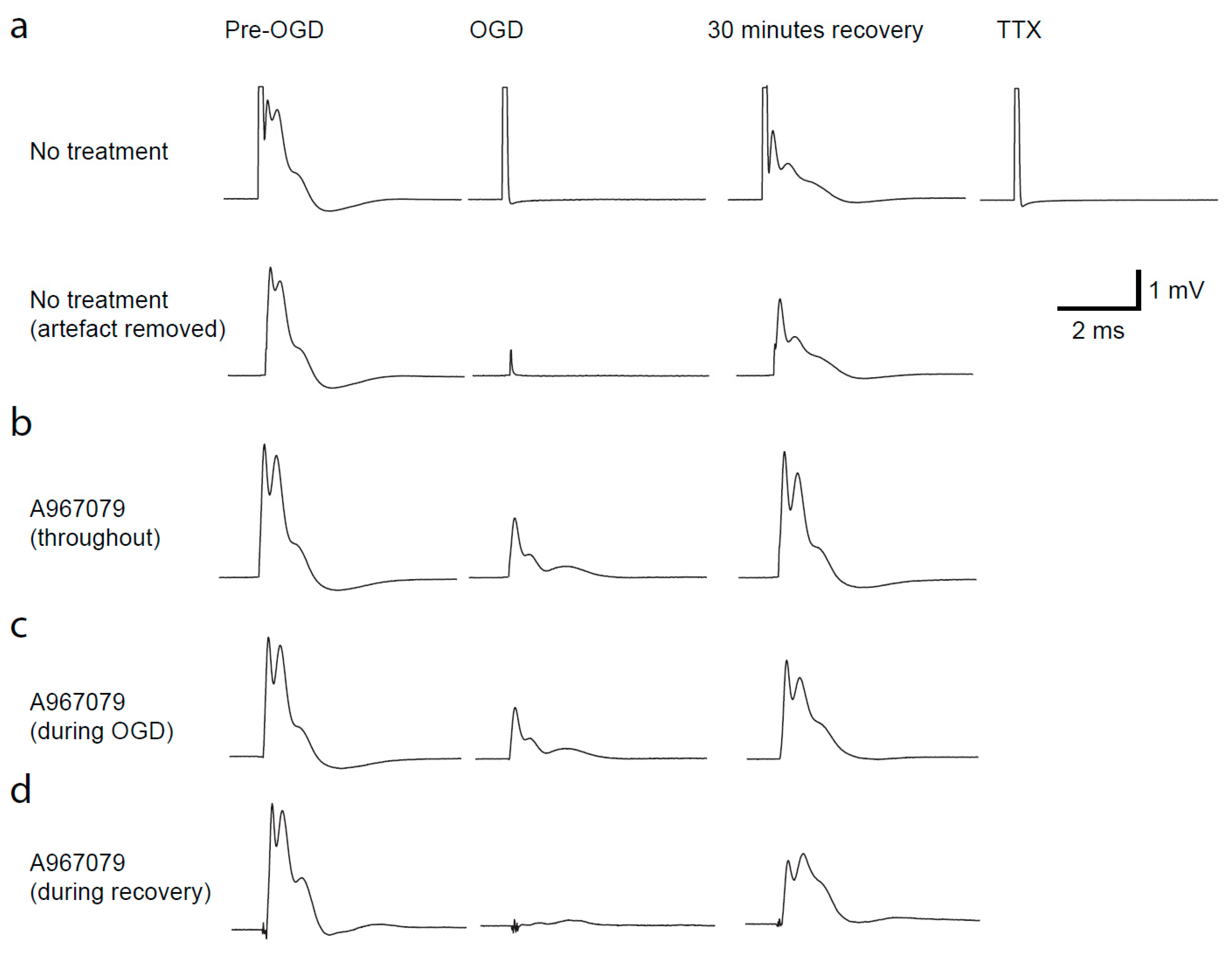
2.3. TRPA1 Block Protects against Loss of the Compound Action Potential during OGD
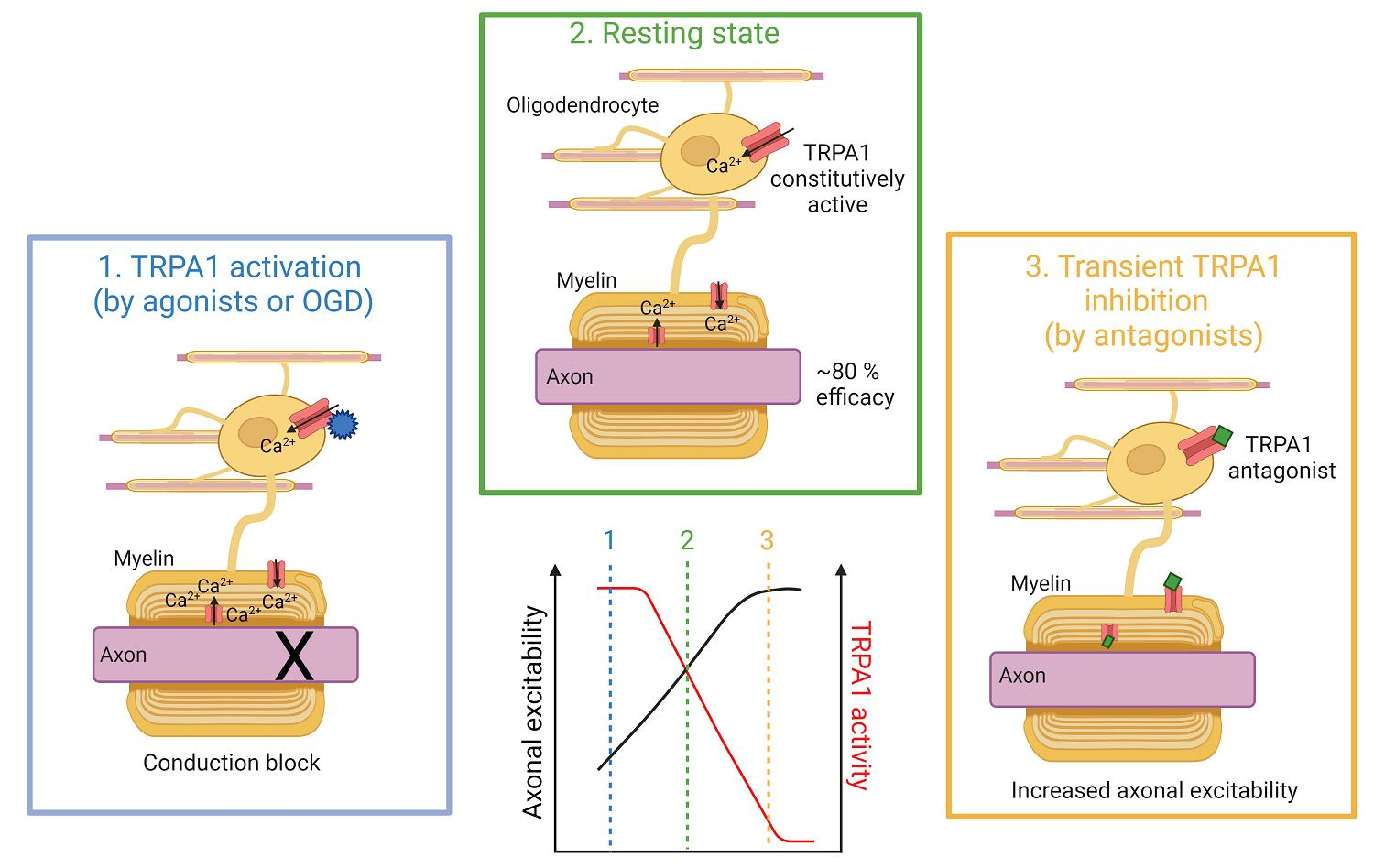
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Brain Slice and Optic Nerve Preparation
4.3. Cell Identification and Electrophysiology
4.4. External Solutions
4.5. Intracellular Solutions
4.6. Single Cell Ion Imaging
4.7. Drug Application
4.8. Compound Action Potential Recording
4.9. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, L.-L.; Huang, Y.-N.; Cui, Z.-T. Clinical features of acute corpus callosum infarction patients. Int. J. Clin. Exp. Pathol. 2014, 7, 5160–5164. [Google Scholar]
- Hamilton, N.B.; Kolodziejczyk, K.; Kougioumtzidou, E.; Attwell, D. Proton-gated Ca2+-permeable TRP channels damage myelin in conditions mimicking ischaemia. Nature 2016, 529, 523–527. [Google Scholar] [CrossRef]
- Kim, D.; Cavanaugh, E.J. Requirement of a soluble intracellular factor for activation of transient receptor potential A1 by pungent chemicals: Role of inorganic polyphosphates. J. Neurosci. 2007, 27, 6500–6509. [Google Scholar] [CrossRef]
- Shigetomi, E.; Tong, X.; Kwan, K.Y.; Corey, D.P.; Khakh, B.S. TRPA1 channels regulate astrocyte resting calcium and inhibitory synapse efficacy through GAT-3. Nat. Neurosci. 2012, 15, 70–80. [Google Scholar] [CrossRef]
- Meents, J.E.; Fischer, M.J.M.; McNaughton, P.A. Agonist-induced sensitisation of the irritant receptor ion channel TRPA1. J. Physiol. 2016, 594, 6643–6660. [Google Scholar] [CrossRef]
- Hu, H.; Tian, J.; Zhu, Y.; Wang, C.; Xiao, R.; Herz, J.M.; Wood, J.D.; Zhu, M.X. Activation of TRPA1 channels by fenamate nonsteroidal anti-inflammatory drugs. Pflugers Arch. 2010, 459, 579–592. [Google Scholar] [CrossRef]
- Andrade, E.L.; Meotti, F.C.; Calixto, J.B. TRPA1 antagonists as potential analgesic drugs. Pharmacol. Ther. 2012, 133, 189–204. [Google Scholar] [CrossRef]
- Ton, H.T.; Phan, T.X.; Abramyan, A.M.; Shi, L.; Ahern, G.P. Identification of a putative binding site critical for general anesthetic activation of TRPA1. Proc. Natl. Acad. Sci. USA 2017, 114, 3762–3767. [Google Scholar] [CrossRef] [PubMed]
- Mathie, K.; Lainer, J.; Spreng, S.; Dawid, C.; Andersson, D.A.; Bevan, S.; Hofmann, T. Structure-Pungency Relationships and TRP Channel Activation of Drimane Sesquiterpenes in Tasmanian Pepper (Tasmannia lanceolata). J. Agric. Food Chem. 2017, 65, 5700–5712. [Google Scholar] [CrossRef] [PubMed]
- Hinman, A.; Chuang, H.-H.; Bautista, D.M.; Julius, D. TRP channel activation by reversible covalent modification. Proc. Natl. Acad. Sci. USA 2006, 103, 19564–19568. [Google Scholar] [CrossRef] [PubMed]
- Takaya, J.; Mio, K.; Shiraishi, T.; Kurokawa, T.; Otsuka, S.; Mori, Y.; Uesugi, M. A Potent and Site-Selective Agonist of TRPA1. J. Am. Chem. Soc. 2015, 137, 15859–15864. [Google Scholar] [CrossRef]
- Micu, I.; Jiang, Q.; Coderre, E.; Ridsdale, A.; Zhang, L.; Woulfe, J.; Yin, X.; Trapp, B.D.; McRory, J.E.; Rehak, R.; et al. NMDA receptors mediate calcium accumulation in myelin during chemical ischaemia. Nature 2006, 439, 988–992. [Google Scholar] [CrossRef]
- Stys, P.K.; Ransom, B.R.; Waxman, S.G. Compound action potential of nerve recorded by suction electrode: A theoretical and experimental analysis. Brain Res. 1991, 546, 18–32. [Google Scholar] [CrossRef]
- Stys, P.K.; Ransom, B.R.; Waxman, S.G.; Davis, P.K. Role of extracellular calcium in anoxic injury of mammalian central white matter. Proc. Natl. Acad. Sci. USA 1990, 87, 4212–4216. [Google Scholar] [CrossRef]
- Bakiri, Y.; Hamilton, N.B.; Káradóttir, R.; Attwell, D. Testing NMDA receptor block as a therapeutic strategy for reducing ischaemic damage to CNS white matter. Glia 2008, 56, 233–240. [Google Scholar] [CrossRef]
- Baltan, S. Age-specific localization of NMDA receptors on oligodendrocytes dictates axon function recovery after ischemia. Neuropharmacology 2016, 110, 626–632. [Google Scholar] [CrossRef][Green Version]
- Bolton, S.; Butt, A.M. The optic nerve: A model for axon-glial interactions. J. Pharmacol. Toxicol. Methods 2005, 51, 221–233. [Google Scholar] [CrossRef]
- Bastian, C.; Brunet, S.; Baltan, S. Ex vivo studies of optic nerve axon electrophysiology. Methods Mol. Biol. 2020, 2143, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.-J.; Hare, W.A. Contribution to ischemic injury of rat optic nerves by intracellular sodium overload. Doc. Ophthalmol. 2005, 110, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Hamner, M.A.; Ye, Z.; Lee, R.V.; Colman, J.R.; Le, T.; Gong, D.C.; Ransom, B.R.; Weinstein, J.R. Ischemic preconditioning in white matter: Magnitude and mechanism. J. Neurosci. 2015, 35, 15599–15611. [Google Scholar] [CrossRef] [PubMed]
- Shigetomi, E.; Jackson-Weaver, O.; Huckstepp, R.T.; O’Dell, T.J.; Khakh, B.S. TRPA1 channels are regulators of astrocyte basal calcium levels and long-term potentiation via constitutive D-serine release. J. Neurosci. 2013, 33, 10143–10153. [Google Scholar] [CrossRef]
- Lee, K.-I.; Lin, H.-C.; Lee, H.-T.; Tsai, F.-C.; Lee, T.-S. Loss of transient receptor potential ankyrin 1 channel deregulates emotion, learning and memory, cognition, and social behavior in mice. Mol. Neurobiol. 2017, 54, 3606–3617. [Google Scholar] [CrossRef] [PubMed]
- Marques, S.; Zeisel, A.; Codeluppi, S.; van Bruggen, D.; Mendanha Falcão, A.; Xiao, L.; Li, H.; Häring, M.; Hochgerner, H.; Romanov, R.A.; et al. Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science 2016, 352, 1326–1329. [Google Scholar] [CrossRef] [PubMed]
- Kriszta, G.; Nemes, B.; Sándor, Z.; Ács, P.; Komoly, S.; Berente, Z.; Bölcskei, K.; Pintér, E. Investigation of Cuprizone-Induced Demyelination in mGFAP-Driven Conditional Transient Receptor Potential Ankyrin 1 (TRPA1) Receptor Knockout Mice. Cells 2019, 9, 81. [Google Scholar] [CrossRef] [PubMed]
- Bölcskei, K.; Kriszta, G.; Sághy, É.; Payrits, M.; Sipos, É.; Vranesics, A.; Berente, Z.; Ábrahám, H.; Ács, P.; Komoly, S.; et al. Behavioural alterations and morphological changes are attenuated by the lack of TRPA1 receptors in the cuprizone-induced demyelination model in mice. J. Neuroimmunol. 2018, 320, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cornillot, M.; Giacco, V.; Hamilton, N.B. The role of TRP channels in white matter function and ischaemia. Neurosci. Lett. 2019, 690, 202–209. [Google Scholar] [CrossRef]
- Ye, W.; Tu, Y.-H.; Cooper, A.J.; Zhang, Z.; Katritch, V.; Liman, E.R. Activation Stoichiometry and Pore Architecture of TRPA1 Probed with Channel Concatemers. Sci. Rep. 2018, 8, 17104. [Google Scholar] [CrossRef] [PubMed]
- Cahoy, J.D.; Emery, B.; Kaushal, A.; Foo, L.C.; Zamanian, J.L.; Christopherson, K.S.; Xing, Y.; Lubischer, J.L.; Krieg, P.A.; Krupenko, S.A.; et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J. Neurosci. 2008, 28, 264–278. [Google Scholar] [CrossRef]
- Papanikolaou, M.; Lewis, A.; Butt, A.M. Store-operated calcium entry is essential for glial calcium signalling in CNS white matter. Brain Struct. Funct. 2017, 222, 2993–3005. [Google Scholar] [CrossRef]
- Djukic, B.; Casper, K.B.; Philpot, B.D.; Chin, L.-S.; McCarthy, K.D. Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J. Neurosci. 2007, 27, 11354–11365. [Google Scholar] [CrossRef]
- Larson, V.A.; Mironova, Y.; Vanderpool, K.G.; Waisman, A.; Rash, J.E.; Agarwal, A.; Bergles, D.E. Oligodendrocytes control potassium accumulation in white matter and seizure susceptibility. Elife 2018, 7, e34829. [Google Scholar] [CrossRef]
- Kofuji, P.; Newman, E.A. Potassium buffering in the central nervous system. Neuroscience 2004, 129, 1045–1056. [Google Scholar] [CrossRef] [PubMed]
- Rash, J.E. Molecular disruptions of the panglial syncytium block potassium siphoning and axonal saltatory conduction: Pertinence to neuromyelitis optica and other demyelinating diseases of the central nervous system. Neuroscience 2010, 168, 982–1008. [Google Scholar] [CrossRef] [PubMed]
- Ransom, C.B.; Ransom, B.R.; Sontheimer, H. Activity-dependent extracellular K+ accumulation in rat optic nerve: The role of glial and axonal Na+ pumps. J. Physiol. 2000, 522, 427–442. [Google Scholar] [CrossRef] [PubMed]
- Salter, M.G.; Fern, R. NMDA receptors are expressed in developing oligodendrocyte processes and mediate injury. Nature 2005, 438, 1167–1171. [Google Scholar] [CrossRef]
- Tekkök, S.B.; Goldberg, M.P. Ampa/kainate receptor activation mediates hypoxic oligodendrocyte death and axonal injury in cerebral white matter. J. Neurosci. 2001, 21, 4237–4248. [Google Scholar] [CrossRef]
- McCarran, W.J.; Goldberg, M.P. White matter axon vulnerability to AMPA/kainate receptor-mediated ischemic injury is developmentally regulated. J. Neurosci. 2007, 27, 4220–4229. [Google Scholar] [CrossRef]
- Tekkök, S.B.; Ye, Z.; Ransom, B.R. Excitotoxic mechanisms of ischemic injury in myelinated white matter. J. Cereb. Blood Flow Metab. 2007, 27, 1540–1552. [Google Scholar] [CrossRef]
- Follett, P.L.; Rosenberg, P.A.; Volpe, J.J.; Jensen, F.E. NBQX attenuates excitotoxic injury in developing white matter. J. Neurosci. 2000, 20, 9235–9241. [Google Scholar] [CrossRef] [PubMed]
- Manning, S.M.; Talos, D.M.; Zhou, C.; Selip, D.B.; Park, H.-K.; Park, C.-J.; Volpe, J.J.; Jensen, F.E. NMDA receptor blockade with memantine attenuates white matter injury in a rat model of periventricular leukomalacia. J. Neurosci. 2008, 28, 6670–6678. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Hamner, M.A.; Brown, A.M.; Evans, R.D.; Ye, Z.-C.; Chen, S.; Ransom, B.R. Novel hypoglycemic injury mechanism: N-methyl-D-aspartate receptor-mediated white matter damage. Ann. Neurol. 2014, 75, 492–507. [Google Scholar] [CrossRef]
- Káradóttir, R.; Cavelier, P.; Bergersen, L.H.; Attwell, D. NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature 2005, 438, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Joshi, S.K.; DiDomenico, S.; Perner, R.J.; Mikusa, J.P.; Gauvin, D.M.; Segreti, J.A.; Han, P.; Zhang, X.-F.; Niforatos, W.; et al. Selective blockade of TRPA1 channel attenuates pathological pain without altering noxious cold sensation or body temperature regulation. Pain 2011, 152, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Earley, S. TRPA1 channels in the vasculature. Br. J. Pharmacol. 2012, 167, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Pires, P.W.; Earley, S. Neuroprotective effects of TRPA1 channels in the cerebral endothelium following ischemic stroke. Elife 2018, 7, e35316. [Google Scholar] [CrossRef]
- Sullivan, M.N.; Gonzales, A.L.; Pires, P.W.; Bruhl, A.; Leo, M.D.; Li, W.; Oulidi, A.; Boop, F.A.; Feng, Y.; Jaggar, J.H.; et al. Localized TRPA1 channel Ca2+ signals stimulated by reactive oxygen species promote cerebral artery dilation. Sci. Signal. 2015, 8, ra2. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TRPA1 Agonist | Covalent Binding (N-Terminal) | Non-Covalent Binding |
|---|---|---|
| Carvacrol | - | Unknown other site [5] |
| Flufenamic Acid (FFA) | - | Unknown other site [6] |
| Menthol | - | TM5 [7] |
| Isoflurane, propofol | - | TM5, TM6 and the pore helix 1 [8] |
| A-967079 (antagonist) | - | TM5, TM6 and the pore helix 1 [8] |
| Polygodial (PolyG) | Lys residues [9] | - |
| AITC (mustard oil) | Cys and Lys residues [10] | - |
| Cinnemaldehyde | Cys and Lys residues [10] | - |
| JT010 | Cys621 only [11] | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lajoso, W.; Flower, G.; Giacco, V.; Kaul, A.; La Mache, C.; Brăban, A.; Roxas, A.; Hamilton, N.B. Transient Receptor Potential Ankyrin-1 (TRPA1) Block Protects against Loss of White Matter Function during Ischaemia in the Mouse Optic Nerve. Pharmaceuticals 2021, 14, 909. https://doi.org/10.3390/ph14090909
Lajoso W, Flower G, Giacco V, Kaul A, La Mache C, Brăban A, Roxas A, Hamilton NB. Transient Receptor Potential Ankyrin-1 (TRPA1) Block Protects against Loss of White Matter Function during Ischaemia in the Mouse Optic Nerve. Pharmaceuticals. 2021; 14(9):909. https://doi.org/10.3390/ph14090909
Chicago/Turabian StyleLajoso, Wendy, Grace Flower, Vincenzo Giacco, Anjuli Kaul, Circe La Mache, Andra Brăban, Angela Roxas, and Nicola B. Hamilton. 2021. "Transient Receptor Potential Ankyrin-1 (TRPA1) Block Protects against Loss of White Matter Function during Ischaemia in the Mouse Optic Nerve" Pharmaceuticals 14, no. 9: 909. https://doi.org/10.3390/ph14090909
APA StyleLajoso, W., Flower, G., Giacco, V., Kaul, A., La Mache, C., Brăban, A., Roxas, A., & Hamilton, N. B. (2021). Transient Receptor Potential Ankyrin-1 (TRPA1) Block Protects against Loss of White Matter Function during Ischaemia in the Mouse Optic Nerve. Pharmaceuticals, 14(9), 909. https://doi.org/10.3390/ph14090909