
A Role of BDNF in the Depression Pathogenesis and a Potential Target as Antidepressant: The Modulator of Stress Sensitivity “Shati/Nat8l-BDNF System” in the Dorsal Striatum


Abstract
:1. Introduction
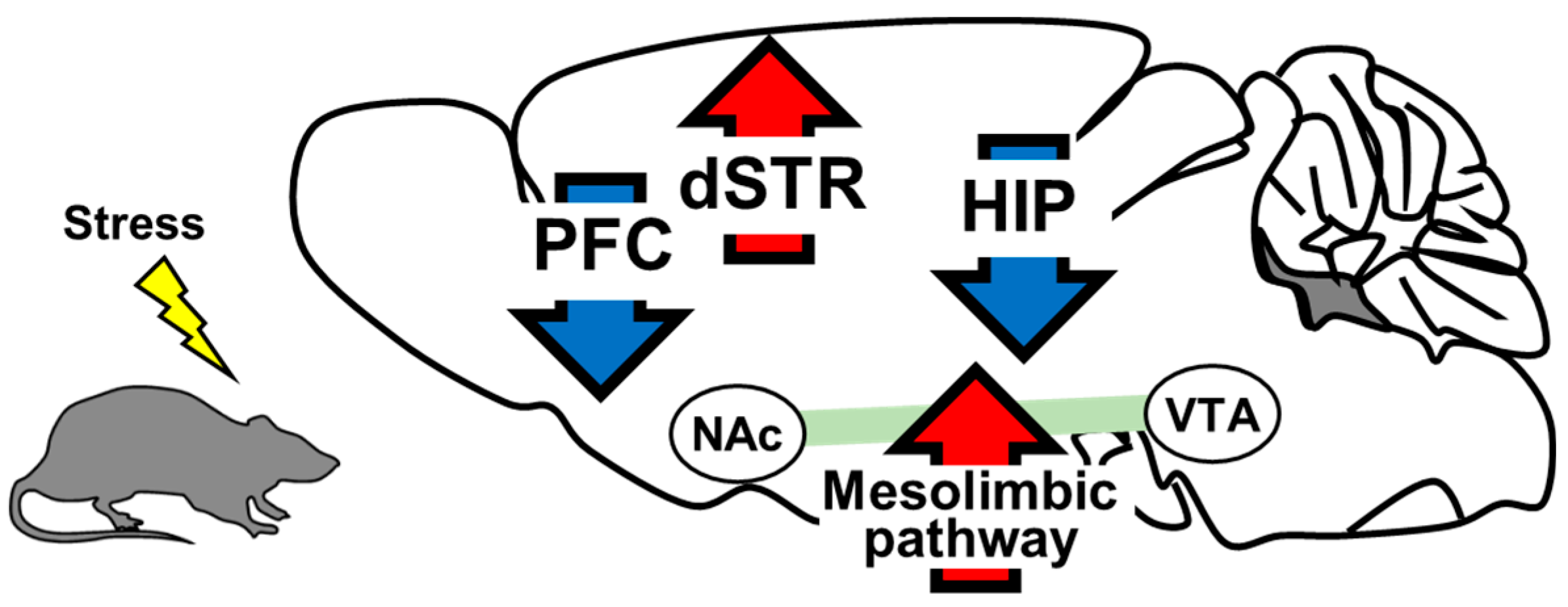
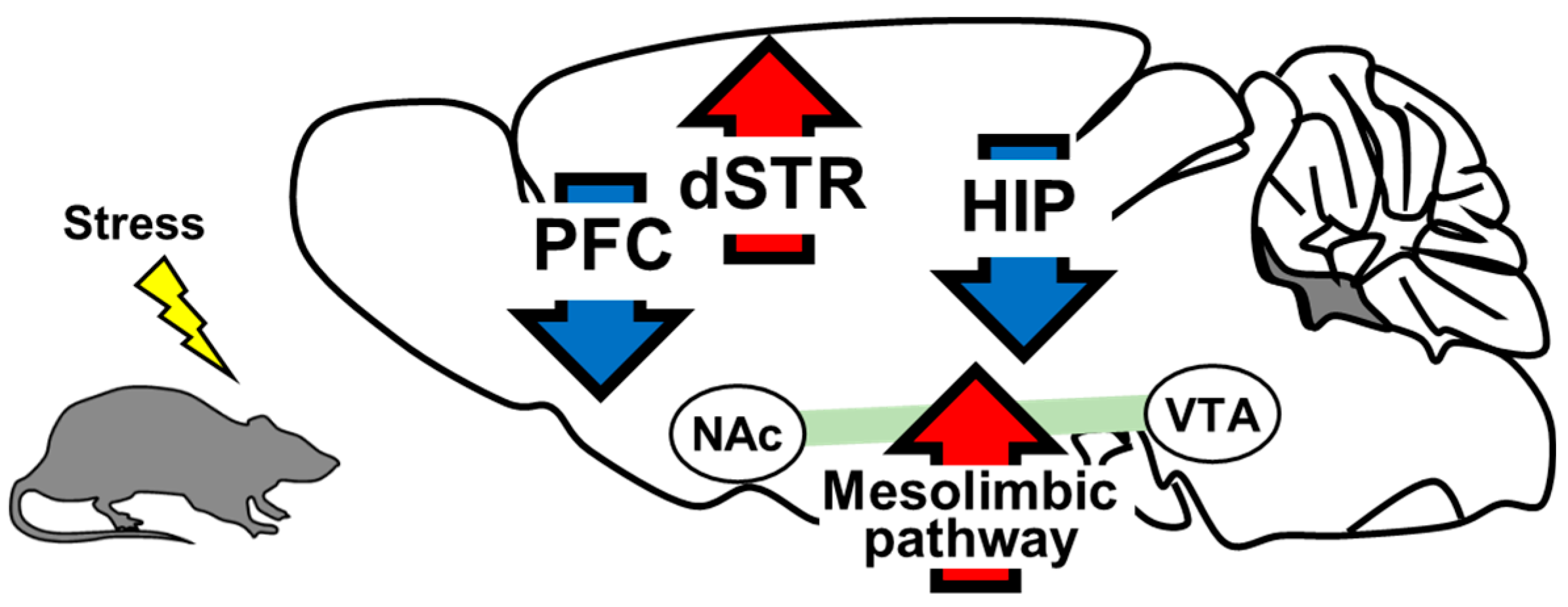
2. The Role of Brain-Derived Neurotrophic Factor in the Pathology of Depression
2.1. Prefrontal Cortex
2.2. Hippocampus
2.3. Mesolimbic Pathway
2.4. Dorsal Striatum
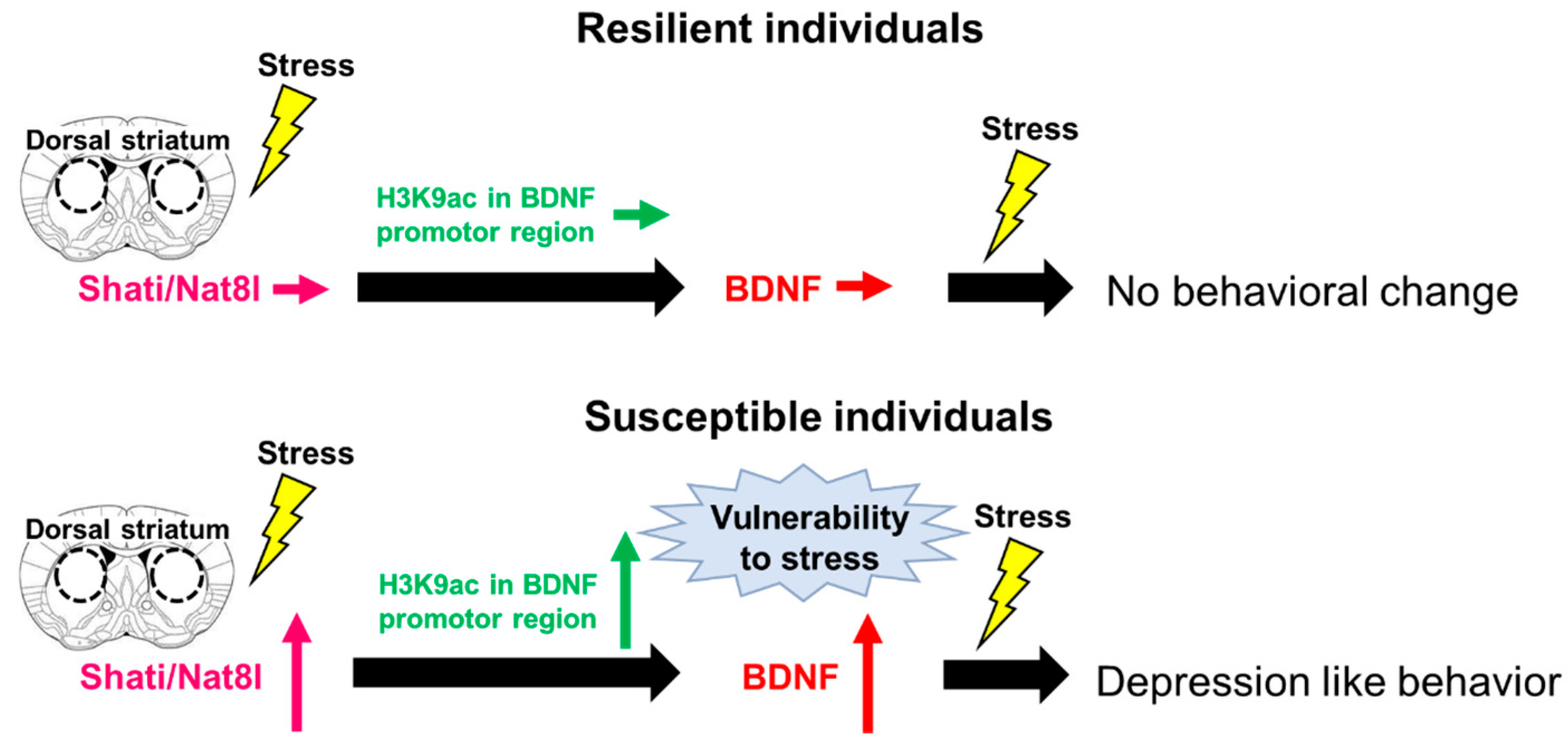
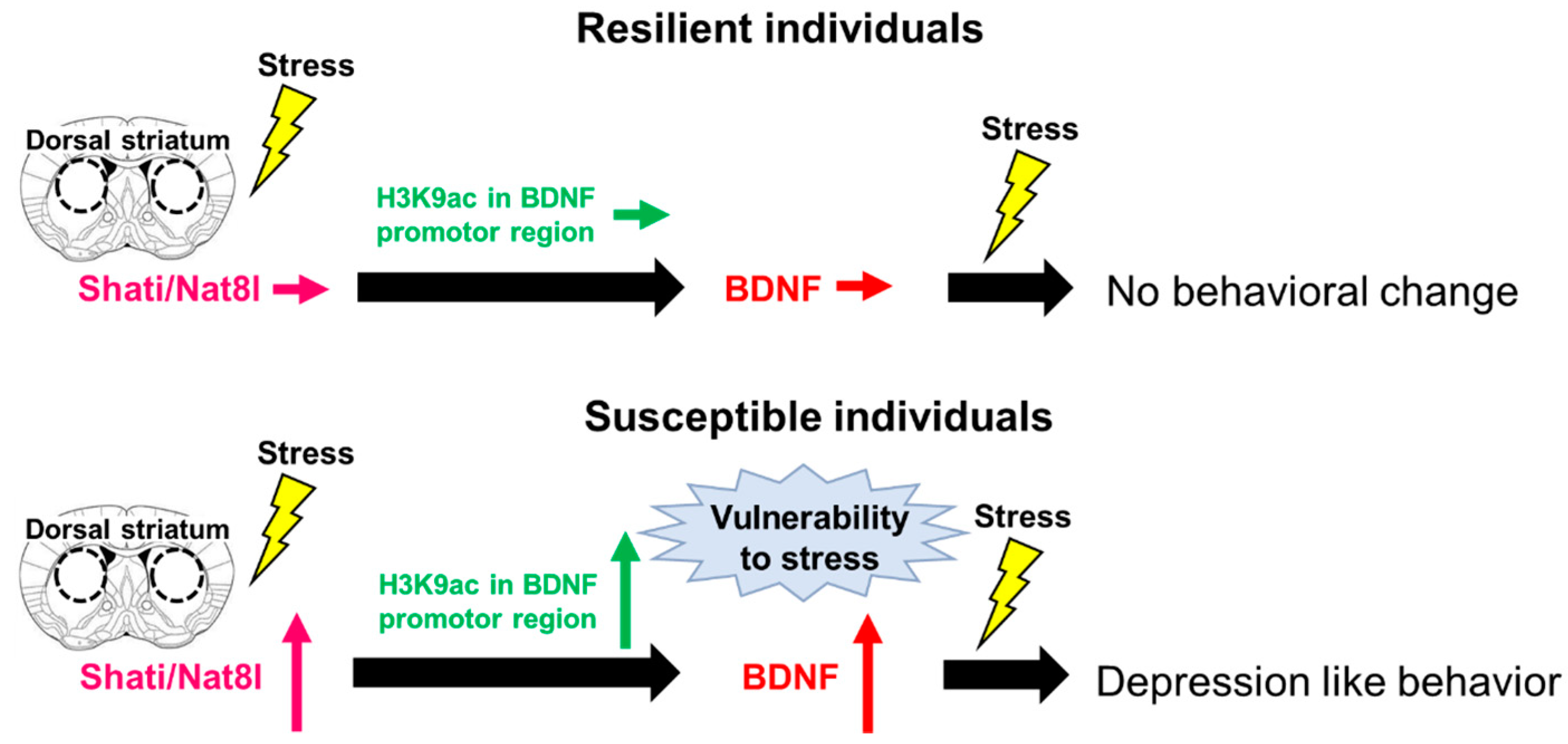
3. Shati/Nat8l
Shati/Nat8l and Depression
4. Future Prospects
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Publishing: Arlingon, VA, USA, 2013. [Google Scholar]
- World Health Organization (WHO). Depression and Other Common Mental Disorders. Available online: https://www.who.int/publications/i/item/depression-global-health-estimates (accessed on 23 July 2021).
- Bromet, E.; Andrade, L.H.; Hwang, I.; A Sampson, N.; Alonso, J.; De Girolamo, G.; De Graaf, R.; Demyttenaere, K.; Hu, C.; Iwata, N.; et al. Cross-national epidemiology of DSM-IV major depressive episode. BMC Med. 2011, 9, 90. [Google Scholar] [CrossRef]
- Dehn, L.B.; Beblo, T. Verstimmt, verzerrt, vergesslich: Das Zusammenwirken emotionaler und kognitiver Dysfunktionen bei Depression. Neuropsychiatrie 2019, 33, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Price, R.B.; Duman, R. Neuroplasticity in cognitive and psychological mechanisms of depression: An integrative model. Mol. Psychiatry 2019, 25, 530–543. [Google Scholar] [CrossRef]
- Grahek, I.; Shenhav, A.; Musslick, S.; Krebs, R.M.; Koster, E.H. Motivation and cognitive control in depression. Neurosci. Biobehav. Rev. 2019, 102, 371–381. [Google Scholar] [CrossRef] [Green Version]
- Lambert, C.; Da Silva, S.; Ceniti, A.K.; Rizvi, S.J.; Foussias, G.; Kennedy, S.H. Anhedonia in depression and schizophrenia: A transdiagnostic challenge. CNS Neurosci. Ther. 2018, 24, 615–623. [Google Scholar] [CrossRef]
- Hawton, K.; Comabella, C.C.; Haw, C.; Saunders, K. Risk factors for suicide in individuals with depression: A systematic review. J. Affect. Disord. 2013, 147, 17–28. [Google Scholar] [CrossRef]
- Ribeiro, J.D.; Huang, X.; Fox, K.R.; Franklin, J.C. Depression and hopelessness as risk factors for suicide ideation, attempts and death: Meta-analysis of longitudinal studies. Br. J. Psychiatry 2018, 212, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.T.; Bronstein, A.C. Selective serotonin reuptake inhibitor exposure. Top. Companion Anim. Med. 2013, 28, 13–17. [Google Scholar] [CrossRef] [Green Version]
- Trivedi, M.H.; Rush, A.; Wisniewski, S.; Nierenberg, A.A.; Warden, D.; Ritz, L.; Norquist, G.; Howland, R.H.; Lebowitz, B.; McGrath, P.; et al. Evaluation of outcomes with citalopram for depression using measurement-based Care in STAR*D: Implications for Clinical Practice. Am. J. Psychiatry 2006, 163, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.B.; Gelenberg, A.J.; Hirschfeld, R.M.A.; Rush, A.J.; Thase, M.E.; Kocsis, J.H.; Markowitz, J.C.; Fawcett, J.A.; Koran, L.M.; Klein, D.N.; et al. The Treatment of Chronic Depression, Part 2: A double-blind, randomized trial of sertraline and imipramine. J. Clin. Psychiatry 1998, 59, 598–607. [Google Scholar] [CrossRef]
- Rush, A.J.; Trivedi, M.H.; Wisniewski, S.R.; Nierenberg, A.A.; Stewart, J.W.; Warden, D.; Niederehe, G.; Thase, M.E.; Lavori, P.W.; Lebowitz, B.; et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: A STAR*D report. Am. J. Psychiatry 2006, 163, 1905–1917. [Google Scholar] [CrossRef] [PubMed]
- Luft, M.J.; Lamy, M.; DelBello, M.P.; McNamara, R.K.; Strawn, J.R. Antidepressant-induced activation in children and adolescents: Risk, recognition and management. Curr. Probl. Pediatr. Adolesc. Health Care 2018, 48, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Hammad, T.A.; Laughren, T.; Racoosin, J. Suicidality in pediatric patients treated with antidepressant drugs. Arch. Gen. Psychiatry 2006, 63, 332–339. [Google Scholar] [CrossRef]
- Park, C.; Rosenblat, J.D.; Brietzke, E.; Pan, Z.; Lee, Y.; Cao, B.; Zuckerman, H.; Kalantarova, A.; McIntyre, R.S. Stress, epigenetics and depression: A systematic review. Neurosci. Biobehav. Rev. 2019, 102, 139–152. [Google Scholar] [CrossRef]
- Sheng, J.; Liu, S.; Wang, Y.; Cui, R.; Zhang, X. The Link between depression and chronic pain: Neural mechanisms in the brain. Neural Plast. 2017, 2017, 9724371. [Google Scholar] [CrossRef] [PubMed]
- Rith-Najarian, L.R.; Boustani, M.M.; Chorpita, B.F. A systematic review of prevention programs targeting depression, anxiety, and stress in university students. J. Affect. Disord. 2019, 257, 568–584. [Google Scholar] [CrossRef]
- Slavich, G.M.; Sacher, J. Stress, sex hormones, inflammation, and major depressive disorder: Extending social signal transduction theory of depression to account for sex differences in mood disorders. Psychopharmacologia 2019, 236, 3063–3079. [Google Scholar] [CrossRef]
- De Kloet, E.R.; Joels, M.; Holsboer, F. Stress and the brain: From adaptation to disease. Nat. Rev. Neurosci. 2005, 6, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Monroe, S.M.; Harkness, K. Life stress, the “kindling” Hypothesis, and the recurrence of depression: Considerations from a life stress perspective. Psychol. Rev. 2005, 112, 417–445. [Google Scholar] [CrossRef] [Green Version]
- Technow, J.R.; Hazel, N.A.; Abela, J.R.Z.; Hankin, B.L. Stress sensitivity interacts with depression history to predict depressive symptoms among youth: Prospective changes following first depression onset. J. Abnorm. Child Psychol. 2014, 43, 489–501. [Google Scholar] [CrossRef]
- Fleshner, M.; Maier, S.F.; Lyons, D.M.; Raskind, M.A. The neurobiology of the stress-resistant brain. Stress 2011, 14, 498–502. [Google Scholar] [CrossRef] [Green Version]
- Bowen, M.; Dass, S.H.; Booth, J.; Suraev, A.; Vyas, A.; McGregor, I.S. Active coping toward predatory stress is associated with lower corticosterone and progesterone plasma levels and decreased methylation in the medial amygdala vasopressin system. Horm. Behav. 2014, 66, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Pearson-Leary, J.; Zhao, C.; Bittinger, K.; Eacret, D.; Luz, S.; Vigderman, A.S.; Dayanim, G.; Bhatnagar, S. The gut microbiome regulates the increases in depressive-type behaviors and in inflammatory processes in the ventral hippocampus of stress vulnerable rats. Mol. Psychiatry 2019, 25, 1068–1079. [Google Scholar] [CrossRef] [PubMed]
- Golden, S.; Iii, H.E.C.; Berton, O.; Russo, S.J. A standardized protocol for repeated social defeat stress in mice. Nat. Protoc. 2011, 6, 1183–1191. [Google Scholar] [CrossRef]
- Shinohara, R.; Taniguchi, M.; Ehrlich, A.T.; Yokogawa, K.; Deguchi, Y.; Cherasse, Y.; Lazarus, M.; Urade, Y.; Ogawa, A.; Kitaoka, S.; et al. Dopamine D1 receptor subtype mediates acute stress-induced dendritic growth in excitatory neurons of the medial prefrontal cortex and contributes to suppression of stress susceptibility in mice. Mol. Psychiatry 2017, 23, 1717–1730. [Google Scholar] [CrossRef] [Green Version]
- Groves, O.J. Is it time to reassess the BDNF hypothesis of depression? Mol. Psychiatry 2007, 12, 1079–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berton, O.; McClung, C.A.; DiLeone, R.J.; Krishnan, V.; Renthal, W.; Russo, S.J.; Graham, D.; Tsankova, N.M.; Bolanos, C.A.; Rios, M.; et al. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science 2006, 311, 864–868. [Google Scholar] [CrossRef] [Green Version]
- Jiang, C.; Lin, W.-J.; Sadahiro, M.; Labonté, B.; Menard, C.; Pfau, M.L.; Tamminga, C.A.; Turecki, G.; Nestler, E.J.; Russo, S.J.; et al. VGF function in depression and antidepressant efficacy. Mol. Psychiatry 2017, 23, 1632–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levi-Montalcini, R.; Hamburger, V. Selective growth stimulating effects of mouse sarcoma on the sensory and sympathetic nervous system of the chick embryo. J. Exp. Zoöl. 1951, 116, 321–361. [Google Scholar] [CrossRef]
- Barde, Y.; Edgar, D.; Thoenen, H. Purification of a new neurotrophic factor from mammalian brain. EMBO J. 1982, 1, 549–553. [Google Scholar] [CrossRef]
- Colucci-D’Amato, L.; Speranza, L.; Volpicelli, F. Neurotrophic factor BDNF, physiological functions and therapeutic potential in depression, neurodegeneration and brain cancer. Int. J. Mol. Sci. 2020, 21, 7777. [Google Scholar] [CrossRef]
- Björkholm, C.; Monteggia, L.M. BDNF—A key transducer of antidepressant effects. Neuropharmacology 2015, 102, 72–79. [Google Scholar] [CrossRef] [Green Version]
- Ernfors, P.; Lee, K.-F.; Jaenisch, R. Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature 1994, 368, 147–150. [Google Scholar] [CrossRef]
- Kojima, M.; Mizui, T. BDNF propeptide: A novel modulator of synaptic plasticity. Vitam. Hom. 2017, 104, 19–28. [Google Scholar] [CrossRef]
- Leal, G.; Bramham, C.; Duarte, C. BDNF and hippocampal synaptic plasticity. Vitam. Horm. 2017, 104, 153–195. [Google Scholar] [CrossRef]
- Soppet, D.; Escandon, E.; Maragos, J.; Middlemas, D.S.; Raid, S.W.; Blair, J.; Burton, L.E.; Stanton, B.R.; Kaplan, D.R.; Hunter, T.; et al. The neurotrophic factors brain-derived neurotrophic factor and neurotrophin-3 are ligands for the trkB tyrosine kinase receptor. Cell 1991, 65, 895–903. [Google Scholar] [CrossRef]
- Liang, J.; Deng, G.; Huang, H. The activation of BDNF reduced inflammation in a spinal cord injury model by TrkB/p38 MAPK signaling. Exp. Ther. Med. 2018, 17, 1688–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rai, S.N.; Dilnashin, H.; Birla, H.; Singh, S.S.; Zahra, W.; Rathore, A.; Singh, B.K.; Singh, S. The role of PI3K/Akt and ERK in neurodegenerative disorders. Neurotox. Res. 2019, 35, 775–795. [Google Scholar] [CrossRef] [PubMed]
- Gudasheva, T.A.; Logvinov, I.O.; Nikolaev, S.V.; Antipova, T.A.; Povarnina, P.Y.; Seredenin, S.B. Dipeptide mimetics of different NGF and BDNF loops activate PLC-γ1. Dokl. Biochem. Biophys. 2020, 494, 244–247. [Google Scholar] [CrossRef] [PubMed]
- Pruunsild, P.; Kazantseva, A.; Aid, T.; Palm, K.; Timmusk, T. Dissecting the human BDNF locus: Bidirectional transcription, complex splicing, and multiple promoters. Genomics 2007, 90, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-C.; Huang, T.-L. Brain-derived neurotrophic factor and mental disorders. Biomed. J. 2020, 43, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Timmusk, T.; Palm, K.; Metsis, M.; Reintam, T.; Paalme, V.; Saarma, M.; Persson, H. Multiple promoters direct tissue-specific expression of the rat BDNF gene. Neuron 1993, 10, 475–489. [Google Scholar] [CrossRef]
- Seo, M.K.; Ly, N.N.; Lee, C.H.; Cho, H.Y.; Choi, C.M.; Nhu, L.H.; Lee, J.G.; Lee, B.J.; Kim, G.-M.; Yoon, B.J.; et al. Early life stress increases stress vulnerability through BDNF gene epigenetic changes in the rat hippocampus. Neuropharmacology 2016, 105, 388–397. [Google Scholar] [CrossRef]
- Tsankova, N.M.; Berton, O.; Renthal, W.; Kumar, A.; Neve, R.L.; Nestler, E.J. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat. Neurosci. 2006, 9, 519–525. [Google Scholar] [CrossRef]
- Tadić, A.; Müller-Engling, L.; Schlicht, K.F.; Kotsiari, A.; Dreimüller, N.; Kleimann, A.; Bleich, S.; Lieb, K.; Frieling, H. Methylation of the promoter of brain-derived neurotrophic factor exon IV and antidepressant response in major depression. Mol. Psychiatry 2013, 19, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Borba, E.M.; Duarte, J.A.; Bristot, G.; Scotton, E.; Camozzato, A.L.; Chaves, M.L.F. Brain-derived neurotrophic factor serum levels and hippocampal volume in mild cognitive impairment and dementia due to Alzheimer disease. Dement. Geriatr. Cogn. Disord. Extra 2016, 6, 559–567. [Google Scholar] [CrossRef]
- De Pins, B.; Cifuentes-Díaz, C.; Farah, A.T.; López-Molina, L.; Montalban, E.; Sancho-Balsells, A.; López, A.; Ginés, S.; Delgado-García, J.M.; Alberch, J.; et al. Conditional BDNF delivery from astrocytes rescues memory deficits, spine density and synaptic properties in the 5xFAD mouse model of Alzheimer disease. J. Neurosci. 2019, 39, 2441–2458. [Google Scholar] [CrossRef] [Green Version]
- Kim, O.Y.; Song, J. The importance of BDNF and RAGE in diabetes-induced dementia. Pharmacol. Res. 2020, 160, 105083. [Google Scholar] [CrossRef] [PubMed]
- Armeanu, R.; Mokkonen, M.; Crespi, B. Meta-Analysis of BDNF Levels in Autism. Cell. Mol. Neurobiol. 2016, 37, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Chen, S.; Li, C.; Lu, N.; Yue, Y.; Yin, Y.; Zhang, Y.; Zhi, X.; Zhang, D.; Yuan, Y. The serum protein levels of the tPA–BDNF pathway are implicated in depression and antidepressant treatment. Transl. Psychiatry 2017, 7, e1079. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Fang, X.; Fan, W.; Tang, W.; Cai, J.; Song, L.; Zhang, C. Interaction between BDNF and TNF-α genes in schizophrenia. Psychoneuroendocrinology 2017, 89, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, T.; Shi, C.; Yang, Y.; Li, X.; Wu, Y.; Xu, Z.-Q.D. Inhibition of GALR1 in PFC alleviates depressive-like behaviors in postpartum depression rat model by upregulating CREB-BNDF and 5-HT levels. Front. Psychiatry 2018, 9, 588. [Google Scholar] [CrossRef]
- Fukumoto, K.; Fogaça, M.V.; Liu, R.-J.; Duman, C.H.; Li, X.-Y.; Chaki, S.; Duman, R.S. Medial PFC AMPA receptor and BDNF signaling are required for the rapid and sustained antidepressant-like effects of 5-HT1A receptor stimulation. Neuropsychopharmacology 2020, 45, 1725–1734. [Google Scholar] [CrossRef]
- Li, M.; Li, C.; Yu, H.; Cai, X.; Shen, X.; Sun, X.; Wang, J.; Zhang, Y.; Wang, C. Lentivirus-mediated interleukin-1β (IL-1β) knock-down in the hippocampus alleviates lipopolysaccharide (LPS)-induced memory deficits and anxiety- and depression-like behaviors in mice. J. Neuroinflammation 2017, 14, 190. [Google Scholar] [CrossRef] [Green Version]
- Jiang, N.; Lv, J.-W.; Wang, H.-X.; Lu, C.; Wang, Q.; Xia, T.-J.; Bao, Y.; Li, S.-S.; Liu, X.-M. Dammarane sapogenins alleviates depression-like behaviours induced by chronic social defeat stress in mice through the promotion of the BDNF signalling pathway and neurogenesis in the hippocampus. Brain Res. Bull. 2019, 153, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Nestler, E.J.; Carlezon, W.A. The mesolimbic dopamine reward circuit in depression. Biol. Psychiatry 2006, 59, 1151–1159. [Google Scholar] [CrossRef] [PubMed]
- Hiser, J.; Koenigs, M. The multifaceted role of the ventromedial prefrontal cortex in emotion, decision making, social cognition, and psychopathology. Biol. Psychiatry 2018, 83, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Gholaminejad, A.; Gholamipour-Badie, H.; Nasehi, M.; Naghdi, N. Prelimbic of medial prefrontal cortex GABA modulation through testosterone on spatial learning and memory. Iran. J. Pharm. Res. 2019, 18, 1429–1444. [Google Scholar] [PubMed]
- Palm, U.; Hasan, A.; Strube, W.; Padberg, F. tDCS for the treatment of depression: A comprehensive review. Eur. Arch. Psychiatry Clin. Neurosci. 2016, 266, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kar, S.K. How electroconvulsive therapy works? Understanding the neurobiological mechanisms. Clin. Psychopharmacol. Neurosci. 2017, 15, 210–221. [Google Scholar] [CrossRef] [Green Version]
- Daskalakis, Z.J.; Dimitrova, J.; McClintock, S.M.; Sun, Y.; Voineskos, D.; Rajji, T.K.; Goldbloom, D.S.; Wong, A.H.C.; Knyahnytska, Y.; Mulsant, B.H.; et al. Magnetic seizure therapy (MST) for major depressive disorder. Neuropsychopharmacology 2019, 45, 276–282. [Google Scholar] [CrossRef]
- De Risio, L.; Borgi, M.; Pettorruso, M.; Miuli, A.; Ottomana, A.M.; Sociali, A.; Martinotti, G.; Nicolò, G.; Macrì, S.; Di Giannantonio, M.; et al. Recovering from depression with repetitive transcranial magnetic stimulation (rTMS): A systematic review and meta-analysis of preclinical studies. Transl. Psychiatry 2020, 10, 393. [Google Scholar] [CrossRef]
- Drobisz, D.; Damborská, A. Deep brain stimulation targets for treating depression. Behav. Brain Res. 2018, 359, 266–273. [Google Scholar] [CrossRef]
- Hare, B.D.; Duman, R.S. Prefrontal cortex circuits in depression and anxiety: Contribution of discrete neuronal populations and target regions. Mol. Psychiatry 2020, 25, 2742–2758. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, C.G.; Averill, C.L.; Salas, R.; Averill, L.A.; Baldwin, P.R.; Krystal, J.H.; Mathew, S.J.; Mathalon, D.H. Prefrontal Connectivity and glutamate transmission: Relevance to depression pathophysiology and ketamine treatment. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2017, 2, 566–574. [Google Scholar] [CrossRef]
- Phillips, J.; Norris, S.; Talbot, J.; Birmingham, M.; Hatchard, T.; Ortiz, A.; Owoeye, O.; Batten, L.A.; Blier, P. Single, repeated, and maintenance ketamine infusions for treatment-resistant depression: A randomized controlled trial. Am. J. Psychiatry 2019, 176, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, Y.; Rizavi, H.S.; Conley, R.R.; Roberts, R.C.; Tamminga, C.A.; Pandey, G.N. Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects. Arch. Gen. Psychiatry 2003, 60, 804–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karege, F.; Vaudan, G.; Schwald, M.; Perroud, N.; La Harpe, R. Neurotrophin levels in postmortem brains of suicide victims and the effects of antemortem diagnosis and psychotropic drugs. Mol. Brain Res. 2005, 136, 29–37. [Google Scholar] [CrossRef]
- Qi, X.-R.; Zhao, J.; Liu, J.; Fang, H.; Swaab, D.; Zhou, J.-N. Abnormal retinoid and TrkB signaling in the prefrontal cortex in mood disorders. Cereb. Cortex 2013, 25, 75–83. [Google Scholar] [CrossRef]
- Tripp, A.; Oh, H.; Guilloux, J.-P.; Martinowich, K.; Lewis, D.A.; Sibille, E. Brain-derived neurotrophic factor signaling and subgenual anterior cingulate cortex dysfunction in major depressive disorder. Am. J. Psychiatry 2012, 169, 1194–1202. [Google Scholar] [CrossRef] [Green Version]
- Duman, R.S.; Aghajanian, G.K.; Sanacora, G.; Krystal, J.H. Synaptic plasticity and depression: New insights from stress and rapid-acting antidepressants. Nat. Med. 2016, 22, 238–249. [Google Scholar] [CrossRef] [Green Version]
- Duman, C.H.; Duman, R.S. Spine synapse remodeling in the pathophysiology and treatment of depression. Neurosci. Lett. 2015, 601, 20–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wohleb, E.S.; Terwilliger, R.; Duman, C.H.; Duman, R.S. Stress-induced neuronal colony stimulating factor 1 provokes microglia-mediated neuronal remodeling and depressive-like behavior. Biol. Psychiatry 2017, 83, 38–49. [Google Scholar] [CrossRef]
- Xu, H.; Zhang, Y.; Zhang, F.; Yuan, S.-N.; Shao, F.; Wang, W. Effects of duloxetine treatment on cognitive flexibility and BDNF expression in the mPFC of adult male mice exposed to social stress during adolescence. Front. Mol. Neurosci. 2016, 9, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Zhang, J.-C.; Han, M.; Yao, W.; Yang, C.; Ren, Q.; Ma, M.; Chen, Q.-X.; Hashimoto, K. Comparison of R-ketamine and rapastinel antidepressant effects in the social defeat stress model of depression. Psychopharmacology 2016, 233, 3647–3657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.-J.; Lee, F.S.; Li, X.-Y.; Bambico, F.; Duman, R.S.; Aghajanian, G.K. Brain-derived neurotrophic factor Val66Met allele impairs basal and ketamine-stimulated synaptogenesis in prefrontal cortex. Biol. Psychiatry 2012, 71, 996–1005. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Wang, D.-D.; Wang, Y.; Liu, T.; Lee, F.S.; Chen, Z.-Y. Variant Brain-Derived Neurotrophic Factor Val66Met Polymorphism Alters Vulnerability to Stress and Response to Antidepressants. J. Neurosci. 2012, 32, 4092–4101. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.-C.; Wu, J.; Fujita, Y.; Yao, W.; Ren, Q.; Yang, C.; Li, S.-X.; Shirayama, Y.; Hashimoto, K. Antidepressant effects of TrkB ligands on depression-like behavior and dendritic changes in mice after inflammation. Int. J. Neuropsychopharmacol. 2015, 18, pyu077. [Google Scholar] [CrossRef]
- Kato, T.; Fogaça, M.V.; Deyama, S.; Li, X.-Y.; Fukumoto, K.; Duman, R.S. BDNF release and signaling are required for the antidepressant actions of GLYX-13. Mol. Psychiatry 2017, 23, 2007. [Google Scholar] [CrossRef]
- Duman, R.S.; Sanacora, G.; Krystal, J.H. Altered connectivity in depression: GABA and glutamate neurotransmitter deficits and reversal by novel treatments. Neuron 2019, 102, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Deyama, S.; Duman, R.S. Neurotrophic mechanisms underlying the rapid and sustained antidepressant actions of ketamine. Pharmacol. Biochem. Behav. 2019, 188, 172837. [Google Scholar] [CrossRef] [PubMed]
- Lepack, A.E.; Fuchikami, M.; Dwyer, J.M.; Banasr, M.; Duman, R.S. BDNF release is required for the behavioral actions of ketamine. Int. J. Neuropsychopharmacol. 2014, 18, pyu033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepack, A.E.; Bang, E.; Lee, B.; Dwyer, J.M.; Duman, R.S. Fast-acting antidepressants rapidly stimulate ERK signaling and BDNF release in primary neuronal cultures. Neuropharmacology 2016, 111, 242–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Leary, O.F.; Cryan, J. A ventral view on antidepressant action: Roles for adult hippocampal neurogenesis along the dorsoventral axis. Trends Pharmacol. Sci. 2014, 35, 675–687. [Google Scholar] [CrossRef]
- Liu, W.; Ge, T.; Leng, Y.; Pan, Z.; Fan, J.; Yang, W.; Cui, R. The role of neural plasticity in depression: From hippocampus to prefrontal cortex. Neural Plast. 2017, 2017, 6871089. [Google Scholar] [CrossRef] [Green Version]
- Gatt, J.; Nemeroff, C.B.; Dobson-Stone, C.; Paul, R.H.; Bryant, R.; Schofield, P.; Gordon, E.; Kemp, A.; Williams, L.M. Interactions between BDNF Val66Met polymorphism and early life stress predict brain and arousal pathways to syndromal depression and anxiety. Mol. Psychiatry 2009, 14, 681–695. [Google Scholar] [CrossRef] [Green Version]
- Cagni, F.C.; Campêlo, C.L.D.C.; Coimbra, D.G.; Barbosa, M.R.; Júnior, L.G.O.; Neto, A.B.S.; Ribeiro, A.M.; Júnior, C.D.O.G.; de Andrade, T.; Silva, R.H. Association of BDNF Val66MET polymorphism with Parkinson’s disease and depression and anxiety symptoms. J. Neuropsychiatry Clin. Neurosci. 2017, 29, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Youssef, M.M.; Underwood, M.D.; Huang, Y.-Y.; Hsiung, S.-C.; Liu, Y.; Simpson, N.R.; Bakalian, M.J.; Rosoklija, G.B.; Dwork, A.J.; Arango, V.; et al. Association of BDNF Val66Met polymorphism and brain BDNF levels with major depression and suicide. Int. J. Neuropsychopharmacol. 2018, 21, 528–538. [Google Scholar] [CrossRef]
- Chiaruttini, C.; Vicario, A.; Li, Z.; Baj, G.; Braiuca, P.; Wu, Y.; Lee, F.S.; Gardossi, L.; Baraban, J.M.; Tongiorgi, E. Dendritic trafficking of BDNF mRNA is mediated by translin and blocked by the G196A (Val66Met) mutation. Proc. Natl. Acad. Sci. USA 2009, 106, 16481. [Google Scholar] [CrossRef] [Green Version]
- Egan, M.F.; Kojima, M.; Callicott, J.; Goldberg, T.E.; Kolachana, B.S.; Bertolino, A.; Zaitsev, E.; Gold, B.; Goldman, D.; Dean, M.; et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 2003, 112, 257–269. [Google Scholar] [CrossRef] [Green Version]
- Pezawas, L.; Verchinski, B.A.; Mattay, V.S.; Callicott, J.; Kolachana, B.S.; Straub, R.E.; Egan, M.F.; Meyer-Lindenberg, A.; Weinberger, D.R. The brain-derived neurotrophic factor val66met polymorphism and variation in human cortical morphology. J. Neurosci. 2004, 24, 10099–10102. [Google Scholar] [CrossRef]
- Frodl, T.; Schüle, C.; Schmitt, G.; Born, C.; Baghai, T.; Zill, P.; Bottlender, R.; Rupprecht, R.; Bondy, B.; Reiser, M.; et al. Association of the brain-derived neurotrophic factor Val66Met polymorphism with reduced hippocampal volumes in major depression. Arch. Gen. Psychiatry 2007, 64, 410–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anacker, C.; Luna, V.M.; Stevens, G.S.; Millette, A.; Shores, R.; Jimenez, J.C.; Chen, B.; Hen, R. Hippocampal neurogenesis confers stress resilience by inhibiting the ventral dentate gyrus. Nature 2018, 559, 98–102. [Google Scholar] [CrossRef]
- Jiang, N.; Wang, H.; Li, C.; Zeng, G.; Lv, J.; Wang, Q.; Chen, Y.; Liu, X. The antidepressant-like effects of the water extract of panax ginseng and polygala tenuifolia are mediated via the BDNF-TrkB signaling pathway and neurogenesis in the hippocampus. J. Ethnopharmacol. 2020, 267, 113625. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, B.L.; van Praag, H.; Gage, F.H. Adult brain neurogenesis and psychiatry: A novel theory of depression. Mol. Psychiatry 2000, 5, 262–269. [Google Scholar] [CrossRef]
- Rossi, C.; Angelucci, A.; Costantin, L.; Braschi, C.; Mazzantini, M.; Babbini, F.; Fabbri, M.E.; Tessarollo, L.; Maffei, L.; Berardi, N.; et al. Brain-derived neurotrophic factor (BDNF) is required for the enhancement of hippocampal neurogenesis following environmental enrichment. Eur. J. Neurosci. 2006, 24, 1850–1856. [Google Scholar] [CrossRef] [PubMed]
- Bremner, J.D.; Narayan, M.; Anderson, E.R.; Staib, L.; Miller, H.L.; Charney, D.S. Hippocampal volume reduction in major depression. Am. J. Psychiatry 2000, 157, 115–118. [Google Scholar] [CrossRef]
- Barch, D.M.; Tillman, R.; Kelly, D.; Whalen, D.; Gilbert, K.; Luby, J.L. Hippocampal volume and depression among young children. Psychiatry Res. Neuroimaging 2019, 288, 21–28. [Google Scholar] [CrossRef]
- Duman, R.S.; Monteggia, L.M. A Neurotrophic model for stress-related mood disorders. Biol. Psychiatry 2006, 59, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Nibuya, M.; Takahashi, M.; Russell, D.S.; Duman, R.S. Repeated stress increases catalytic TrkB mRNA in rat hippocampus. Neurosci. Lett. 1999, 267, 81–84. [Google Scholar] [CrossRef]
- Ma, M.; Ren, Q.; Yang, C.; Zhang, J.-C.; Yao, W.; Dong, C.; Ohgi, Y.; Futamura, T.; Hashimoto, K. Adjunctive treatment of brexpiprazole with fluoxetine shows a rapid antidepressant effect in social defeat stress model: Role of BDNF-TrkB signaling. Sci. Rep. 2016, 6, 39209. [Google Scholar] [CrossRef] [PubMed]
- Taliaz, D.; Stall, N.; E Dar, D.; Zangen, A. Knockdown of brain-derived neurotrophic factor in specific brain sites precipitates behaviors associated with depression and reduces neurogenesis. Mol. Psychiatry 2009, 15, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Garza, A.A.; Ha, T.G.; Garcia, C.; Chen, M.J.; A Russo-Neustadt, A. Exercise, antidepressant treatment, and BDNF mRNA expression in the aging brain. Pharmacol. Biochem. Behav. 2003, 77, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Shirayama, Y.; Chen, A.C.-H.; Nakagawa, S.; Russell, D.S.; Duman, R.S. Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J. Neurosci. 2002, 22, 3251–3261. [Google Scholar] [CrossRef] [Green Version]
- Mattson, M.P.; Maudsley, S.; Martin, B. BDNF and 5-HT: A dynamic duo in age-related neuronal plasticity and neurodegenerative disorders. Trends Neurosci. 2004, 27, 589–594. [Google Scholar] [CrossRef]
- Erickson, K.I.; Miller, D.L.; Roecklein, K.A. The aging hippocampus: Interactions between exercise, depression, and BDNF. Neuroscientist 2011, 18, 82–97. [Google Scholar] [CrossRef]
- Diniz, D.; Calabrese, F.; Brivio, P.; Riva, M.; Grandjean, J.; Homberg, J. BDNF overexpression in the ventral hippocampus promotes antidepressant- and anxiolytic-like activity in serotonin transporter knockout rats. Int. J. Mol. Sci. 2021, 22, 5040. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.W.; Chaudhury, D.; Han, M.-H.; Nestler, E.J. Role of mesolimbic brain-derived neurotrophic factor in depression. Biol. Psychiatry 2019, 86, 738–748. [Google Scholar] [CrossRef]
- Krishnan, V.; Han, M.-H.; Graham, D.L.; Berton, O.; Renthal, W.; Russo, S.; LaPlant, Q.; Graham, A.; Lutter, M.; Lagace, D.C.; et al. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell 2007, 131, 391–404. [Google Scholar] [CrossRef] [Green Version]
- Taliaz, D.; Nagaraj, V.; Haramati, S.; Chen, A.; Zangen, A. Altered brain-derived neurotrophic factor expression in the ventral tegmental area, but not in the hippocampus, is essential for antidepressant-like effects of electroconvulsive therapy. Biol. Psychiatry 2012, 74, 305–312. [Google Scholar] [CrossRef]
- Eisch, A.; A Bolaños, C.; de Wit, J.; Simonak, R.D.; Pudiak, C.M.; Barrot, M.; Verhaagen, J.; Nestler, E.J. Brain-derived neurotrophic factor in the ventral midbrain–nucleus accumbens pathway: A role in depression. Biol. Psychiatry 2003, 54, 994–1005. [Google Scholar] [CrossRef] [PubMed]
- Seroogy, K.B.; Lundgren, K.H.; Tran, T.M.D.; Guthrie, K.M.; Isackson, P.J.; Gall, C.M. Dopaminergic neurons in rat ventral midbrain express brain-derived neurotrophic factor and neurotrophin-3 mRNAs. J. Comp. Neurol. 1994, 342, 321–334. [Google Scholar] [CrossRef]
- Numan, S.; Seroogy, K.B. Expression of trkB and trkC mRNAs by adult midbrain dopamine neurons: A double-label in situ hybridization study. Comp. Neurol. 1999, 403, 295–308. [Google Scholar] [CrossRef]
- Berhow, M.; Russell, D.; Terwilliger, R.; Beitner-Johnson, D.; Self, D.; Lindsay, R.; Nestler, E. Influence of neurotrophic factors on morphine- and cocaine-induced biochemical changes in the mesolimbic dopamine system. Neuroscience 1995, 68, 969–979. [Google Scholar] [CrossRef]
- Kramar, C.; Loureiro, M.; Renard, J.; LaViolette, S.R. Palmitoylethanolamide modulates GPR55 receptor signaling in the ventral hippocampus to regulate mesolimbic dopamine activity, social interaction, and memory processing. Cannabis Cannabinoid Res. 2017, 2, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Supekar, K.; Kochalka, J.; Schaer, M.; Wakeman, H.; Qin, S.; Padmanabhan, A.; Menon, V. Deficits in mesolimbic reward pathway underlie social interaction impairments in children with autism. Brain 2018, 141, 2795–2805. [Google Scholar] [CrossRef] [Green Version]
- Papakostas, G.I. Happiness and treatment outcome in resistant depression. J. Clin. Psychiatry 2020, 81, 20com13636. [Google Scholar] [CrossRef]
- Nestler, E.J. Role of the brain’s reward circuitry in depression: Transcriptional mechanics. Int. Rev. Neurobiol. 2015, 124, 151–170. [Google Scholar] [CrossRef] [Green Version]
- Chaudhury, D.; Walsh, J.; Friedman, A.K.; Juarez, B.; Ku, S.M.; Koo, J.W.; Ferguson, D.; Tsai, H.-C.; Pomeranz, L.; Christoffel, D.; et al. Rapid regulation of depression-related behaviours by control of midbrain dopamine neurons. Nature 2012, 493, 532–536. [Google Scholar] [CrossRef]
- Koo, J.W.; Labonté, B.; Engmann, O.; Calipari, E.; Juarez, B.; Lorsch, Z.; Walsh, J.; Friedman, A.K.; Yorgason, J.T.; Han, M.-H.; et al. Essential role of mesolimbic brain-derived neurotrophic factor in chronic social stress–induced depressive behaviors. Biol. Psychiatry 2015, 80, 469–478. [Google Scholar] [CrossRef] [Green Version]
- Melief, E.J.; McKinley, J.W.; Lam, J.Y.; Whiteley, N.M.; Gibson, A.W.; Neumaier, J.F.; Henschen, C.W.; Palmiter, R.D.; Bamford, N.S.; Darvas, M. Loss of glutamate signaling from the thalamus to dorsal striatum impairs motor function and slows the execution of learned behaviors. NPJ Park. Dis. 2018, 4, 23. [Google Scholar] [CrossRef]
- Kawashima, S.; Ueki, Y.; Kato, T.; Ito, K.; Matsukawa, N. Reduced striatal dopamine release during motor skill acquisition in Parkinson’s disease. PLoS ONE 2018, 13, e0196661. [Google Scholar] [CrossRef]
- Hintiryan, H.; Foster, N.; Bowman, I.; Bay, M.; Song, M.Y.; Gou, L.; Yamashita, S.; Bienkowski, M.; Zingg, B.; Zhu, M.; et al. The mouse cortico-striatal projectome. Nat. Neurosci. 2016, 19, 1100–1114. [Google Scholar] [CrossRef] [PubMed]
- Klein, T.A.; Ullsperger, M.; Jocham, G. Learning relative values in the striatum induces violations of normative decision making. Nat. Commun. 2017, 8, 16033. [Google Scholar] [CrossRef] [Green Version]
- Bang, D.; Kishida, K.T.; Lohrenz, T.; White, J.P.; Laxton, A.W.; Tatter, S.B.; Fleming, S.M.; Montague, P.R. Sub-second dopamine and serotonin signaling in human striatum during perceptual decision-making. Neuron 2020, 108, 999–1010. [Google Scholar] [CrossRef]
- Kang, S.; Hong, S.-I.; Lee, J.; Peyton, L.; Baker, M.; Choi, S.; Kim, H.; Chang, S.-Y.; Choi, D.-S. Activation of astrocytes in the dorsomedial striatum facilitates transition from habitual to goal-directed reward-seeking behavior. Biol. Psychiatry 2020, 88, 797–808. [Google Scholar] [CrossRef]
- Balleine, B.W.; Delgado, M.R.; Hikosaka, O. The Role of the Dorsal Striatum in Reward and Decision-Making. J. Neurosci. 2007, 27, 8161–8165. [Google Scholar] [CrossRef] [Green Version]
- Nadel, J.A.; Pawelko, S.S.; Copes-Finke, D.; Neidhart, M.; Howard, C.D. Lesion of striatal patches disrupts habitual behaviors and increases behavioral variability. PLoS ONE 2020, 15, e0224715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muranishi, M.; Inokawa, H.; Yamada, H.; Ueda, Y.; Matsumoto, N.; Nakagawa, M.; Kimura, M. Inactivation of the putamen selectively impairs reward history-based action selection. Exp. Brain Res. 2011, 209, 235–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nonomura, S.; Nishizawa, K.; Sakai, Y.; Kawaguchi, Y.; Kato, S.; Uchigashima, M.; Watanabe, M.; Yamanaka, K.; Enomoto, K.; Chiken, S.; et al. Monitoring and updating of action selection for goal-directed behavior through the striatal direct and indirect pathways. Neuron 2018, 99, 1302–1314.e5. [Google Scholar] [CrossRef] [Green Version]
- Lago, T.; Davis, A.; Grillon, C.; Ernst, M. Striatum on the anxiety map: Small detours into adolescence. Brain Res. 2016, 1654, 177–184. [Google Scholar] [CrossRef] [Green Version]
- Oosterwijk, C.S.; Vriend, C.; Berendse, H.W.; van der Werf, Y.; van den Heuvel, O.A. Anxiety in Parkinson’s disease is associated with reduced structural covariance of the striatum. J. Affect. Disord. 2018, 240, 113–120. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Iegaki, N.; Fu, K.; Ishikawa, Y.; Sumi, K.; Azuma, S.; Uno, K.; Muramatsu, S.-I.; Nitta, A. Striatal n-acetylaspartate synthetase Shati/Nat8l regulates depression-like behaviors via mGluR3-mediated serotonergic suppression in mice. Int. J. Neuropsychopharmacol. 2017, 20, 1027–1035. [Google Scholar] [CrossRef] [Green Version]
- Miyanishi, H.; Muramatsu, S.-I.; Nitta, A. Striatal Shati/Nat8l–BDNF pathways determine the sensitivity to social defeat stress in mice through epigenetic regulation. Neuropsychopharmacology 2021, 46, 1594–1605. [Google Scholar] [CrossRef] [PubMed]
- Dias-Ferreira, E.; Sousa, J.C.; Melo, I.; Morgado, P.; Mesquita, A.R.; Cerqueira, J.J.; Costa, R.M.; Sousa, N. Chronic stress causes frontostriatal reorganization and affects decision-making. Science 2009, 325, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, I.I.; Hanson, D.R. Human development: Biological and genetic processes. Annu. Rev. Psychol. 2005, 56, 263–286. [Google Scholar] [CrossRef] [PubMed]
- Heim, C.; Binder, E.B. Current research trends in early life stress and depression: Review of human studies on sensitive periods, gene–environment interactions, and epigenetics. Exp. Neurol. 2012, 233, 102–111. [Google Scholar] [CrossRef]
- Niwa, M.; Nitta, A.; Mizoguchi, H.; Ito, Y.; Noda, Y.; Nagai, T.; Nabeshima, T. A novel molecule “Shati” is involved in methamphetamine-induced hyperlocomotion, sensitization, and conditioned place preference. J. Neurosci. 2007, 27, 7604–7615. [Google Scholar] [CrossRef] [Green Version]
- Ariyannur, P.S.; Moffett, J.R.; Manickam, P.; Pattabiraman, N.; Arun, P.; Nitta, A.; Nabeshima, T.; Madhavarao, C.N.; Namboodiri, A.M. Methamphetamine-induced neuronal protein NAT8L is the NAA biosynthetic enzyme: Implications for specialized acetyl coenzyme A metabolism in the CNS. Brain Res. 2010, 1335, 1–13. [Google Scholar] [CrossRef]
- Becker, I.; Lodder, J.; Gieselmann, V.; Eckhardt, M. Molecular characterization of N-acetylaspartylglutamate synthetase. J. Biol. Chem. 2010, 285, 29156–29164. [Google Scholar] [CrossRef] [Green Version]
- Neale, J.H.; Bzdega, T.; Wroblewska, B. N-acetylaspartylglutamate: The most abundant peptide neurotransmitter in the mammalian central nervous system. J. Neurochem. 2002, 75, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Neale, J.H.; Olszewski, R.T.; Zuo, D.; Janczura, K.J.; Profaci, C.; Lavin, K.; Madore, J.C.; Bzdega, T. Advances in understanding the peptide neurotransmitter NAAG and appearance of a new member of the NAAG neuropeptide family. J. Neurochem. 2011, 118, 490–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bzdega, T.; Turi, T.; Wroblewska, B.; She, D.; Chung, H.S.; Kim, H.; Neale, J.H. Molecular cloning of a peptidase against N-acetylaspartylglutamate from a rat hippocampal cDNA library. J. Neurochem. 2002, 69, 2270–2277. [Google Scholar] [CrossRef]
- Singhal, N.K.; Huang, H.; Li, S.; Clements, R.; Gadd, J.; Daniels, A.; Kooijman, E.E.; Bannerman, P.; Burns, T.; Guo, F.; et al. The neuronal metabolite NAA regulates histone H3 methylation in oligodendrocytes and myelin lipid composition. Exp. Brain Res. 2016, 235, 279–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, L.M.; Reynolds, G. Differential regional N-acetylaspartate deficits in postmortem brain in schizophrenia, bipolar disorder and major depressive disorder. J. Psychiatr. Res. 2011, 45, 54–59. [Google Scholar] [CrossRef]
- Haddar, M.; Uno, K.; Hamatani, K.; Muramatsu, S.; Nitta, A. Regulatory system of mGluR group II in the nucleus accumbens for methamphetamine-induced dopamine increase by the medial prefrontal cortex. Neuropsychopharmacol. Rep. 2019, 39, 209–216. [Google Scholar] [CrossRef]
- Haddar, M.; Uno, K.; Azuma, K.; Muramatsu, S.; Nitta, A. Inhibitory effects of Shati/Nat8l overexpression in the medial prefrontal cortex on methamphetamine-induced conditioned place preference in mice. Addict. Biol. 2019, 25, e12749. [Google Scholar] [CrossRef] [Green Version]
- Nitta, A.; Noike, H.; Sumi, K.; Miyanishi, H.; Tanaka, T.; Takaoka, K.; Nagakura, M.; Iegaki, N.; Kaji, J.-I.; Miyamoto, Y.; et al. Shati/Nat8l and N-acetylaspartate (NAA) have important roles in regulating nicotinic acetylcholine receptors in neuronal and psychiatric diseases in animal models and humans. In Nicotinic Acetylcholine Receptor Signaling in Neuroprotection [Internet]; Springer: Singapore, 2018; pp. 89–111. [Google Scholar] [CrossRef]
- Miyanishi, H.; Uno, K.; Iwata, M.; Kikuchi, Y.; Yamamori, H.; Yasuda, Y.; Ohi, K.; Hashimoto, R.; Hattori, K.; Yoshida, S.; et al. Investigating DNA methylation of SHATI/NAT8L promoter sites in blood of unmedicated patients with major depressive disorder. Biol. Pharm. Bull. 2020, 43, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Uno, K.; Miyanishi, H.; Sodeyama, K.; Fujiwara, T.; Miyazaki, T.; Muramatsu, S.-I.; Nitta, A. Vulnerability to depressive behavior induced by overexpression of striatal Shati/Nat8l via the serotonergic neuronal pathway in mice. Behav. Brain Res. 2019, 376, 112227. [Google Scholar] [CrossRef]
- Furukawa-Hibi, Y.; Nitta, A.; Fukumitsu, H.; Somiya, H.; Toriumi, K.; Furukawa, S.; Nabeshima, T.; Yamada, K. Absence of SHATI/Nat8l reduces social interaction in mice. Neurosci. Lett. 2012, 526, 79–84. [Google Scholar] [CrossRef]
- Costa, G.; Serra, M.; Marongiu, J.; Morelli, M.; Simola, N. Influence of dopamine transmission in the medial prefrontal cortex and dorsal striatum on the emission of 50-kHz ultrasonic vocalizations in rats treated with amphetamine: Effects on drug-stimulated and conditioned calls. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 97, 109797. [Google Scholar] [CrossRef] [PubMed]
- Sachs, B.D.; Ni, J.R.; Caron, M.G. Brain 5-HT deficiency increases stress vulnerability and impairs antidepressant responses following psychosocial stress. Proc. Natl. Acad. Sci. USA 2015, 112, 2557–2562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Wu, W.; Li, Y.; Li, L. Influences of the gut microbiota on DNA methylation and histone modification. Dig. Dis. Sci. 2017, 62, 1155–1164. [Google Scholar] [CrossRef]
- Wang, Y.; Yuan, Q.; Xie, L. Histone modifications in aging: The underlying mechanisms and implications. Curr. Stem Cell Res. Ther. 2018, 13, 125–135. [Google Scholar] [CrossRef]
- Bogdanović, O.; Lister, R. DNA methylation and the preservation of cell identity. Curr. Opin. Genet. Dev. 2017, 46, 9–14. [Google Scholar] [CrossRef]
- Xu, H.; Wang, J.; Zhang, K.; Zhao, M.; Ellenbroek, B.; Shao, F.; Wang, W. Effects of adolescent social stress and antidepressant treatment on cognitive inflexibility and Bdnf epigenetic modifications in the mPFC of adult mice. Psychoneuroendocrinology 2017, 88, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Dong, E.; Tueting, P.; Matrisciano, F.; Grayson, D.R.; Guidotti, A. Behavioral and molecular neuroepigenetic alterations in prenatally stressed mice: Relevance for the study of chromatin remodeling properties of antipsychotic drugs. Transl. Psychiatry 2016, 6, e711. [Google Scholar] [CrossRef]
- Toriumi, K.; Mamiya, T.; Song, Z.; Honjo, T.; Watanabe, H.; Tanaka, J.; Kondo, M.; Mouri, A.; Kim, H.-C.; Nitta, A.; et al. Deletion of SHATI/NAT8L decreases the N-acetylaspartate content in the brain and induces behavioral deficits, which can be ameliorated by administering N-acetylaspartate. Eur. Neuropsychopharmacol. 2015, 25, 2108–2117. [Google Scholar] [CrossRef]
- Zarate, C.A.; Singh, J.B.; Carlson, P.J.; Brutsche, N.E.; Ameli, R.; Luckenbaugh, D.A.; Charney, D.S.; Manji, H.K. A Randomized Trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch. Gen. Psychiatry 2006, 63, 856–864. [Google Scholar] [CrossRef]
- Fukumoto, K.; Iijima, M.; Chaki, S. The antidepressant effects of an mGlu2/3 receptor antagonist and ketamine require AMPA receptor stimulation in the mPFC and subsequent activation of the 5-HT neurons in the DRN. Neuropsychopharmacology 2015, 41, 1046–1056. [Google Scholar] [CrossRef]
- Shin, C.; Kim, Y.-K. Ketamine in major depressive disorder: Mechanisms and future perspectives. Psychiatry Investig. 2020, 17, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Brain region | Speceie | Trials | Effect to Depression Pathogenesis | Alteration of Expression Levels in Depression Individuals |
|---|---|---|---|---|
| PFC | Human | [69,70,71,72] | pro-depressant | increasing |
| Animal | [76,77,78,79,80,81] | |||
| Hippocampus | Human | [93,94,101,108] | pro-depressant | increasing |
| Animal | [30,102,103,104,105,106,109] | |||
| Mesolimbic pathway | Human | [111] | anti-depressant | decreasing |
| Animal | [29,58,110,112,113,122] | |||
| Dorsal striatum | Human | None | anti-depressant | decreasing |
| Animal | [136] | |||
| PFC: Prefrontal cortex | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miyanishi, H.; Nitta, A. A Role of BDNF in the Depression Pathogenesis and a Potential Target as Antidepressant: The Modulator of Stress Sensitivity “Shati/Nat8l-BDNF System” in the Dorsal Striatum. Pharmaceuticals 2021, 14, 889. https://doi.org/10.3390/ph14090889
Miyanishi H, Nitta A. A Role of BDNF in the Depression Pathogenesis and a Potential Target as Antidepressant: The Modulator of Stress Sensitivity “Shati/Nat8l-BDNF System” in the Dorsal Striatum. Pharmaceuticals. 2021; 14(9):889. https://doi.org/10.3390/ph14090889
Chicago/Turabian StyleMiyanishi, Hajime, and Atsumi Nitta. 2021. "A Role of BDNF in the Depression Pathogenesis and a Potential Target as Antidepressant: The Modulator of Stress Sensitivity “Shati/Nat8l-BDNF System” in the Dorsal Striatum" Pharmaceuticals 14, no. 9: 889. https://doi.org/10.3390/ph14090889
APA StyleMiyanishi, H., & Nitta, A. (2021). A Role of BDNF in the Depression Pathogenesis and a Potential Target as Antidepressant: The Modulator of Stress Sensitivity “Shati/Nat8l-BDNF System” in the Dorsal Striatum. Pharmaceuticals, 14(9), 889. https://doi.org/10.3390/ph14090889