
Lignosulfonic Acid Sodium Is a Noncompetitive Inhibitor of Human Factor XIa



Abstract
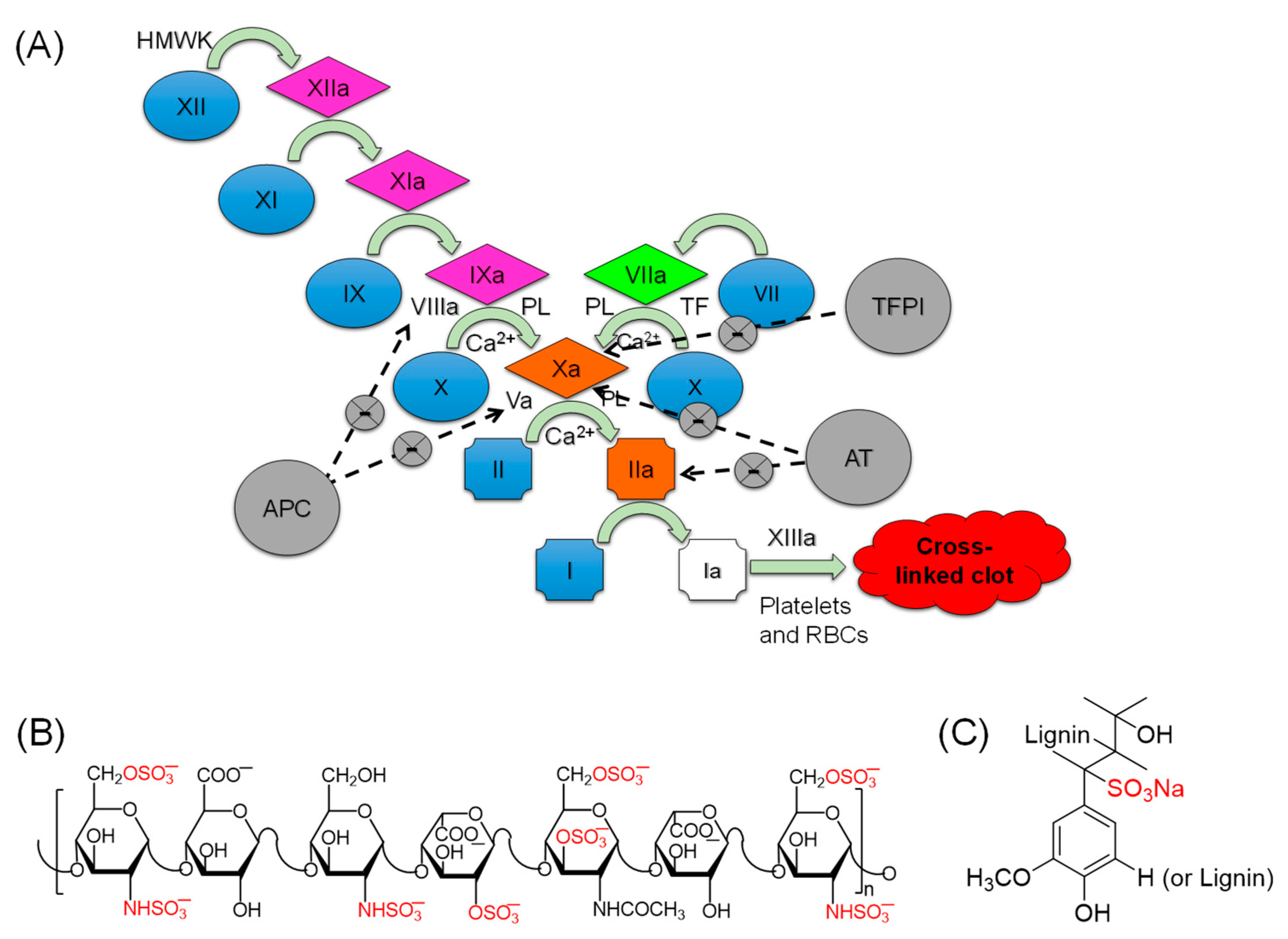
:1. Introduction
2. Results and Discussion
2.1. Effects of LSAS on Clotting Times of Normal and Deficient Human Plasmas
2.2. Inhibition of Human FXIa by LSAS in Chromogenic Substrate Assay
2.3. Selectivity Studies: Inhibition Potential of LSAS toward Other Coagulation Proteins
2.4. Selectivity Studies: Inhibition Potential of LSAS toward Other Serine Protease Important for Fibrinolysis, Digestion, and Inflammation
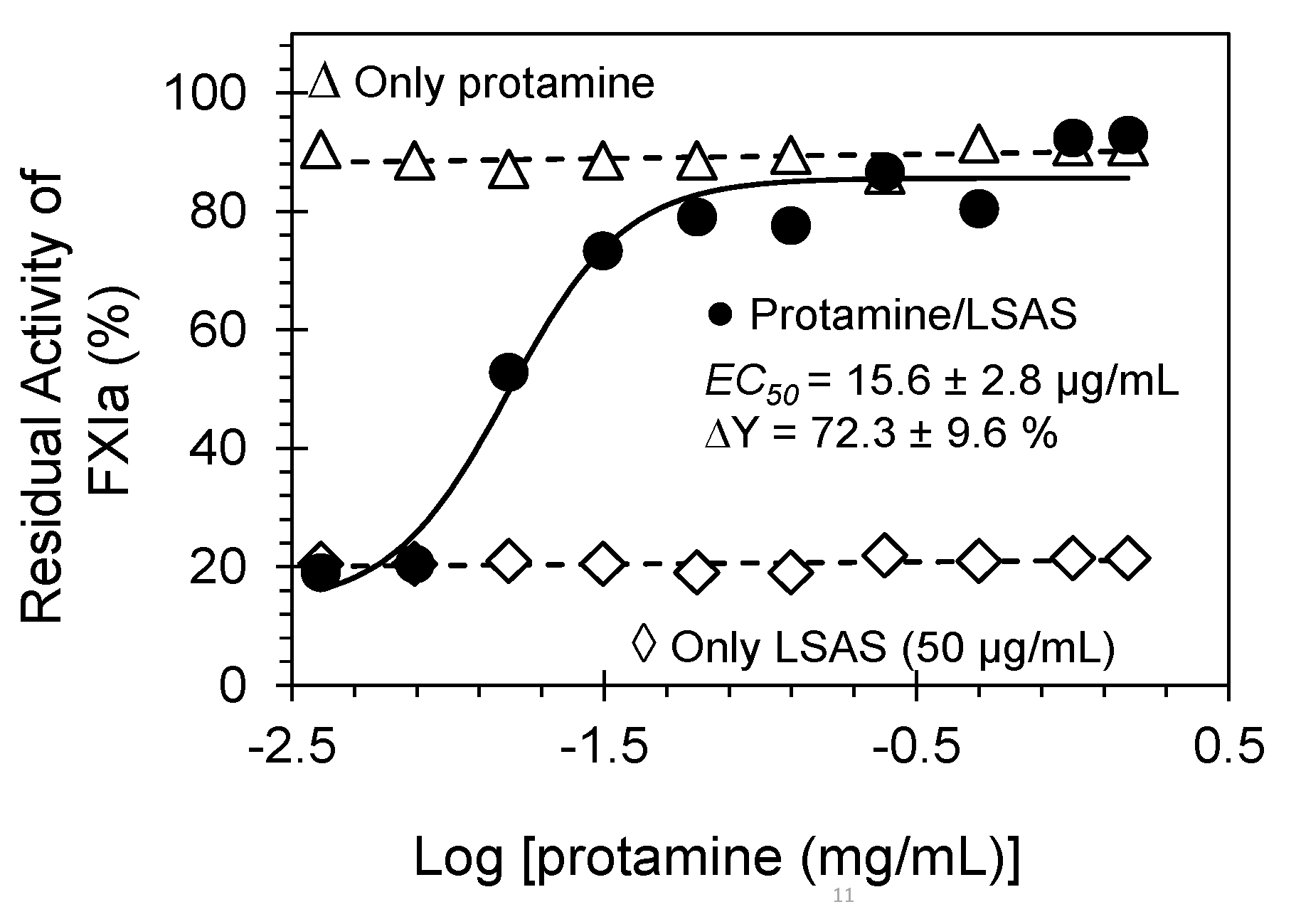
2.5. Reversibility of FXIa Inhibition by LSAS. Protamine as a Reversal Agent
2.6. Effect of LSAS on the Physiological Function of FXIa, i.e., Activation of FIX
2.7. Effect of LSAS on FXIIIa-Mediated Polymerization of Fibrin(ogen)
2.8. Effect of LSAS on FXIa Interaction with Macromolecules
2.9. Competition Studies with UFH
2.10. Mechanism of FXIa Inhibition by LSAS: Michaelis–Menten Kinetics
2.11. Effects of Acetylation and Desulfonation on the Activity of LSAS towards FXIa
3. Materials and Methods
3.1. Materials
3.2. Effect of LSAS on Clotting Times of Deficient and Normal Human Plasmas
3.3. Inhibition of Human FXIa in Chromogenic Tripeptide Substrate Hydrolysis Assay by LSAS
3.4. Effect of LSAS on Other Enzymes in the Coagulation Process
3.5. Effect of LSAS on Fibrinolysis, Digestive, and Inflammatory Serine Proteases
3.6. Reversibility of FXIa Inhibition by LSAS
3.7. Effect of LSAS on the Physiological Function of Human FXIa, i.e., FIX Activation
3.8. Effect of LSAS on FXIIIa-Mediated Polymerization of Fibrin(ogen)
3.9. Effect of LSAS Polymer on FXIa–AT Complex Formation
3.10. Effect of LSAS Polymer on Thrombin-Mediated Activation of FXI
3.11. Effect of LSAS Polymer on FXI Activation by FXIIa-Mediated Activation of FXI
3.12. Competition Studies of LSAS with UFH
3.13. Michaelis–Menten Kinetics for S-2366 Hydrolysis by Human FXIa in the Presence of LSAS
3.14. Effects of Acetylation and Desulfonation on the Activity of LSAS towards FXIa
4. Conclusions and Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Al-Horani, R.A.; Afosah, D.K. Recent advances in the discovery and development of factor XI/XIa inhibitors. Med. Res. Rev. 2018, 38, 1974–2023. [Google Scholar] [CrossRef]
- Mulloy, B.; Hogwood, J.; Gray, E.; Lever, R.; Page, C.P. Pharmacology of Heparin and Related Drugs. Pharmacol. Rev. 2016, 68, 76–141. [Google Scholar] [CrossRef]
- Holbrook, A.; Schulman, S.; Witt, D.M.; Vandvik, P.O.; Fish, J.; Kovacs, M.J.; Svensson, P.J.; Veenstra, D.L.; Crowther, M.; Guyatt, G.H. Evidence-based management of anticoagulant therapy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians EvidenceBased Clinical Practice Guidelines. Chest 2012, 141 (Suppl. 2), e152S–e184S. [Google Scholar] [CrossRef] [Green Version]
- House Hearing, 110 Congress. From the U.S. Government Printing Office. The Heparin Disaster: Chinese Counterfeits and American Failures. Available online: https://www.govinfo.gov/content/pkg/CHRG110hhrg53183/html/CHRG-110hhrg53183.htm (accessed on 1 July 2021).
- Vilanova, E.; Tovar, A.M.F.; Mourão, P.A.S. Imminent risk of a global shortage of heparin caused by the African Swine Fever afflicting the Chinese pig herd. J. Thromb. Haemost. 2019, 17, 254–256. [Google Scholar] [CrossRef] [PubMed]
- Alban, S. Adverse effects of heparin. Handb. Exp. Pharmacol. 2012, 207, 211–263. [Google Scholar]
- Kar, S.; Mottamal, M.; Al-Horani, R.A. Discovery of Benzyl Tetraphosphonate Derivative as Inhibitor of Human Factor XIa. ChemistryOpen 2020, 9, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Al-Horani, R.A.; Clemons, D.; Mottamal, M. The In Vitro Effects of Pentamidine Isethionate on Coagulation and Fibrinolysis. Molecules 2019, 24, 2146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obaidullah, A.J.; Al-Horani, R.A. Discovery of Chromen-7-yl Furan-2-Carboxylate as a Potent and Selective Factor XIa Inhibitor. Cardiovasc. Hematol. Agents Med. Chem. 2017, 15, 40–48. [Google Scholar] [CrossRef]
- Argade, M. Discovery and Biophysical Characterization of Allosteric Inhibitors of Factor XIa (FXIa). Master’s Thesis, Virginia Commonwealth University, Richmond, VA, USA, 2012. [Google Scholar] [CrossRef]
- Al-Horani, R.A.; Mehta, A.Y.; Desai, U.R. Potent direct inhibitors of factor Xa based on the tetrahydroisoquinoline scaffold. Eur. J. Med. Chem. 2012, 54, 771–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Horani, R.A.; Karuturi, R.; Lee, M.; Afosah, D.K.; Desai, U.R. Allosteric Inhibition of Factor XIIIa. Non-Saccharide Glycosaminoglycan Mimetics, but Not Glycosaminoglycans, Exhibit Promising Inhibition Profile. PLoS ONE 2016, 11, e0160189. [Google Scholar] [CrossRef] [Green Version]
- Al-Horani, R.A.; Aliter, K.F.; Kar, S.; Mottamal, M. Sulfonated Nonsaccharide Heparin Mimetics Are Potent and Noncompetitive Inhibitors of Human Neutrophil Elastase. ACS Omega 2021, 6, 12699–12710. [Google Scholar] [CrossRef] [PubMed]
- Al-Horani, R.A.; Gailani, D.; Desai, U.R. Allosteric inhibition of factor XIa. Sulfated non-saccharide glycosaminoglycan mimetics as promising anticoagulants. Thromb. Res. 2015, 136, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Al-Horani, R.A.; Ponnusamy, P.; Mehta, A.Y.; Gailani, D.; Desai, U.R. Sulfated pentagalloylglucoside is a potent, allosteric, and selective inhibitor of factor XIa. J. Med. Chem. 2013, 56, 867–878. [Google Scholar] [CrossRef] [Green Version]
- Gailani, D.; Geng, Y.; Verhamme, I.; Sun, M.-F.; Bajaj, S.P.; Messer, A.; Emsley, J. The mechanism underlying activation of factor IX by factor XIa. Thromb. Res. 2014, 133, S48–S51. [Google Scholar] [CrossRef] [Green Version]
- Fredenburgh, J.C.; Weitz, J.I. Overview of Hemostasis and Thrombosis. In Hematology, 7th ed.; Hoffman, R., Benz, E.J., Silberstein, L.E., Heslop, H.E., Weitz, J.I., Anastasi, J., Salama, M.E., Abutalib, S.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 1831–1842. [Google Scholar] [CrossRef]
- Yang, L.; Sun, M.F.; Gailani, D.; Rezaie, A.R. Characterization of a heparin-binding site on the catalytic domain of factor XIa: Mechanism of heparin acceleration of factor XIa inhibition by the serpins antithrombin and C1-inhibitor. Biochemistry 2009, 48, 1517–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, Y.; Verhamme, I.M.; Smith, S.B.; Sun, M.-F.; Matafonov, A.; Cheng, Q.; Smith, S.A.; Morrissey, J.H.; Gailani, D. The dimeric structure of factor XI and zymogen activation. Blood 2013, 121, 3962–3969. [Google Scholar] [CrossRef] [Green Version]
- Scott, C.F.; Colman, R.W. Fibrinogen blocks the autoactivation and thrombin-mediated activation of factor XI on dextran sulfate. Proc. Natl. Acad. Sci. USA 1992, 89, 11189–11193. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.H.; Smith, S.A.; Morrissey, J.H. Polyphosphate is a cofactor for the activation of factor XI by thrombin. Blood 2011, 118, 6963–6970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naito, K.; Fujikawa, K. Activation of human blood coagulation factor XI independent of factor XII. Factor XI is activated by thrombin and factor XIa in the presence of negatively charged surfaces. J. Biol. Chem. 1991, 266, 7353–7358. [Google Scholar] [CrossRef]
- Gailani, D.; Broze, G.J., Jr. Factor XI activation in a revised model of blood coagulation. Science 1991, 253, 909–912. [Google Scholar] [CrossRef]
- Gailani, D.; Broze, G.J., Jr. Effects of glycosaminoglycans on factor XI activation by thrombin. Blood Coagul. Fibrinolysis 1993, 4, 15–20. [Google Scholar] [CrossRef]
- Gailani, D.; Broze, G.J., Jr. Factor XII-independent activation of factor XI in plasma: Effects of sulfatides on tissue factor-induced coagulation. Blood 1993, 82, 813–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Horani, R.A.; Desai, U.R. Factor XIa inhibitors: A review of the patent literature. Expert Opin. Ther. Pat. 2016, 26, 323–345. [Google Scholar] [CrossRef] [PubMed]
- Donkor, D.; Bhakta, V.; Eltringham-Smith, L.J.; Stafford, A.R.; Weitz, J.I.; Sheffield, W.P. Selection and characterization of a DNA aptamer inhibiting coagulation factor XIa. Sci. Rep. 2017, 7, 2102. [Google Scholar] [CrossRef]
- Chang, J.; Jin, J.; Lollar, P.; Bode, W.; Brandstetter, J.; Hamaguchi, N.; Straight, D.L.; Stafford, D.W. Changing Residue 338 in Human Factor IX from Arginine to Alanine Causes an Increase in Catalytic Activity. J. Biol. Chem. 1998, 273, 12089–12094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnaswamy, S. Exosite-driven substrate specificity and function in coagulation. J. Thromb. Haemost. 2005, 3, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; Macgillivray, R.T.; Di Cera, E. Determinants of specificity in coagulation proteases. J. Thromb. Haemost. 2005, 3, 2401–2408. [Google Scholar] [CrossRef]
- Abdel Aziz, M.H.; Mosier, P.D.; Desai, U.R. Identification of the site of binding of sulfated, low molecular weight lignins on thrombin. Biochem. Biophys. Res. Commun. 2011, 413, 348–352. [Google Scholar] [CrossRef] [Green Version]
- Safety Data Sheet, Sigma Aldrich. Available online: https: //www.sigmaaldrich.com/US/en/sds/aldrich/471038 (accessed on 20 August 2021).
- Alamneh, E.A.; Chalmers, L.; Bereznicki, L.R. Suboptimal Use of Oral Anticoagulants in Atrial Fibrillation: Has the Introduction of Direct Oral Anticoagulants Improved Prescribing Practices? Am. J. Cardiovasc. Drugs 2016, 16, 183–200. [Google Scholar] [CrossRef]
- Barra, M.E.; Fanikos, J.; Connors, J.M.; Sylvester, K.W.; Piazza, G.; Goldhaber, S.Z. Evaluation of Dose Reduced Direct Oral Anticoagulant Therapy. Am. J. Med. 2016, 129, 1198–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black-Maier, E.; Piccini, J.P. Oral anticoagulation in end-stage renal disease and atrial fibrillation: Is it time to just say no to drugs? Heart 2017, 103, 807–808. [Google Scholar] [CrossRef]
- Hughes, S.; Szeki, I.; Nash, M.J.; Thachil, J. Anticoagulation in chronic kidney disease patients-the practical aspects. Clin. Kidney J. 2014, 7, 442–449. [Google Scholar] [CrossRef] [Green Version]
- Lutz, J.; Jurk, K.; Schinzel, H. Direct oral anticoagulants in patients with chronic kidney disease: Patient selection and special considerations. Int. J. Nephrol. Renov. Dis. 2017, 10, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Shatzel, J.J.; DeLoughery, E.P.; Lorentz, C.U.; Tucker, E.I.; Aslan, J.E.; Hinds, M.T.; Gailani, D.; Weitz, J.I.; McCarty, O.J.T.; Gruber, A. The contact activation system as a potential therapeutic target in patients with COVID-19. Res. Pract. Thromb. Haemost. 2020, 4, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Silasi, R.; Keshari, R.S.; Lupu, C.; Van Rensburg, W.J.; Chaaban, H.; Regmi, G.; Shamanaev, A.; Shatzel, J.J.; Puy, C.; Lorentz, C.U.; et al. Inhibition of contact-mediated activation of factor XI protects baboons against S aureus-induced organ damage and death. Blood Adv. 2019, 3, 658–669. [Google Scholar] [CrossRef]
- Raghunathan, V.; Zilberman-Rudenko, J.; Olson, S.R.; Lupu, F.; McCarty, O.J.T.; Shatzel, J.J. The contact pathway and sepsis. Res. Pract. Thromb. Haemost. 2019, 3, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Tucker, E.I.; Verbout, N.G.; Leung, P.Y.; Hurst, S.; McCarty, O.J.; Gailani, D.; Gruber, A. Inhibition of factor XI activation attenuates inflammation and coagulopathy while improving the survival of mouse polymicrobial sepsis. Blood 2012, 119, 4762–47628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosikowska, P.; Lesner, A. Inhibitors of cathepsin G: A patent review (2005 to present). Expert Opin. Ther. Pat. 2013, 23, 1611–1624. [Google Scholar] [CrossRef]
- Allen, D.H.; Tracy, P.B. Human coagulation factor V is activated to the functional cofactor by elastase and cathepsin G expressed at the monocyte surface. J. Biol. Chem. 1995, 270, 1408–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouchard, B.A.; Tracy, P.B. The participation of leukocytes in coagulant reactions. J. Thromb. Haemost. 2003, 1, 464–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gale, A.J.; Rozenshteyn, D. Cathespsin G, a leukocyte protease, activates coagulation factor VIII. Thromb. Hemost. 2008, 99, 44–51. [Google Scholar]
- Qiu, X.; Kong, Q.; Zhou, M.; Yang, D. Aggregation behavior of sodium lignosulfonate in water solution. J. Phys. Chem. B 2010, 114, 15857–15861. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Human Plasma | (LSAS) (µg/mL) to Double Clotting Time |
|---|---|
| APTT assay | APTTEC×2 |
| Normal | 308.8 b ± 23.9 c |
| Deficient of AT | 275.0 ± 62.0 |
| Deficient of FVII | 329.1 ± 58.7 |
| Deficient of FIX | 254.4 ± 27.5 |
| Deficient of FXI | 428.9 ± 45.6 |
| Deficient of FXII | 282.2 ± 119.8 |
| PT assay | PTEC×2 |
| Normal | 980.1 ± 145.0 |
| TT assay | TTEC×2 |
| Normal | >>500 |
| Enzyme | IC50 (µg/mL) | HS | ∆Y (%) |
|---|---|---|---|
| Thrombin | >125 | ND c | ND |
| FXa | >125 | ND | ND |
| FIXa | > 673 | ND | ND |
| FXIa | 7.9 ± 0.9 b | 0.9 ± 0.2 | 98.1 ± 4.7 |
| FXIIa | 714.0 ± 107.5 | 2.4 ± 0.5 | 92.6 ± 4.5 |
| FXIIIa | >12.5 | ND | ND |
| Plasmin | 212.5 ± 25.8 | 2.6 ± 0.8 | 112.4 ± 6.1 |
| Trypsin | >600 | ND | ND |
| Chymotrypsin | >2000 | ND | ND |
| HNE | 4.7 ± 0.2 | 1.8 ± 0.2 | 104.9 ± 2.1 |
| Cathepsin G | 0.73 ± 0.11 | 1.3 ± 0.3 | 89.7 ± 4.0 |
| UFH (µM) | IC50 (µg/mL) | HS | ∆Y (%) |
|---|---|---|---|
| 0 | 11.0 ± 1.8 b | 1.1 ± 0.2 | 99.8 ± 5.4 |
| 0.5 | 24.6 ± 3.7 | 0.9 ± 0.1 | 95.6 ± 4.5 |
| 5 | 28.9 ± 5.6 | 0.8 ± 0.1 | 102.1 ± 5.2 |
| 50 | 70.6 ± 11.6 | 1.1 ± 0.2 | 82.0 ± 4.5 |
| 150 | 91.8 ± 21.3 | 0.7 ± 0.1 | 90.6 ± 5.3 |
| 250 | 131.6 ± 22.7 | 0.8 ± 0.1 | 89.3 ± 2.9 |
| [1] (µg/mL) | S-2366 KM (mM) | VMAX (mAU/min) |
|---|---|---|
| 0 | 0.36 ± 0.03 b | 37.7 ± 1.0 |
| 1 | 0.29 ± 0.03 | 26.9 ± 0.7 |
| 10 | 0.31 ± 0.05 | 24.6 ± 1.2 |
| 25 | 0.31 ± 0.07 | 13.3 ± 0.9 |
| 50 | 0.43 ± 0.04 | 9.0 ± 0.3 |
| 150 | 0.30 ± 0.14 | 4.9 ± 0.7 |
| Polymer | FXIa IC50 (µg/mL) | Thrombin IC50 (µg/mL) | FXa IC50 (µg/mL) |
|---|---|---|---|
| LSAS | 7.9 ± 0.9 b | >125 | >125 |
| LSAS acetate | 0.39 ± 0.1 | 0.73 ± 0.04 | 0.48 ± 0.09 |
| Desulfonated | 53.9 ± 16.3 | ND c | ND |
| Desulfonated Polymer (µg/mL) | S-2366 KM (mM) | VMAX (mAU/min) |
|---|---|---|
| 0 | 0.25 ± 0.05 b | 32.1 ± 2.1 |
| 10 | 0.35 ± 0.04 | 29.7 ± 1.1 |
| 50 | 0.99 ± 0.24 | 17.8 ± 2.0 |
| 250 | 0.70 ± 0.12 | 10.6 ± 0.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kar, S.; Bankston, P.; Afosah, D.K.; Al-Horani, R.A. Lignosulfonic Acid Sodium Is a Noncompetitive Inhibitor of Human Factor XIa. Pharmaceuticals 2021, 14, 886. https://doi.org/10.3390/ph14090886
Kar S, Bankston P, Afosah DK, Al-Horani RA. Lignosulfonic Acid Sodium Is a Noncompetitive Inhibitor of Human Factor XIa. Pharmaceuticals. 2021; 14(9):886. https://doi.org/10.3390/ph14090886
Chicago/Turabian StyleKar, Srabani, Page Bankston, Daniel K. Afosah, and Rami A. Al-Horani. 2021. "Lignosulfonic Acid Sodium Is a Noncompetitive Inhibitor of Human Factor XIa" Pharmaceuticals 14, no. 9: 886. https://doi.org/10.3390/ph14090886
APA StyleKar, S., Bankston, P., Afosah, D. K., & Al-Horani, R. A. (2021). Lignosulfonic Acid Sodium Is a Noncompetitive Inhibitor of Human Factor XIa. Pharmaceuticals, 14(9), 886. https://doi.org/10.3390/ph14090886