
Combined Treatment with Acalabrutinib and Rapamycin Inhibits Glioma Stem Cells and Promotes Vascular Normalization by Downregulating BTK/mTOR/VEGF Signaling

 , ,
, ,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Drugs
2.2. Cells and Cell Cultures
2.3. Western Blotting
2.4. Sulforhodamine B Cytotoxicity Assay
2.5. Colony-Formation Assay
2.6. Tumorsphere-Formation Assay
2.7. VEGF Enzyme-Linked Immunosorbent Assay (ELISA)
2.8. Endothelial Migration Assays
2.9. Endothelial Tube-Formation Assays
2.10. Histology and Immunostaining
2.11. Statistical Analysis
3. Results
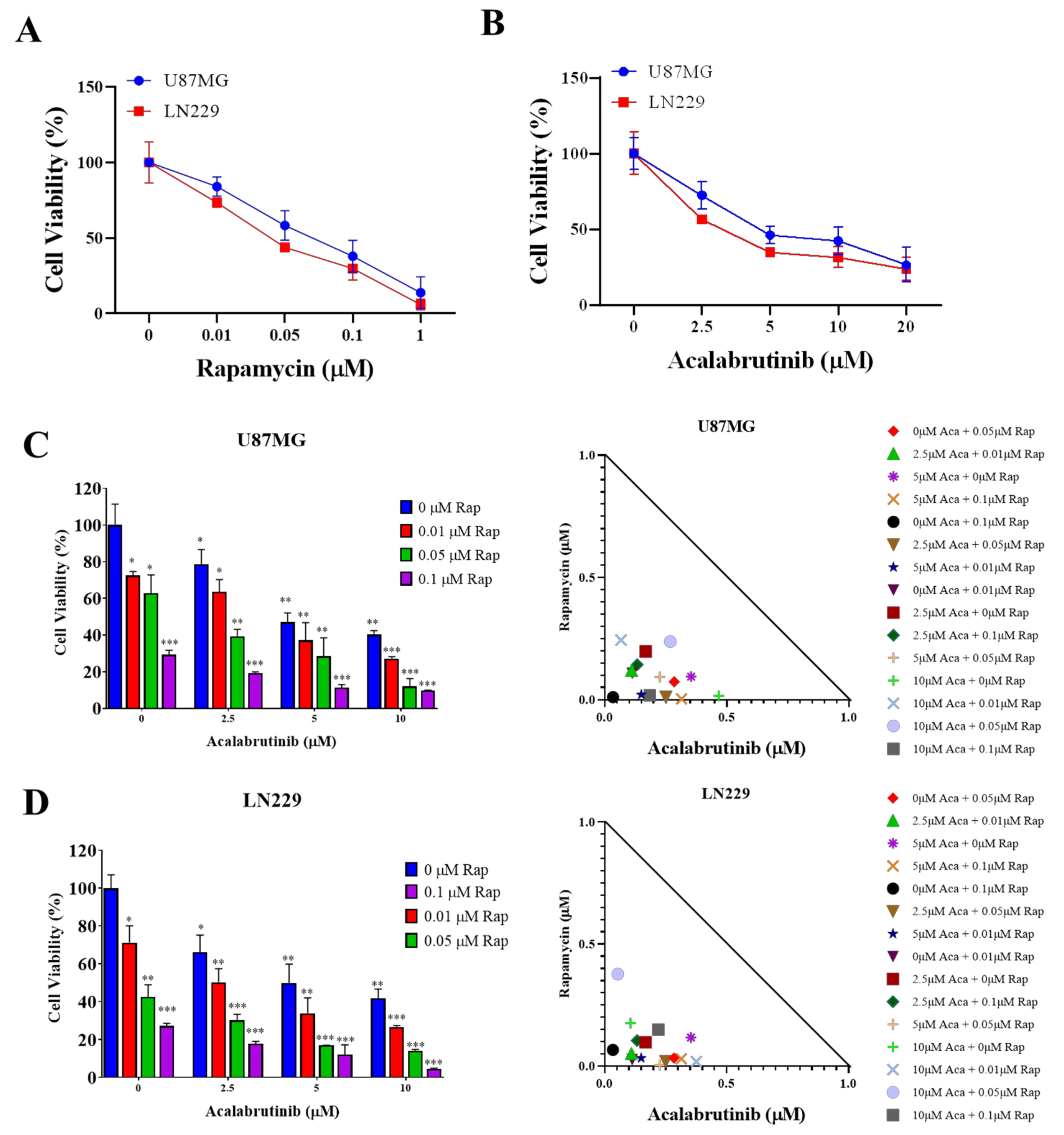
3.1. Rapamycin and Acalabrutinib Effectively Reduced the Viability of GBM Cell Lines and Exerted a Synergistic Antiproliferation Effect
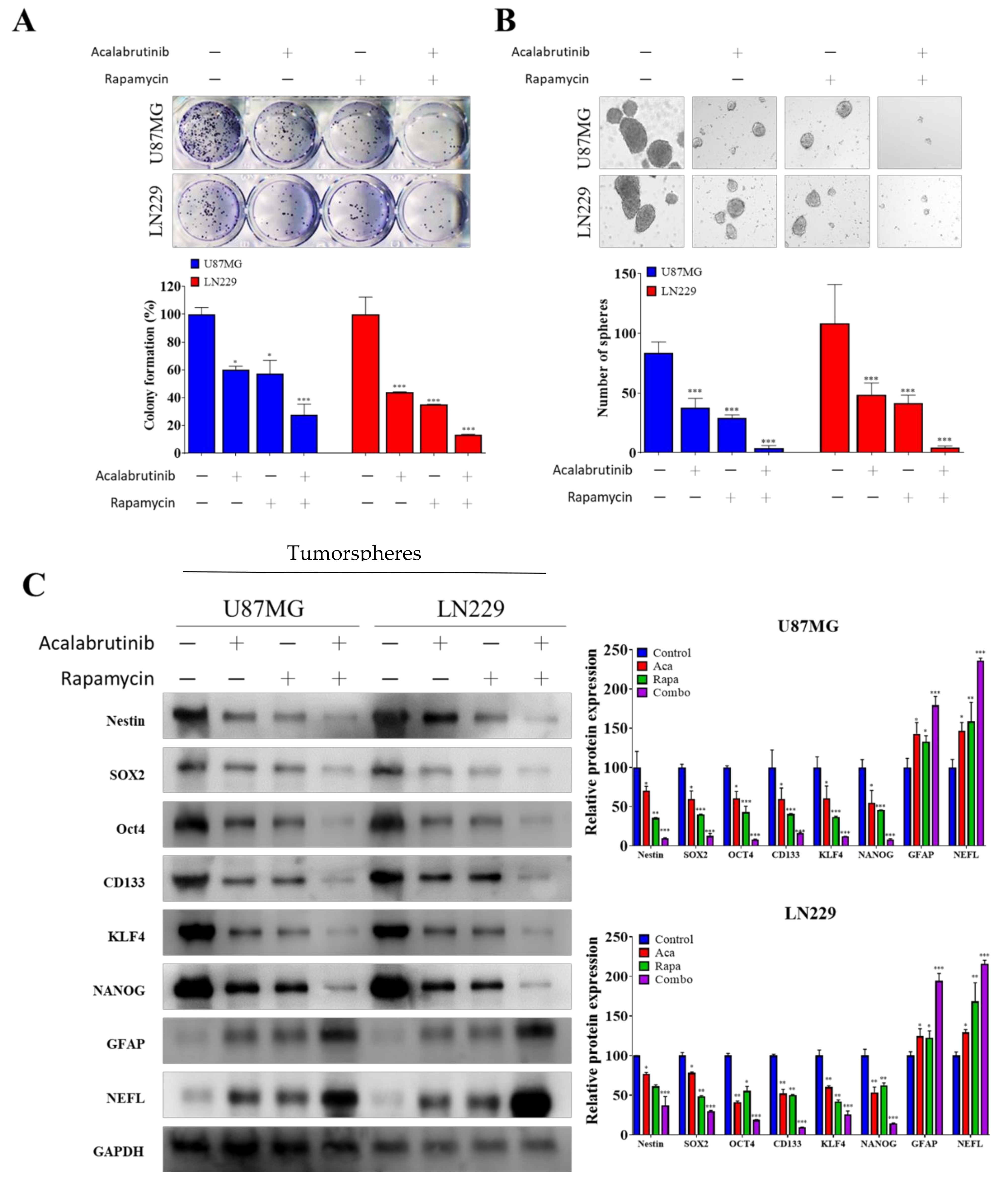
3.2. Combination Therapy of Acalabrutinib and Rapamycin Considerably Reduced the Tumorsphere-Formation Potential of GBM Cells
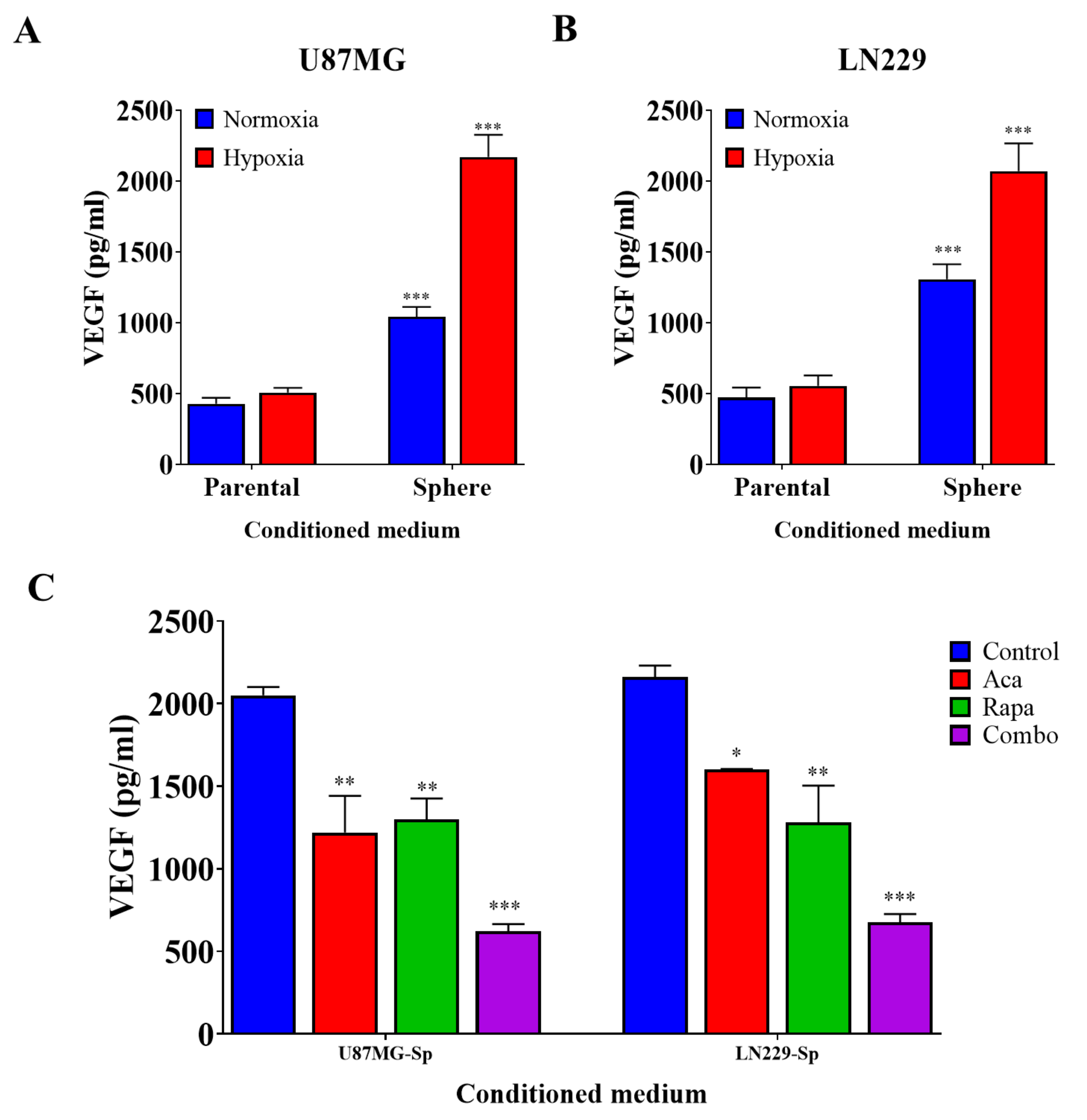
3.3. GBM CSCs Consistently Secreted Markedly Elevated Levels of Vascular Endothelial Growth Factor
3.4. Targeting VEGF Inhibited the Effects of GBM CSCs on Endothelial Cells
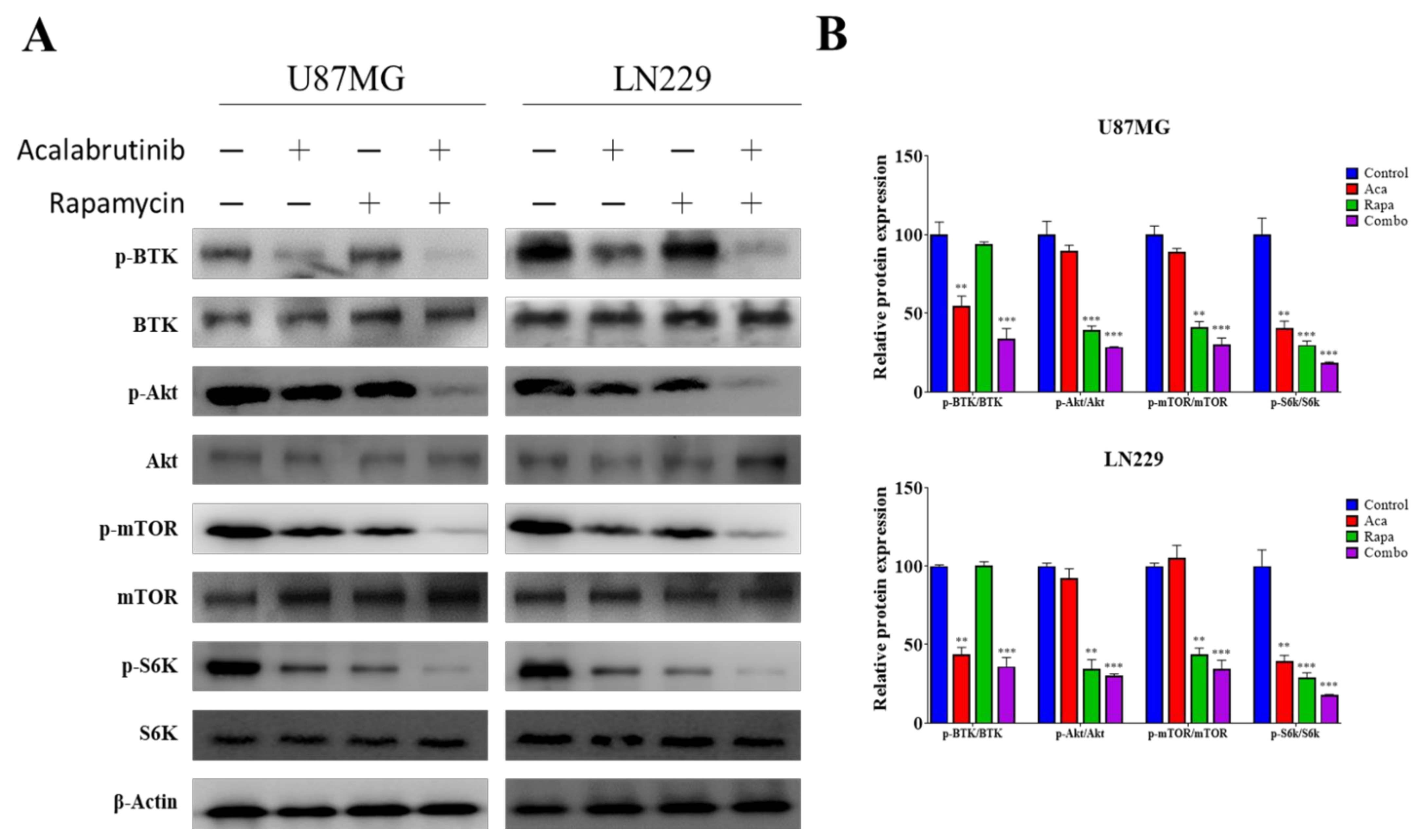
3.5. Acalabrutinib Inhibited BTK, Akt, and ERK Signaling, Enhancing Rapamycin Inhibition of mTOR Activation
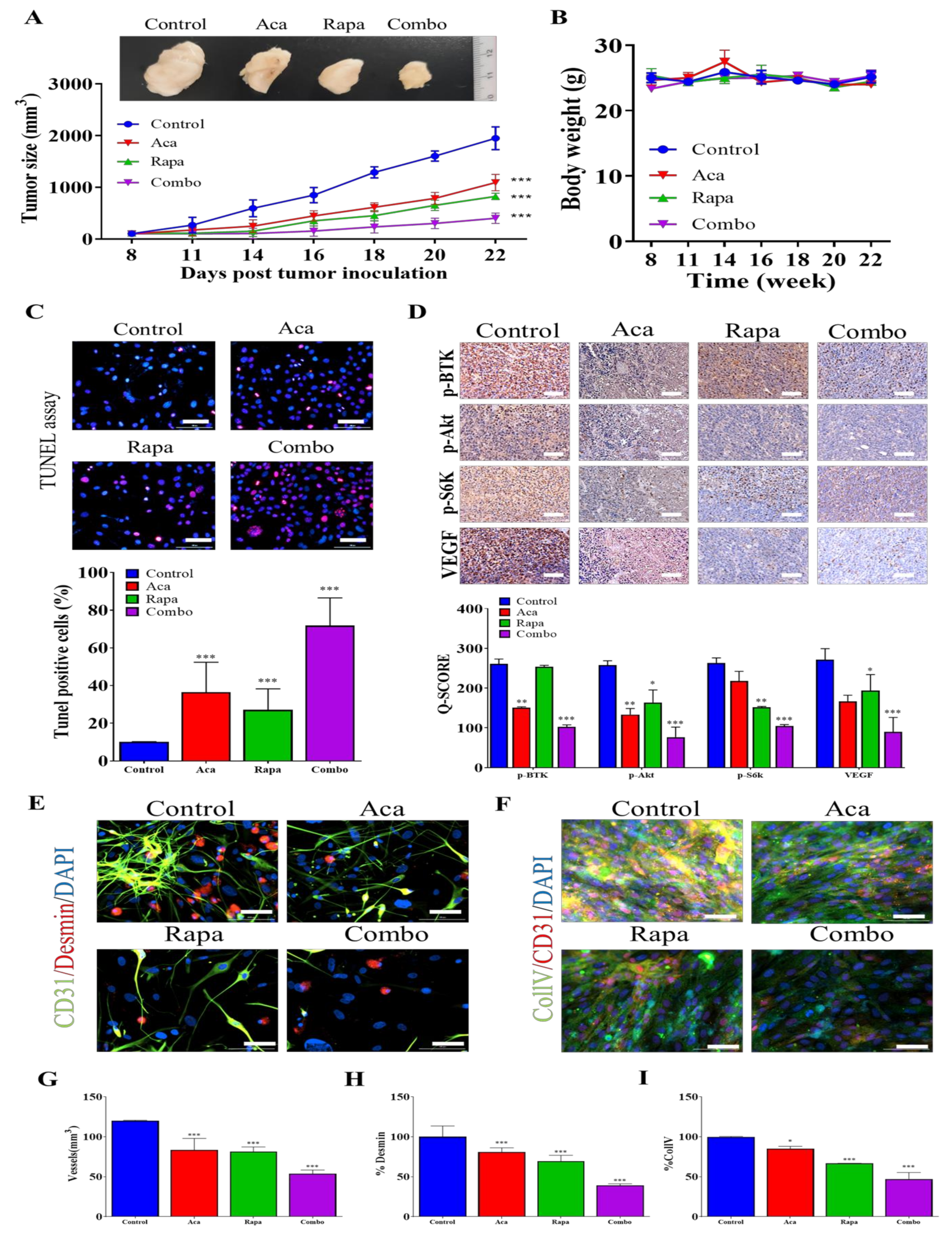
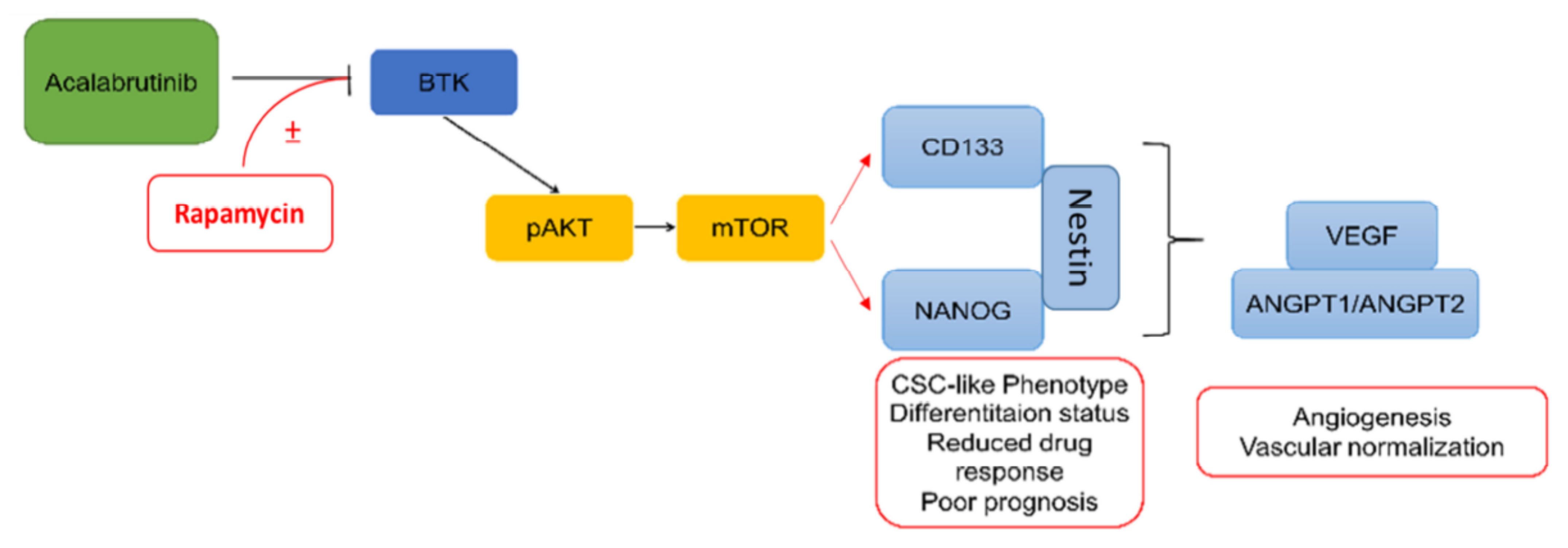
3.6. Combination Treatment with BTK Inhibitor Acalabrutinib and mTOR Inhibitor Rapamycin Reduced In Vivo Tumorigenesis and Angiogenesis through the Inhibition of the BTK/Akt/mTOR Signaling Axis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wrensch, M.; Minn, Y.; Chew, T.; Bondy, M.; Berger, M.S. Epidemiology of primary brain tumors: Current concepts and review of the literature. Neuro Oncol. 2002, 4, 278–299. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Lopez, P.D.; Corrales-Garcia, E.M. Survival in glioblastoma: A review on the impact of treatment modalities. Clin. Transl. Oncol. 2016, 18, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Li, H.; Ren, G. Epithelial-mesenchymal transition and drug resistance in breast cancer (Review). Int. J. Oncol. 2015, 47, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Iser, I.C.; Pereira, M.B.; Lenz, G.; Wink, M.R. The Epithelial-to-Mesenchymal Transition-Like Process in Glioblastoma: An Updated Systematic Review and In Silico Investigation. Med. Res. Rev. 2017, 37, 271–313. [Google Scholar] [CrossRef] [PubMed]
- Skovierova, H.; Okajcekova, T.; Strnadel, J.; Vidomanova, E.; Halasova, E. Molecular regulation of epithelial-to-mesenchymal transition in tumorigenesis (Review). Int. J. Mol. Med. 2018, 41, 1187–1200. [Google Scholar] [CrossRef]
- Heddleston, J.M.; Hitomi, M.; Venere, M.; Flavahan, W.A.; Yang, K.; Kim, Y.; Minhas, S.; Rich, J.N.; Hjelmeland, A.B. Glioma stem cell maintenance: The role of the microenvironment. Curr. Pharm. Des. 2011, 17, 2386–2401. [Google Scholar] [CrossRef] [PubMed]
- Dey, M.; Ulasov, I.V.; Tyler, M.A.; Sonabend, A.M.; Lesniak, M.S. Cancer stem cells: The final frontier for glioma virotherapy. Stem. Cell Rev. Rep. 2011, 7, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Gursel, D.B.; Shin, B.J.; Burkhardt, J.K.; Kesavabhotla, K.; Schlaff, C.D.; Boockvar, J.A. Glioblastoma stem-like cells-biology and therapeutic implications. Cancers 2011, 3, 2655–2666. [Google Scholar] [CrossRef]
- Kawamura, Y.; Takouda, J.; Yoshimoto, K.; Nakashima, K. New aspects of glioblastoma multiforme revealed by similarities between neural and glioblastoma stem cells. Cell Biol. Toxicol. 2018, 34, 425–440. [Google Scholar] [CrossRef]
- Qiu, Y.; Kung, H.J. Signaling network of the Btk family kinases. Oncogene 2000, 19, 5651–5661. [Google Scholar] [CrossRef]
- Corneth, O.B.J.; Klein Wolterink, R.G.J.; Hendriks, R.W. BTK Signaling in B Cell Differentiation and Autoimmunity. Curr. Top. Microbiol. Immunol. 2016, 393, 67–105. [Google Scholar] [CrossRef]
- Ormsby, T.; Schlecker, E.; Ferdin, J.; Tessarz, A.S.; Angelisova, P.; Koprulu, A.D.; Borte, M.; Warnatz, K.; Schulze, I.; Ellmeier, W.; et al. Btk is a positive regulator in the TREM-1/DAP12 signaling pathway. Blood 2011, 118, 936–945. [Google Scholar] [CrossRef] [PubMed]
- Melcher, M.; Unger, B.; Schmidt, U.; Rajantie, I.A.; Alitalo, K.; Ellmeier, W. Essential roles for the Tec family kinases Tec and Btk in M-CSF receptor signaling pathways that regulate macrophage survival. J. Immunol. 2008, 180, 8048–8056. [Google Scholar] [CrossRef] [PubMed]
- Dobrovolsky, D.; Wang, E.S.; Morrow, S.; Leahy, C.; Faust, T.; Nowak, R.P.; Donovan, K.A.; Yang, G.; Li, Z.; Fischer, E.S.; et al. Bruton tyrosine kinase degradation as a therapeutic strategy for cancer. Blood 2019, 133, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Pal Singh, S.; Dammeijer, F.; Hendriks, R.W. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer 2018, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Gunderson, A.J.; Kaneda, M.M.; Tsujikawa, T.; Nguyen, A.V.; Affara, N.I.; Ruffell, B.; Gorjestani, S.; Liudahl, S.M.; Truitt, M.; Olson, P.; et al. Bruton Tyrosine Kinase-Dependent Immune Cell Cross-talk Drives Pancreas Cancer. Cancer Discov. 2016, 6, 270–285. [Google Scholar] [CrossRef]
- Yang, Y.; Shi, J.; Gu, Z.; Salama, M.E.; Das, S.; Wendlandt, E.; Xu, H.; Huang, J.; Tao, Y.; Hao, M.; et al. Bruton tyrosine kinase is a therapeutic target in stem-like cells from multiple myeloma. Cancer Res. 2015, 75, 594–604. [Google Scholar] [CrossRef]
- Wei, L.; Su, Y.K.; Lin, C.M.; Chao, T.Y.; Huang, S.P.; Huynh, T.T.; Jan, H.J.; Whang-Peng, J.; Chiou, J.F.; Wu, A.T.; et al. Preclinical investigation of ibrutinib, a Bruton’s kinase tyrosine (Btk) inhibitor, in suppressing glioma tumorigenesis and stem cell phenotypes. Oncotarget 2016, 7, 69961–69975. [Google Scholar] [CrossRef] [PubMed]
- Zucha, M.A.; Wu, A.T.; Lee, W.H.; Wang, L.S.; Lin, W.W.; Yuan, C.C.; Yeh, C.T. Bruton’s tyrosine kinase (Btk) inhibitor ibrutinib suppresses stem-like traits in ovarian cancer. Oncotarget 2015, 6, 13255–13268. [Google Scholar] [CrossRef] [PubMed]
- Zippo, A.; De Robertis, A.; Bardelli, M.; Galvagni, F.; Oliviero, S. Identification of Flk-1 target genes in vasculogenesis: Pim-1 is required for endothelial and mural cell differentiation in vitro. Blood 2004, 103, 4536–4544. [Google Scholar] [CrossRef]
- Qiu, Q.; Li, C.; Song, Y.; Shi, T.; Luo, X.; Zhang, H.; Hu, L.; Yan, X.; Zheng, H.; Liu, M.; et al. Targeted delivery of ibrutinib to tumor-associated macrophages by sialic acid-stearic acid conjugate modified nanocomplexes for cancer immunotherapy. Acta Biomater. 2019, 92, 184–195. [Google Scholar] [CrossRef]
- Li, J.; Wang, X.; Xie, Y.; Ying, Z.; Liu, W.; Ping, L.; Zhang, C.; Pan, Z.; Ding, N.; Song, Y.; et al. The mTOR kinase inhibitor everolimus synergistically enhances the anti-tumor effect of the Bruton’s tyrosine kinase (BTK) inhibitor PLS-123 on Mantle cell lymphoma. Int. J. Cancer 2018, 142, 202–213. [Google Scholar] [CrossRef]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; Sathornsumetee, S.; Hao, Y.; Li, Z.; Hjelmeland, A.B.; Shi, Q.; McLendon, R.E.; Bigner, D.D.; Rich, J.N. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006, 66, 7843–7848. [Google Scholar] [CrossRef] [PubMed]
- Lindvall, J.; Islam, T.C. Interaction of Btk and Akt in B cell signaling. Biochem. Biophys. Res. Commun. 2002, 293, 1319–1326. [Google Scholar] [CrossRef]
- Lin, C.H.; Elkholy, K.H.; Wani, N.A.; Li, D.; Hu, P.; Barajas, J.M.; Yu, L.; Zhang, X.; Jacob, S.T.; Khan, W.N.; et al. Ibrutinib potentiates anti-hepatocarcinogenic efficacy of sorafenib by targeting EGFR in tumor cells and BTK in immune cells in the stroma. Mol. Cancer Ther. 2020, 19, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kinoshita, T.; Sukbuntherng, J.; Chang, B.Y.; Elias, L. Ibrutinib Inhibits ERBB Receptor Tyrosine Kinases and HER2-Amplified Breast Cancer Cell Growth. Mol. Cancer Ther. 2016, 15, 2835–2844. [Google Scholar] [CrossRef]
- Atashzar, M.R.; Baharlou, R.; Karami, J.; Abdollahi, H.; Rezaei, R.; Pourramezan, F.; Zoljalali Moghaddam, S.H. Cancer stem cells: A review from origin to therapeutic implications. J. Cell Physiol. 2020, 235, 790–803. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wei, B.; Han, X.; Zheng, Z.; Huang, J.; Liu, J.; Huang, Y.; Wei, H. LGR5 is required for the maintenance of spheroid-derived colon cancer stem cells. Int. J. Mol. Med. 2014, 34, 35–42. [Google Scholar] [CrossRef]
- Chikamatsu, K.; Ishii, H.; Murata, T.; Sakakura, K.; Shino, M.; Toyoda, M.; Takahashi, K.; Masuyama, K. Alteration of cancer stem cell-like phenotype by histone deacetylase inhibitors in squamous cell carcinoma of the head and neck. Cancer Sci. 2013, 104, 1468–1475. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, Q. Cancer stem cells and tumor metastasis (Review). Int. J. Oncol. 2014, 44, 1806–1812. [Google Scholar] [CrossRef]
- Rohlenova, K.; Veys, K.; Miranda-Santos, I.; De Bock, K.; Carmeliet, P. Endothelial Cell Metabolism in Health and Disease. Trends Cell Biol. 2018, 28, 224–236. [Google Scholar] [CrossRef]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-induced angiogenesis: Good and evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef]
- Lopez, A.; Harada, K.; Vasilakopoulou, M.; Shanbhag, N.; Ajani, J.A. Targeting Angiogenesis in Colorectal Carcinoma. Drugs 2019, 79, 63–74. [Google Scholar] [CrossRef]
- Wang, Y.; Xing, D.; Zhao, M.; Wang, J.; Yang, Y. The Role of a Single Angiogenesis Inhibitor in the Treatment of Recurrent Glioblastoma Multiforme: A Meta-Analysis and Systematic Review. PLoS ONE 2016, 11, e0152170. [Google Scholar] [CrossRef] [PubMed]
- Eyler, C.E.; Rich, J.N. Survival of the fittest: Cancer stem cells in therapeutic resistance and angiogenesis. J. Clin. Oncol. 2008, 26, 2839–2845. [Google Scholar] [CrossRef] [PubMed]
- Shan, F.; Miao, R.; Xue, K.; Li, Z.; Li, Z.; Bu, Z.; Wu, A.; Zhang, L.; Wu, X.; Zong, X.; et al. Controlling angiogenesis in gastric cancer: A systematic review of anti-angiogenic trials. Cancer Lett. 2016, 380, 598–607. [Google Scholar] [CrossRef]
- Sooriakumaran, P.; Kaba, R. Angiogenesis and the tumour hypoxia response in prostate cancer: A review. Int. J. Surg. 2005, 3, 61–67. [Google Scholar] [CrossRef][Green Version]
- Huang, Y.; Goel, S.; Duda, D.G.; Fukumura, D.; Jain, R.K. Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res. 2013, 73, 2943–2948. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.; van Beijnum, J.R.; Bonvin, D.; Jichlinski, P.; Dyson, P.J.; Griffioen, A.W.; Nowak-Sliwinska, P. Low-dose angiostatic tyrosine kinase inhibitors improve photodynamic therapy for cancer: Lack of vascular normalization. J. Cell Mol. Med. 2014, 18, 480–491. [Google Scholar] [CrossRef]
- Park, J.S.; Kim, I.K.; Han, S.; Park, I.; Kim, C.; Bae, J.; Oh, S.J.; Lee, S.; Kim, J.H.; Woo, D.C.; et al. Normalization of Tumor Vessels by Tie2 Activation and Ang2 Inhibition Enhances Drug Delivery and Produces a Favorable Tumor Microenvironment. Cancer Cell 2017, 31, 157–158. [Google Scholar] [CrossRef]
- Di Tacchio, M.; Macas, J.; Weissenberger, J.; Sommer, K.; Bahr, O.; Steinbach, J.P.; Senft, C.; Seifert, V.; Glas, M.; Herrlinger, U.; et al. Tumor Vessel Normalization, Immunostimulatory Reprogramming, and Improved Survival in Glioblastoma with Combined Inhibition of PD-1, Angiopoietin-2, and VEGF. Cancer Immunol. Res. 2019, 7, 1910–1927. [Google Scholar] [CrossRef]
- Xiao, Q.; Yang, S.; Ding, G.; Luo, M. Anti-vascular endothelial growth factor in glioblastoma: A systematic review and meta-analysis. Neurol. Sci. 2018, 39, 2021–2031. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.V.; Bergers, G. Mechanisms of evasive resistance to anti-VEGF therapy in glioblastoma. CNS Oncol. 2013, 2, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Kiss, E.A.; Saharinen, P. Anti-angiogenic Targets: Angiopoietin and Angiopoietin Receptors. Tumor Angiogen. 2019, 139, 173–250. [Google Scholar]
- Sie, M.; Wagemakers, M.; Molema, G.; Mooij, J.J.; de Bont, E.S.; den Dunnen, W.F. The angiopoietin 1/angiopoietin 2 balance as a prognostic marker in primary glioblastoma multiforme. J. Neurosurg. 2009, 110, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Kienast, Y.; Klein, C.; Scheuer, W. Ang-2-VEGF-A CrossMab, a novel bispecific human IgG1 antibody blocking VEGF-A and Ang-2 functions simultaneously, mediates potent antitumor, antiangiogenic, and antimetastatic efficacy. Clin. Cancer Res. 2013, 19, 6730–6740. [Google Scholar] [CrossRef] [PubMed]
- Australia, T.G.A. Australian Public Assessment Report for Acalabrutinib. Health Do. 2020. Available online: https://www.tga.gov.au/auspar/auspar-acalabrutinib (accessed on 10 July 2021).
- Dittus, C. LCCC1841: A Phase 2 Trial of Acalabrutinib in Relapsed/Refractory Primary and Secondary CNS Lymphomas. ClinicalTrials.gov. 2020. Available online: https://clinicaltrials.gov/ct2/show/results/NCT04548648?view=results (accessed on 10 July 2021).
- Mehta-Shah, N. Acalabrutinib and Durvalumab in Primary and Secondary Central Nervous System Lymphoma. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT04688151 (accessed on 10 July 2021).
- Caitlin Meeks, A.V. Robert Robey, Michael Gottesman, Sadhana Jackson. EXTH-19. Ibrutinib Modulates Blood-Brain Barrier Integrity To Enhance Chemotherapy Permeability. Neuro Oncol. 2019, 21, vi86. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Image Name | N0. Nodes | N0. Junctions | N0. Master Junction | N0. Master Segments | Tot. Master Segments Lenght | N0. Segments | N0. Branches | Tot. Branching Length | Tot. Segments Length | Tot. Branches Length | Branching Interval |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Parental_ Control | 14 | 5 | 0 | 1 | 224 | 4 | 7 | 875 | 181 | 694 | 25.857 |
| Sphere_ Control | 28 | 9 | 1 | 3 | 523 | 10 | 7 | 1003 | 489 | 514 | 69.857 |
| Parental_ Aca | 6 | 2 | 1 | 3 | 523 | 1 | 4 | 315 | 14 | 301 | 3.5 |
| Sphere_ Aca | 19 | 5 | 1 | 2 | 121 | 4 | 7 | 626 | 102 | 524 | 14.571 |
| Parental_ Rapa | 0 | 0 | 1 | 2 | 121 | 0 | 0 | 0 | 0 | 0 | 0 |
| Sphere_ Rapa | 26 | 7 | 3 | 4 | 339 | 6 | 9 | 948 | 300 | 648 | 33.333 |
| Parental_ Combo | 0 | 0 | 3 | 4 | 339 | 0 | 0 | 0 | 0 | 0 | 0 |
| Sphere_ Combo | 0 | 0 | 3 | 4 | 339 | 0 | 0 | 0 | 0 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, Y.-K.; Bamodu, O.A.; Su, I.-C.; Pikatan, N.W.; Fong, I.-H.; Lee, W.-H.; Yeh, C.-T.; Chiu, H.-Y.; Lin, C.-M. Combined Treatment with Acalabrutinib and Rapamycin Inhibits Glioma Stem Cells and Promotes Vascular Normalization by Downregulating BTK/mTOR/VEGF Signaling. Pharmaceuticals 2021, 14, 876. https://doi.org/10.3390/ph14090876
Su Y-K, Bamodu OA, Su I-C, Pikatan NW, Fong I-H, Lee W-H, Yeh C-T, Chiu H-Y, Lin C-M. Combined Treatment with Acalabrutinib and Rapamycin Inhibits Glioma Stem Cells and Promotes Vascular Normalization by Downregulating BTK/mTOR/VEGF Signaling. Pharmaceuticals. 2021; 14(9):876. https://doi.org/10.3390/ph14090876
Chicago/Turabian StyleSu, Yu-Kai, Oluwaseun Adebayo Bamodu, I-Chang Su, Narpati Wesa Pikatan, Iat-Hang Fong, Wei-Hwa Lee, Chi-Tai Yeh, Hsiao-Yean Chiu, and Chien-Min Lin. 2021. "Combined Treatment with Acalabrutinib and Rapamycin Inhibits Glioma Stem Cells and Promotes Vascular Normalization by Downregulating BTK/mTOR/VEGF Signaling" Pharmaceuticals 14, no. 9: 876. https://doi.org/10.3390/ph14090876
APA StyleSu, Y.-K., Bamodu, O. A., Su, I.-C., Pikatan, N. W., Fong, I.-H., Lee, W.-H., Yeh, C.-T., Chiu, H.-Y., & Lin, C.-M. (2021). Combined Treatment with Acalabrutinib and Rapamycin Inhibits Glioma Stem Cells and Promotes Vascular Normalization by Downregulating BTK/mTOR/VEGF Signaling. Pharmaceuticals, 14(9), 876. https://doi.org/10.3390/ph14090876