
Two Antibody-Guided Lactic-co-Glycolic Acid-Polyethylenimine (LGA-PEI) Nanoparticle Delivery Systems for Therapeutic Nucleic Acids

 ,
,  ,
,
Abstract
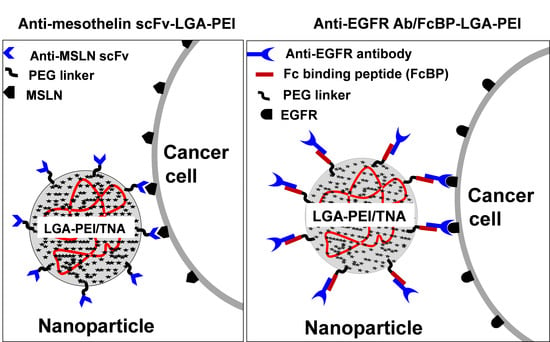
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
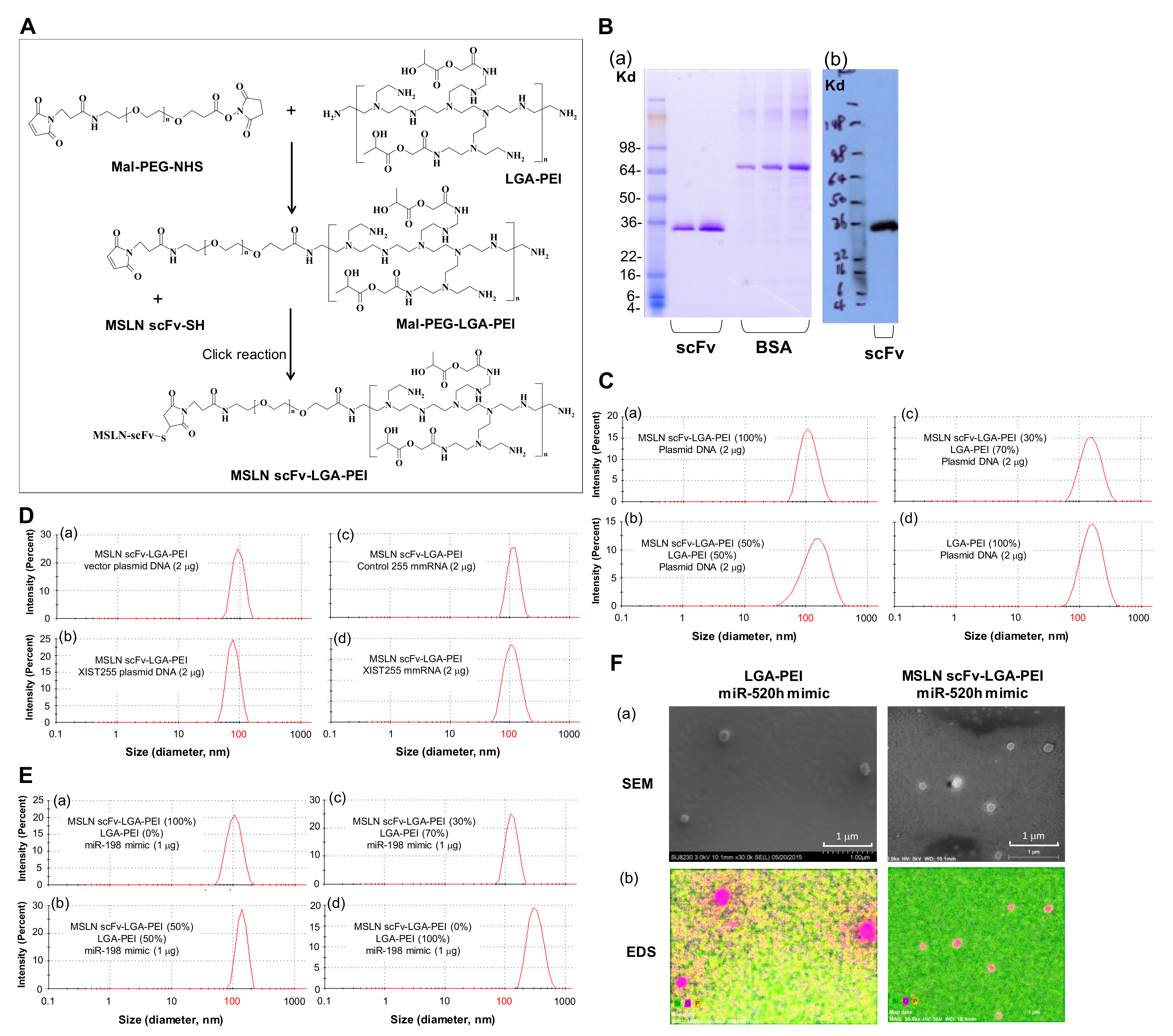
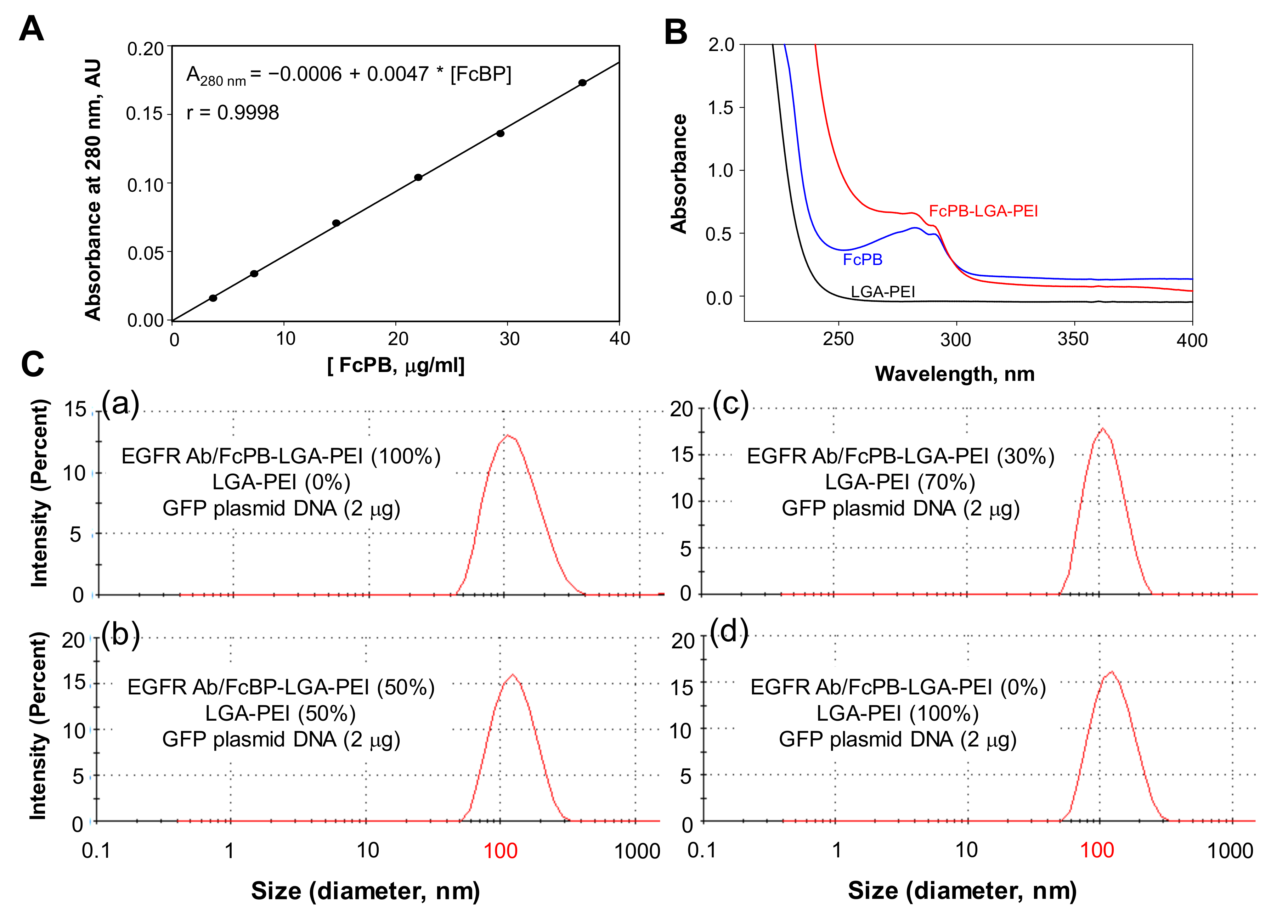
2.1. MSLN scFv-LGA-PEI and Anti-EGFR Ab/FcBP-LGA-PEI Polymers Effectively Load NAs and Form Functional NPs
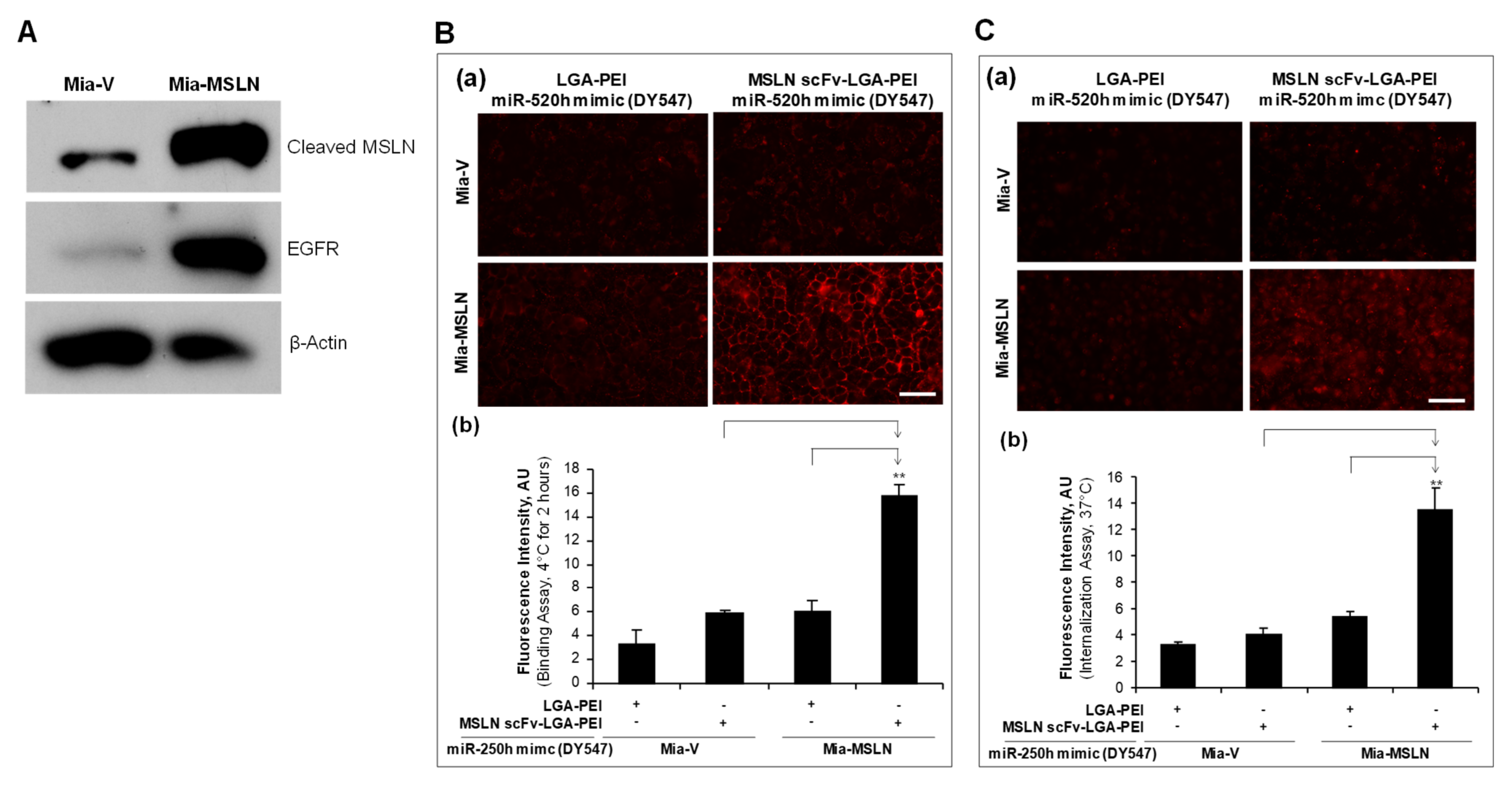
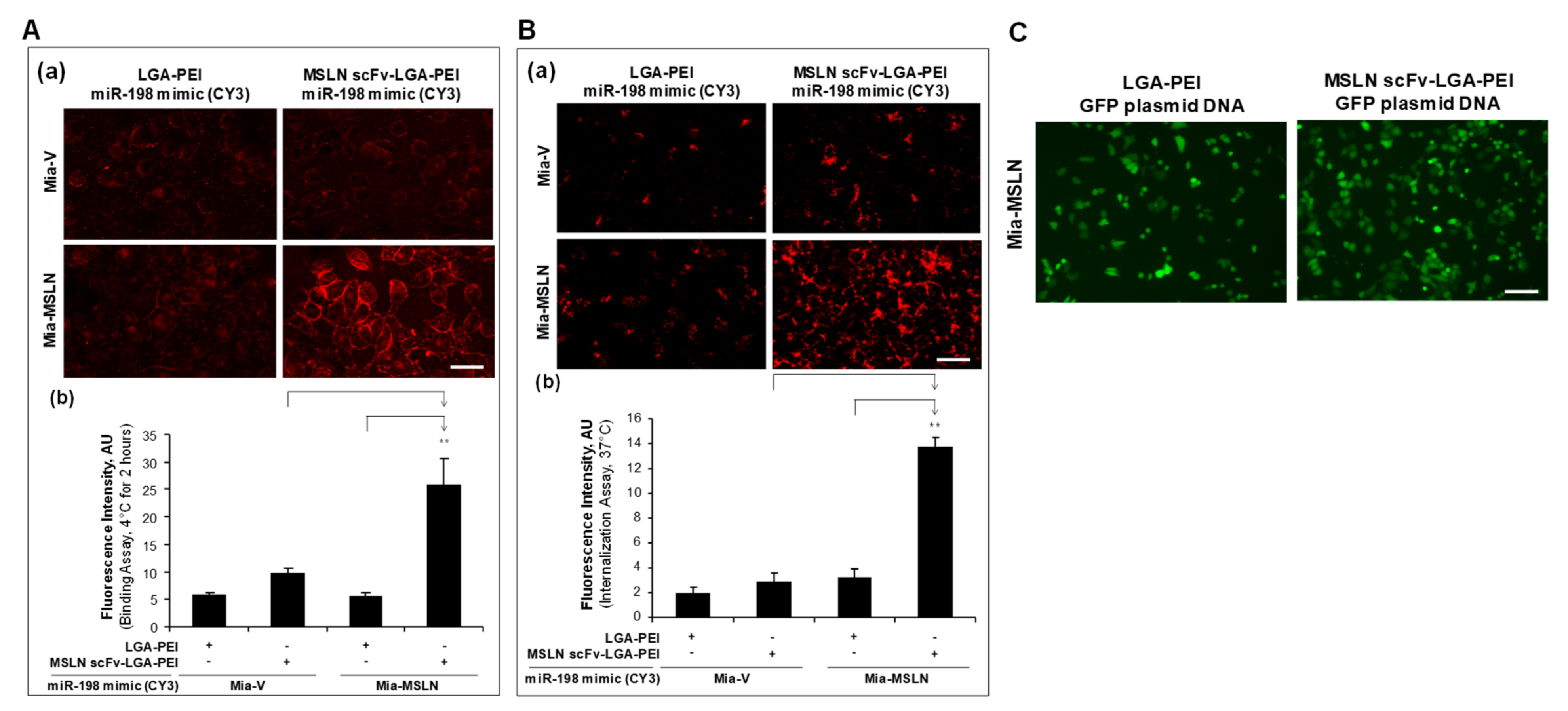
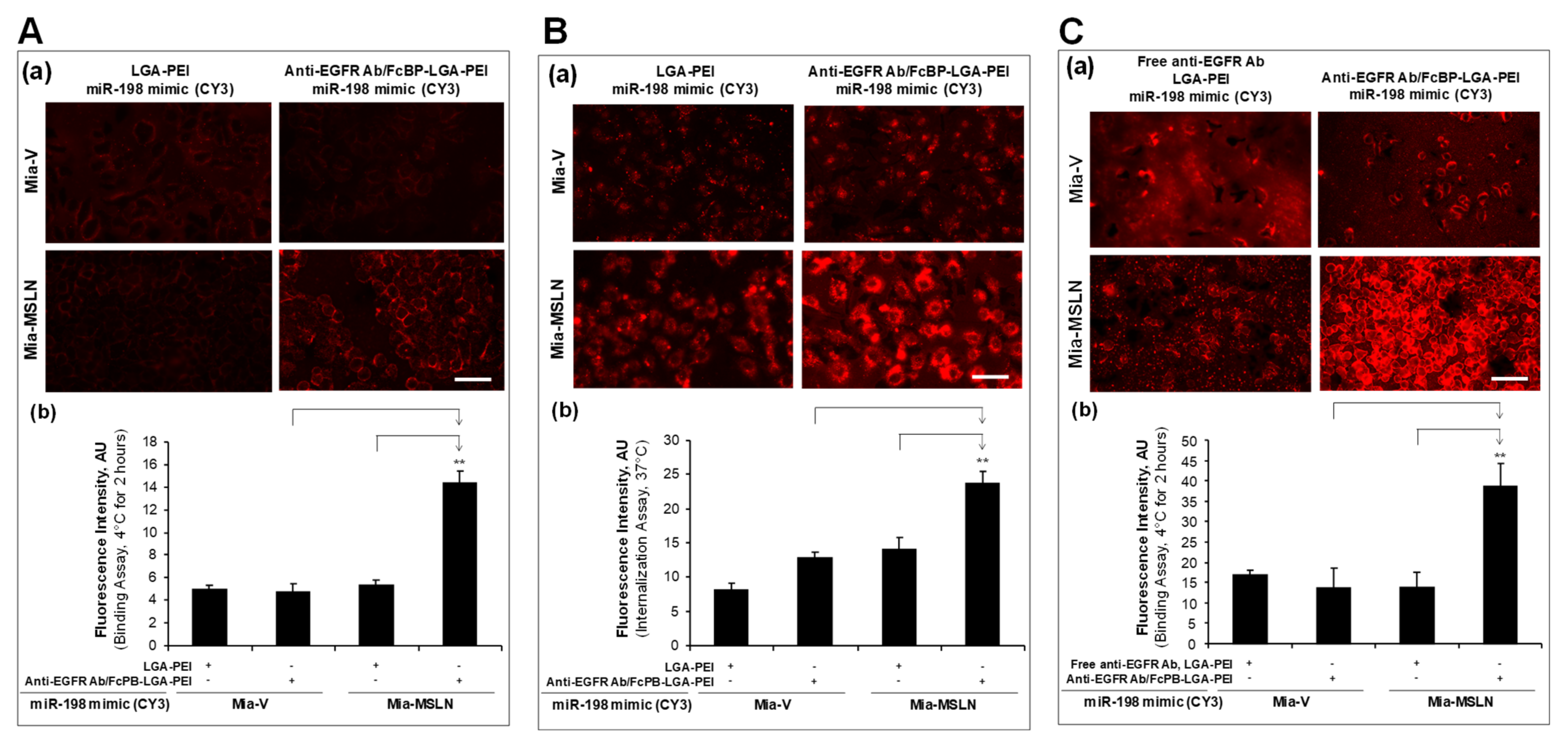
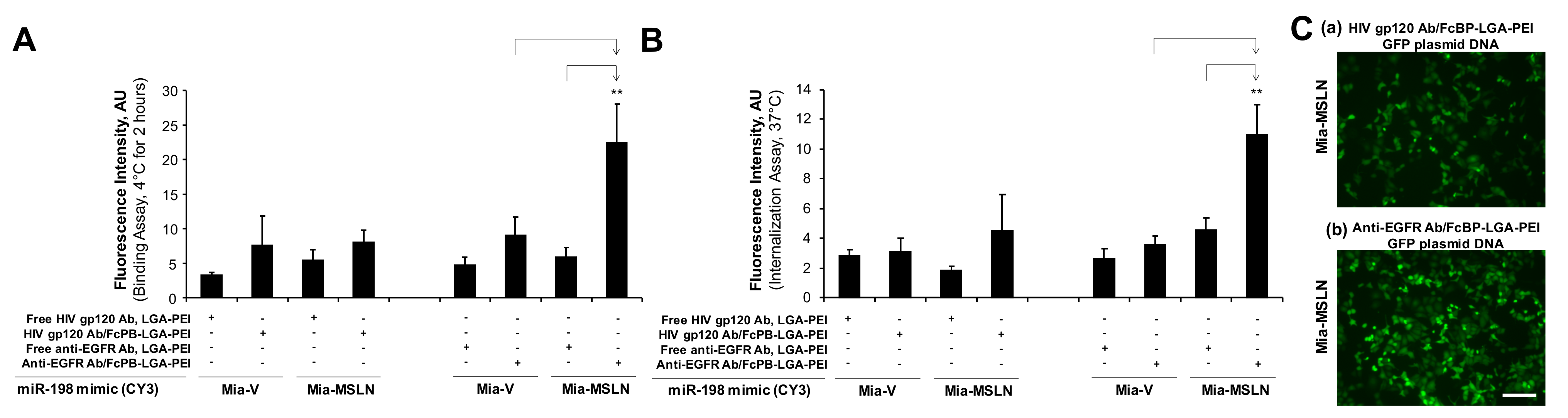
2.2. MSLN scFv-LGA-PEI and Anti-EGFR Ab/FcBP-LGA-PEI NPs with NAs Specifically Enhance Their Binding and Internalization into Targeting PC Cell Lines
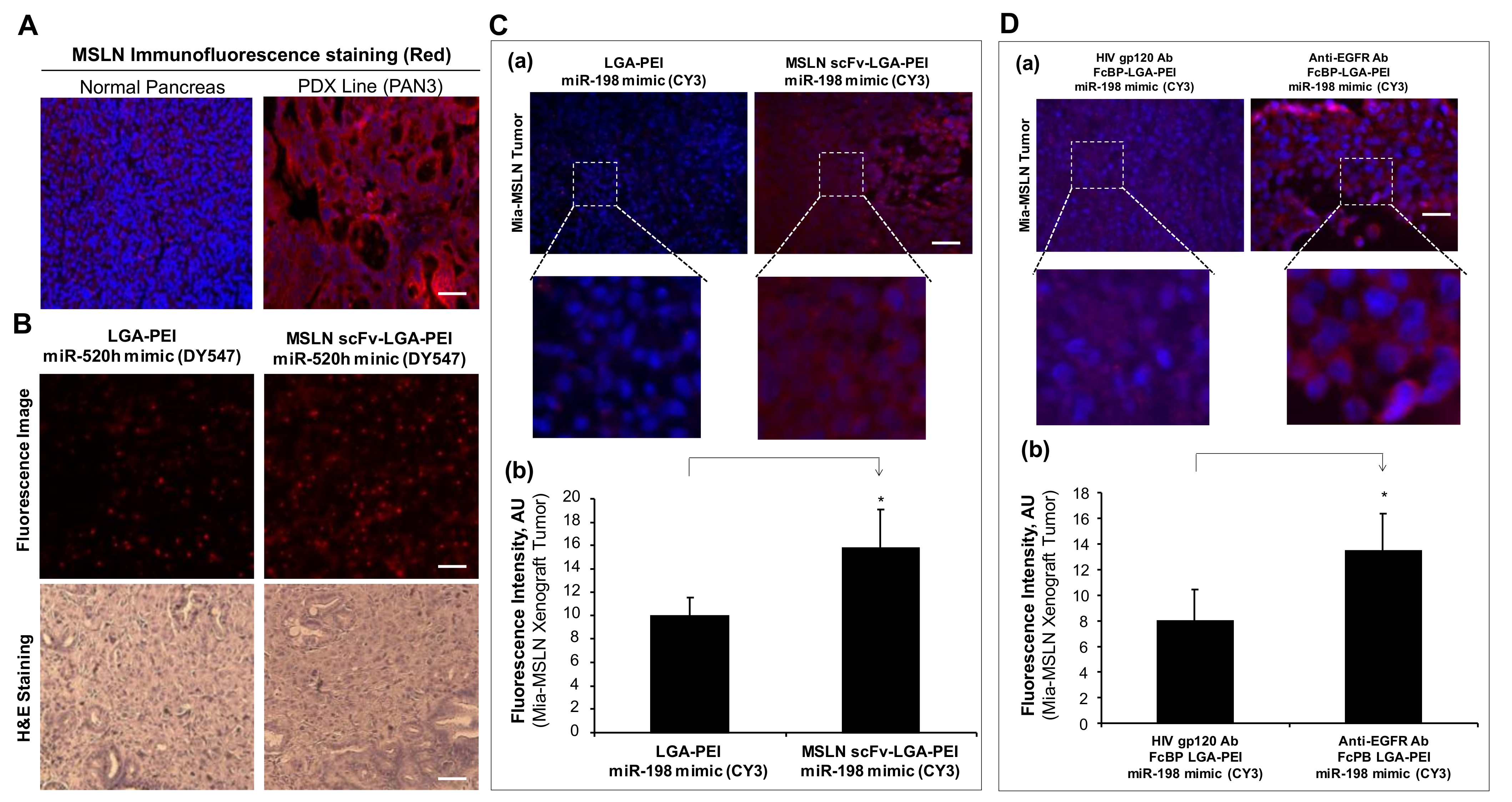
2.3. MSLN scFv-LGA-PEI and Anti-EGFR Ab/FcBP-LGA-PEI NPs Specifically Deliver NAs into Targeting PC Tumor Tissues in Mouse Models
3. Discussion
4. Material and Methods
4.1. Materials
4.2. Preparation of MSLN scFv-LGA-PEI and FcBP-LGA-PEI
4.3. MSLN scFv-LGA-PEI or FcBP-LGA-PEI Polymer and NA Loading
4.4. Particle Size and Zeta Potential Measurements
4.5. Cell Binding and Internalization Assays
4.6. Delivery of GFP Gene In Vitro
4.7. Delivery of Nucleic Acids in Mouse Models
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xu, J.; Murphy, S.L.; Kochanek, K.D.; Arias, E. Mortality in the United States, 2018. NCHS Data Brief. 2020, 355, 1–8. [Google Scholar]
- Harding, M.C.; Sloan, C.D.; Merrill, R.M.; Harding, T.M.; Thacker, B.J.; Thacker, E.L. Transitions from heart disease to cancer as the leading cause of death in US states, 1999–2016. Prev. Chronic. Dis. 2018, 15, E158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Gibby, C.C.; Chan, W.; Abbruzzese, J.L.; Xiong, H.Q.; Ho, L.; Evans, D.B.; Varadhachary, G.; Bhat, S.; Wolff, R.A.; Crane, C. Patterns of self-reported symptoms in pancreatic cancer patients receiving chemoradiation. J. Pain Symptom Manag. 2007, 34, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Löhr, M. Is it possible to survive pancreatic cancer? Nat. Clin. Pract. Gastroenterol. Hepatol. 2006, 3, 236–237. [Google Scholar] [CrossRef]
- Stephenson, M.L.; Zamecnik, P.C. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 285–288. [Google Scholar] [CrossRef] [Green Version]
- Weng, Y.; Huang, Q.; Li, C.; Yang, Y.; Wang, X.; Yu, J.; Huang, Y.; Liang, X.J. Improved nucleic acid therapy with advanced Nanoscale Biotechnology. Mol. Ther. Nucleic Acids 2020, 19, 581–601. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. A Service of the U.S. National Institutes of Health. Available online: https://clinicaltrials.gov/ (accessed on 24 July 2020).
- Pastor, F.; Berraondo, P.; Etxeberria, I.; Frederick, J.; Sahin, U.; Gilboa, E.; Melero, I. An RNA toolbox for cancer immunotherapy. Nat. Rev. Drug Discov. 2018, 17, 751–767. [Google Scholar] [CrossRef]
- Kanasty, R.; Dorkin, J.R.; Vegas, A.; Anderson, D. Delivery materials for siRNA therapeutics. Nat. Mater. 2013, 12, 967–977. [Google Scholar] [CrossRef]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef]
- Greish, K. Enhanced permeability and retention (EPR) effect for anticancer nanomedicine drug targeting. Methods Mol. Biol. 2010, 624, 25–37. [Google Scholar] [PubMed]
- Kalyane, D.; Raval, N.; Maheshwari, R.; Tambe, V.; Kalia, K.; Tekade, R.K. Employment of enhanced permeability and retention effect (EPR): Nanoparticle-based precision tools for targeting of therapeutic and diagnostic agent in cancer. Mater. Sci. Eng. C Mater. Biol. Appl. 1999, 98, 1252–1276. [Google Scholar] [CrossRef] [PubMed]
- Kircheis, R.; Wightman, L.; Wagner, E. Design and gene delivery activity of modified polyethylenimines. Adv. Drug Deliv. Rev. 2001, 53, 341–358. [Google Scholar] [CrossRef]
- Godbey, W.T.; Wu, K.K.; Mikos, A.G. Size matters: Molecular weight affects the efficiency of poly(ethylenimine) as a gene delivery vehicle. J. Biomed. Mater. Res. 1999, 45, 268–275. [Google Scholar] [CrossRef]
- Suh, J.; Paik, H.J.; Hwang, B.K. Ionization of poly(ethylenimine) and poly(allylamine) at various PH’s. Bioorg. Chem. 1994, 22, 318–327. [Google Scholar] [CrossRef]
- Tang, M.X.; Szoka, F.C. The influence of polymer structure on the interactions of cationic polymers with DNA and morphology of the resulting complexes. Gene Ther. 1997, 4, 823–832. [Google Scholar] [CrossRef] [Green Version]
- Kichler, A.; Leborgne, C.; Coeytaux, E.; Danos, O. Polyethylenimine-mediated gene delivery: A mechanistic study. J. Gene Med. 2001, 3, 135–144. [Google Scholar] [CrossRef]
- Akinc, A.; Thomas, M.; Klibanov, A.M.; Langer, R. Exploring polyethylenimine-mediated DNA transfection and the proton sponge hypothesis. J. Gene Med. 2005, 7, 657–663. [Google Scholar] [CrossRef]
- Jere, D.; Jiang, H.L.; Arote, R.; Kim, Y.K.; Choi, Y.J.; Cho, M.H.; Akaike, T.; Cho, C.S. Degradable polyethylenimines as DNA and small interfering RNA carriers. Expert Opin. Drug Deliv. 2009, 6, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, C.K.; Roy, S. Structural and dynamical properties of polyethylenimine in explicit water at different protonation states: A molecular dynamics study. Soft Matter. 2013, 9, 2269–2281. [Google Scholar] [CrossRef]
- Grandinetti, G.; Smith, A.E.; Reineke, T.M. Membrane and nuclear permeabilization by polymeric pDNA vehicles: Efficient method for gene delivery or mechanism of cytotoxicity? Mol. Pharm. 2012, 9, 523–538. [Google Scholar] [CrossRef]
- Guo, X.; Huang, L. Recent advances in nonviral vectors for gene delivery. Acc. Chem. Res. 2012, 45, 971–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moghimi, S.M.; Symonds, P.; Murray, J.C.; Hunter, A.C.; Debska, G.; Szewczyk, A. A two-stage poly(ethylenimine)-mediated cytotoxicity: Implications for gene transfer/therapy. Mol. Ther. 2005, 11, 990–995. [Google Scholar] [CrossRef]
- Godbey, W.T.; Wu, K.K.; Mikos, A.G. Poly(ethylenimine) and its role in gene delivery. J. Control. Release 1999, 60, 149–160. [Google Scholar] [CrossRef]
- Neuberg, P.; Kichler, A. Recent developments in nucleic acid delivery with polyethylenimines. Adv. Genet. 2014, 88, 263–288. [Google Scholar] [PubMed]
- Pandey, A.P.; Sawant, K.K. Polyethylenimine: A versatile, multifunctional non-viral vector for nucleic acid delivery. Mater. Sci. Eng. C Mater. Biol. Appl. 2016, 68, 904–918. [Google Scholar] [CrossRef]
- Lü, J.M.; Liang, Z.; Wang, X.; Gu, J.; Yao, Q.; Chen, C. New polymer of lactic-co-glycolic acid-modified polyethylenimine for nucleic acid delivery. Nanomedicine 2016, 11, 1971–1991. [Google Scholar] [CrossRef] [Green Version]
- Biffi, S.; Voltan, R.; Bortot, B.; Zauli, G.; Secchiero, P. Actively targeted nanocarriers for drug delivery to cancer cells. Expert Opin. Drug Deliv. 2019, 16, 481–496. [Google Scholar] [CrossRef]
- Muhamad, N.; Plengsuriyakarn, T.; Na-Bangchang, K. Application of active targeting nanoparticle delivery system for chemotherapeutic drugs and traditional/herbal medicines in cancer therapy: A systematic review. Int. J. Nanomed. 2018, 13, 3921–3935. [Google Scholar] [CrossRef] [Green Version]
- Strehblow, C.; Schuster, M.; Moritz, T.; Kirch, H.C.; Opalka, B.; Petri, J.B. Monoclonal antibody-polyethyleneimine conjugates targeting her-2/neu or CD90 allow cell typespecific nonviral gene delivery. J. Control. Release 2005, 102, 737–747. [Google Scholar] [CrossRef]
- Li, M.; Bharadwaj, U.; Zhang, R.; Zhang, S.; Mu, H.; Fisher, W.E.; Brunicardi, F.C.; Chen, C.; Yao, Q. Mesothelin is a malignant factor and therapeutic vaccine target for pancreatic cancer. Mol. Cancer Ther. 2008, 7, 286–296. [Google Scholar] [CrossRef] [Green Version]
- Argani, P.; Iacobuzio-Donahue, C.; Ryu, B.; Rosty, C.; Goggins, M.; Wilentz, R.E.; Murugesan, S.R.; Leach, S.D.; Jaffee, E.; Yeo, C.J.; et al. Mesothelin is overexpressed in the vast majority of ductal adenocarcinomas of the pancreas: Identification of a new pancreatic cancer marker by serial analysis of gene expression (SAGE). Clin. Cancer Res. 2001, 7, 3862–3868. [Google Scholar]
- Chang, K.; Pastan, I. Molecular cloning of mesothelin, a differentiation antigen present on mesothelium, mesotheliomas, and ovarian cancers. Proc. Natl. Acad. Sci. USA 1996, 3, 136–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baba, K.; Ishigami, S.; Arigami, T.; Uenosono, Y.; Okumura, H.; Matsumoto, M.; Kurahara, H.; Uchikado, Y.; Kita, Y.; Kijima, Y.; et al. Mesothelin expression correlates with prolonged patient survival in gastric cancer. J. Surg. Oncol. 2012, 105, 195–199. [Google Scholar] [CrossRef]
- Tchou, J.; Wang, L.C.; Selven, B.; Zhang, H.; Conejo-Garcia, J.; Borghaei, H.; Kalos, M.; Vondeheide, R.H.; Albelda, S.M.; June, C.H.; et al. Mesothelin, a novel immunotherapy target for triple negative breast cancer. Breast Cancer Res. Treat. 2012, 133, 799–804. [Google Scholar] [CrossRef] [Green Version]
- Dainty, L.A.; Risinger, J.I.; Morrison, C.; Chandramouli, G.V.; Bidus, M.A.; Zahn, C.; Rose, G.S.; Fowler, J.; Berchuck, A.; Maxwell, G.L. Overexpression of folate binding protein and mesothelin are associated with uterine serous carcinoma. Gynecol. Oncol. 2007, 105, 563–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinbach, D.; Onda, M.; Voigt, A.; Dawczynski, K.; Wittig, S.; Hassan, R.; Gruhn, B.; Pastan, I. Mesothelin, a possible target for immunotherapy, is expressed in primary AML cells. Eur. J. Haematol. 2007, 79, 281–286. [Google Scholar] [CrossRef]
- Chang, K.; Pastan, I.; Willingham, M.C. Isolation and characterization of a monoclonal antibody, K1, reactive with ovarian cancers and normal mesothelium. Int. J. Cancer 1992, 50, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.J.; Sharon, E.; Pastan, I.; Hassan, R. Mesothelin-targeted agents in clinical trials and in preclinical development. Mol. Cancer Ther. 2012, 11, 517–525. [Google Scholar] [CrossRef] [Green Version]
- Scales, S.J.; Gupta, N.; Pacheco, G.; Firestein, R.; French, D.M.; Koeppen, H.; Rangell, L.; Barry-Hamilton, V.; Luis, E.; Chuh, J.; et al. An antimesothelin-monomethyl auristatin e conjugate with potent antitumor activity in ovarian, pancreatic, and mesothelioma models. Mol. Cancer Ther. 2014, 13, 2630–2640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, K.; Sasaki, T.; Takenaka, F.; Yakushiji, H.; Fujii, Y.; Kishi, Y.; Kita, S.; Shen, L.; Kumon, H.; Matsuura, E. A novel PET imaging using ⁶⁴Cu-labeled monoclonal antibody against mesothelin commonly expressed on cancer cells. J. Immunol. Res. 2015, 2015, 268172. [Google Scholar] [CrossRef] [Green Version]
- Lv, J.; Li, P. Mesothelin as a biomarker for targeted therapy. Biomark. Res. 2019, 7, 18. [Google Scholar] [CrossRef] [Green Version]
- Ordonez, N.G. Value of mesothelin immunostaining in the diagnosis of mesothelioma. Mod. Pathol. 2003, 16, 192–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ordonez, N.G. Application of mesothelin immunostaining in tumor diagnosis. Am. J. Surg. Pathol. 2003, 27, 1418–1428. [Google Scholar] [CrossRef]
- Hassan, R.; Laszik, Z.G.; Lerner, M.; Raffeld, M.; Postier, R.; Brackett, D. Mesothelin is overexpressed in pancreaticobiliary adenocarcinomas but not in normal pancreas and chronic pancreatitis. Am. J. Clin. Pathol. 2005, 124, 838–845. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, M.H.; Stashwick, C.; Plesa, G.; Tanyi, J.L. Overcoming barriers of car T-cell therapy in patients with mesothelin-expressing cancers. Immunotherapy 2017, 9, 767–780. [Google Scholar] [CrossRef]
- Chang, K.; Pastan, I.; Willingham, M.C. Frequent expression of the tumor antigen CAK1 in squamous-cell carcinomas. Int. J. Cancer 1992, 51, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Rizk, N.P.; Servais, E.L.; Tang, L.H.; Sima, C.S.; Gerdes, H.; Fleisher, M.; Rusch, V.W.; Adusumilli, P.S. Tissue and serum mesothelin are potential markers of neoplastic progression in Barrett’s associated esophageal adenocarcinoma. Cancer Epidemiol. Biomark. Prev. 2012, 21, 482–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morello, A.; Sadelain, M.; Adusumilli, P.S. Mesothelin-targeted CARs: Driving T cells to solid tumors. Cancer Discov. 2016, 6, 133–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichetti, F.; Marra, A.; Corti, F.; Guidi, A.; Raimondi, A.; Prinzi, N.; de Braud, F.; Pusceddu, S. The Role of Mesothelin as a Diagnostic and Therapeutic Target in Pancreatic Ductal Adenocarcinoma: A Comprehensive Review. Target. Oncol. 2018, 13, 333–351. [Google Scholar] [CrossRef]
- DeLano, W.L.; Ultsch, M.H.; de Vos, A.M.; Wells, J.A. Convergent solutions to binding at a protein-protein interface. Science 2000, 287, 1279–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poteet, E.; Liu, D.; Liang, Z.; Van Buren, G.; Chen, C.; Yao, Q. Mesothelin and TGF-α predict pancreatic cancer cell sensitivity to EGFR inhibitors and effective combination treatment with trametinib. PLoS ONE 2019, 14, e0213294. [Google Scholar] [CrossRef] [PubMed]
- Grapa, C.M.; Mocan, T.; Gonciar, D.; Zdrehus, C.; Mosteanu, O.; Pop, T.; Mocan, L. Epidermal growth factor receptor and its role in pancreatic cancer treatment mediated by nanoparticles. Int. J. Nanomed. 2019, 14, 9693–9706. [Google Scholar] [CrossRef] [Green Version]
- Shetty, S.; Yeeravalli, R.; Bera, T.; Das, A. Recent advances on epidermal growth factor receptor as a molecular target for breast cancer therapeutics. Anticancer. Agents Med. Chem. 2020, 21, 1783–1792. [Google Scholar] [CrossRef]
- Saadeh, F.S.; Mahfouz, R.; Assi, H.I. EGFR as a clinical marker in glioblastomas and other gliomas. Int. J. Biol. Markers 2018, 33, 22–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foroughi, S.; Tie, J.; Gibbs, P.; Burgess, A.W. Epidermal growth factor receptor ligands: Targets for optimizing treatment of metastatic colorectal cancer. Growth Factors 2019, 37, 209–225. [Google Scholar] [CrossRef]
- Marin-Muller, C.; Li, D.; Bharadwaj, U.; Li, M.; Chen, C.; Hodges, S.E.; Fisher, W.E.; Mo, Q.; Hung, M.C.; Yao, Q. A tumorigenic factor interactome connected through tumor suppressor microRNA-198 in human pancreatic cancer. Clin. Cancer Res. 2013, 19, 5901–5913. [Google Scholar] [CrossRef] [Green Version]
- Quan, M.; Zeng, L.; Lv, C.; Shen, C.; Gong, W.; Wu, J.; Chen, X.; Hu, W.; Lv, X.; Si, W.; et al. miR-198 inhibits the progression of renal cell carcinoma by targeting BIRC5. Cancer Cell Int. 2021, 21, 390. [Google Scholar]
- Xiao, H.; Zheng, Y.; Chen, J.; Shen, H. MiR-198 inhibits proliferation, invasion and migration of ovarian cancer cells by regulating the PI3K/Akt signaling pathway. Acta Biochim. Pol. 2021. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, X.; Yang, C.; Xu, S. MicroRNA-198-5p inhibits the migration and invasion of non-small lung cancer cells by targeting fucosyltransferase 8. Clin. Exp. Pharmacol. Physiol. 2019, 46, 955–967. [Google Scholar] [CrossRef]
- Ray, J.; Hoey, C.; Huang, X.; Jeon, J.; Taeb, S.; Downes, M.R.; Boutros, P.C.; Liu, S.K. MicroRNA-198 suppresses prostate tumorigenesis by targeting MIB1. Oncol. Rep. 2019, 42, 1047–1056. [Google Scholar]
- Wang, F.; Xue, X.; Wei, J.; An, Y.; Yao, J.; Cai, H.; Wu, J.; Dai, C.; Qian, Z.; Xu, Z.; et al. hsa-miR-520h downregulates ABCG2 in pancreatic cancer cells to inhibit migration, invasion, and side populations. Br. J. Cancer 2010, 103, 567–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, X.; Ma, R.; Yang, S.; Jiang, L.; Wang, Z.; Zhu, Z.; Li, H. miR-520g and miR-520h overcome bortezomib resistance in multiple myeloma via suppressing APE1. Cell Cycle 2019, 18, 1660–1669. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Yao, Q.; Sun, J.; Feng, L.; Lu, H.; Ma, Y.; Liu, L.; Wang, F.; Li, J.; Yue, Y.; et al. Downregulation of histone deacetylase 1 by microRNA-520h contributes to the chemotherapeutic effect of doxorubicin. FEBS Lett. 2014, 588, 184–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parodi, S. Xist noncoding RNA could act as a tumor suppressor gene in patients with. J. Cancer Res. Ther. 2020, 16, 7–12. [Google Scholar] [CrossRef]
- Sun, K.; Jia, Z.; Duan, R.; Yan, Z.; Jin, Z.; Yan, L.; Li, Q.; Yang, J. Long non-coding RNA XIST regulates miR-106b-5p/P21 axis to suppress tumor progression in renal cell carcinoma. Biochem. Biophys. Res. Commun. 2019, 510, 416–420. [Google Scholar] [CrossRef]
- Zheng, R.; Lin, S.; Guan, L.; Yuan, H.; Liu, K.; Liu, C.; Ye, W.; Liao, Y.; Jia, J.; Zhang, R. Long non-coding RNA XIST inhibited breast cancer cell growth, migration, and invasion via miR-155/CDX1 axis. Biochem. Biophys. Res. Commun. 2018, 498, 1002–1008. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Weng, X.D.; Wang, L.; Liu, X.H.; Zhu, H.C.; Guo, J.; Ning, J.Z.; Xiao, C.C. LncRNA XIST acts as a tumor suppressor in prostate cancer through sponging miR-23a to modulate RKIP expression. Oncotarget 2017, 8, 94358–94370. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, P.S.; Viner, J.L.; Beers, R.; Pastan, I. Isolation of a high-affinity stable single-chain Fv specific for mesothelin from DNA-immunized mice by phage display and construction of a recombinant immunotoxin with anti-tumor activity. Proc. Natl. Acad. Sci. USA 1998, 95, 669–674. [Google Scholar] [CrossRef] [Green Version]
- Pastan, I.; Chowdhury, P.S. Ant-Mesothelin Antibodes Having High Binding Affinity. U.S. Patent No 7,081,518 B1, 25 July 2006. [Google Scholar]
- Hassan, R.; Thomas, A.; Alewine, C.; Le, D.T.; Jaffee, E.M.; Pastan, I. Mesothelin Immunotherapy for Cancer: Ready for Prime Time? J. Clin. Oncol. 2016, 34, 4171–4179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastan, I.; Hassan, R. Discovery of mesothelin and exploiting it as a target for immunotherapy. Cancer Res. 2014, 74, 2907–2912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, R.; Bullock, S.; Premkumar, A.; Kreitman, R.J.; Kindler, H.; Willingham, M.C.; Pastan, I. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin. Cancer Res. 2007, 13, 5144–5149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Tang, W.K.; Esser, L.; Pastan, I.; Xia, D. Characterization of crystals of an antibody-recognition fragment of the cancer differentiation antigen mesothelin in complex with the therapeutic antibody MORAb-009. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2012, 68, 950–953. [Google Scholar] [CrossRef]
- Beatty, G.L.; Haas, A.R.; Maus, M.V.; Torigian, D.A.; Soulen, M.C.; Plesa, G.; Chew, A.; Zhao, Y.; Levine, B.L.; Albelda, S.M.; et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol. Res. 2014, 2, 112–120. [Google Scholar] [CrossRef] [Green Version]
- Kichler, A.; Chillo, M.; Leborgne, C.; Danos, O.; Frisch, B. Intranasal gene delivery with a polyethylenimine-PEG conjugate. J. Control. Release 2002, 81, 379–388. [Google Scholar] [CrossRef]
- Petersen, H.; Fechner, P.M.; Martin, A.L.; Kunath, K.; Stolnik, S.; Roberts, C.J.; Fischer, D.; Davies, M.C.; Kissel, T. Polyethylenimine-graft-poly(ethylene glycol) copolymers: Influence of copolymer block structure on DNA complexation and biological activities as gene delivery system. Bioconjug. Chem. 2002, 13, 845–854. [Google Scholar] [CrossRef]
- Sadeqzadeh, E.; Rahbarizadeh, F.; Ahmadvand, D.; Rasaee, M.J.; Parhamifar, L.; Moghimi, S.M. Combined MUC1-specific nanobody-tagged PEG-polyethylenimine polyplex targeting and transcriptional targeting of tBid transgene for directed killing of MUC1 over-expressing tumour cells. J. Control. Release 2011, 156, 85–91. [Google Scholar] [CrossRef]
- Germershau, O.; Merdan, T.; Bakowsky, U.; Behe, M.; Kissel, T. Trastuzumab-polyethylenimine-polyethylene glycol conjugates for targeting Her2-expressing tumors. Bioconjug. Chem. 2006, 17, 1190–1199. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, H.; Cheng, L.; Zhu, S.; Cai, C.; Yang, T.; Yang, L.; Ding, P. Thiol Michael addition reaction: A facile tool for introducing peptides into polymer-based gene delivery systems. Polym. Int. 2018, 67, 25–31. [Google Scholar] [CrossRef]
- Leung, M.K.M.; Hagemeyer, C.E.; Johnston, A.P.R.; Gonzales, C.; Kamphuis, M.M.J.; Ardipradja, K.G.K.; Peter, K.; Caruso, F. Bio-click chemistry: Enzymatic functionalization of PEGylated capsules for targeting applications. Angew. Chem. Int. Ed. Engl. 2012, 51, 7132–7136. [Google Scholar] [CrossRef] [Green Version]
- Holliger, P.; Hudson, P.J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol. 2005, 23, 1126–1136. [Google Scholar] [CrossRef]
- Ketola, T.M.; Hanzlíková, M.; Leppänen, L.; Raviña, M.; Bishop, C.J.; Green, J.J.; Urtti, A.; Lemmetyinen, H.; Yliperttula, M.; Vuorimaa-Laukkanen, E. Independent versus Cooperative Binding in Polyethylenimine–DNA and Poly(L-lysine)–DNA Polyplexes. J. Phys. Chem. B 2013, 117, 10405–10413. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Kang, H.J.; Lee, J.M.; Jung, S.O.; Yun, W.S.; Chung, S.J.; Chung, B.H. Controlled antibody immobilization onto immunoanalytical platforms by synthetic peptide. Anal. Biochem. 2008, 374, 99–105. [Google Scholar] [CrossRef]
- Shim, G.; Kim, D.; Lee, S.; Chang, R.S.; Byun, J.; Oh, Y.K. Staphylococcus aureus-mimetic control of antibody orientation on nanoparticles. Nanomedicine 2019, 16, 267–277. [Google Scholar] [CrossRef]
- Sockolosky, J.T.; Kivimäe, S.; Szoka, F.C. Fusion of a short peptide that binds immunoglobulin G to a recombinant protein substantially increases its plasma half-life in mice. PLoS ONE 2014, 9, e102566. [Google Scholar] [CrossRef]
- Sakamoto, K.; Ito, Y.; Hatanaka, T.; Soni, P.B.; Mori, T.; Sugimura, K. Discovery and characterization of a peptide motif that specifically recognizes a non-native conformation of human IgG induced by acidic pH conditions. J. Biol. Chem. 2009, 284, 9986–9993. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.J.; Kang, Y.J.; Lee, Y.M.; Shin, H.H.; Chung, S.J.; Kang, S. Developing an antibody-binding protein cage as a molecular recognition drug modular nanoplatform. Biomaterials 2012, 33, 5423–5430. [Google Scholar] [CrossRef]
- Moon, H.; Lee, J.; Kim, H.; Heo, S.; Min, J.; Kang, S. Genetically engineering encapsulin protein cage nanoparticle as a SCC-7 cell targeting optical nanoprobe. Biomater. Res. 2014, 18, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Jang, K.T.; Ki, C.S.; Lim, T.; Park, Y.S.; Lim, H.Y.; Choi, D.W.; Kang, W.K.; Park, K.; Park, J.O. Impact of epidermal growth factor receptor (EGFR) kinase mutations, EGFR gene amplifications, and KRAS mutations on survival of pancreatic adenocarcinoma. Cancer 2007, 109, 1561–1569. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, C.W.; Frolov, A.; Frolova, N.; Jhala, N.C.; Howard, J.H.; Buchsbaum, D.J.; Vickers, S.M.; Heslin, M.J.; Arnoletti, J.P. Epidermal growth factor receptor (EGFR) is highly conserved in pancreatic cancer. Surgery 2007, 141, 464–469. [Google Scholar] [CrossRef]
- Bloomston, M.; Bhardwaj, A.; Ellison, E.C.; Frankel, W.L. Epidermal growth factor receptor expression in pancreatic carcinoma using tissue microarray technique. Dig. Surg. 2006, 23, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, N.R.; Hughes, C.M.; Barton, C.M.; Poulsom, R.; Jeffery, R.E.; Klöppel, G.; Hall, P.A.; Gullick, W.J. The epidermal growth factor receptor in human pancreatic cancer. J. Pathol. 1992, 166, 7–12. [Google Scholar] [CrossRef]
- Nedaeinia, R.; Avan, A.; Manian, M.; Salehi, R.; Ghayour-Mobarhan, M. EGFR as a potential target for the treatment of pancreatic cancer: Dilemma and controversies. Curr. Drug Targets 2014, 15, 1293–1301. [Google Scholar] [CrossRef]
- Valsecchi, M.E.; McDonald, M.; Brody, J.R.; Hyslop, T.; Freydin, B.; Yeo, C.J.; Solomides, C.; Peiper, S.C.; Witkiewicz, A.K. Epidermal growth factor receptor and insulinlike growth factor 1 receptor expression predict poor survival in pancreatic ductal adenocarcinoma. Cancer 2012, 118, 3484–3493. [Google Scholar] [CrossRef]
- Ueda, S.; Ogata, S.; Tsuda, H.; Kawarabayashi, N.; Kimura, M.; Sugiura, Y.; Tamai, S.; Matsubara, O.; Hatsuse, K.; Mochizuki, H. The correlation between cytoplasmic overexpression of epidermal growth factor receptor and tumor aggressiveness: Poor prognosis in patients with pancreatic ductal adenocarcinoma. Pancreas 2004, 29, e1–e8. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Wei, Q.; Zhou, Y.; Wang, J.; Liu, Q.; Xu, H. A systematic analysis of FDA-approved anticancer drugs. BMC Syst. Biol. 2017, 11, 87. [Google Scholar] [CrossRef] [PubMed]
- Deepagan, V.G.; Sarmento, B.; Menon, D.; Nascimento, A.; Jayasree, A.; Sreeranganathan, M.; Koyakutty, M.; Nair, S.V.; Rangasamy, J. In vitro targeted imaging and delivery of camptothecin using cetuximab-conjugated multifunctional PLGA-ZnS nanoparticles. Nanomedicine 2012, 7, 507–519. [Google Scholar] [CrossRef]
- Raoof, M.; Corr, S.J.; Kaluarachchi, W.D.; Massey, K.L.; Briggs, K.; Zhu, C.; Cheney, M.A.; Wilson, L.J.; Curley, S.A. Stability of antibody-conjugated gold nanoparticles in the endolysosomal nanoenvironment: Implications for noninvasive radiofrequency-based cancer therapy. Nanomedicine 2012, 8, 1096–1105. [Google Scholar] [CrossRef] [Green Version]
- Biscaglia, F.; Rajendran, S.; Conflitti, P.; Benna, C.; Sommaggio, R.; Litti, L.; Mocellin, S.; Bocchinfuso, G.; Rosato, A.; Palleschi, A.; et al. Enhanced EGFR Targeting Activity of Plasmonic Nanostructures with Engineered GE11 Peptide. Adv. Healthc. Mater. 2017, 6, 23. [Google Scholar] [CrossRef]
- Höbel, S.; Vornicescu, D.; Bauer, M.; Fischer, D.; Keusgen, M.; Aigner, A. A novel method for the assessment of targeted PEI-based nanoparticle binding based on a static surface plasmon resonance system. Anal. Chem. 2014, 86, 6827–6835. [Google Scholar] [CrossRef]
- Rejman, J.; Bragonzi, A.; Conese, M. Role of clathrin- and caveolae-mediated endocytosis in gene transfer mediated by lipo- and polyplexes. Mol. Ther. 2005, 12, 468–474. [Google Scholar] [CrossRef]
- Le Roy, C.; Wrana, J.L. Clathrin- and non-clathrinmediated endocytic regulation of cell signalling. Nat. Rev. Mol. Cell Biol. 2005, 6, 112–126. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.; Tchistiakova, L.; Scott, N. Implications of receptor-mediated endocytosis and intracellular trafficking dynamics in the development of antibody drug conjugates. MAbs 2013, 5, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Takei, K.; Haucke, V. Clathrin-mediated endocytosis: Membrane factors pull the trigger. Trends. Cell. Biol. 2001, 11, 385–391. [Google Scholar] [CrossRef]
- Yoo, J.; Park, C.; Yi, G.; Lee, D.; Koo, H. Active Targeting Strategies Using Biological Ligands for Nanoparticle Drug Delivery Systems. Cancers 2019, 11, 640. [Google Scholar] [CrossRef] [Green Version]
- Voskoglou-Nomikos, T.; Pater, J.L.; Seymour, L. Clinical predictive value of the in vitro cell line, human xenograft, and mouse allograft preclinical cancer models. Clin. Cancer Res. 2003, 9, 4227–4239. [Google Scholar] [PubMed]
- Tentler, J.J.; Tan, A.C.; Weekes, C.D.; Jimeno, A.; Leong, S.; Pitts, T.M.; Arcaroli, J.J.; Messersmith, W.A.; Eckhardt, S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 2012, 9, 338–350. [Google Scholar] [CrossRef] [Green Version]
- Rubio-Viqueira, B.; Jimeno, A.; Cusatis, G.; Zhang, X.; Iacobuzio-Donahue, C.; Karikari, C.; Shi, C.; Danenberg, K.; Danenberg, P.V.; Kuramochi, H.; et al. An in vivo platform for translational drug development in pancreatic cancer. Clin. Cancer Res. 2006, 12, 4652–4661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.P.; Evans, D.B.; Wang, H.; Abbruzzese, J.L.; Fleming, J.B.; Gallick, G.E. Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice. Nat. Protoc. 2009, 4, 1670–1680. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.; Bruckheimer, E.; Rajeshkumar, N.V.; Garrido-Laguna, I.; De Oliveira, E.; Rubio-Viqueira, B.; Strawn, S.; Wick, M.J.; Martell, J.; Sidransky, D. A pilot clinical study of treatment guided by personalized tumorgrafts in patients with advanced cancer. Mol. Cancer Ther. 2011, 10, 1311–1316. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.L.; Fehling, S.C.; Garcia, P.L.; Gamblin, T.L.; Council, L.N.; van Waardenburg, R.C.A.M.; Yang, E.S.; Bradner, J.E.; Yoon, K.J. The BET inhibitor JQ1 attenuates double-strand break repair and sensitizes models of pancreatic ductal adenocarcinoma to PARP inhibitors. EBioMedicine 2019, 44, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Fiebig, H.H.; Maier, A.; Burger, A.M. Clonogenic assay with established human tumour xenografts: Correlation of in vitro to in vivo activity as a basis for anticancer drug discovery. Eur. J. Cancer 2004, 40, 802–820. [Google Scholar] [CrossRef]
- Ahmadzada, T.; Reid, G.; McKenzie, D.R. Fundamentals of siRNA and miRNA therapeutics and a review of targeted nanoparticle delivery systems in breast cancer. Biophys. Rev. 2018, 10, 69–86. [Google Scholar] [CrossRef]
- Liu, J.F.; Lan, Z.; Ferrari, C.; Stein, J.M.; Higbee-Dempsey, E.; Yan, L.; Amirshaghaghi, A.; Cheng, Z.; Issadore, D.; Tsourkas, A. Use of oppositely polarized external magnets to improve the accumulation and penetration of magnetic nanocarriers into solid tumors. ACS Nano 2020, 14, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Hasanzadeh Davarani, F.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M.R. Impact of particle size and polydispersity index on the clinical applications of lipidic nanocarrier systems. Pharmaceutics 2018, 10, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badran, M. Formulation and in vitro evaluation of flufenamic acid loaded deformable liposome for improved skin delivery. Digest J. Nanomater. Biostruct. 2014, 9, 83–91. [Google Scholar]
- Chen, M.; Liu, X.; Fahr, A. Skin penetration and deposition of carboxyfluorescein and temoporfin from different lipid vesicular systems: In vitro study with finite and infinite dosage application. Int. J. Pharm. 2011, 408, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Putri, D.C.; Dwiastuti, R.; Marchaban, M.; Nugroho, A.K. Optimization of mixing temperature and sonication duration in liposome preparation. J. Pharm. Sci. Commun. 2017, 14, 79–85. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lü, J.-M.; Liang, Z.; Liu, D.; Zhan, B.; Yao, Q.; Chen, C. Two Antibody-Guided Lactic-co-Glycolic Acid-Polyethylenimine (LGA-PEI) Nanoparticle Delivery Systems for Therapeutic Nucleic Acids. Pharmaceuticals 2021, 14, 841. https://doi.org/10.3390/ph14090841
Lü J-M, Liang Z, Liu D, Zhan B, Yao Q, Chen C. Two Antibody-Guided Lactic-co-Glycolic Acid-Polyethylenimine (LGA-PEI) Nanoparticle Delivery Systems for Therapeutic Nucleic Acids. Pharmaceuticals. 2021; 14(9):841. https://doi.org/10.3390/ph14090841
Chicago/Turabian StyleLü, Jian-Ming, Zhengdong Liang, Dongliang Liu, Bin Zhan, Qizhi Yao, and Changyi Chen. 2021. "Two Antibody-Guided Lactic-co-Glycolic Acid-Polyethylenimine (LGA-PEI) Nanoparticle Delivery Systems for Therapeutic Nucleic Acids" Pharmaceuticals 14, no. 9: 841. https://doi.org/10.3390/ph14090841
APA StyleLü, J.-M., Liang, Z., Liu, D., Zhan, B., Yao, Q., & Chen, C. (2021). Two Antibody-Guided Lactic-co-Glycolic Acid-Polyethylenimine (LGA-PEI) Nanoparticle Delivery Systems for Therapeutic Nucleic Acids. Pharmaceuticals, 14(9), 841. https://doi.org/10.3390/ph14090841