
JAK-STAT Pathway Regulation of Intestinal Permeability: Pathogenic Roles and Therapeutic Opportunities in Inflammatory Bowel Disease


Abstract
:1. Introduction
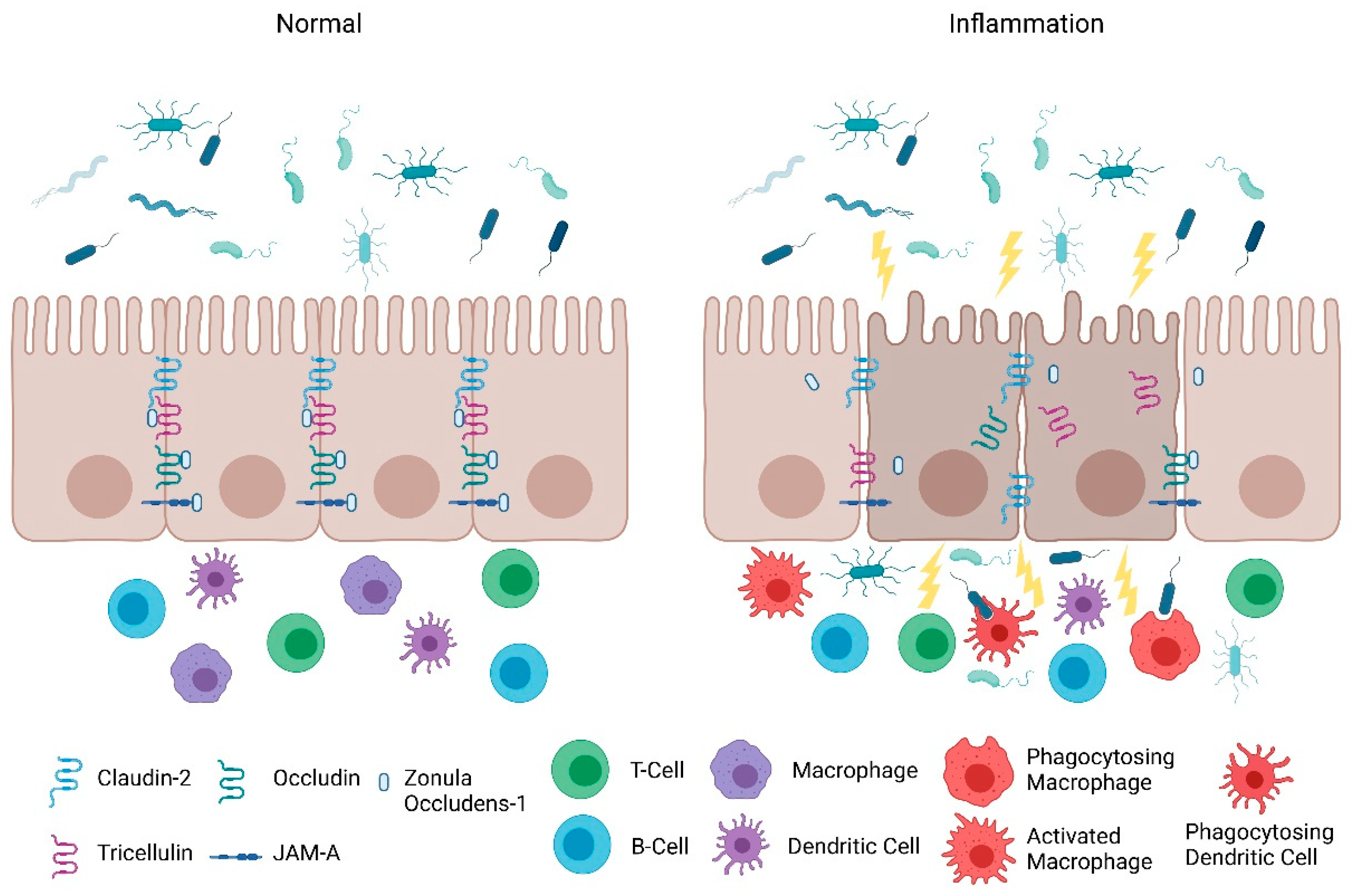
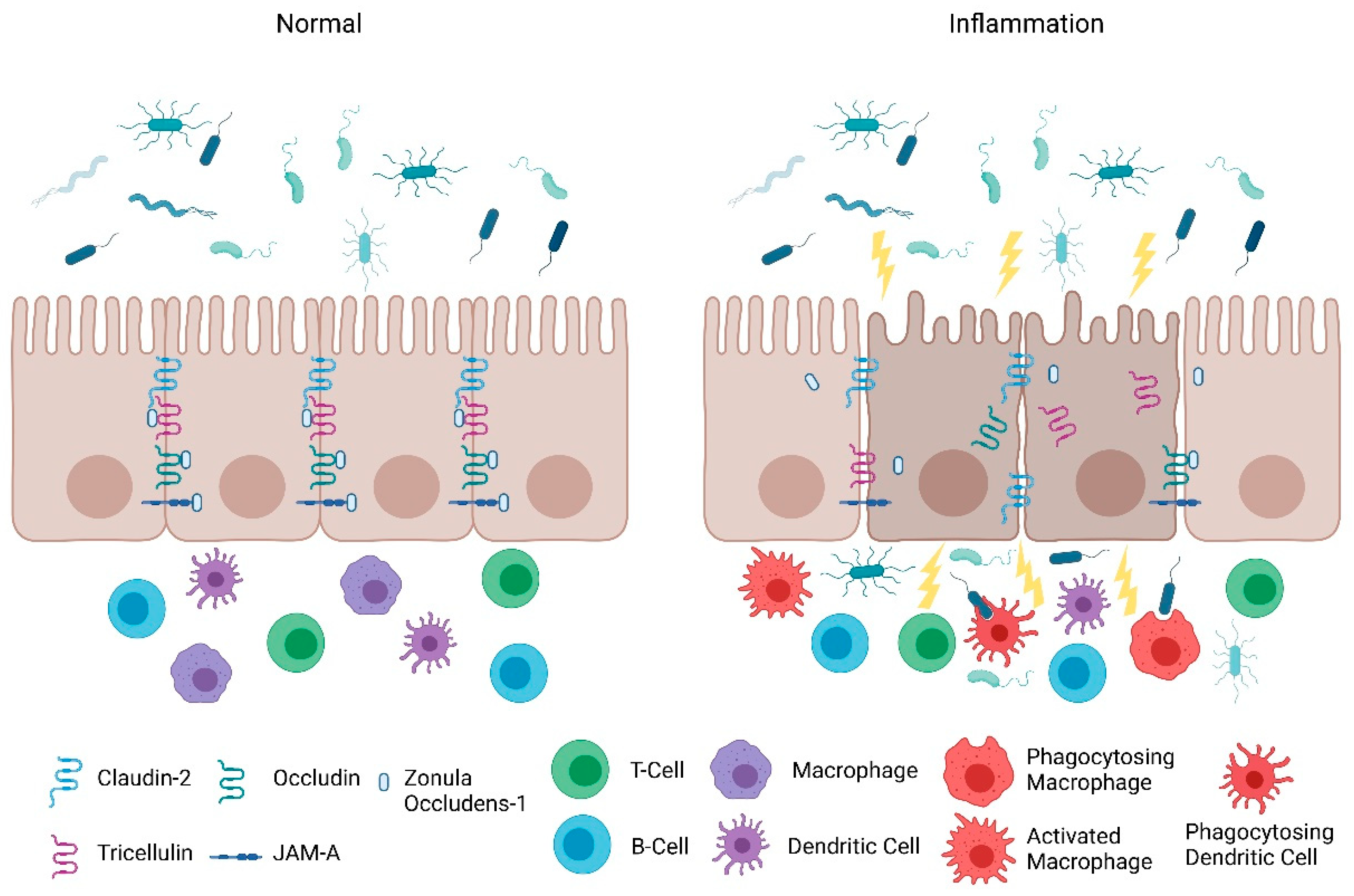
2. Intestinal Permeability in IBD
2.1. Intestinal Epithelial Barrier
2.2. Intestinal Permeability: Maintenance and Its Role in Inflammation
3. Protection of the Intestinal Epithelial Barrier
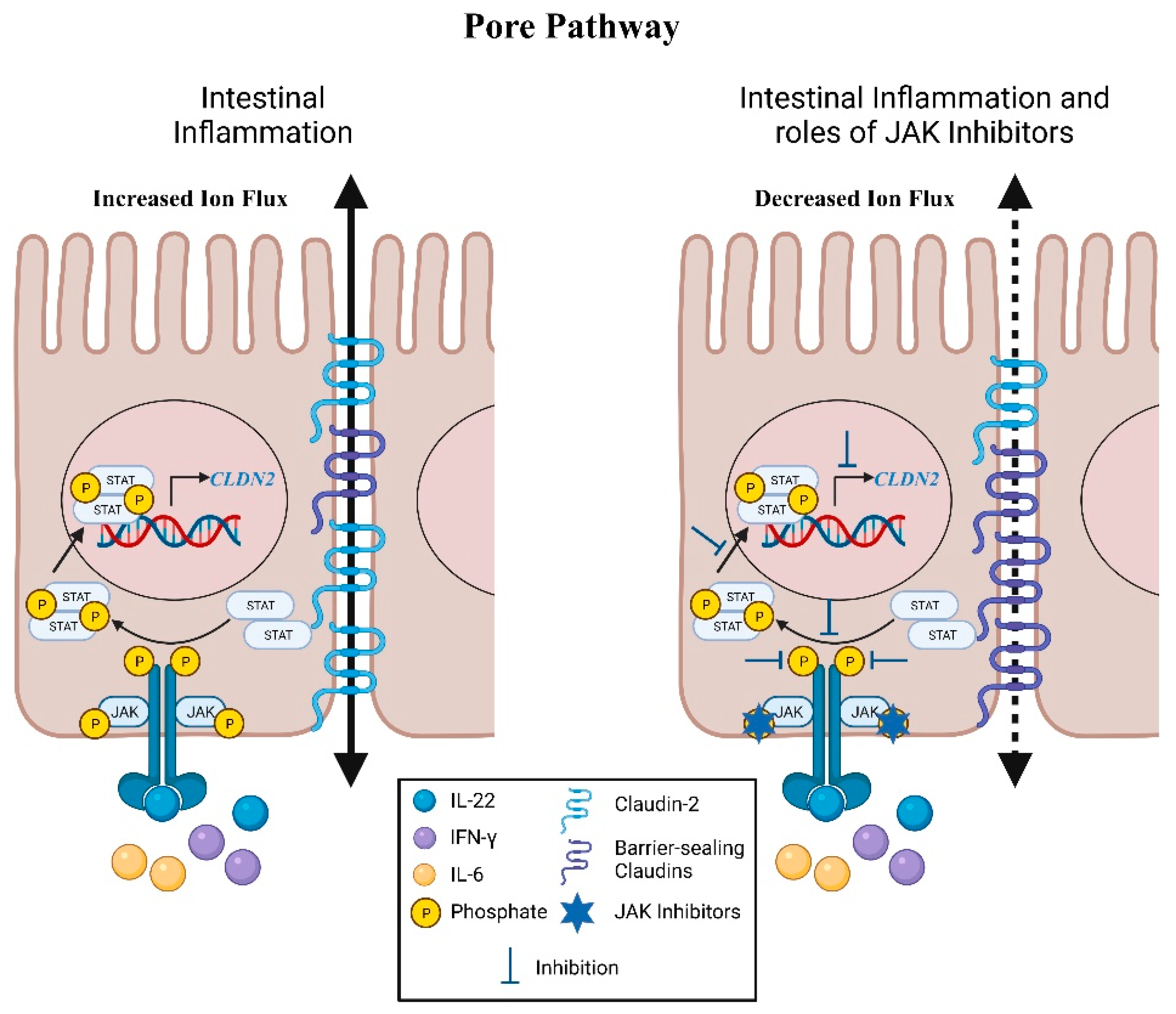
4. Pore Pathway: Electrolyte Flux
Claudins
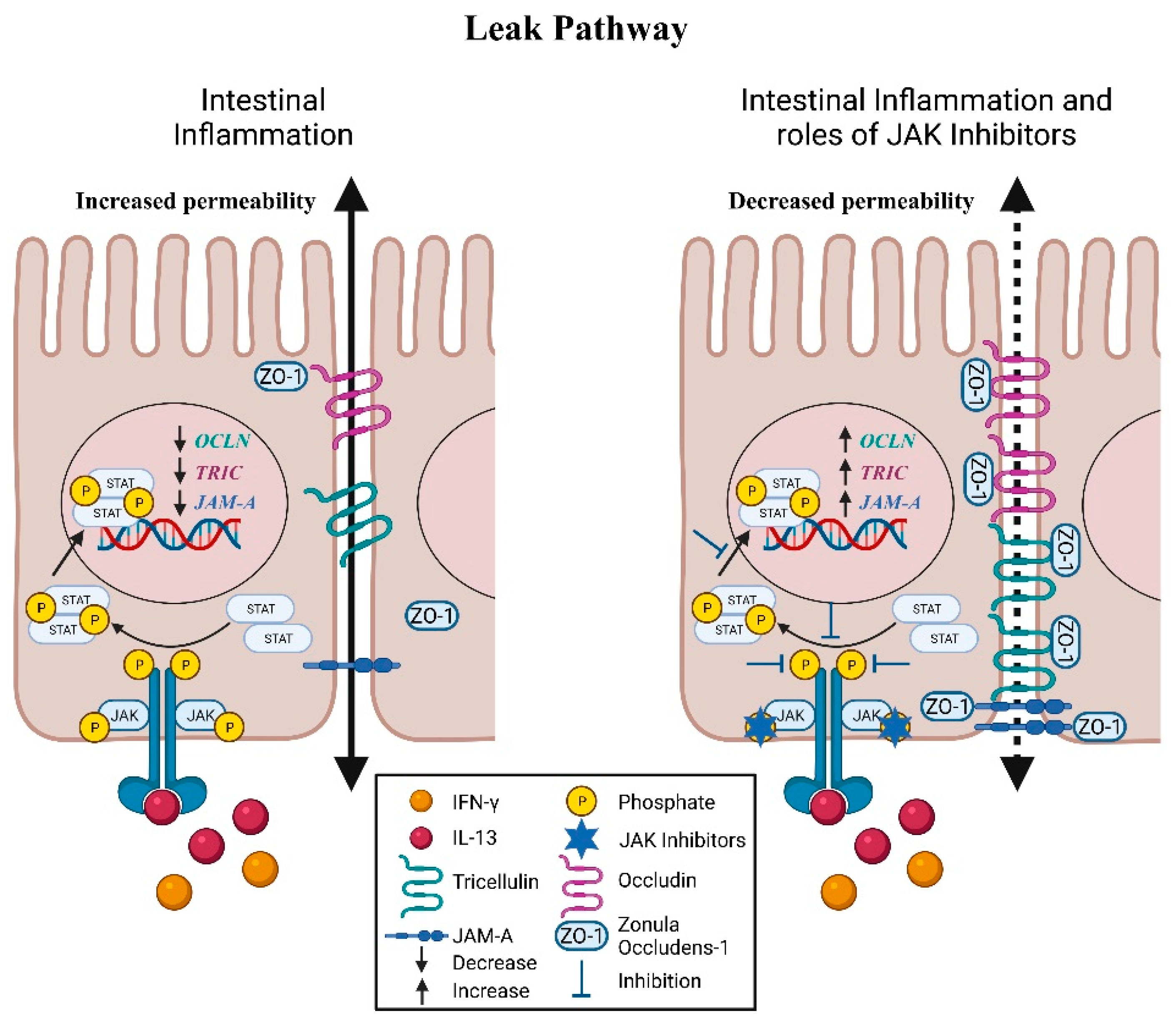
5. Leak Pathway: Molecular Mediators
5.1. Zonula Occludens
5.2. Occludin
5.3. Tricellulin
5.4. JAM-A
6. JAK-STAT Signaling in Apoptosis and Necroptosis of Intestinal Epithelial Cells
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lichtenstein, G.R.; Loftus, E.; Isaacs, K.L.; Regueiro, M.D.; Gerson, L.B.; Sands, B.E. ACG Clinical Guideline: Management of Crohn’s Disease in Adults. Am. J. Gastroenterol. 2018, 113, 481–517. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.; Bonovas, S.; Doherty, G.; Kucharzik, T.; Gisbert, J.P.; Raine, T.; Adamina, M.; Armuzzi, A.; Bachmann, O.; Bager, P.; et al. ECCO Guidelines on Therapeutics in Crohn’s Disease: Medical Treatment. J. Crohn’s Colitis 2019, 14, 4–22. [Google Scholar] [CrossRef]
- Rubin, D.T.; Ananthakrishnan, A.N.; Siegel, C.A.; Sauer, B.G.; Long, M.D. ACG Clinical Guideline: Ulcerative Colitis in Adults. Am. J. Gastroenterol. 2019, 114, 384–413. [Google Scholar] [CrossRef]
- Maaser, C.; Sturm, A.; Vavricka, S.R.; Kucharzik, T.; Fiorino, G.; Annese, V.; Calabrese, E.; Baumgart, D.C.; Bettenworth, D.; Borralho Nunes, P.; et al. ECCO-ESGAR guideline for diagnostic assessment in IBD part 1: Initial diagnosis, monitoring of known IBD, detection of complications. J. Crohn’s Colitis 2019, 13, 144–164. [Google Scholar] [CrossRef] [Green Version]
- Langner, C.; Magro, F.; Driessen, A.; Ensari, A.; Mantzaris, G.J.; Villanacci, V.; Becheanu, G.; Nunes, P.B.; Cathomas, G.; Fries, W.; et al. The histopathological approach to inflammatory bowel disease: A practice guide. Virchows Arch. 2014, 464, 511–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magro, F.; Langner, C.; Driessen, A.; Ensari, A.; Geboes, K.; Mantzaris, G.; Villanacci, V.; Becheanu, G.; Nunes, P.B.; Cathomas, G.; et al. European consensus on the histopathology of inflammatory bowel disease. J. Crohn’s Colitis 2013, 7, 827–851. [Google Scholar] [CrossRef] [Green Version]
- Alatab, S.; Sepanlou, S.G.; Ikuta, K.; Vahedi, H.; Bisignano, C.; Safiri, S.; Sadeghi, A.; Nixon, M.R.; Abdoli, A.; Abolhassani, H.; et al. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Spalinger, M.R.; Sayoc-Becerra, A.; Ordookhanian, C.; Canale, V.; Santos, A.N.; King, S.J.; Krishnan, M.; Nair, M.G.; Scharl, M.; McCole, D.F. The JAK Inhibitor Tofacitinib Rescues Intestinal Barrier Defects Caused by Disrupted Epithelial-macrophage Interactions. J. Crohn’s Colitis 2021, 15, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Sayoc-Becerra, A.; Krishnan, M.; Fan, S.; Jimenez, J.; Hernandez, R.; Gibson, K.; Preciado, R.; Butt, G.; McCole, D.F. The JAK-Inhibitor Tofacitinib Rescues Human Intestinal Epithelial Cells and Colonoids from Cytokine-Induced Barrier Dysfunction. Inflamm. Bowel Dis. 2020, 26, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Michielan, A.; D’Incà, R. Intestinal Permeability in Inflammatory Bowel Disease: Pathogenesis, Clinical Evaluation, and Therapy of Leaky Gut. Mediat. Inflamm. 2015, 2015, 628157. [Google Scholar] [CrossRef] [Green Version]
- Turpin, W.; Lee, S.H.; Raygoza Garay, J.A.; Madsen, K.L.; Meddings, J.B.; Bedrani, L.; Power, N.; Espin-Garcia, O.; Xu, W.; Smith, M.I.; et al. Increased Intestinal Permeability Is Associated with Later Development of Crohn’s Disease. Gastroenterology 2020, 159, 2092–2100.e5. [Google Scholar] [CrossRef]
- Rogler, G. Efficacy of JAK inhibitors in Crohn’s Disease. J. Crohn’s Colitis 2019, 14 (Suppl. S2), S746–S754. [Google Scholar] [CrossRef]
- Perez, T.; Tyler, C.J.; Boyer, J.D.; Karuppuchamy, T.; Yarur, A.; Giles, D.A.; Yeasmin, S.; Lundborg, L.; Sandborn, W.J.; Patel, D.R.; et al. Targeting Cytokine Signaling and Lymphocyte Traffic via Small Molecules in Inflammatory Bowel Disease: JAK Inhibitors and S1PR Agonists. Front. Pharmacol. 2019, 10, 212. [Google Scholar] [CrossRef]
- Panés, J.; Sandborn, W.J.; Schreiber, S.; Sands, B.E.; Vermeire, S.; D’Haens, G.; Panaccione, R.; Higgins, P.D.R.; Colombel, J.-F.; Feagan, B.G.; et al. Tofacitinib for induction and maintenance therapy of Crohn’s disease: Results of two phase IIb randomised placebo-controlled trials. Gut 2017, 66, 1049–1059. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, F.; Parigi, T.L.; Fiorino, G.; Peyrin-Biroulet, L.; Danese, S. Tofacitinib in the treatment of ulcerative colitis: Efficacy and safety from clinical trials to real-world experience. Ther. Adv. Gastroenterol. 2019, 12, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nash, P.; Kerschbaumer, A.; Dörner, T.; Dougados, M.; Fleischmann, R.M.; Geissler, K.; McInnes, I.; E Pope, J.; Van Der Heijde, D.; Stoffer-Marx, M.; et al. Points to consider for the treatment of immune-mediated inflammatory diseases with Janus kinase inhibitors: A consensus statement. Ann. Rheum. Dis. 2021, 80, 71–87. [Google Scholar] [CrossRef]
- Odenwald, M.A.; Turner, J.R. The intestinal epithelial barrier: A therapeutic target? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Chelakkot, C.; Ghim, J.; Ryu, S.H. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp. Mol. Med. 2018, 50, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neal, M.D.; Leaphart, C.; Levy, R.; Prince, J.; Billiar, T.R.; Watkins, S.; Li, J.; Cetin, S.; Ford, H.; Schreiber, A.; et al. Enterocyte TLR4 Mediates Phagocytosis and Translocation of Bacteria Across the Intestinal Barrier. J. Immunol. 2006, 176, 3070–3079. [Google Scholar] [CrossRef] [PubMed]
- McCole, D.F. IBD Candidate Genes and Intestinal Barrier Regulation. Inflamm. Bowel Dis. 2014, 20, 1829–1849. [Google Scholar] [CrossRef] [Green Version]
- Martini, E.; Krug, S.; Siegmund, B.; Neurath, M.F.; Becker, C. Mend Your Fences. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Turpin, W.; Espin-Garcia, O.; Bedrani, L.; Madsen, K.; Meddings, J.B.; Garay, J.A.R.; Silverberg, M.S.; Smith, M.I.; Griffiths, A.M.; Moayyedi, P.; et al. Analysis of Genetic Association of Intestinal Permeability in Healthy First-degree Relatives of Patients with Crohn’s Disease. Inflamm. Bowel Dis. 2019, 25, 1796–1804. [Google Scholar] [CrossRef] [PubMed]
- Hollander, D.; Vadheim, C.M.; Brettholz, E.; Peterson, G.M.; Delahunty, T.; Rotter, J. Increased Intestinal Permeability m Patients with Crohn’ s Disease and Their Relatives. Ann. Intern. Med. 1986, 105, 883–885. [Google Scholar] [CrossRef] [PubMed]
- May, G.R.; Sutherland, L.R.; Meddings, J.B. Is small intestinal permeability really increased in relatives of patients with Crohn’s disease? Gastroenterology 1993, 104, 1627–1632. [Google Scholar] [CrossRef]
- Irvine, E.; Marshall, J. Increased intestinal permeability precedes the onset of Crohn’s disease in a subject with familial risk. Gastroenterology 2000, 119, 1740–1744. [Google Scholar] [CrossRef]
- Olson, T.S.; Reuter, B.K.; Scott, K.G.-E.; Morris, M.A.; Wang, X.-M.; Hancock, L.N.; Burcin, T.L.; Cohn, S.M.; Ernst, P.; Cominelli, F.; et al. The primary defect in experimental ileitis originates from a nonhematopoietic source. J. Exp. Med. 2006, 203, 541–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Resta-Lenert, S.; Smitham, J.; Barrett, K.E. Epithelial dysfunction associated with the development of colitis in conventionally housed mdr1a−/− mice. Am. J. Physiol. Liver Physiol. 2005, 289, G153–G162. [Google Scholar] [CrossRef] [Green Version]
- Madsen, K.; Cornish, A.; Soper, P.; McKaigney, C.; Jijon, H.; Yachimec, C.; Doyle, J.; Jewell, L.; De Simone, C. Probiotic bacteria enhance murine and human intestinal epithelial barrier function. Gastroenterology 2001, 121, 580–591. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, H.; Takechi, M.; Kiyonari, H.; Shioi, G.; Tamura, A.; Tsukita, S. Intestinal deletion of Claudin-7 enhances paracellular organic solute flux and initiates colonic inflammation in mice. Gut 2015, 64, 1529–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeissig, S.; Bürgel, N.; Günzel, D.; Richter, J.; Mankertz, J.; Wahnschaffe, U.; Kroesen, A.J.; Zeitz, M.; Fromm, M.; Schulzke, J.D. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 2007, 56, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Shen, L.; Clayburgh, D.; Nalle, S.C.; Sullivan, E.A.; Meddings, J.B.; Abraham, C.; Turner, J.R. Targeted Epithelial Tight Junction Dysfunction Causes Immune Activation and Contributes to Development of Experimental Colitis. Gastroenterology 2009, 136, 551–563. [Google Scholar] [CrossRef] [Green Version]
- Laukoetter, M.G.; Nava, P.; Lee, W.Y.; Severson, E.A.; Capaldo, C.T.; Babbin, B.A.; Williams, I.R.; Koval, M.; Peatman, E.; Campbell, J.A.; et al. JAM-A regulates permeability and inflammation in the intestine in vivo. J. Exp. Med. 2007, 204, 3067–3076. [Google Scholar] [CrossRef]
- Vetrano, S.; Rescigno, M.; Cera, M.R.; Correale, C.; Rumio, C.; Doni, A.; Fantini, M.; Sturm, A.; Borroni, E.; Repici, A.; et al. Unique Role of Junctional Adhesion Molecule-A in Maintaining Mucosal Homeostasis in Inflammatory Bowel Disease. Gastroenterology 2008, 135, 173–184. [Google Scholar] [CrossRef] [Green Version]
- Tsai, P.-Y.; Zhang, B.; He, W.; Zha, J.-M.; Odenwald, M.A.; Singh, G.; Tamura, A.; Shen, L.; Sailer, A.; Yeruva, S.; et al. IL-22 Upregulates Epithelial Claudin-2 to Drive Diarrhea and Enteric Pathogen Clearance. Cell Host Microbe 2017, 21, 671–681.e4. [Google Scholar] [CrossRef] [Green Version]
- Spalinger, M.R.; Sayoc-Becerra, A.; Santos, A.N.; Shawki, A.; Canale, V.; Krishnan, M.; Niechcial, A.; Obialo, N.; Scharl, M.; Li, J.; et al. PTPN2 Regulates Interactions Between Macrophages and Intestinal Epithelial Cells to Promote Intestinal Barrier Function. Gastroenterology 2020, 159, 1763–1777.e14. [Google Scholar] [CrossRef] [PubMed]
- Lameris, A.L.; Huybers, S.; Kaukinen, K.; Mäkelä, T.H.; Bindels, R.J.; Hoenderop, J.G.; Nevalainen, P.I. Expression profiling of claudins in the human gastrointestinal tract in health and during inflammatory bowel disease. Scand. J. Gastroenterol. 2012, 48, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Barmeyer, C.; Schulzke, J.D.; Fromm, M. Claudin-related intestinal diseases. Semin. Cell Dev. Biol. 2015, 42, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Mingrino, R.; Kaukinen, K.; Hayes, K.L.; Powell, R.M.; Macdonald, T.T.; Collins, J. Inflammatory processes have differential effects on claudins 2, 3 and 4 in colonic epithelial cells. Lab. Investig. 2005, 85, 1139–1162. [Google Scholar] [CrossRef]
- Li, E.; Ajuwon, K.M. Mechanism of endocytic regulation of intestinal tight junction remodeling during nutrient starvation in jejunal IPEC-J2 cells. FASEB J. 2021, 35, e21356. [Google Scholar] [CrossRef]
- Rosenthal, R.; Milatz, S.; Krug, S.; Oelrich, B.; Schulzke, J.-D.; Amasheh, S.; Günzel, D.; Fromm, M. Claudin-2, a component of the tight junction, forms a paracellular water channel. J. Cell Sci. 2010, 123, 1913–1921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Mumm, J.B.; Herbst, R.; Kolbeck, R.; Wang, Y. IL-22 Increases Permeability of Intestinal Epithelial Tight Junctions by Enhancing Claudin-2 Expression. J. Immunol. 2017, 199, 3316–3325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, M.; Penrose, H.; Shah, N.N.; Marchelletta, R.R.; McCole, D.F. VSL#3 Probiotic Stimulates T-cell Protein Tyrosine Phosphatase–mediated Recovery of IFN-γ–induced Intestinal Epithelial Barrier Defects. Inflamm. Bowel Dis. 2016, 22, 2811–2823. [Google Scholar] [CrossRef]
- Suzuki, T.; Yoshinaga, N.; Tanabe, S. Interleukin-6 (IL-6) Regulates Claudin-2 Expression and Tight Junction Permeability in Intestinal Epithelium. J. Biol. Chem. 2011, 286, 31263–31271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, M.; McCole, D.F. T cell protein tyrosine phosphatase prevents STAT1 induction of claudin-2 expression in intestinal epithelial cells. Ann. N. Y. Acad. Sci. 2017, 1405, 116–130. [Google Scholar] [CrossRef] [PubMed]
- Marchelletta, R.R.; McCole, D.F. T-Cell Protein Tyrosine Phosphatase Protects Intestinal Barrier Function by Restricting Epithelial Tight Junction Remodeling. J. Clin. Investig. 2021, in press. [Google Scholar]
- The Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nat. Cell Biol. 2007, 447, 661–678. [Google Scholar] [CrossRef] [Green Version]
- Simoncic, P.D.; Lee-Loy, A.; Barber, D.L.; Tremblay, M.L.; McGlade, C. The T Cell Protein Tyrosine Phosphatase Is a Negative Regulator of Janus Family Kinases 1 and 3. Curr. Biol. 2002, 12, 446–453. [Google Scholar] [CrossRef] [Green Version]
- Scharl, M.; Hruz, P.; McCole, D.F. Protein Tyrosine Phosphatase non-Receptor Type 2 regulates IFN-γ-induced cytokine signaling in THP-1 monocytes. Inflamm. Bowel Dis. 2010, 16, 2055–2064. [Google Scholar] [CrossRef] [PubMed]
- Glas, J.; Wagner, J.; Seiderer, J.; Olszak, T.; Wetzke, M.; Beigel, F.; Tillack, C.; Stallhofer, J.; Friedrich, M.; Steib, C.; et al. PTPN2 Gene Variants Are Associated with Susceptibility to Both Crohn’s Disease and Ulcerative Colitis Supporting a Common Genetic Disease Background. PLoS ONE 2012, 7, e33682. [Google Scholar] [CrossRef]
- Buzza, M.S.; Netzel-Arnett, S.; Shea-Donohue, T.; Zhao, A.; Lin, C.-Y.; List, K.; Szabo, R.; Fasano, A.; Bugge, T.H.; Antalis, T.M. Membrane-anchored serine protease matriptase regulates epithelial barrier formation and permeability in the intestine. Proc. Natl. Acad. Sci. USA 2010, 107, 4200–4205. [Google Scholar] [CrossRef] [Green Version]
- Fanning, A.S.; Anderson, J.M. PDZ domains: Fundamental building blocks in the organization of protein complexes at the plasma membrane. J. Clin. Investig. 1999, 103, 767–772. [Google Scholar] [CrossRef]
- McNeil, E.; Capaldo, C.T.; Macara, I.G. Zonula Occludens-1 Function in the Assembly of Tight Junctions in Madin-Darby Canine Kidney Epithelial Cells. Mol. Biol. Cell 2006, 17, 1922–1932. [Google Scholar] [CrossRef] [Green Version]
- González-Mariscal, L.; Betanzos, A.; Nava, P.; Jaramillo, B. Tight junction proteins. Prog. Biophys. Mol. Biol. 2003, 81, 1–44. [Google Scholar] [CrossRef]
- Itoh, M.; Furuse, M.; Morita, K.; Kubota, K.; Saitou, M.; Tsukita, S. Direct Binding of Three Tight Junction-Associated Maguks, Zo-1, Zo-2, and Zo-3, with the Cooh Termini of Claudins. J. Cell Biol. 1999, 147, 1351–1363. [Google Scholar] [CrossRef] [Green Version]
- Ebnet, K.; Schulz, C.U.; zu Brickwedde, M.-K.M.; Pendl, G.G.; Vestweber, D. Junctional Adhesion Molecule Interacts with the PDZ Domain-containing Proteins AF-6 and ZO-1. J. Biol. Chem. 2000, 275, 27979–27988. [Google Scholar] [CrossRef] [Green Version]
- Oshitani, N.; Watanabe, K.; Nakamura, S.; Fujiwara, Y.; Higuchi, K.; Arakawa, T. Dislocation of tight junction proteins without F-actin disruption in inactive Crohn’s disease. Int. J. Mol. Med. 2005, 15, 407–410. [Google Scholar] [CrossRef]
- Gassler, N.; Rohr, C.; Schneider, A.; Kartenbeck, J.; Bach, A.; Obermüller, N.; Otto, H.F.; Autschbach, F. Inflammatory bowel disease is associated with changes of enterocytic junctions. Am. J. Physiol. Liver Physiol. 2001, 281, G216–G228. [Google Scholar] [CrossRef]
- Das, P.; Goswami, P.; Das, T.K.; Nag, T.; Sreenivas, V.; Ahuja, V.; Panda, S.K.; Gupta, S.D.; Makharia, G.K. Comparative tight junction protein expressions in colonic Crohn’s disease, ulcerative colitis, and tuberculosis: A new perspective. Virchows Arch. 2012, 460, 261–270. [Google Scholar] [CrossRef]
- Nusrat, A.; Chen, J.A.; Foley, C.S.; Liang, T.W.; Tom, J.; Cromwell, M.; Quan, C.; Mrsny, R.J. The Coiled-coil Domain of Occludin Can Act to Organize Structural and Functional Elements of the Epithelial Tight Junction. J. Biol. Chem. 2000, 275, 29816–29822. [Google Scholar] [CrossRef] [Green Version]
- Furuse, M.; Hirase, T.; Itoh, M.; Nagafuchi, A.; Yonemura, S.; Tsukita, S. Occludin: A novel integral membrane protein localizing at tight junctions. J. Cell Biol. 1993, 123, 1777–1788. [Google Scholar] [CrossRef]
- Heller, F.; Florian, P.; Bojarski, C.; Richter, J.; Christ, M.; Hillenbrand, B.; Mankertz, J.; Gitter, A.H.; Bürgel, N.; Fromm, M. Interleukin-13 Is the Key Effector Th2 Cytokine in Ulcerative Colitis That Affects Epithelial Tight Junctions, Apoptosis, and Cell Restitution. Gastroenterology 2005, 129, 550–564. [Google Scholar] [CrossRef]
- Poritz, L.S.; Harris, L.R.; Kelly, A.A.; Koltun, W.A. Increase in the Tight Junction Protein Claudin-1 in Intestinal Inflammation. Dig. Dis. Sci. 2011, 56, 2802–2809. [Google Scholar] [CrossRef] [Green Version]
- Krug, S.M.; Amasheh, S.; Richter, J.F.; Milatz, S.; Günzel, D.; Westphal, J.K.; Huber, O.; Schulzke, J.D.; Fromm, M. Tricellulin Forms a Barrier to Macromolecules in Tricellular Tight Junctions without Affecting Ion Permeability. Mol. Biol. Cell 2009, 20, 3713–3724. [Google Scholar] [CrossRef] [Green Version]
- Krug, S.M.; Bojarski, C.; Fromm, A.; Schulzke, J.D.; Fromm, M. Tricellulin in Crohn’s disease and ulcerative colitis. FASEB J. 2010, 24, 998.1. [Google Scholar] [CrossRef]
- Hu, J.-C.E.; Weiß, F.; Bojarski, C.; Branchi, F.; Schulzke, J.-D.; Fromm, M.; Krug, S.M. Expression of tricellular tight junction proteins and the paracellular macromolecule barrier are recovered in remission of ulcerative colitis. BMC Gastroenterol. 2021, 21, 141. [Google Scholar] [CrossRef]
- Krug, S.; Bojarski, C.; Fromm, A.; Lee, I.M.; Dames, P.; Richter, J.F.; Turner, J.R.; Fromm, M.; Schulzke, J.-D. Tricellulin is regulated via interleukin-13-receptor α2, affects macromolecule uptake, and is decreased in ulcerative colitis. Mucosal Immunol. 2018, 11, 345–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.-C.E.; Bojarski, C.; Branchi, F.; Fromm, M.; Krug, S.M. Leptin Downregulates Angulin-1 in Active Crohn’s Disease via STAT3. Int. J. Mol. Sci. 2020, 21, 7824. [Google Scholar] [CrossRef] [PubMed]
- Eder, P.; Adler, M.; Dobrowolska, A.; Kamhieh-Milz, J.; Witowski, J. The Role of Adipose Tissue in the Pathogenesis and Therapeutic Outcomes of Inflammatory Bowel Disease. Cells 2019, 8, 628. [Google Scholar] [CrossRef] [Green Version]
- Martìn-Padura, I.; Lostaglio, S.; Schneemann, M.; Williams, L.; Romano, M.; Fruscella, P.; Panzeri, C.; Stoppacciaro, A.; Ruco, L.; Villa, A.; et al. Junctional Adhesion Molecule, a Novel Member of the Immunoglobulin Superfamily That Distributes at Intercellular Junctions and Modulates Monocyte Transmigration. J. Cell Biol. 1998, 142, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Kucharzik, T.; Walsh, S.V.; Chen, J.; Parkos, C.A.; Nusrat, A. Neutrophil Transmigration in Inflammatory Bowel Disease Is Associated with Differential Expression of Epithelial Intercellular Junction Proteins. Am. J. Pathol. 2001, 159, 2001–2009. [Google Scholar] [CrossRef] [Green Version]
- Fan, S.; Weight, C.M.; Luissint, A.-C.; Hilgarth, R.S.; Brazil, J.C.; Ettel, M.; Nusrat, A.; Parkos, C.A. Role of JAM-A tyrosine phosphorylation in epithelial barrier dysfunction during intestinal inflammation. Mol. Biol. Cell 2019, 30, 566–578. [Google Scholar] [CrossRef]
- Marchiando, A.M.; Shen, L.; Graham, W.; Edelblum, K.L.; Duckworth, C.; Guan, Y.; Montrose, M.H.; Turner, J.R.; Watson, A.J. The Epithelial Barrier Is Maintained by In Vivo Tight Junction Expansion during Pathologic Intestinal Epithelial Shedding. Gastroenterology 2011, 140, 1208–1218.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Günther, C.; Ruder, B.; Stolzer, I.; Dorner, H.; He, G.-W.; Chiriac, M.T.; Aden, K.; Strigli, A.; Bittel, M.; Zeissig, S.; et al. Interferon Lambda Promotes Paneth Cell Death Via STAT1 Signaling in Mice and Is Increased in Inflamed Ileal Tissues of Patients with Crohn’s Disease. Gastroenterology 2019, 157, 1310–1322.e13. [Google Scholar] [CrossRef] [PubMed]
- Stolzer, I.; Dressel, A.; Chiriac, M.T.; Neurath, M.F.; Günther, C. An IFN-STAT Axis Augments Tissue Damage and Inflammation in a Mouse Model of Crohn’s Disease. Front. Med. 2021, 8, 667. [Google Scholar] [CrossRef]
- Richmond, C.A.; Rickner, H.; Shah, M.S.; Ediger, T.; Deary, L.; Zhou, F.; Tovaglieri, A.; Carlone, D.L.; Breault, D.T. JAK/STAT-1 Signaling Is Required for Reserve Intestinal Stem Cell Activation during Intestinal Regeneration Following Acute Inflammation. Stem Cell Rep. 2018, 10, 17–26. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
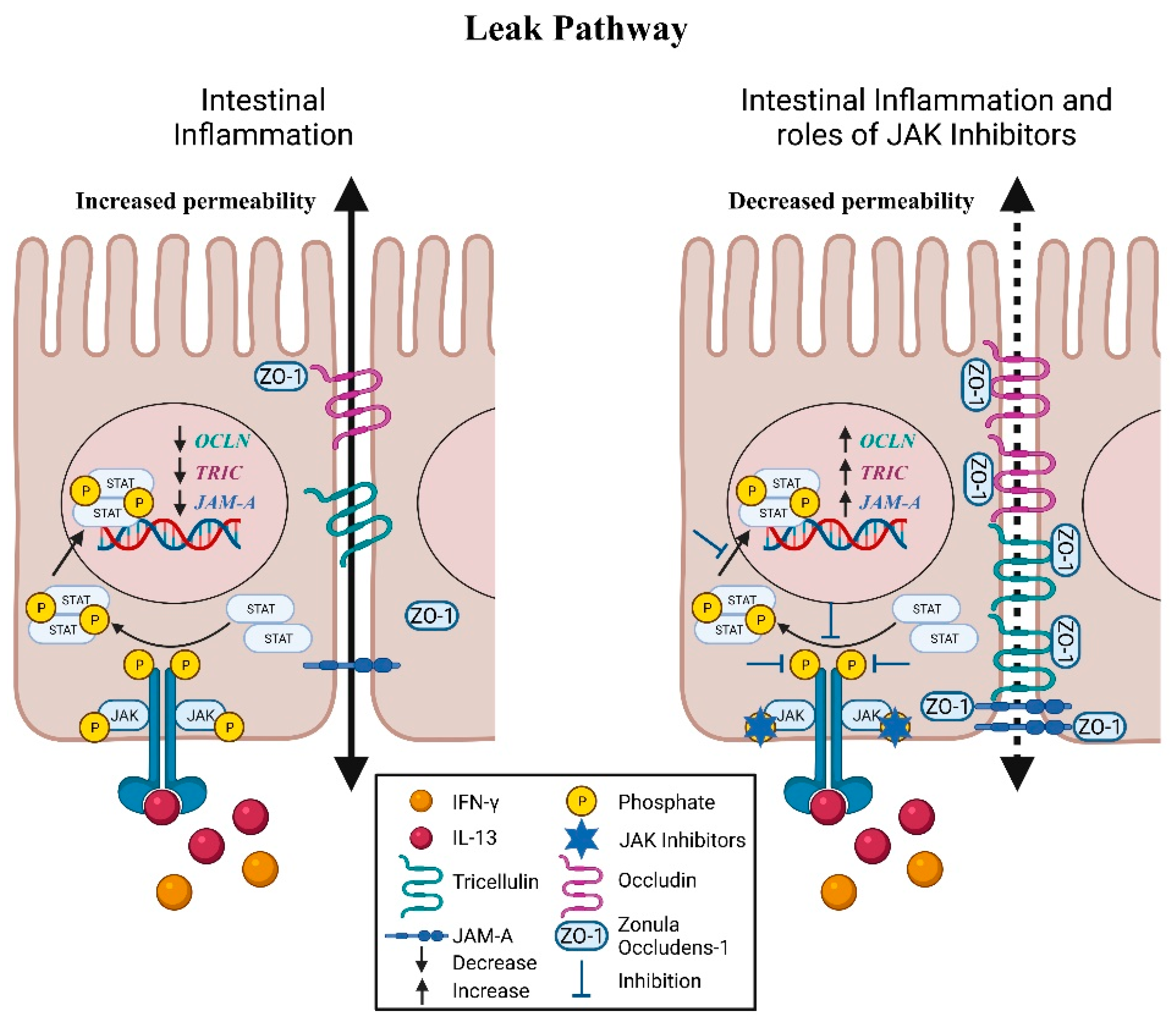{kind=link}
| JAK Inhibitors | Target | Study | Administration | Phase |
|---|---|---|---|---|
| SHR0302 | JAK1 | A Phase II Study in Patients With Moderate to Severe Active Crohn’s Disease | N/A | Phase 2 |
| TD-1473 | Pan-JAK | Efficacy and Safety of TD-1473 in Crohn’s Disease | Oral | Phase 2 |
| TD-1473 | Pan-JAK | TD-1473 Long-Term Safety (LTS) Ulcerative Colitis (UC) Study | Oral | Phase 2/Phase 3 |
| TD-1473 | Pan-JAK | Efficacy and Safety of TD-1473 in Ulcerative Colitis | Oral | Phase 2B/Phase 3 |
| Upadacitinib | JAK1 | A Maintenance and Long-Term Extension Study of the Efficacy and Safety of Upadacitinib (ABT-494) in Participants with Crohn’s Disease Who Completed the Studies M14-431 or M14-433 | Oral | Phase 3 |
| Upadacitinib | JAK1 | A Study to Evaluate the Long-Term Safety and Efficacy of Upadacitinib (ABT-494) in Participants with Ulcerative Colitis (UC) | Oral | Phase 3 |
| Upadacitinib | JAK1 | A Study of the Efficacy and Safety of Upadacitinib (ABT-494) in Participants with Moderately to Severely Active Crohn’s Disease Who Have Inadequately Responded to or Are Intolerant to Biologic Therapy | Oral | Phase 3 |
| Upadacitinib | JAK1 | A Study to Evaluate the Safety and Efficacy of Upadacitinib (ABT-494) for Induction and Maintenance Therapy in Participants With Moderately to Severely Active Ulcerative Colitis (UC) | Oral | Phase 3 |
| Upadacitinib | JAK1 | A Study of the Efficacy and Safety of Upadacitinib (ABT-494) in Participants With Moderately to Severely Active Crohn’s Disease Who Have Inadequately Responded to or Are Intolerant to Conventional and/or Biologic Therapies | Oral | Phase 3 |
| Upadacitinib | JAK1 | A Study to Evaluate the Long-Term Efficacy, Safety, and Tolerability of Repeated Administration of Upadacitinib (ABT-494) in Participants with Crohn’s Disease | Oral | Phase 2 |
| Tofacitinib | Pan-JAK | A Study of Tofacitinib in Patients with Ulcerative Colitis in Stable Remission | Oral | Phase 4 |
| JAK-STATs | Activators in IBD | Effect on TJ Proteins | Effect on Permeability | JAK/STAT Inhibitors | References |
|---|---|---|---|---|---|
| STAT1 and STAT3 | IL-22 | Increased claudin-2 expression | Increased paracellular permeability to ionic solutes; Reduced TEER | JAK Inhibitor 1 and AZD1480 (Inhibited the STAT3-dependent gene, SOCS3) | [41] |
| JAK1-STAT1/STAT3 | IFN-γ | Increased claudin-2 expression | Increased paracellular permeability; Reduced TEER, Increased FD4 permeability | Tofacitinib | [9] |
| STAT3 | IL-6 | Increases claudin-2 expression | Increases paracellular permeability to ionic solutes; Reduced TEER | AG490, STAT3 siRNA | [35,43] |
| Undetermined | Presumably IFN-γ | Decreased JAM-A expression; Possible redistribution of JAM-A | Increased paracellular permeability to macromolecules; Presumably reduces TEER and increases FD4 permeability | Tofacitinib | [8] |
| Undetermined | IFN-γ | Decreased occludin expression; Redistribution of occludin | Increased paracellular permeability to larger macromolecules | Tofacitinib | [8,9] |
| JAK1/JAK2 | IL-13, IFN-γ | Downregulation of tricellulin; Redistribution of tricellulin | Increased uptake of macromolecules through the paracellular space | Baricitinib, Tofacitinib | [8,9,66] |
| STAT3 and JAK2 | Leptin | Downregulation of angulin-1 | Tricellulin localization is altered; Increased intestinal permeability | Stattic, WP1006, and partially by AG490 | [66,67,68] |
| Undetermined | IFN-γ | Downregulation and redistribution of ZO-1 | Increased paracellular permeability to macromolecules | Tofacitinib | [8,9] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lei, H.; Crawford, M.S.; McCole, D.F. JAK-STAT Pathway Regulation of Intestinal Permeability: Pathogenic Roles and Therapeutic Opportunities in Inflammatory Bowel Disease. Pharmaceuticals 2021, 14, 840. https://doi.org/10.3390/ph14090840
Lei H, Crawford MS, McCole DF. JAK-STAT Pathway Regulation of Intestinal Permeability: Pathogenic Roles and Therapeutic Opportunities in Inflammatory Bowel Disease. Pharmaceuticals. 2021; 14(9):840. https://doi.org/10.3390/ph14090840
Chicago/Turabian StyleLei, Hillmin, Meli’sa S. Crawford, and Declan F. McCole. 2021. "JAK-STAT Pathway Regulation of Intestinal Permeability: Pathogenic Roles and Therapeutic Opportunities in Inflammatory Bowel Disease" Pharmaceuticals 14, no. 9: 840. https://doi.org/10.3390/ph14090840
APA StyleLei, H., Crawford, M. S., & McCole, D. F. (2021). JAK-STAT Pathway Regulation of Intestinal Permeability: Pathogenic Roles and Therapeutic Opportunities in Inflammatory Bowel Disease. Pharmaceuticals, 14(9), 840. https://doi.org/10.3390/ph14090840