1. Introduction
The diagnosis and quantification of various physiological and pathophysiological processes in vivo by position emission tomography (PET) have become increasingly crucial in medical research [
1]. The high resolution and sensitivity of PET allow the detection of changes in cellular function or receptor densities during disease development using molecular tracers, most frequently labeled with fluorine-18. Radiolabeled biological and pharmaceutical active molecules carrying
18F are of increasing importance for preclinical, clinical, and nuclear medical research due to the unique properties of
18F, such as low β
+-energy, long half-life (109.77 min), and the easy accessibility of no-carrier-added [
18F]fluoride [
2]. The nuclear reaction of
18O(p,n)
18F in a small-scale cyclotron is the commonly applied process for the production of [
18F]fluoride, which involves the bombardment of H
2[
18O]O with highly accelerated protons, and is the basis of the supply of radiopharmaceutical facilities. The extraction of [
18F]fluoride from water uses anion-exchange columns, particularly quaternary methylammonium (QMA) cartridges, which allow the removal and recovery of H
2[
18O]O. The trapped [
18F]fluoride on the cartridge is commonly eluted using an aqueous MeCN solution of a base and phase transfer catalyst (PTC), such as Kryptofix
®2.2.2 or tetraalkylammonium bicarbonates, which are part of a conserved protocol. In the reaction vial, the water is finally removed by azeotropic drying under heating and gas flow to provide the reactive and dry [
18F]fluoride-PTC reagent for nucleophilic labeling of target precursors [
3,
4].
The nucleophilic substitution (S
N2) is a widely adopted method in
18F chemistry for the introduction of [
18F]fluoride to provide important radiotracers, such as [
18F]FP-CIT [
5], [
18F]FDG [
6], [
18F]FMISO [
7], [
18F]F-tryptophan derivatives [
8], and many others. This strategy has gained significant importance in the radiopharmaceutical field, as it is not limited to aliphatic systems, but can also be applied to aromatic systems, whereas [
18F]fluoride is always introduced to the reaction by taking advantage of the eluting agent consisting of an appropriate solvent and the PTC.
Numerous PTCs have been developed to improve radiosynthetic processes [
9,
10]; among them, tetraalkylammonium salts have been studied most widely due to their counter anion exchanging ability with [
18F]fluoride and fair solubility in most organic solvents including CH
3CN and alcohols. The S
NAr radiofluorination with phosphonium borane modified cartridges and tetrabutylammonium cyanide (TBACN) as an eluting agent was reported, whereas tetrabutylammonium cations serve as PTCs and the cyanide counter anion displaces [
18F]fluoride from the cartridge [
11]. Radiochemists have made efforts to skip the typical aqueous elution of [
18F]fluoride and the following azeotropic drying steps. Recently, Aerts et al. developed the
n-tetradecyltrimethylammonium cation (TDTMA) to avoid the azeotropic drying step before radiofluorination [
12]. Tetraethylammonium (TEA) hydrogen carbonate, tosylate, or perchlorate salts were developed by Inkster et al. to avoid basic reaction conditions as well as azeotropic drying. Such PTC salts were efficiently used for both aliphatic and aromatic
18F-fluorination reactions [
10]. Apart from tetraalkylammonium salts, various arylonium salts, such as quaternary anilinium, diaryliodonium, and triarylsulfonium, were developed to minimalize the
18F-labeling approach [
13]. The aforementioned onium salts act as PTC as well as promoters for radiofluorinations of aliphatic and aromatic compounds. However, radiolabeling using quaternary alkylammonium [
18F]fluoride is much faster at >80 °C; the reaction conditions typically produce by-products, such as alcohols or alkenes, due to strongly basic PTC–solvent systems.
Recent reports on alcohol-containing solvent systems for
18F-fluorination [
14,
15] or
tert-alcohol coordinated tetraalkylammonium fluoride [
16,
17] demonstrated the favorable properties of such reagents in terms of reactivity, nucleophilicity and solubility in organic solvents. Furthermore,
tert-alcohol functionalized tri-
tert-butanolamine was reported as a promoter of S
N2 fluorination reactions; these accelerate the nucleophilic aliphatic substitution due to the coordination between hydroxyl (-OH) and amine functionalities and fluoride [
18]. Importantly, nucleophilic properties of fluorine dominate over the basicity in favor of S
N2 substitutions [
19].
In this work, we aimed to develop tri-(
tert-butanol)-methylammonium fluoride as a PTC for radiofluorinations, offering reduced basicity of fluoride by hydrogen-bonding coordination. Herein, we assessed the reactivity and [
18F]fluoride elution efficiency of tri-(
tert-butanol)amine (tri-(
t-BuOH)A) and quaternary ammonium salt tri-(
tert-butanol)-methylammonium iodide (TBMA-I,
Scheme 1) as PTC in the
18F-fluorination of 1,3-ditosylpropane and aromatic boronic acid pinacol esters as model precursors.
2. Results and Discussion
Initially, we examined the application of known tri-(
t-BuOH)A [
18] on the recovery of [
18F]fluoride from QMA cartridges (
Table 1). Entry 2 of
Table 1 illustrates that the recovery of [
18F]fluoride was very poor, at 11%. Most of the [
18F]fluoride was trapped on the cartridge, similar to an elution without any PTC (entry 1). Thus, tri-(
t-BuOH)A was converted into the corresponding quaternary ammonium form by following a reported procedure with some modifications [
20,
21].
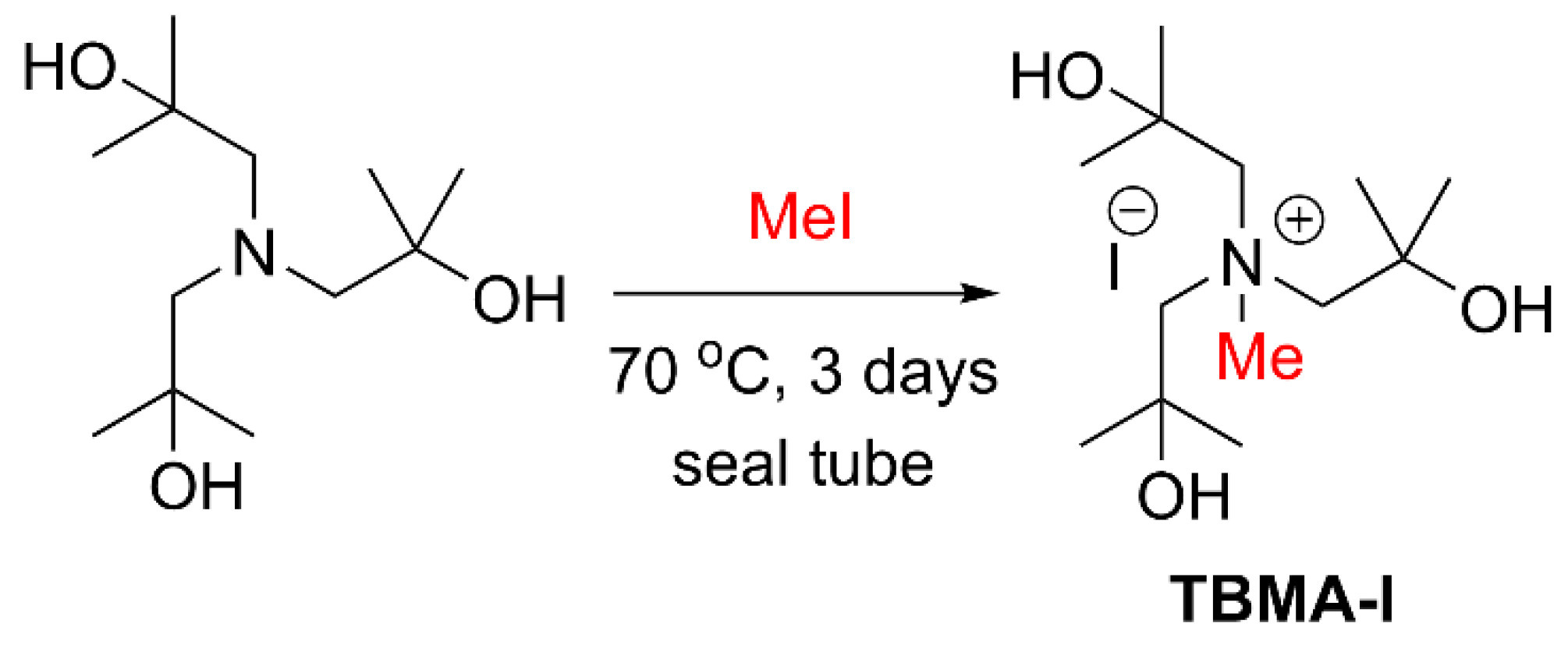
Scheme 1 shows the synthesis of TBMA-I by neat-treating of tri-(
tert-BuOH)A with 1.1 equivalents of MeI under pressure at 70 °C for 3 days. The resulting product was semi-solid and immiscible with DCM, Et
2O, EtOAc, and hexane, but soluble in CH
3CN, H
2O, MeOH, and DMSO. The purity and identity of TBMA-I were confirmed by LC/MS and
1H-NMR analysis, and the reactivity of TBMA-I was studied by performing iodination reactions with 1,3-ditosylpropane using 1.5 equivalents of TBMA-I in CH
3CN at room temperature for 12 h, providing the desired 3-iodo-1-tosylpropane with a yield of 67%.
With TBMA-I, we studied its property as a PTC for [
18F]fluoride elution, as summarized in
Table 1, when different parameters were optimized for the elution of [
18F]fluoride from QMA cartridges using 1 mL of various elution mixtures of PTC. We investigated the elution efficiency of the protic functionalized quaternary ammonium iodide salt TBMA-I in an elution mixture of an aqueous solution (1 mL total volume) of K
2CO
3 (1 M, 15 µL) and CH
3CN (800 µL), which showed excellent eluent properties, with an
18F recovery of 98.8% (entry 3). The same elution in the presence of tri-(
t-BuOH)A resulted in a lower
18F recovery, similar to that in the absence of a PTC (entries 1 and 2). The concentration of TBMA-I both lower and higher than 39 µmol, did not significantly affect the efficiency of [
18F]fluoride elution;
18F-recoveries of greater than 94% were obtained (
Table 1, entries 4 and 5).
Entries 6–8 in
Table 1 show similar elution efficiencies to the TBMA-I salt in protic solvents, such as MeOH and
tert-BuOH, without usage of the aforementioned potassium-carbonate-containing elution solvent, producing adequate
18F-recoveries of 98% (MeOH) and 70% (
tert-BuOH), respectively.
The ability of TBMA-I to facilitate nucleophilic
18F-fluorinations in CH
3CN was assessed using [
18F]fluoropropyl tosylate (
[18F]2a) as a model product (
Table 2). TBMA-[
18F]fluoride was eluted from the QMA cartridge by using an elution mixture of K
2CO
3 (1 M, 15 µL) and H
2O (185 µL) in CH
3CN (800 µL), then azeotropically dried by applying anhydrous CH
3CN (3 × 500 µL) in a gas flow at 85 °C. A solution of precursor 1,3-ditosylpropane (
1, 6.0 mg) in anhydrous organic solvent (500 µL) was added into the reaction vial and heated at 85 °C over 20 min, and the radiochemical yield (RCY) was analyzed by radio-TLC at specific time points.
Entry 1 of
Table 2 was performed using tri-(
tert-BuOH)A, which is known to be a good promoter for fluorinations with alkali metal fluoride salts [
18]. However, as mentioned above, the elution efficacy was poor using pure tri-(
tert-BuOH)A, such that the remaining [
18F]fluoride on the cartridge could be eluted using additional elution mixture of K
2CO
3 (1 M, 100 µL) in H
2O (400 µL). Subsequently, the solvent was azeotropically dried using anhydrous CH
3CN and a solution of precursor 1 containing additional tri-(
tert-BuOH)A in CH
3CN was added, but no reaction was observed. Conversely, the
18F-fluorination applying the conventional potassium carbonate/Kryptofix
®2.2.2 system showed 65% RCY for the desired
[18F]2a, along with a significant amount of the alcohol
[18F]2b (29%) (entry 2). Similarly, the
18F substitution employing tetraethylammonium bicarbonate (TEAB) as PTC resulted in a 60% RCY of
[18F]2a and 7% of the alcohol
[18F]2b after a 20 min reaction time (entry 3). Very interestingly, the selective formation of
[18F]2a was observed when the reaction was performed using TBMA-I in CH
3CN solvent (entry 4). The same reaction applying a combination of both solvents
tert-BuOH and CH
3CN in a 1:4 ratio and a total volume of 500 µL offered an RCY of 50% for the desired product
[18F]2a. Surprisingly, the formation of the hydrolytic byproduct
[18F]2b was suppressed. Changing the solvent ratio of
tert-BuOH and MeCN to 1:9, we found that the RCY increased to 57% of
[18F]2a as a single product (
Table 2, entry 6). Notably, the reaction resulted in the best conversion and chemoselectivity compared to the conventional Kryptofix
®2.2.2-assisted reaction (
Table 2, compare entry 6 with entries 2 and 3). Further evaluation of polar solvents (DMSO, DMF, and THF) indicated poor RCY (entries 7 and 8). In an attempt to find a protic solvent with an effect on the reactivity of TBMA-I,
18F fluorination was performed in
iso-propanol, achieving a decreased RCY of 25% after 20 min. The similar reaction in
tert-BuOH was attempted, but the precursor was insoluble.
These results showed that TBMA-I allows adequate recovery of [18F]fluoride from the QMA cartridge using the classical aqueous solution mixture and significant PTC activity in radiofluorinations.
Organic PTCs were reported for the elution of [
18F]fluoride using MeOH as a solvent [
22]. Importantly, TBMA-I has an excellent solubility in MeOH and could be used for the elution of [
18F]fluoride without employing an additional base. Methanolic TBMA-I solutions showed a good ability to elute [
18F]fluoride (
Table 1). Furthermore, MeOH could be evaporated easily below 100 °C. These conditions are time-saving as they allow skipping azeotropic drying, which is needed after the elution under classical aqueous conditions.
Thus, we studied the reactivity of eluted [
18F]fluoride using TBMA-I in MeOH in the absence of water and potassium salt bases.
Table 3 shows the results of the radiofluorination of
1 using [
18F]fluoride, which was eluted with a solution of methanolic TBMA-I (10.4 µmol, entry 7,
Table 1), in various reaction solvent systems. Entry 1 in
Table 3 was performed in CH
3CN (4.5 mL) with
tert-BuOH (0.5 mL) as a co-solvent when
[18F]2a was afforded with 40% RCY. Varying the solvent to
t-BuOH and to CH
3CN ratio to 1:4 and 4:1, respectively, did not increase the RCY of
[18F]2a (entries 3 and 4). We speculate that the basicity of TBMA-I is less than that of other ammonium PTCs due to the presence of three
tert-OH moieties. Therefore, the reaction was also performed under the addition of 15 µL aqueous K
2CO
3 (1 M), but hardly any radiolabeled product was observed (
Table 3, entry 4). To determine the solvent effect, the reaction was also carried out in pure CH
3CN without any co-solvent, and only 14% RCY was observed for
[18F]2a (
Table 3, entry 5). The results suggest that the evaporation of MeOH before addition of the precursor is not sufficient, as the presence of traces of water might affect the [
18F]fluoride reactivity. However, the elution process and the reaction conditions resulted in a maximum RCY of 40% (
Table 3, entry 1), compared to the conventional
tert-BuOH/MeCN conditions with an RCY of 57% (
Table 2, entry 6), but the benefits of a simple and time-saving procedure and omitting the azeotropic drying step may offer great potential for its use in automated radiosynthesis modules.
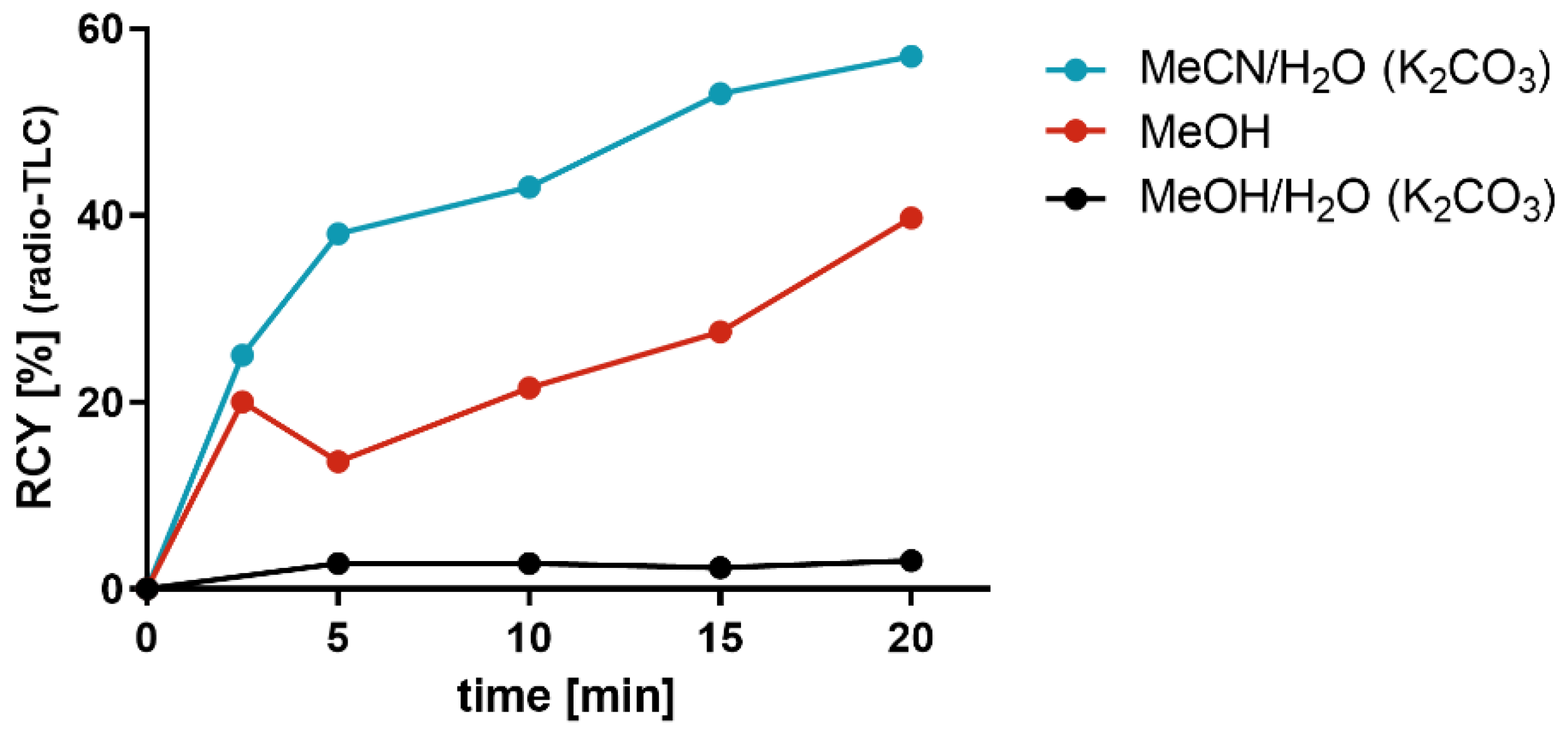
Figure 1 illustrates the comparative results of the RCY for the radiosynthesis of
[18F]2a when using [
18F]fluoride that was obtained by different elution conditions in a time-dependent manner. The reactivity of [
18F]fluoride obtained by the classical elution conditions, i.e., CH
3CN/H
2O containing K
2CO
3, was good, as it provided 57% RCY of
[18F]2a compared to [
18F]fluoride eluted with MeOH or MeOH/H
2O containing K
2CO
3, which produced an RCY of 40% and 3%, respectively. This comparative study more clearly shows the differences in RCY between the reaction starting from [
18F]fluoride after elution with CH
3CN/H
2O and subsequent azeotropic drying and the same reaction starting from [
18F]fluoride after elution with MeOH without azeotropic drying. As mentioned above, it is tempting to speculate that the decrease in RCY from 57% to 40% could be due to the remaining water content in the reaction mixture. Considering the important advantage of TBMA-I that no azeotropic drying is needed to obtain an adequate RCY, we turned our attention to study aromatic radiofluorinations using TBMA-I as the PTC.
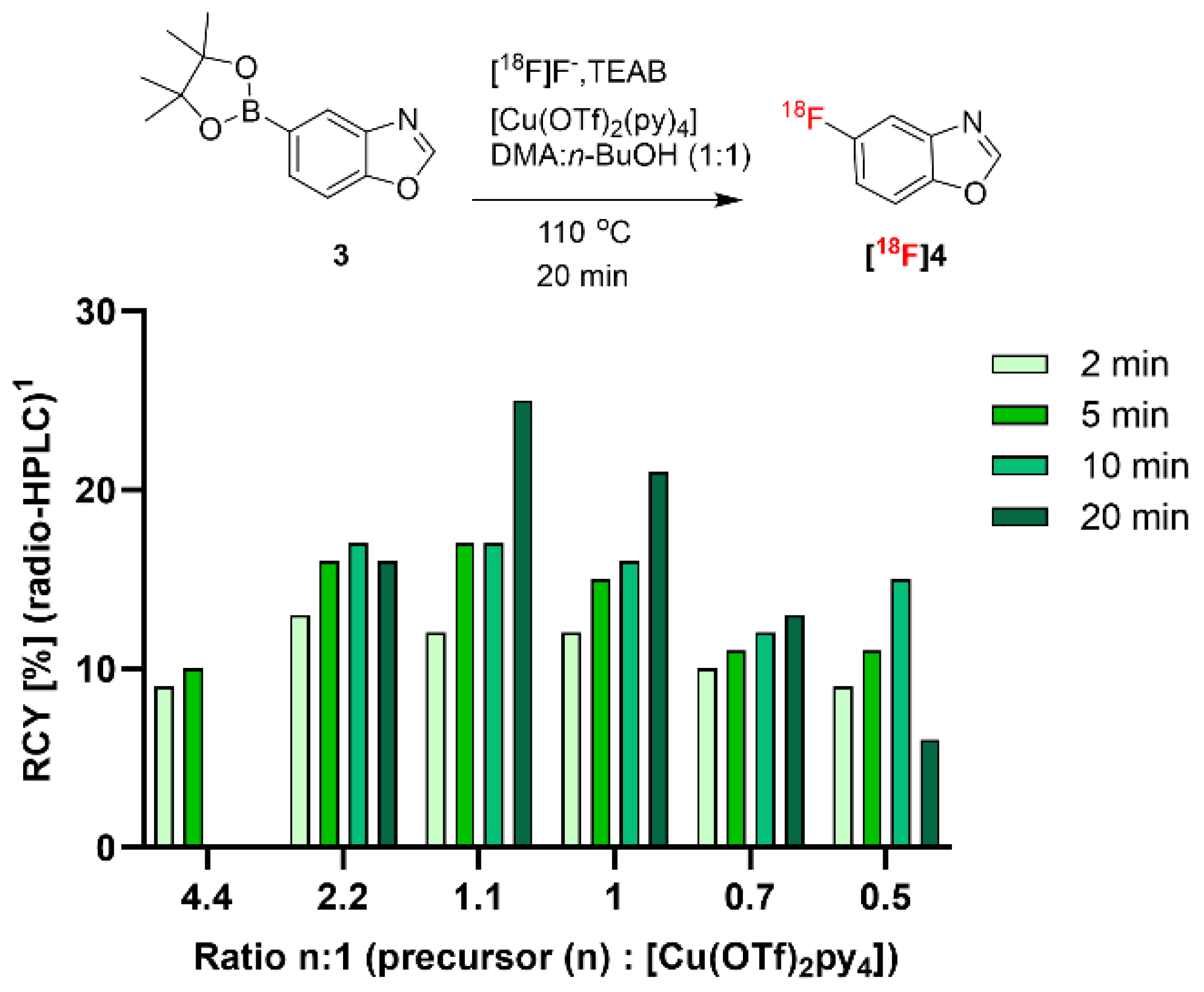
In a first step, starting from the reported reaction conditions of the copper-mediated aromatic
18F substitution of aryl boronic esters utilizing protic solvents in combination with polar aprotic solvents for optimal RCY [
15], we adopted and optimized the precursor-to-[Cu(OTf)
2py
4] ratio for the
18F-labeling of 5-benzoxazole boronic acid pinacol ester (
3), which served as precursor for the radiosynthesis of 5-[
18F]fluorobenzoxazole (
[18F]4;
Figure 2).
Figure 2 shows the optimization approach on the
3-to-[Cu(OTf)
2py
4] ratio, revealing that the reported ratio of 2.2 equivalents provided 16% RCY of
[18F]4 after 20 min. Interestingly, in the case of precursor
3, the ratio of 1.1:1 provided a maximum RCY of 25%. When increasing the ratio to 4.4, the product could not be detected after 10 min and a further decrease in the precursor amount compared to [Cu(OTf)
2py
4] did not improve the RCY. The optimal reaction condition employed a total reaction volume of 1.2 mL (DMA/
n-BuOH, 1:1), and precursor
3 and [Cu(OTf)
2py
4] were added in concentrations of 25 mM and 22.5 mM, respectively.
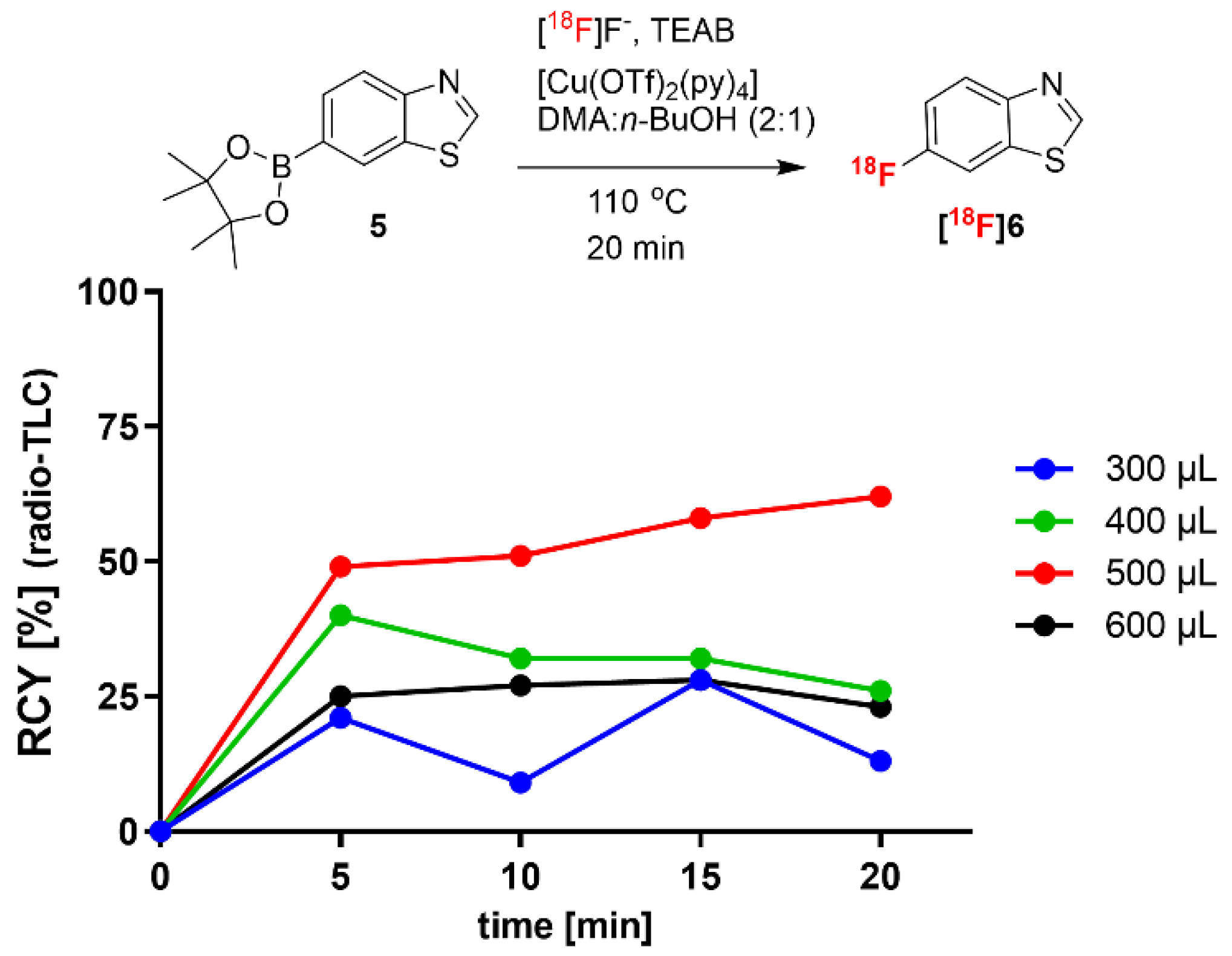
In a second step, we aimed to minimize the reaction volume, as a reduced total amount of boronic acid ester precursor could simplify the purification of
18F-labeled compounds. In
Figure 3, the effect of a reduced reaction volume on the
18F-fluorination of 6-benzothiazole boronic acid pinacol ester (
5) is shown. Elution of [
18F]fluoride with TEAB in MeOH allowed a reduction in the reaction volume to 300–600 µL, while the concentration of precursor
5 and [Cu(OTf)
2py
4] was kept constant (32.2 mM and 29.2 mM, respectively). The highest RCY achieved was 62% for 6-[
18F]fluorobenzothiazole (
[18F]6) after 20 min in a reaction volume of 500 µL, whereas further decrease or increase led to reduced RCYs. The found optimal conditions of the precursor-to-Cu-catalyst ratio (1.1:1), i.e., 16.1 µmol precursor and 14.6 µmol of [Cu(OTf)
2py
4], and a reaction volume of 500 µL, were then applied in the following experiments using TBMA-I as PTC.
Table 4 summarizes the direct comparison of TEAB and TBMA-I as the PTC in nucleophilic S
NAr, applying
18F-fluorination on 5-indoleboronic acid pinacol ester (
7) as a model compound. We evaluated the reactivity of
7 using a total volume of 500 µL of DMA/
n-BuOH (2:1) in the presence of TBMA as PTC, producing the desired
18F-fluorinated indole
[18F]8 with a 55–60% RCY. To compare our results, we performed the same reaction using TEAB as the PTC, which resulted in 10% more RCY of
[18F]8 than using TBMA-I as the PTC.
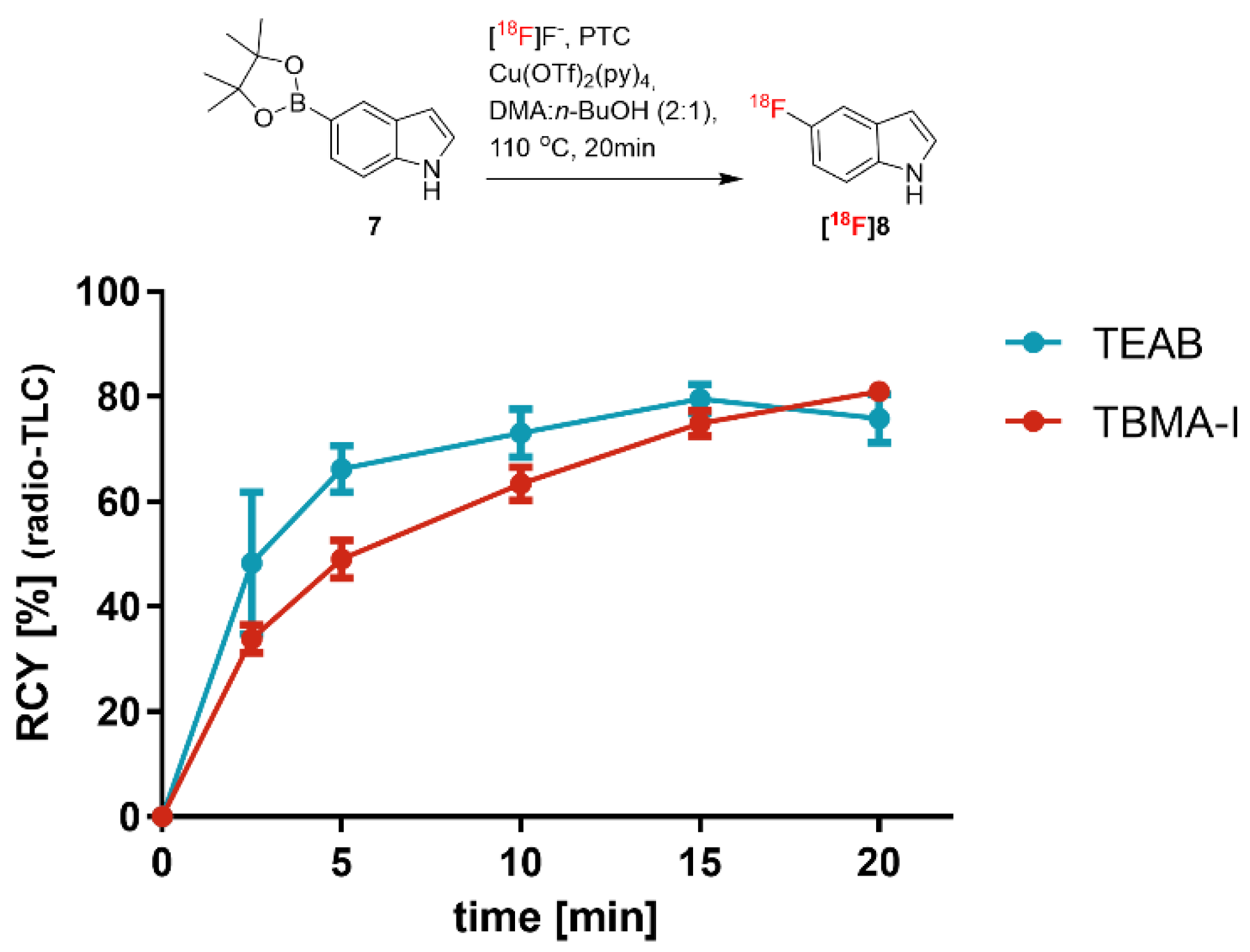
Figure 4 depicts the time dependency of the RCY of
[18F]8 in the presence of either TEAB or TBMA-I. The aromatic
18F substitution using TBMA was initially (up to the reaction time of 2.5 min) about four times slower compared to the reaction using TEAB. The time course of the reaction clearly indicates that the TBMA-
18F intermediate is more stable than the TEAB analog and less reactive, but reaches a comparable RCY of about 80% at 20 min. We suggest that hydrogen bonding between [
18F]fluoride and
tert-OH of TBMA tightly coordinates the [
18F]fluoride. This could be the reason for a relatively slow S
NAr reaction due to the reduced availability of free fluoride anions in the reaction mixture.
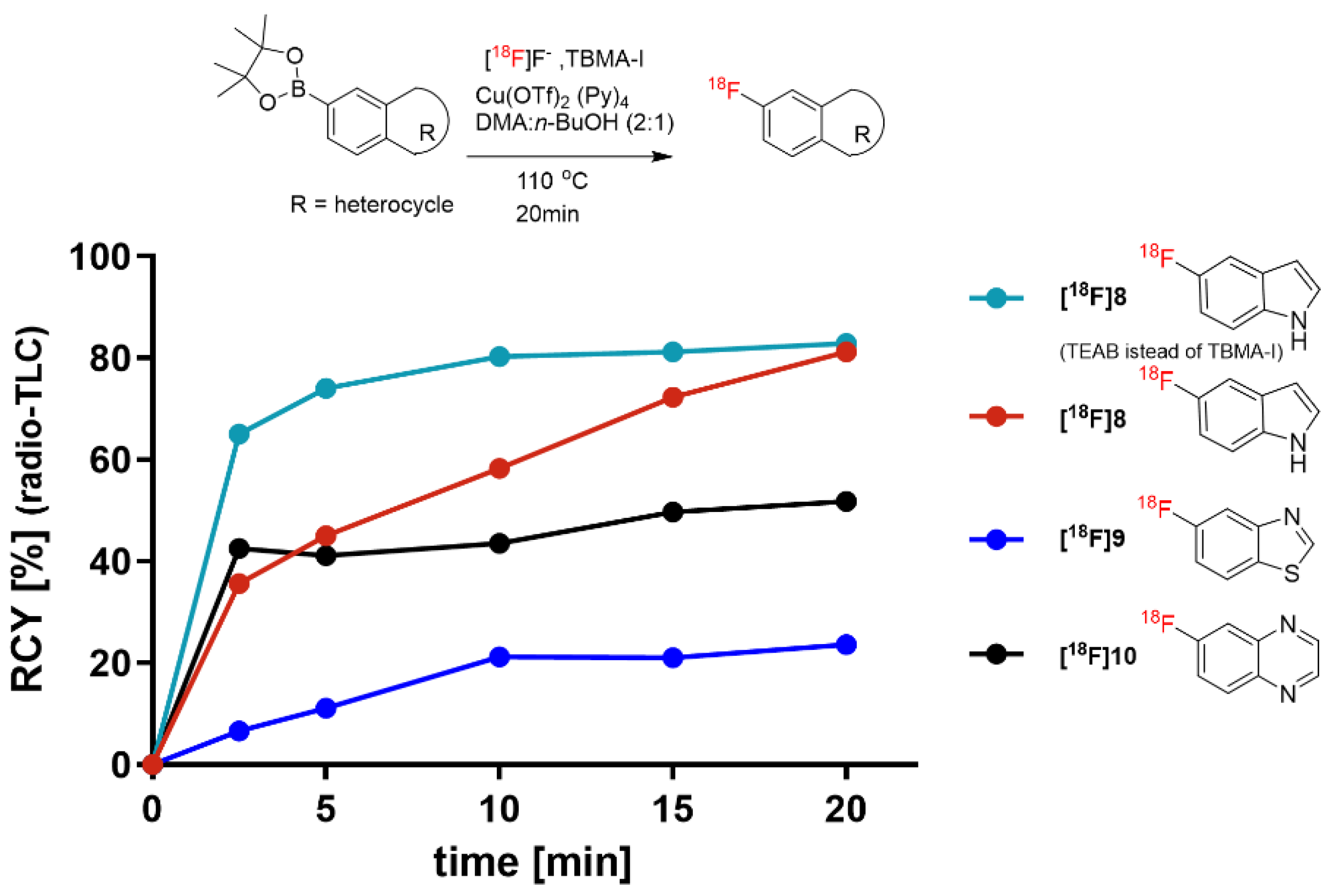
The applicability of TBMA-I as the PTC was further studied with various aryl pinacol boronic esters.
Figure 5 shows the reactivity of TBMA-
18F with commercially available boronic acid pinacol ester derivatives of 5-indole, 6-quinoxaline, and 5-benzothiazole. The found optimal reaction conditions were applied, affording the highest RCY of 80% for the indole
[18F]8. The reactivity of TBMA-
18F with 6-quinoxaline boronic acid pinacol ester and 5-benzothiazole boronic acid pinacol ester was lower, providing 57% and 29% RCY, respectively, of the corresponding
18F-fluorinated products after 20 min.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

