
A Multicomponent Protocol for the Synthesis of Highly Functionalized γ-Lactam Derivatives and Their Applications as Antiproliferative Agents

 ,
,  , ,
, ,  , ,
, ,  ,
,  and
and 
Abstract
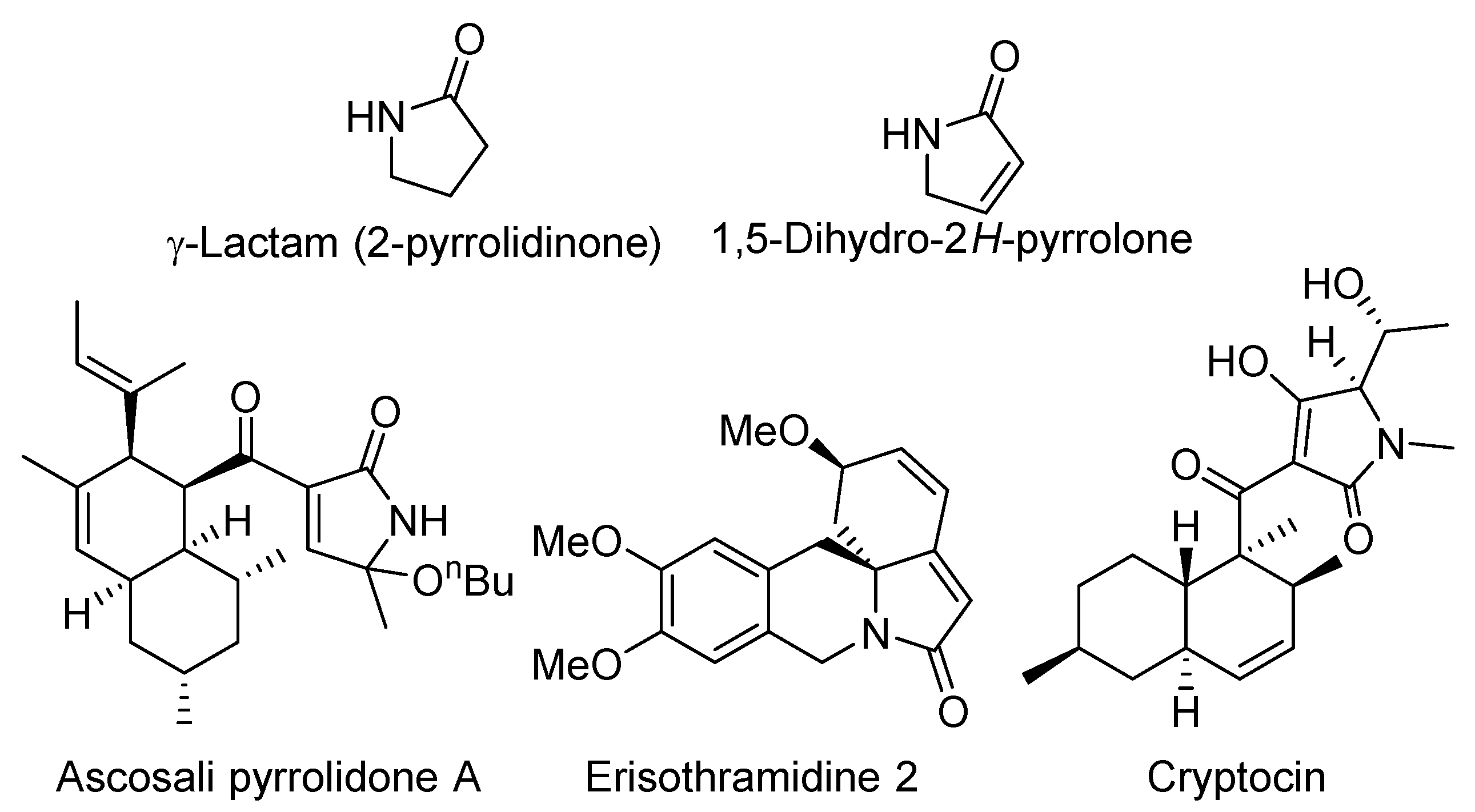
1. Introduction
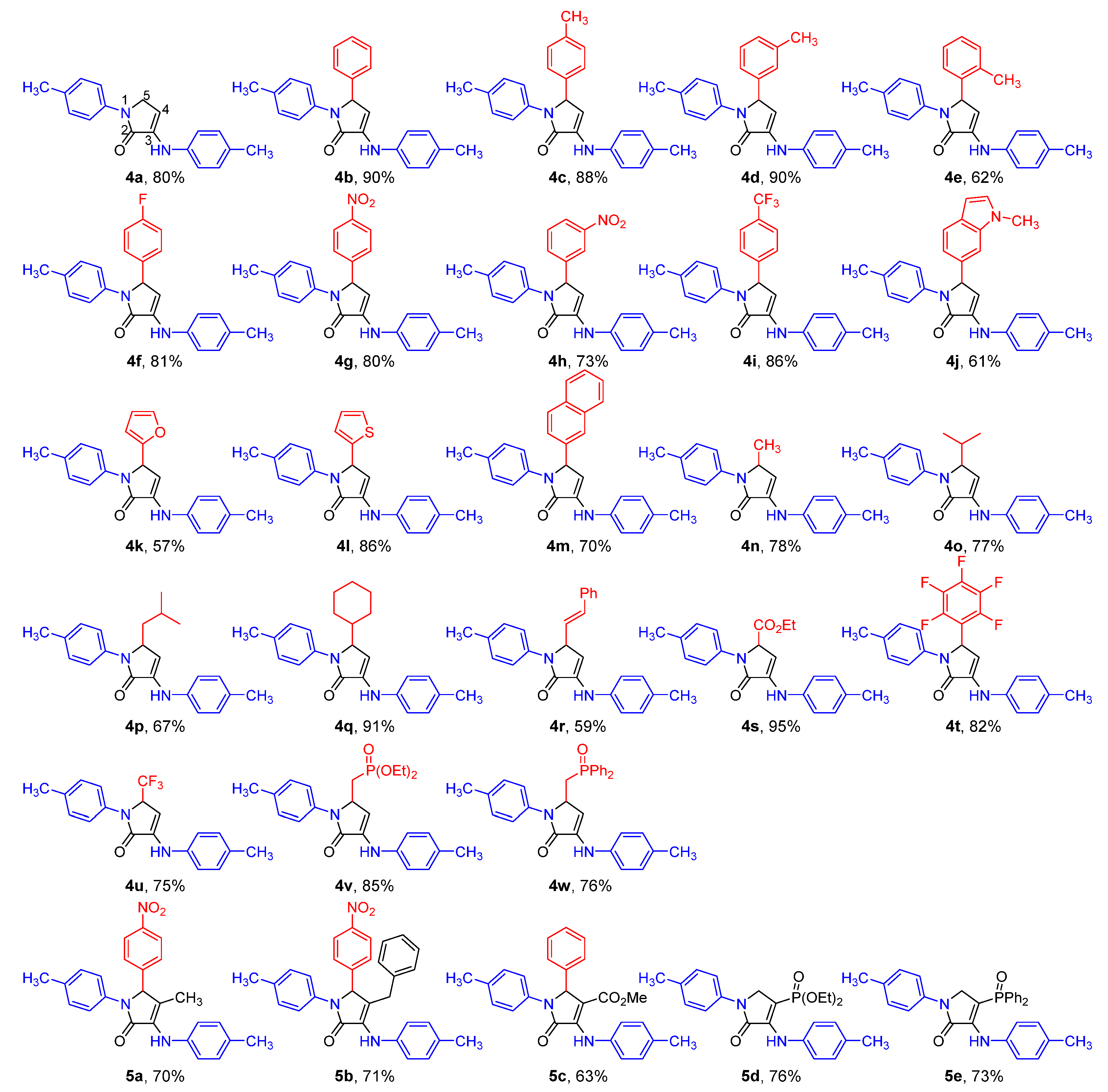
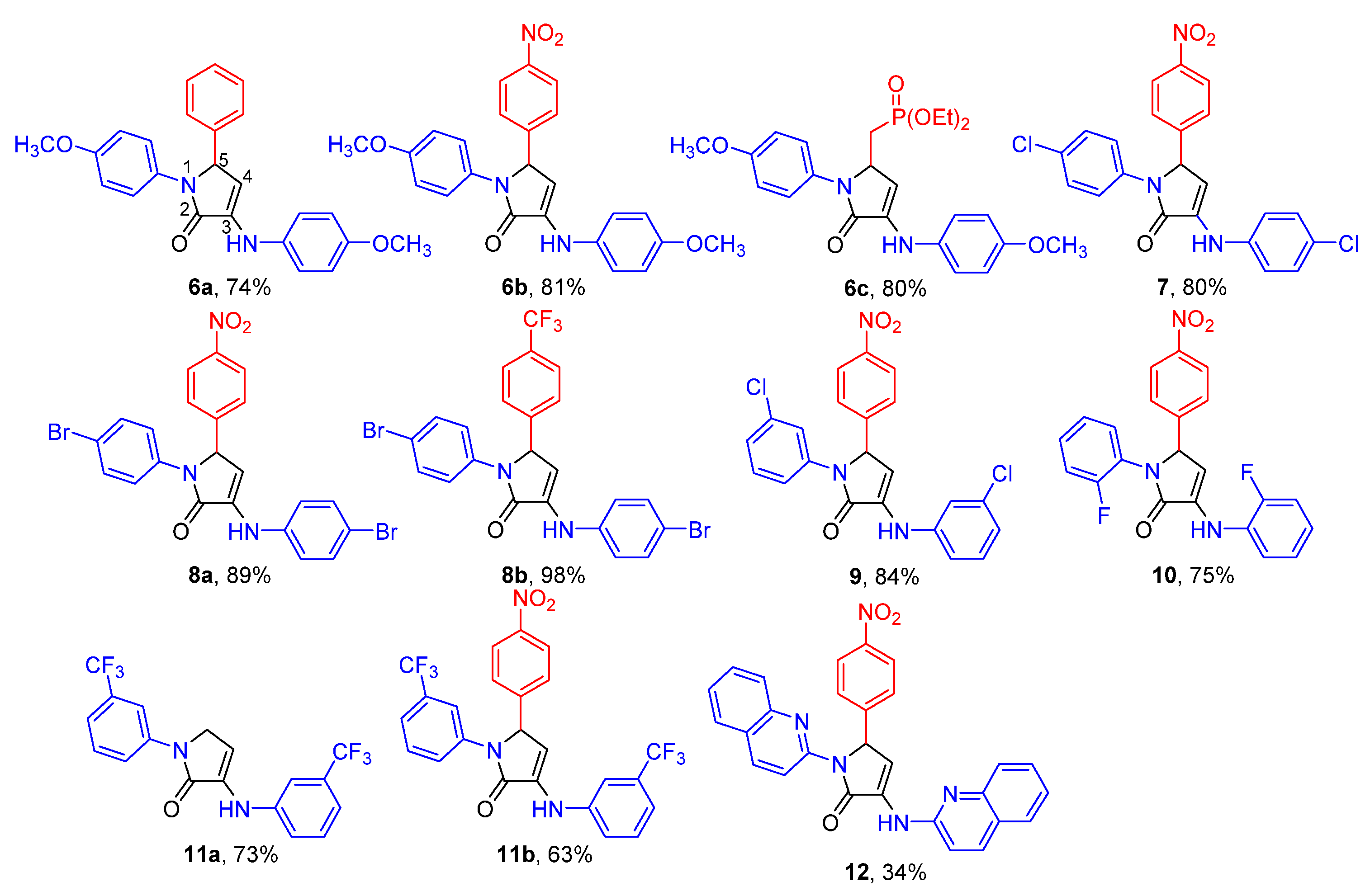
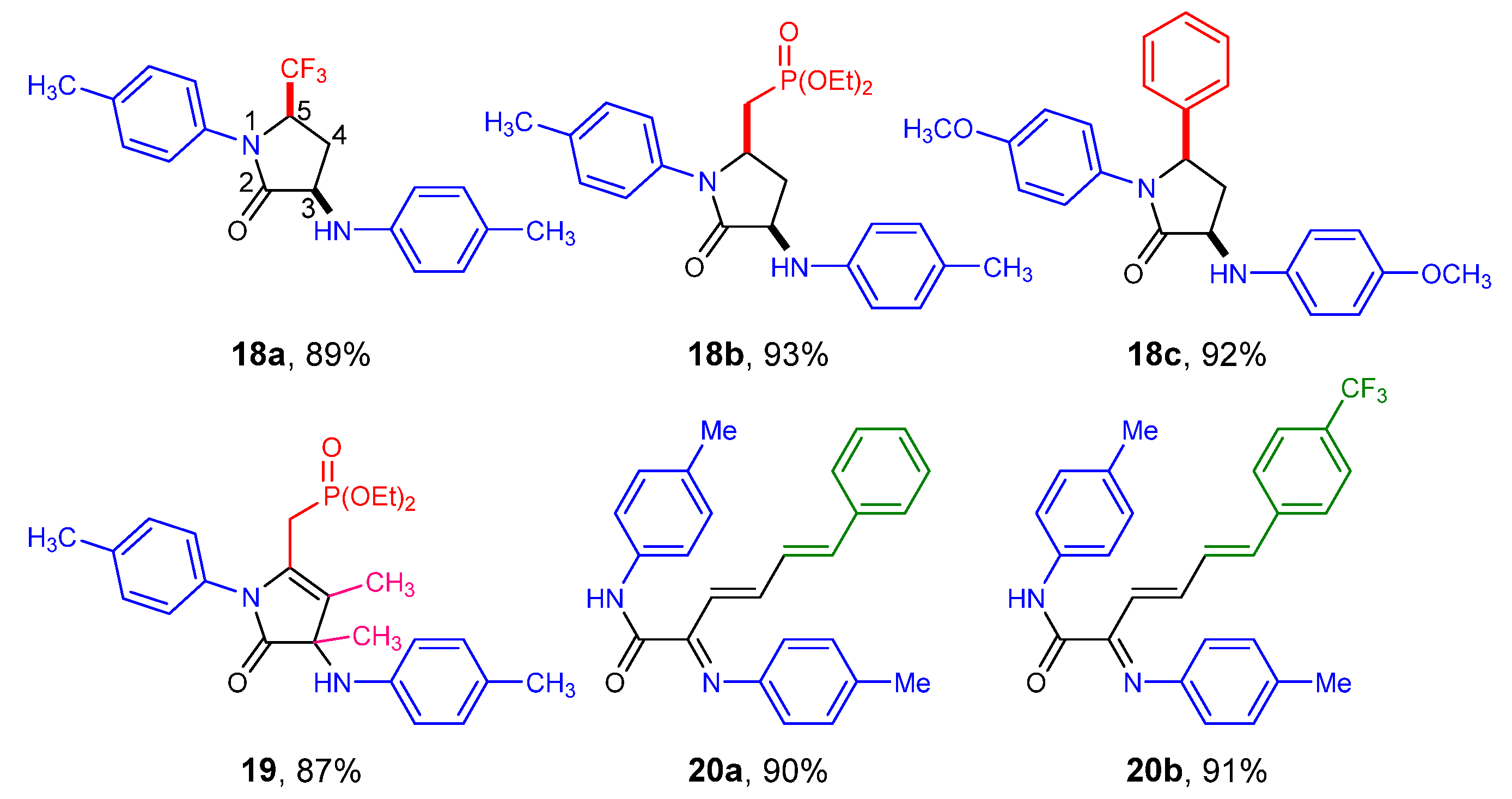
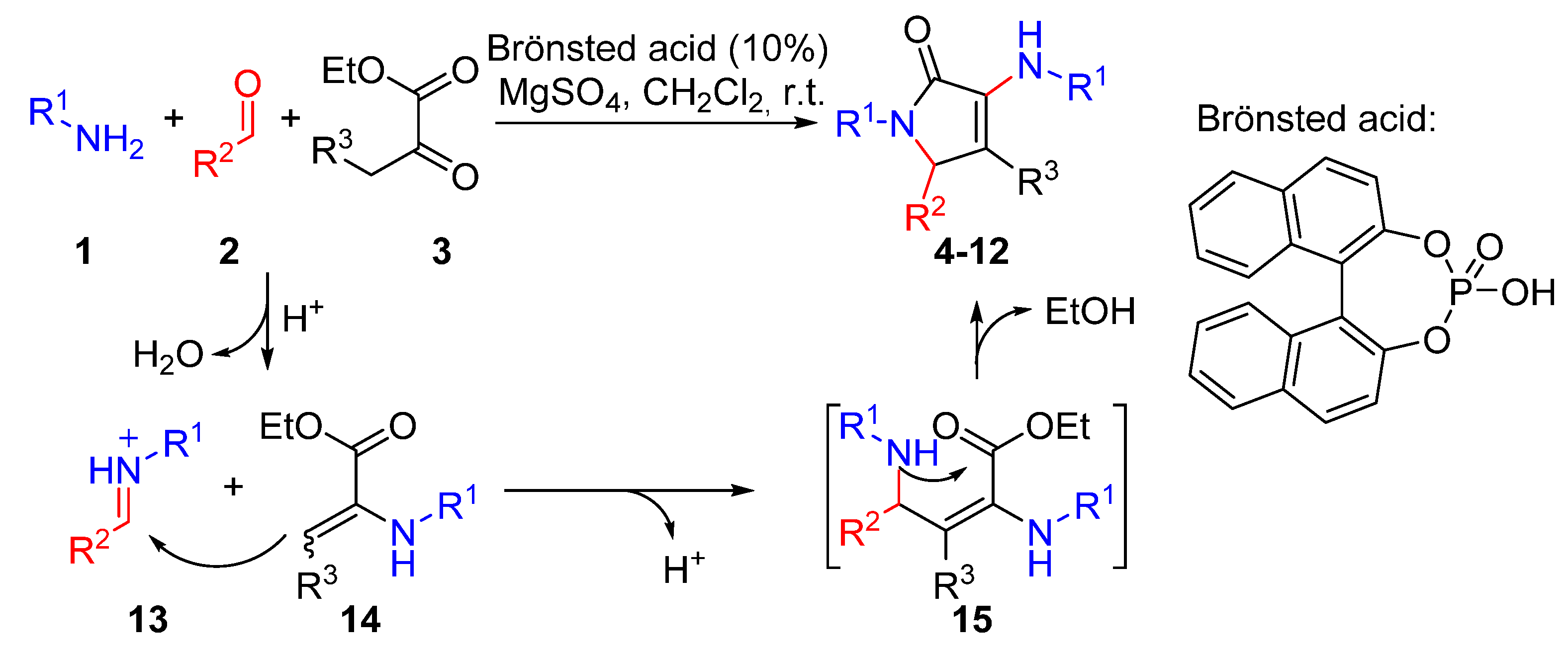
2. Results and Discussion
2.1. Chemistry
2.2. Biological Results
3. Material and Methods
3.1. Chemistry
3.1.1. General Experimental Information
3.1.2. Compounds Purity Analysis
3.1.3. Representative Experimental Procedures and Characterization Data for Compounds 4–12 and 16–21
Representative Procedure for the Multicomponent Reaction of Amines 1, Aldehydes 2, and Pyruvate Derivatives 3
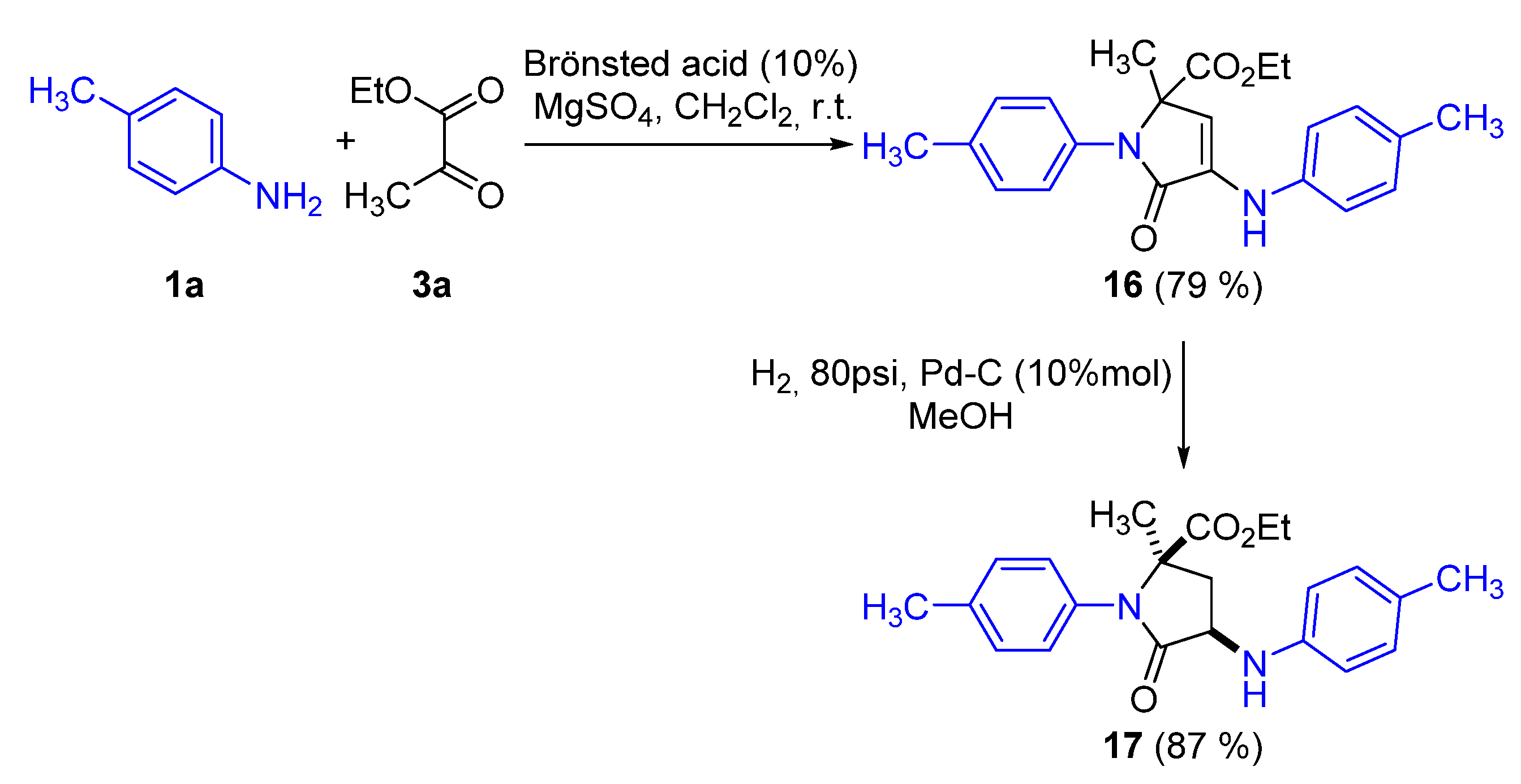
Synthesis of Ethyl 2-methyl-5-oxo-1-(p-tolyl)-4-(p-tolylamino)-2,5-dihydro-1H-pyrrole-2-carboxylate (16)
Representative Procedure for the Hydrogenation of γ-Lactams 4, 6, and 16
Synthesis of Diethyl ((3,4-dimethyl-5-oxo-1-(p-tolyl)-4-(p-tolylamino)-4,5-dihydro-1H-pyrrol-2-yl)methyl)phosphonate (19)
Representative Procedure for the Horner-Wadsworth-Emmons Reactions of γ-Lactam 4v
Synthesis of (Z)-5-phenyl-1-(p-tolyl)-3-(p-tolylimino)-1,3-dihydro-2H-pyrrol-2-one (21)
3.2. Biology
3.2.1. Materials
3.2.2. Cell Culture
3.2.3. Cytotoxicity Assays
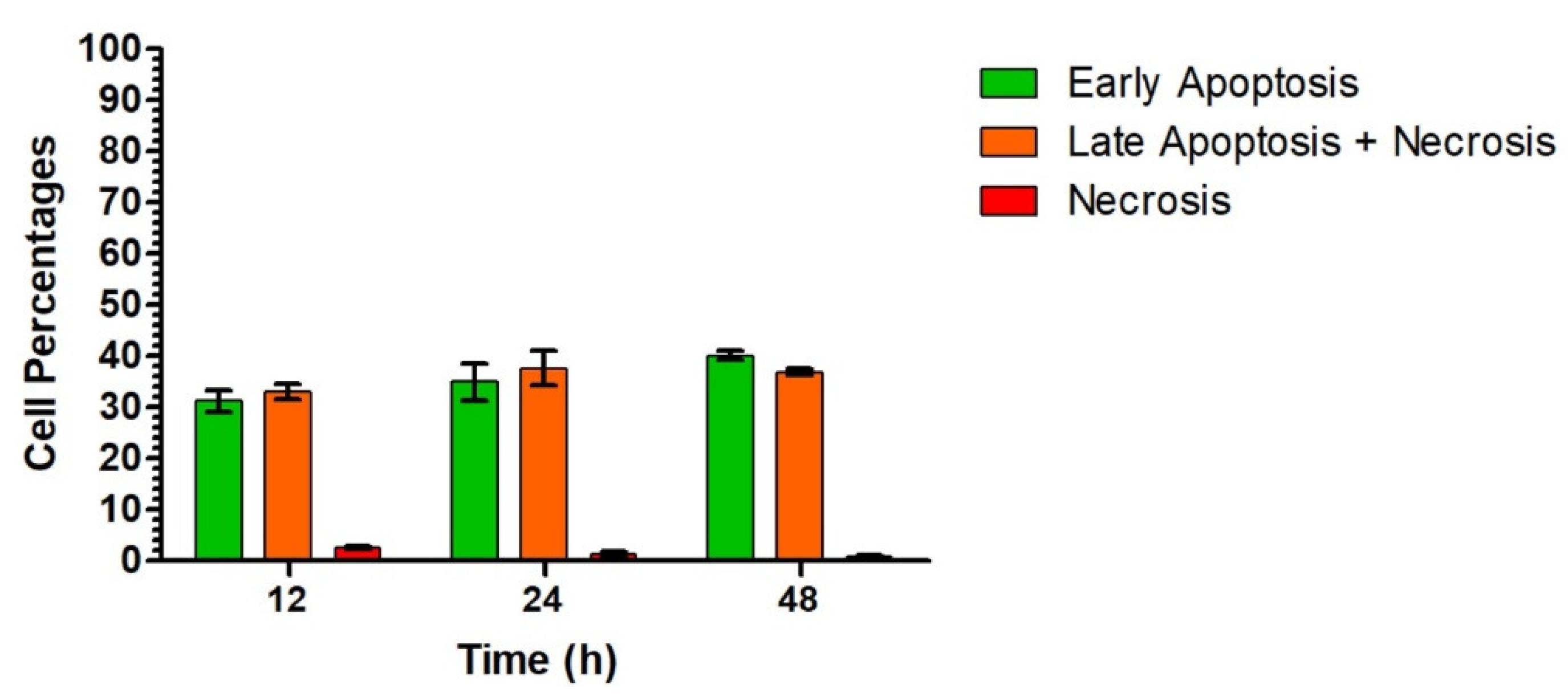
3.2.4. Evaluation of Cytotoxicity Mechanisms
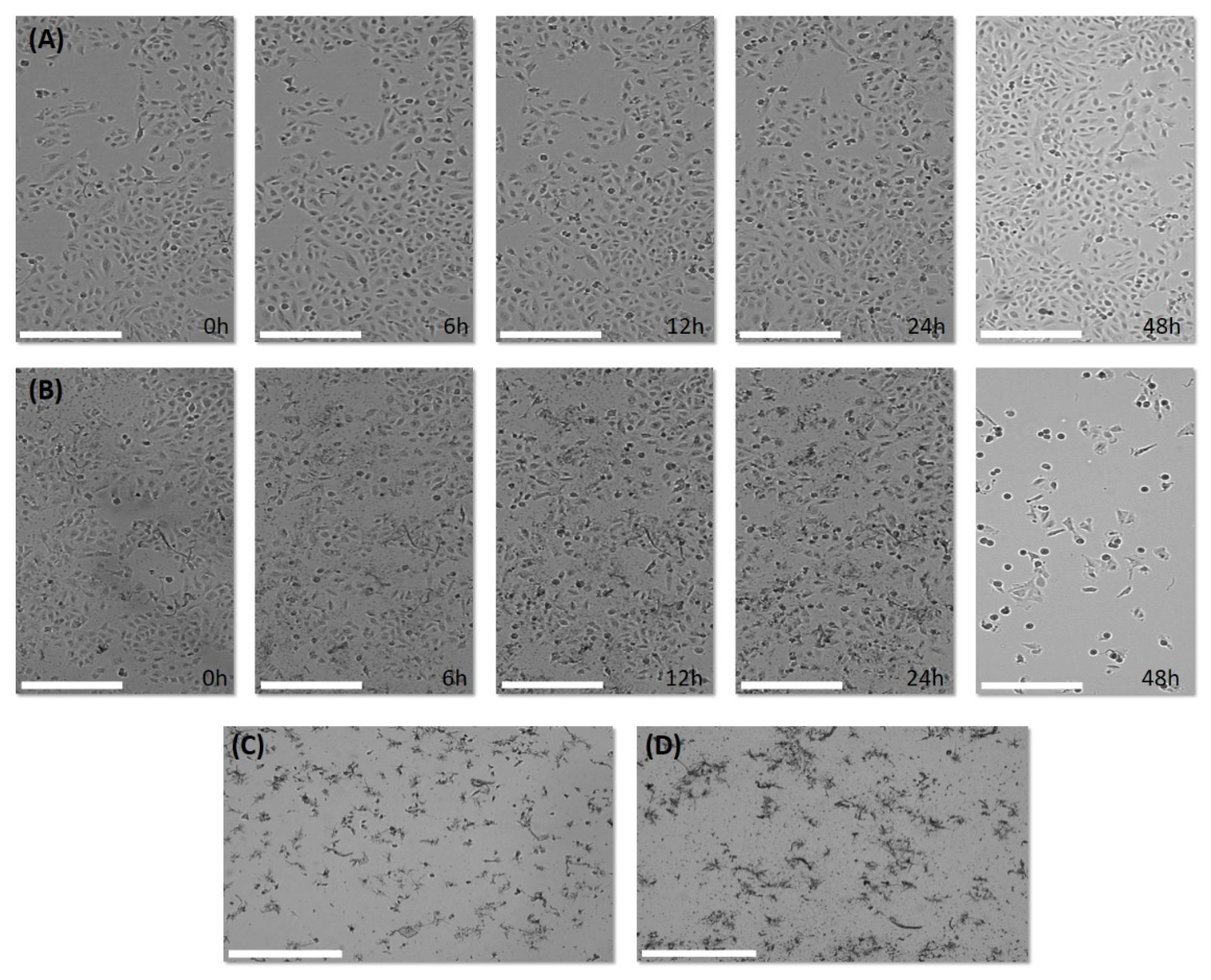
3.2.5. Visualization of Cell Growth and Morphology
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Dickens, E.; Ahmed, S. Principles of cancer treatment by chemotherapy. Surgery 2018, 36, 134–138. [Google Scholar] [CrossRef]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef] [PubMed]
- Caruano, J.; Muccioli, G.G.; Robiette, R. Biologically active γ-lactams: Synthesis and natural sources. Org. Biomol. Chem. 2016, 14, 10134–10156. [Google Scholar] [CrossRef] [PubMed]
- Schümann, J.; Hertweck, C. Molecular Basis of Cytochalasan Biosynthesis in Fungi: Gene Cluster Analysis and Evidence for the Involvement of a PKS-NRPS Hybrid Synthase by RNA Silencing. J. Am. Chem. Soc. 2007, 129, 9564–9565. [Google Scholar] [CrossRef]
- Albrecht, D.; Basler, B.; Bach, T. Preparation and Intramolecular [2+2]-Photocycloaddition of 1,5-Dihydropyrrol-2-ones and 5,6-Dihydro-1H-pyridin-2-ones with C-, N-, and O-linked Alkenyl Side Chains at the 4-Position. J. Org. Chem. 2008, 73, 2345–2356. [Google Scholar] [CrossRef]
- Chatzimpaloglou, A.; Kolosov, M.; Eckols, T.K.; Tweardy, D.J.; Sarli, V. Synthetic and Biological Studies of Phaeosphaerides. J. Org. Chem. 2014, 79, 4043–4054. [Google Scholar] [CrossRef]
- Melekhina, V.G.; Komogortsev, A.N.; Lichitsky, B.V.; Mityanov, V.S.; Fakhrutdinov, A.N.; Dudinov, A.A.; Migulin, V.A.; Nelyubina, Y.V.; Melnikova, E.K.; Krayushkin, M.M. One-pot synthesis of substituted pyrrolo [3,4-b]pyridine-4,5-diones based on the reaction of N-(1-(4-hydroxy-6-methyl-2-oxo-2H-pyran-3-yl)-2-oxo-2-arylethyl)acetamide with amines. Beilstein J. Org. Chem. 2019, 15, 2840–2846. [Google Scholar] [CrossRef]
- Peifer, C.; Selig, R.; Kinkel, K.; Ott, D.; Totzke, F.; Schaechtele, C.; Heidenreich, R.; Roecken, M.; Schollmeyer, D.; Laufer, S. Design, Synthesis and Biological Evaluation of Novel 3-Aryl-4-(1H-indole-3yl)-1,5-dihydro-2H-pyrrole-2-ones as Vascular Endothelial Growth Factor Receptor (VEGF-R) Inhibitors. J. Med. Chem. 2008, 51, 3814–3824. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Wang, P.; Fu, W.; Wan, X.; Zhou, L.; Chu, Y.; Ye, D. Rational design of 2-pyrrolinones as inhibitors of HIV-1 integrase. Bioorg. Med. Chem. Lett. 2011, 21, 6724–6727. [Google Scholar] [CrossRef]
- Kirpotina, L.N.; Schepetkin, I.A.; Khlebnikov, A.I.; Ruban, O.I.; Ge, Y.; Ye, R.D.; Kominsky, D.J.; Quinn, M.T. 4-Aroyl-3-hydroxy-5-phenyl-1H-pyrrol-2(5H)-ones as N-formyl peptide receptor 1 (FPR1) antagonists. Biochem. Pharmacol. 2017, 142, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Osterhage, C.; Kaminski, R.; König, G.M.; Wright, A.D. Ascosalipyrrolidinone A, an Antimicrobial Alkaloid, from the Obligate Marine Fungus Ascochyta Salicorniae. J. Org. Chem. 2000, 65, 6412–6417. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Strobel, G.; Harper, J.; Lobkovsky, E.; Clardy, J. Cryptocin, a Potent Tetramic Acid Antimycotic from the Endophytic Fungus Cryptosporiopsis cf. quercina. Org. Lett. 2000, 2, 767–770. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Tanaka, T.; Etoh, H. Two New Erythrinan Alkaloids from Erythrina x bidwillii. Heterocycles 1999, 51, 2759–2764. [Google Scholar] [CrossRef]
- Wink, M. Evolution of secondary metabolites in legumes (Fabaceae). S. Afr. J. Bot. 2013, 89, 164–175. [Google Scholar] [CrossRef]
- Zhuang, C.; Miao, Z.; Zhu, L.; Dong, G.; Guo, Z.; Wang, S.; Zhang, Y.; Wu, Y.; Yao, J.; Sheng, C.; et al. Discovery, Synthesis, and Biological Evaluation of Orally Active Pyrrolidone Derivatives as Novel Inhibitors of p53–MDM2 Protein–Protein Interaction. J. Med. Chem. 2012, 55, 9630–9642. [Google Scholar] [CrossRef]
- Huang, W.; Dong, Z.; Wang, F.; Peng, H.; Liu, J.-Y.; Zhang, J.-T. A Small Molecule Compound Targeting STAT3 DNA-Binding Domain Inhibits Cancer Cell Proliferation, Migration, and Invasion. ACS Chem. Biol. 2014, 9, 1188–1196. [Google Scholar] [CrossRef]
- Koz’minykh, V.O.; Igidov, N.M.; Zykova, S.S.; Kolla, V.E.; Shuklina, N.S.; Odegova, T. Synthesis and pharmacological activity of 3-hydroxy-1, 5-diaryl-4-pivaloyl-2, 5-dihydro-2-pyrrolone. Pharm. Chem. J. 2002, 36, 188–191. [Google Scholar] [CrossRef]
- Tarnavsky, S.S.; Dubinina, G.G.; Golovach, S.M.; Yarmoluk, S.M. Antitumor activity among derivatives of the 3-chloro-4-(3-hydroxyanilino)-2,5-dihydropyrrole-2,5-dione. Biopolym. Cell 2003, 19, 548–552. [Google Scholar] [CrossRef][Green Version]
- Sánchez, C.; Zhu, L.; Braña, A.F.; Salas, A.P.; Rohr, J.; Méndez, C.; Salas, J.A. Combinatorial biosynthesis of antitumor indolocarbazole compounds. Proc. Natl. Acad. Sci. USA 2005, 102, 461–466. [Google Scholar] [CrossRef]
- Cao, W.; Wang, Z.; Han, X.; Liu, J.; Wang, W. In vitro cytotoxicity screening to identify novel anti-osteosarcoma therapeutics targeting pyruvate dehydrogenase kinase 2. Bioorg. Med. Chem. Lett. 2019, 29, 126665. [Google Scholar] [CrossRef] [PubMed]
- Joksimović, N.; Petronijević, J.; Janković, N.; Baskić, D.; Popović, S.; Todorović, D.; Matić, S.; Bogdanović, G.A.; Vraneš, M.; Tot, A.; et al. Synthesis, characterization, anticancer evaluation and mechanisms of cytotoxic activity of novel 3-hydroxy-3-pyrrolin-2-ones bearing thenoyl fragment: DNA, BSA interactions and molecular docking study. Bioorg. Chem. 2019, 88, 102954–102968. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Huang, C.; Liao, H.; Zhang, H.; Wu, S.; Zhu, Q.; Zhou, Z.-Z. Discovery of Dihydropyrrol-2-ones as Novel G0/G1-Phase Arresting Agents Inducing Apoptosis. ACS Omega 2019, 4, 17556–17560. [Google Scholar] [CrossRef]
- Palacios, F.; Vicario, J.; Aparicio, D. An efficient synthesis of achiral and chiral cyclic dehydro-α-amino acid derivatives through nucleophilic addition of Amines to β,γ-unsaturated α-keto esters. Eur. J. Org. Chem. 2006, 2843–2850. [Google Scholar] [CrossRef]
- Li, X.; Deng, H.; Luo, S.; Cheng, J.-P. Organocatalytic Three-Component Reactions of Pyruvate, Aldehyde and Aniline by Hydrogen-Bonding Catalysts. Eur. J. Org. Chem. 2008, 4350–4356. [Google Scholar] [CrossRef]
- Ghashang, M. 1-Butyl-1-methylpyrrolidinium hydrogen sulfate-promoted preparation of 1,5-diaryl-3-(arylamino)-1H-pyrrol-2(5H)-one derivatives. Res. Chem. Intermed. 2013, 39, 2187–2195. [Google Scholar] [CrossRef]
- del Corte, X.; Maestro, A.; Vicario, J.; Martínez de Marigorta, E.; Palacios, F. Brönsted-Acid-Catalyzed Asymmetric Three-Component Reaction of Amines, Aldehydes, and Pyruvate Derivatives. Enantioselective Synthesis of Highly Functionalized γ-Lactam Derivatives. Org. Lett. 2018, 20, 317–320. [Google Scholar] [CrossRef]
- Gao, H.; Sun, J.; Yan, C.-G. Synthesis of functionalized 2-pyrrolidinones via domino reactions of arylamines, ethyl glyoxylate and acetylenedicarboxylates. Tetrahedron 2013, 69, 589–594. [Google Scholar] [CrossRef]
- del Corte, X.; Martinez de Marigorta, E.; Palacios, F.; Vicario, J. A Brønsted Acid-Catalyzed Multicomponent Reaction for the Synthesis of Highly Functionalized γ-Lactam Derivatives. Molecules 2019, 24, 2951. [Google Scholar] [CrossRef]
- Gein, V.L.; Popov, A.V.; Kolla, V.E.; Popova, N.A. Synthesis and biological activity of 1,5-diaryl-3-alkylamino-4-carboxymethyl-2,5-dihydropyrrol-2-ones and 1,5-diaryl-4-carboxymethyl-tetrahydropyrrol-2,3-diones. Pharmazie 1993, 48, 107–109. [Google Scholar]
- Gein, V.L.; Popov, A.V.; Kolla, V.E.; Popova, N.A.; Potemkin, K.D. Synthesis and biological activity of 1,5-diaryl-3-arylamino-4-carboxymethyl-2,5-dihydro-2-pyrrolones and 1,5-diaryl-4-carboxymethyltetrahydropyrrole-2,3-diones. Pharm. Chem. J. 1993, 27, 343–346. [Google Scholar] [CrossRef]
- Khalaf, A.I.; Waigh, R.D.; Drummond, A.J.; Pringle, B.; McGroarty, I.; Skellern, G.G.; Suckling, C.J. Distamycin Analogues with Enhanced Lipophilicity: Synthesis and Antimicrobial Activity. J. Med. Chem. 2004, 47, 2133–2156. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Fang, F.; Li, Y. Synthesis and anti-biofilm activities of dihydro-pyrrol-2-one derivatives on Pseudomonas aeruginosa. Bioorg. Med. Chem. Lett. 2015, 25, 597–601. [Google Scholar] [CrossRef]
- Zhu, Q.; Gao, L.; Chen, Z.; Zheng, S.; Shu, H.; Li, J.; Jiang, H.; Liu, S. A novel class of small-molecule caspase-3 inhibitors prepared by multicomponent reactions. Eur. J. Med. Chem. 2012, 54, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wever, W.J.; Walsh, C.T.; Bowers, A.A. Dithiolopyrrolones: Biosynthesis, synthesis, and activity of a unique class of disulfide-containing antibiotics. Nat. Prod. Rep. 2014, 31, 905–923. [Google Scholar] [CrossRef] [PubMed]
- Knapp, J.M.; Kurth, M.J.; Shaw, J.T.; Younai, A. Diversity-Oriented Synthesis; Trabocchi, A., Ed.; Willey: New York, NY, USA, 2013; pp. 29–57. [Google Scholar]
- Schreiber, S.L. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science 2000, 287, 1964–1969. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.G.; Rybak, T.; Verdelet, T. Multicomponent Hetero-[4 + 2] Cycloaddition/Allylboration Reaction: From Natural Product Synthesis to Drug Discovery. Acc. Chem. Res. 2016, 49, 2489–2500. [Google Scholar] [CrossRef]
- de Moliner, F.; Kielland, N.; Lavilla, R.; Vendrell, M. Modern Synthetic Avenues for the Preparation of Functional Fluorophores. Angew. Chem. Int. Ed. 2017, 56, 3758–3769. [Google Scholar] [CrossRef]
- del Corte, X.; López-Francés, A.; Maestro, A.; Martinez de Marigorta, E.; Palacios, F.; Vicario, J. Brönsted Acid Catalyzed Multicomponent Synthesis of Phosphorated and Fluorinated γ-Lactam Derivatives. J. Org. Chem. 2020, 85, 14369–14383. [Google Scholar] [CrossRef]
- Palacios, F.; Ochoa de Retana, A.M.; Oyarzabal, J.; Pascual, S.; Fernández de Trocóniz, G. Synthesis of Fluoroalkylated β-Aminophosphonates and Pyridines from Primary β-Enamino-phosphonates. J. Org. Chem. 2008, 73, 4568–4574. [Google Scholar] [CrossRef]
- Alonso, C.; González, M.; Fuertes, M.; Rubiales, G.; Ezpeleta, J.M.; Palacios, F. Synthesis of Fluorinated β-Aminophosphonates and γ-Lactams. J. Org. Chem. 2013, 78, 3858–3866. [Google Scholar] [CrossRef]
- Fernández de Trocóniz, G.; Ochoa de Retana, A.M.; Rubiales, G.; Palacios, F. Fluoroalkylated α,β-Unsaturated Imines as Synthons for the Preparation of Fluorinated Triazinane-2,4-diones and Dihydropyrimidin-2(1H)-ones. J. Org. Chem. 2014, 79, 5173–5181. [Google Scholar] [CrossRef]
- Alonso, C.; Martínez de Marigorta, E.; Rubiales, G.; Palacios, F. Carbon Trifluoromethylation Reactions of Hydrocarbon Derivatives and Heteroarenes. Chem. Rev. 2015, 115, 1847–1935. [Google Scholar] [CrossRef]
- Palacios, F.; Alonso, C.; de los Santos, J.M. Synthesis of β-Aminophosphonates and -Phosphinates. Chem. Rev. 2005, 105, 899–932. [Google Scholar] [CrossRef]
- Palacios, F.; Olszewski, T.K.; Vicario, J. Diastereoselective hydrophosphonylation of imines using (R,R)-TADDOL phosphite. Asymmetric synthesis of α-aminophosphonic acid derivatives. Org. Biomol. Chem. 2010, 8, 4255–4258. [Google Scholar] [CrossRef] [PubMed]
- Vicario, J.; Ezpeleta, J.M.; Palacios, F. Asymmetric Cyanation of α-Ketiminophosphonates Catalyzed by Cinchona Alkaloids: Enantioselective Synthesis of Tetrasubstituted α-Aminophosphonic Acid Derivatives from Trisubstituted α-Amino- phosphonates. Adv. Synth. Catal. 2012, 354, 2641–2647. [Google Scholar] [CrossRef]
- Vicario, J.; Ortiz, P.; Ezpeleta, J.M.; Palacios, F. Asymmetric Synthesis of Functionalized Tetrasubstituted α-Aminophosphonates through Enantioselective Aza-Henry Reaction of Phosphorylated Ketimines. J. Org. Chem. 2015, 80, 156–164. [Google Scholar] [CrossRef]
- Maestro, A.; Martinez de Marigorta, E.; Palacios, F.; Vicario, J. Enantioselective α-aminophosphonate functionalization of indole ring through an organocatalyzed Friedel- Crafts reaction. J. Org. Chem. 2019, 84, 1094–1102. [Google Scholar] [CrossRef]
- Alonso, C.; Fuertes, M.; Martin-Encinas, E.; Selas, A.; Rubiales, G.; Tesauro, C.; Knudssen, B.R.; Palacios, F. Novel topoisomerase I inhibitors. Syntheses and biological evaluation of phosphorus substituted quinoline derivates with antiproliferative activity. Eur. J. Med. Chem. 2018, 149, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Maestro, A.; Martín-Encinas, E.; Martínez de Marigorta, E.; Alonso, C.; Rubiales, G.; Vicario, J.; Palacios, F. Synthesis of novel antiproliferative hybrid bis-(3-indolyl)methane phosphonate derivatives. Eur. J. Med. Chem. 2018, 158, 874–883. [Google Scholar] [CrossRef]
- Carramiñana, V.; Ochoa de Retana, A.M.; Vélez del Burgo, A.; de los Santos, J.M.; Palacios, F. Synthesis and biological evaluation of cyanoaziridine phosphine oxides and phosphonates with antiproliferative activity. Eur. J. Med. Chem. 2019, 163, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Tejería, A.; Pérez-Pertejo, Y.; Reguera, R.M.; Carbajo-Andrés, R.; Balaña-Fouce, R.; Alonso, C.; Martín-Encinas, E.; Selas, A.; Rubiales, G.; Palacios, F. Antileishmanial activity of new hybrid tetrahydroquinoline and quinoline derivatives with phosphorus substituents. Eur. J. Med. Chem. 2019, 162, 18–31. [Google Scholar] [CrossRef]
- Martín-Encinas, E.; Rubiales, G.; Knudsen, B.R.; Palacios, F.; Alonso, C. Straightforward synthesis and biological evaluation as topoisomerase I inhibitors and antiproliferative agents of hybrid chromeno[4,3-b][1,5]naphthyridines and chromeno[4,3-b][1,5]naphthyridin-6(5H)-ones. Eur. J. Med. Chem. 2019, 178, 752–766. [Google Scholar] [CrossRef] [PubMed]
- Carramiñana, V.; Ochoa de Retana, A.M.; de los Santos, J.M.; Palacios, F. First synthesis of merged hybrids phosphorylated azirino[2,1-b]benzo[e][1,3]oxazine derivatives as anticancer agents. Eur. J. Med. Chem. 2020, 185, 111771–111781. [Google Scholar] [CrossRef] [PubMed]
- Martín-Encinas, E.; Selas, A.; Rubiales, G.; Tesauro, C.; Knudsen, B.R.; Palacios, F.; Alonso, C. Synthesis of novel hybrid quinolino[4,3-b][1,5]naphthyridines and quinolino[4,3-b][1,5]naphthyridin-6(5H)-one derivatives and biological evaluation as topoisomerase I inhibitors and antiproliferatives. Eur. J. Med. Chem. 2020, 195, 112292–112313. [Google Scholar] [CrossRef]
- Recio, R.; Vengut-Climent, E.; Mouillac, B.; Orcel, H.; López-Lázaro, M.; Calderón-Montaño, J.M.; Álvarez, E.; Khiar, N.; Fernández, I. Design, synthesis and biological studies of a library of NK1-Receptor Ligands Based on a 5-arylthiosubstituted 2-amino-4,6-diaryl-3-cyano-4H-pyran core: Switch from antagonist to agonist effect by chemical modification. Eur. J. Med. Chem. 2017, 138, 644–660. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, E.J.; Kümmerle, A.E.; Fraga, C.A.M. The Methylation Effect in Medicinal Chemistry. Chem. Rev. 2011, 111, 5215–5246. [Google Scholar] [CrossRef] [PubMed]
- Schönherr, H.; Cernak, T. Profound methyl effects in drug discovery and a call for new C-H methylation reactions. Angew. Chem. Int. Ed. 2013, 52, 12256–12267. [Google Scholar] [CrossRef] [PubMed]
- Müller, K.; Faeh, C.; Diederich, F. Fluorine in pharmaceuticals: Looking beyond intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef]
- Shah, P.; Westwell, A.D. The role of fluorine in medicinal chemistry. J. Enzym. Inhib. Med. Chem. 2007, 22, 527–540. [Google Scholar] [CrossRef]
- Bégué, J.P.; Bonnet-Delpon, D. Bioorganic and Medicinal Chemistry of Fluorine; Wiley: Hoboken, NJ, USA, 2007; ISBN 978-0-470-27830-7. [Google Scholar]
- Kafarski, P.; Lejczak, B. Biological activity of aminophosphonic acids. Phosphorus Sulfur Silicon Relat. Elem. 1991, 63, 193–215. [Google Scholar] [CrossRef]
- Engel, R. Handbook of Organophosphorus Chemistry; Dekker M. Inc.: New York, NY, USA, 1992. [Google Scholar] [CrossRef]
- Karl, D.M. Phosphorus, the Staff of Life. Nature 2000, 406, 31–33. [Google Scholar] [CrossRef] [PubMed]
- Diaz, G.; Miranda, I.L.; Diaz, M.A.N. Quinolines, Isoquinolines, Angustureine, and Congeneric Alkaloids-Occurrence, Chemistry, and Biological Activity. In Phytochemicals-Isolation, Characterisation and Role in Human Health; InTech: London, UK, 2015. [Google Scholar] [CrossRef]
- Shang, X.-F.; Morris-Natschke, S.L.; Liu, Y.-Q.; Guo, X.; Xu, X.-S.; Goto, M.; Li, J.-C.; Yang, G.-Z.; Lee, K.-H. Biologically active quinoline and quinazoline alkaloids part I. Med. Res. Rev. 2018, 775–828. [Google Scholar] [CrossRef]
- Afzal, O.; Kumar, S.; Haider, R.; Ali, R.; Kumar, R.; Jaggi, M.; Bawa, S. A review on anticancer potential of bioactive heterocycle quinolone. Eur. J. Med. Chem. 2015, 97, 871–910. [Google Scholar] [CrossRef]
- Liu, Y.Q.; Li, W.Q.; Morris-Natschke, S.L.; Qian, K.; Yang, L.; Zhu, G.-X.; Wu, X.-B.; Chen, A.-L.; Zhang, S.-Y.; Nan, X.; et al. Perspectives on biologically active camptothecin derivatives. Med. Res. Rev. 2015, 35, 753–789. [Google Scholar] [CrossRef]
- Aboujaoude, E.; Collignon, N.; Savignac, P. A simple synthesis of dialkyl 1-formylalkanephosphonates. Synthesis 1983, 8, 364. [Google Scholar] [CrossRef]
- Varlet, J.-M.; Savignac, P. Synthesis and reductive amination of phosphonopyruvates: Preparation of 2-amino-2-carboxyalkylphosphonic acids (β-phosphonoalanine). Can. J. Chem. 1979, 57, 3216–3220. [Google Scholar] [CrossRef]
- Palacios, F.; Vicario, J.; Aparicio, D. Efficient synthesis of 1-azadienes derived from α-aminoesters. Regioselective preparation of α-dehydroamino acids, vinylglycines, and α-amino acids. J. Org. Chem. 2006, 20, 7690–7696. [Google Scholar] [CrossRef]
- Sucrow, W.; Grosz, K.-P. A convenient preparation of dimethyl and diethyl oxaloacetate. Synth. Commun. 1979, 7, 603–607. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Cmpd. | IC50 (μM) | |||
|---|---|---|---|---|---|
| A549 | SKOV3 | RKO | MRC5 | ||
| 1 | 4a | 38.25 ± 3.35 | >50 | n.d. | >50 |
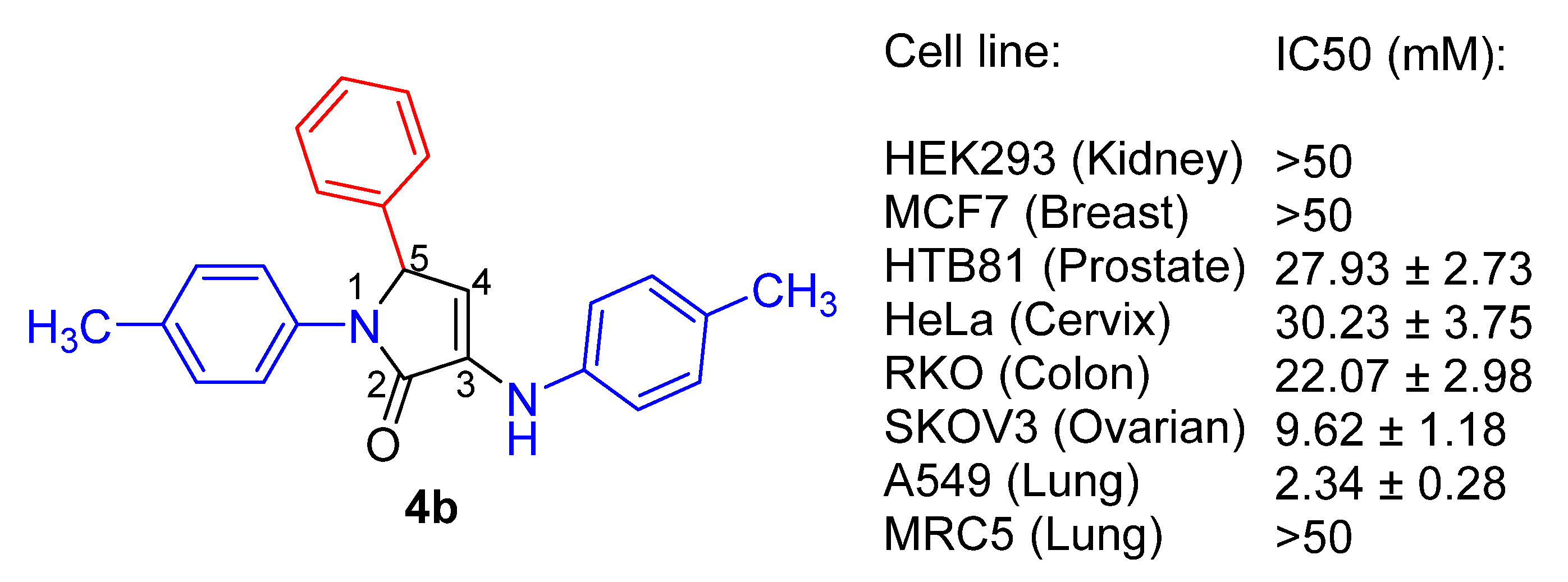
| 2 | 4b | 2.34 ± 0.28 | 9.62 ± 1.18 | 22.07 ± 2.98 | >50 |
| 3 | 4c | 21.02 ± 1.64 | >50 | n.d. | >50 |
| 4 | 4d | 31.16 ± 1.04 | >50 | n.d. | >50 |
| 5 | 4e | 41.28 ± 1.29 | >50 | n.d. | >50 |
| 6 | 4f | 10.72 ± 1.21 | 23.52 ± 0.75 | n.d. | >50 |
| 7 | 4g | 17.08 ± 1.58 | 22.87 ± 0.63 | 23.84 ± 2.53 | 26.55 ± 2.89 |
| 8 | 4h | 7.33 ± 0.57 | 27.65 ± 1.32 | n.d. | 27.23 ± 1.24 |
| 9 | 4i | >50 | >50 | >50 | n.d. |
| 10 | 4j | 19.34 ± 0.7 | >50 | n.d. | >50 |
| 11 | 4k | 11.52 ± 0.85 | >50 | n.d. | >50 |
| 12 | 4l | 12.71 ± 1.07 | 12.65 ± 1.83 | n.d. | >50 |
| 13 | 4m | 12.1 ± 0.74 | 30.52 ± 1.34 | n.d. | 21.29 ± 0.74 |
| 14 | 4n | 15.68 ± 0.92 | 29.16 ± 1.00 | n.d. | >50 |
| 15 | 4o | 11.08 ± 0.71 | >50 | n.d. | >50 |
| 16 | 4p | 20.08 ± 2.00 | >50 | n.d. | 24.05 ± 1.64 |
| 17 | 4q | 10.99 ± 0.90 | >50 | 23.79 ± 1.32 | >50 |
| 18 | 4r | 10.26 ± 0.8 | 9.59 ± 0.82 | n.d. | 18.24 ± 0.81 |
| 19 | 4s | >50 | >50 | n.d. | n.d. |
| 20 | 4t | 33.50 ± 1.68 | >50 | >50 | >50 |
| 21 | 4u | >50 | >50 | >50 | n.d. |
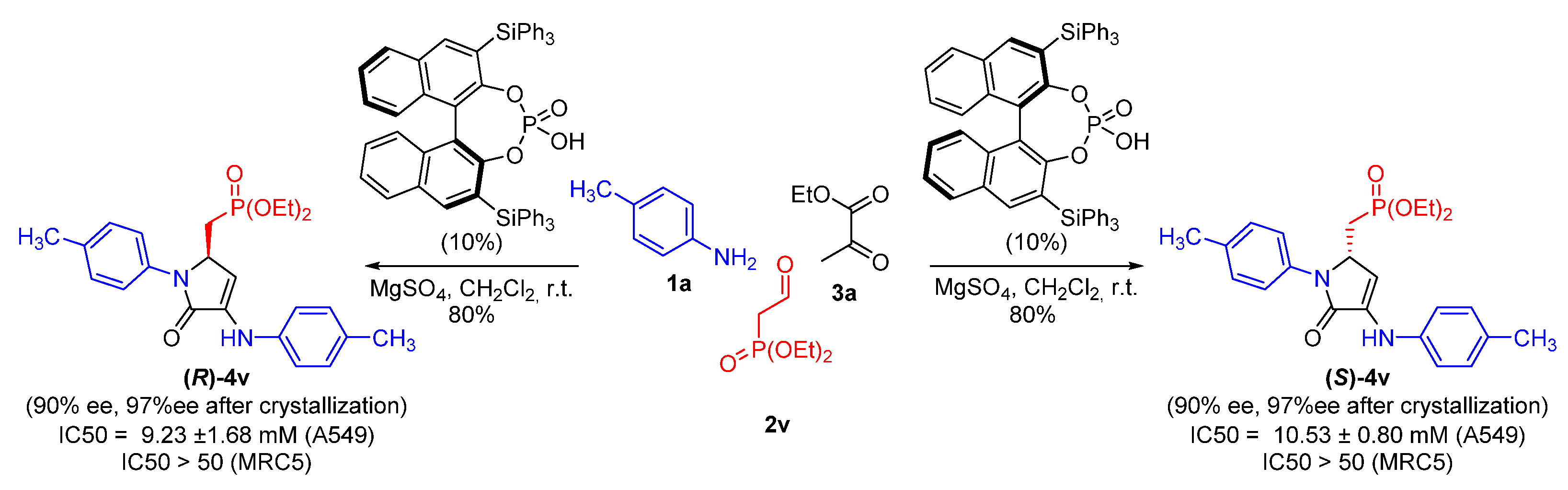
| 22 | 4v | 11.29 ± 1.80 | >50 | >50 | >50 |
| 23 | 4w | 5.67 ± 0.9 | 10.76 ± 0.88 | 28.14 ± 1.47 | 30.10 ± 0.49 |
| 24 | 5a | 2.05 ± 0.23 | >50 | n.d. | >50 |
| 25 | 5b | 9.92 ± 1.15 | >50 | n.d. | 47.56 ± 1.67 |
| 26 | 5c | 1.67 ± 0.49 | >50 | n.d. | >50 |
| 27 | 5d | >50 | >50 | >50 | >50 |
| 28 | 5e | n.d. | n.d. | n.d. | n.d. |
| 29 | Doxorubicin | <0.1 | 0.13 ± 0.098 | <0.1 | >50 |
| Entry | Cmpd. | IC50 (μM) | |||
|---|---|---|---|---|---|
| A549 | SKOV3 | RKO | MRC5 | ||
| 1 | 6a | 12.02 ± 1.96 | 6.84 ± 0.59 | n.d. | >50 |
| 2 | 6b | 31.48 ± 1.37 | >50 | >50 | >50 |
| 3 | 6c | 6.84 ± 0.22 | >50 | n.d. | >50 |
| 4 | 7 | 17.16 ± 2.10 | >50 | n.d. | >50 |
| 5 | 8a | >50 | >50 | n.d. | n.d. |
| 6 | 8b | >50 | >50 | >50 | n.d. |
| 7 | 9 | 8.37 ± 1.27 | 16.13 ± 0.81 | n.d. | 23.81 ± 1.75 |
| 8 | 10 | 4.36 ± 0.51 | 5.55 ± 0.62 | n.d. | >50 |
| 9 | 11a | 22.94 ± 1.13 | >50 | n.d. | >50 |
| 10 | 11b | 2.00 ± 0.78 | 8.50 ± 0.54 | n.d. | 44.57 ± 1.16 |
| 11 | 12a | 9.25 ± 1.49 | >50 | n.d. | >50 |
| 12 | Doxorubicin | <0.1 | 0.13 ± 0.098 | <0.1 | >50 |
| Entry | Cmpd. | IC50 (μM) | |||
|---|---|---|---|---|---|
| A549 | SKOV3 | RKO | MRC5 | ||
| 1 | 16 | 13.97 ± 1.05 | >50 | >50 | >50 |
| 2 | 17 | >50 | >50 | n.d | n.d. |
| 3 | 18a | 3.27 ± 0.65 | >50 | n.d | >50 |
| 4 | 18b | 4.25 ± 0.65 | >50 | >50 | >50 |
| 5 | 18c | 19.45 ± 0.34 | >50 | n.d | >50 |
| 6 | 19 | 3.65 ± 0.25 | >50 | >50 | >50 |
| 7 | 20a | 6.90 ± 0.73 | >50 | n.d | >50 |
| 8 | 20b | 9.77 ± 0.17 | >50 | n.d | >50 |
| 9 | 21 | 0.60 ± 0.08 | 16.72 ± 1.1 | n.d | 6.12 ± 0.76 |
| 10 | Doxorubicin | <0.1 | 0.13 ± 0.098 | <0.1 | >50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
del Corte, X.; López-Francés, A.; Maestro, A.; Villate-Beitia, I.; Sainz-Ramos, M.; Martínez de Marigorta, E.; Pedraz, J.L.; Palacios, F.; Vicario, J. A Multicomponent Protocol for the Synthesis of Highly Functionalized γ-Lactam Derivatives and Their Applications as Antiproliferative Agents. Pharmaceuticals 2021, 14, 782. https://doi.org/10.3390/ph14080782
del Corte X, López-Francés A, Maestro A, Villate-Beitia I, Sainz-Ramos M, Martínez de Marigorta E, Pedraz JL, Palacios F, Vicario J. A Multicomponent Protocol for the Synthesis of Highly Functionalized γ-Lactam Derivatives and Their Applications as Antiproliferative Agents. Pharmaceuticals. 2021; 14(8):782. https://doi.org/10.3390/ph14080782
Chicago/Turabian Styledel Corte, Xabier, Adrián López-Francés, Aitor Maestro, Ilia Villate-Beitia, Myriam Sainz-Ramos, Edorta Martínez de Marigorta, José Luis Pedraz, Francisco Palacios, and Javier Vicario. 2021. "A Multicomponent Protocol for the Synthesis of Highly Functionalized γ-Lactam Derivatives and Their Applications as Antiproliferative Agents" Pharmaceuticals 14, no. 8: 782. https://doi.org/10.3390/ph14080782
APA Styledel Corte, X., López-Francés, A., Maestro, A., Villate-Beitia, I., Sainz-Ramos, M., Martínez de Marigorta, E., Pedraz, J. L., Palacios, F., & Vicario, J. (2021). A Multicomponent Protocol for the Synthesis of Highly Functionalized γ-Lactam Derivatives and Their Applications as Antiproliferative Agents. Pharmaceuticals, 14(8), 782. https://doi.org/10.3390/ph14080782