Synthesis, Biological Evaluation, and In Silico Modeling of N-Substituted Quinoxaline-2-Carboxamides

 , , , ,
, , , ,  and
and 
Abstract
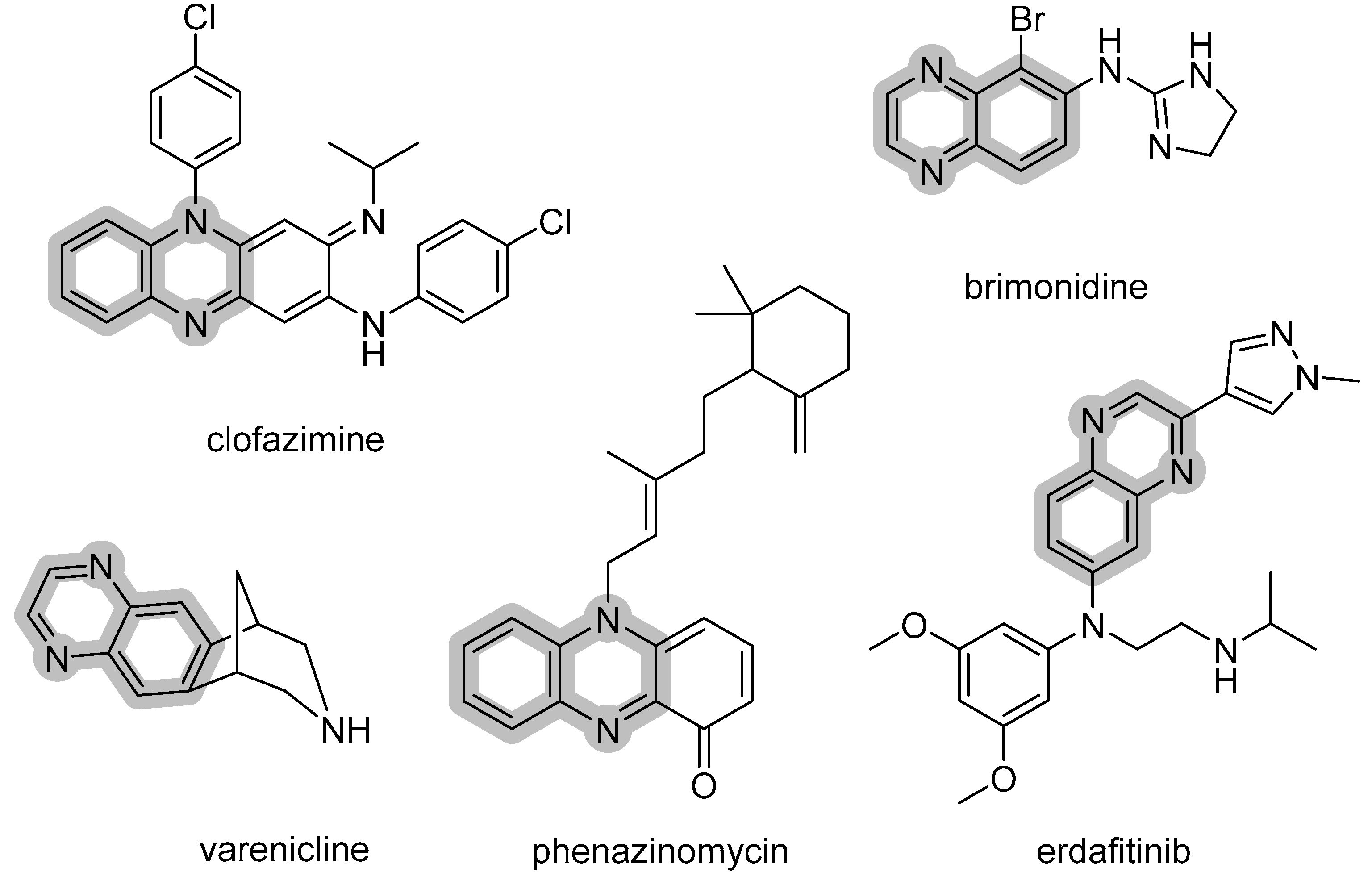
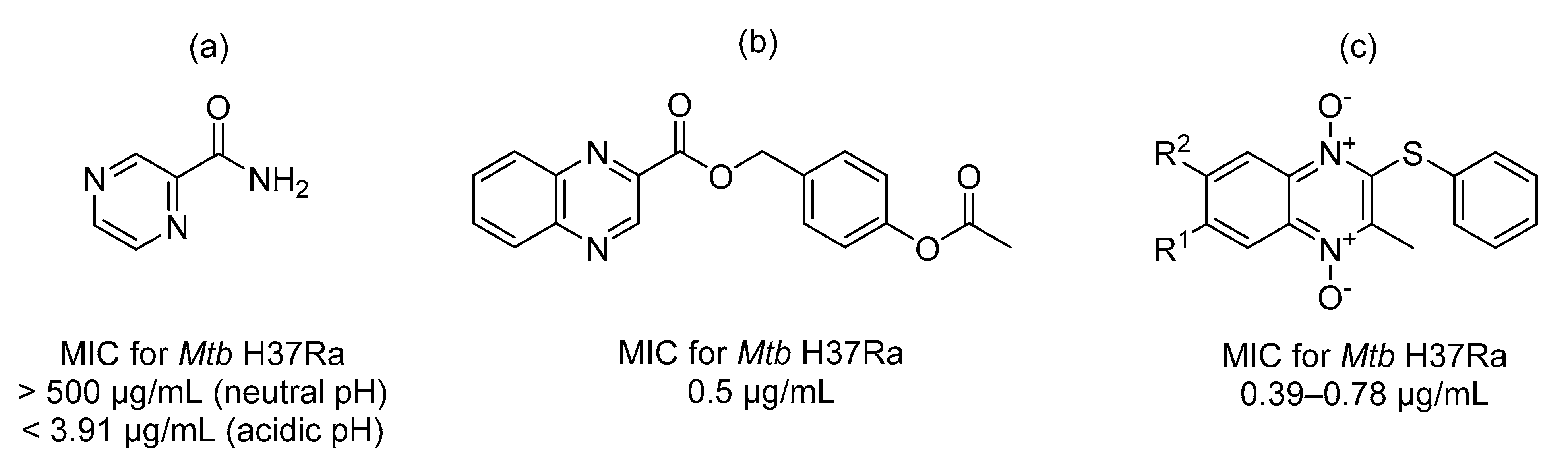
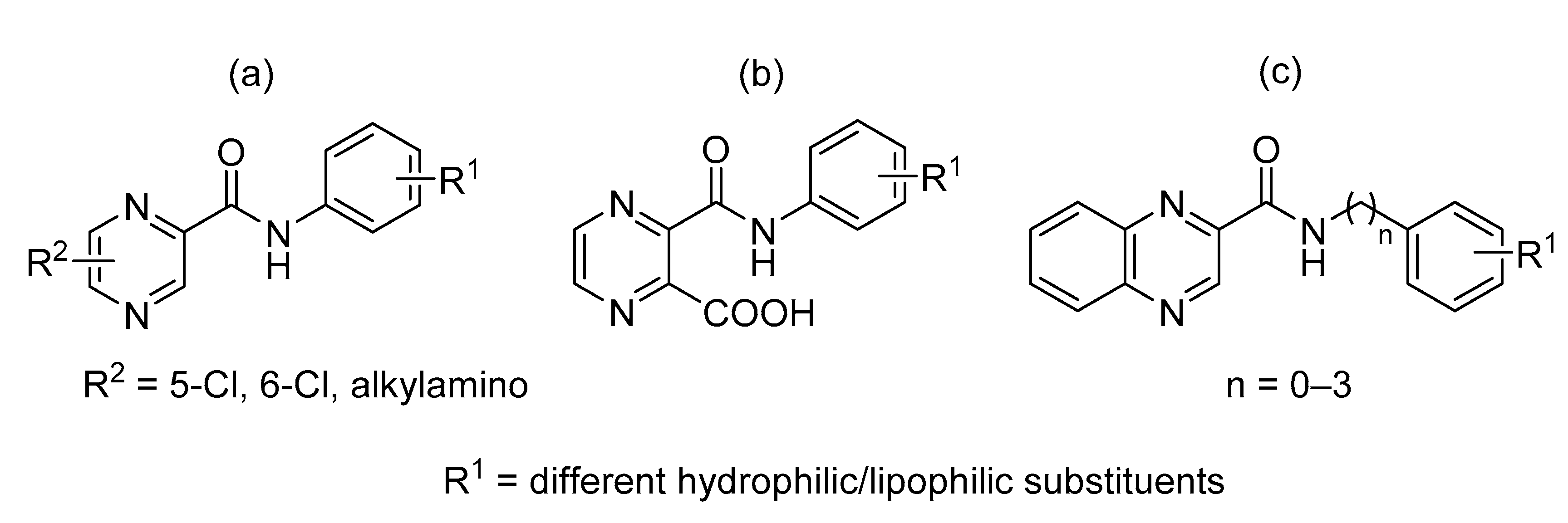
:1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Evaluation of Biological Activities
2.2.1. In Vitro Antimycobacterial Activity
2.2.2. In Vitro Cytotoxicity
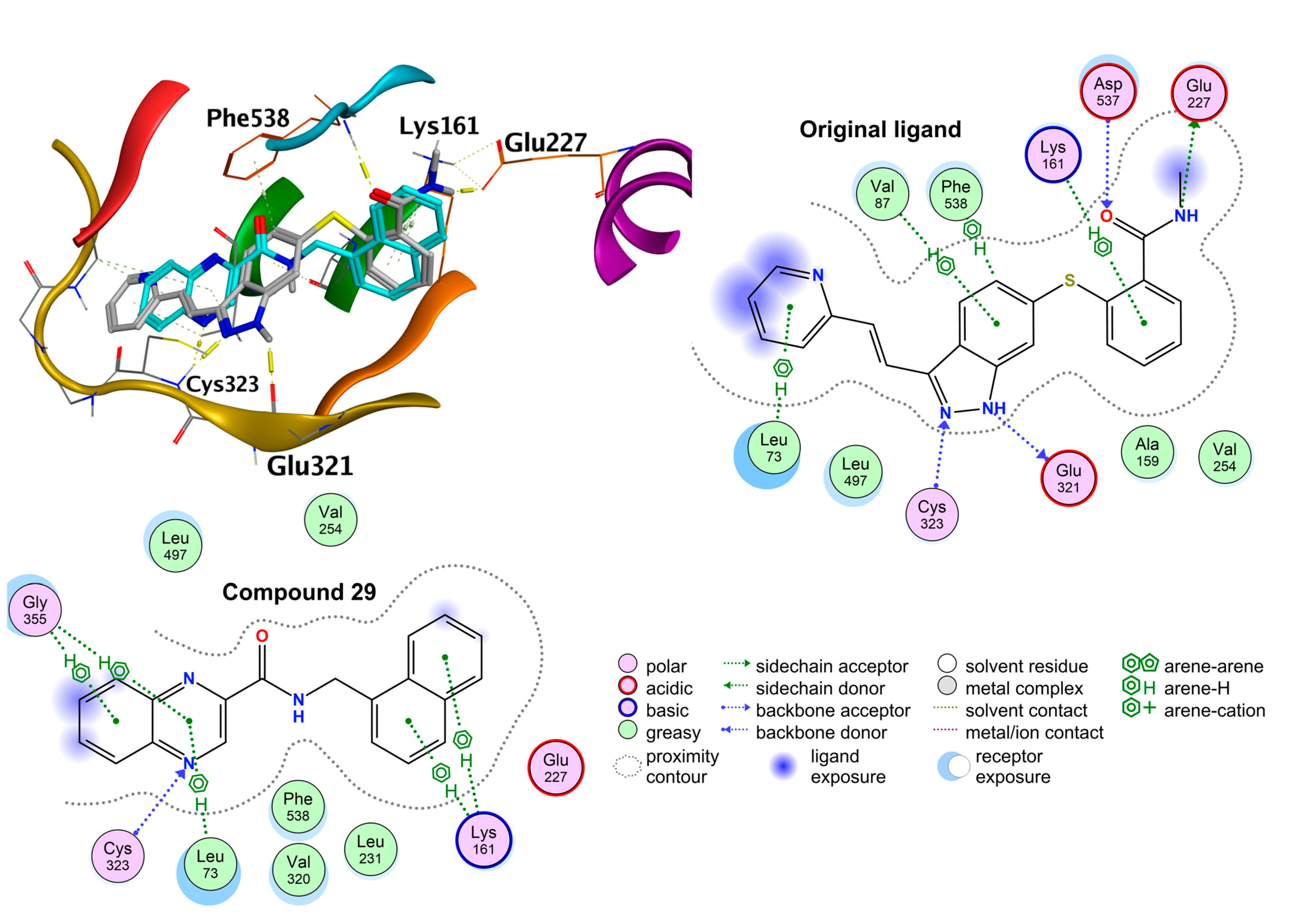
2.3. In Silico Modeling - Molecular Docking Studies
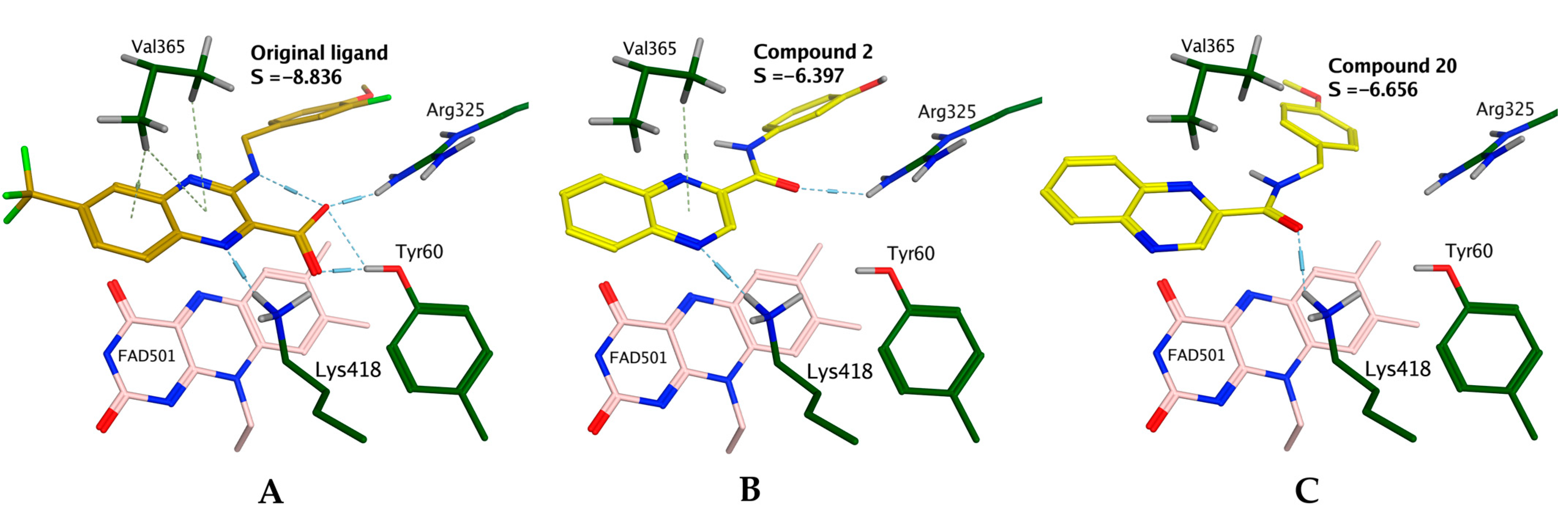
2.3.1. Exploring the Mechanism of Antimycobacterial Activity by In Silico Methods
2.3.2. Exploring the Mechanism of In Vitro Cytotoxicity by in Silico Methods
3. Materials and Methods
3.1. General Information
3.2. Chemistry
3.3. Analytical Data
3.4. Evaluation of Biological Activities
3.4.1. In Vitro Antimycobacterial Activity
3.4.2. In Vitro Antibacterial Activity
3.4.3. In Vitro Antifungal Activity
3.4.4. In Vitro Cytotoxicity
3.5. In Silico Modeling—Molecular Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scorpio, A.; Zhang, Y. Mutations in pncA, a gene encoding pyrazinamidase/nicotinamidase, cause resistance to the antituberculous drug pyrazinamide in tubercle bacillus. Nat. Med. 1996, 2, 662–667. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Tuberculosis Report 2020. Available online: www.who.int/tb/publications/global_report/en/ (accessed on 4 January 2021).
- Seitz, L.E.; Suling, W.J.; Reynolds, R.C. Synthesis and Antimycobacterial Activity of Pyrazine and Quinoxaline Derivatives. J. Med. Chem. 2002, 45, 5604–5606. [Google Scholar] [CrossRef] [PubMed]
- Carta, A.; Loriga, M.; Paglietti, G.; Mattana, A.; Fiori, P.L.; Mollicotti, P.; Sechi, L.; Zanetti, S. Synthesis, anti-mycobacterial, anti-trichomonas and anti-candida in vitro activities of 2-substituted-6,7-difluoro-3-methylquinoxaline 1,4-dioxides. Eur. J. Med. Chem. 2004, 39, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Carta, A.; Paglietti, G.; Nikookar, M.E.R.; Sanna, P.; Sechi, L.; Zanetti, S. Novel substituted quinoxaline 1,4-dioxides with in vitro antimycobacterial and anticandida activity. Eur. J. Med. Chem. 2002, 37, 355–366. [Google Scholar] [CrossRef]
- Jampilek, J. Recent Advances in Design of Potential Quinoxaline Anti-Infectives. Curr. Med. Chem. 2014, 21, 4347–4373. [Google Scholar] [CrossRef]
- Servusová, B.; Vobicková, J.; Paterová, P.; Kubíček, V.; Kuneš, J.; Doležal, M.; Zitko, J. Synthesis and antimycobacterial evaluation of N-substituted 5-chloropyrazine-2-carboxamides. Bioorganic Med. Chem. Lett. 2013, 23, 3589–3591. [Google Scholar] [CrossRef] [PubMed]
- Zitko, J.; Servusová, B.; Janoutová, A.; Paterová, P.; Mandíková, J.R.; Garaj, V.; Vejsová, M.; Marek, J.; Doležal, M. Synthesis and antimycobacterial evaluation of 5-alkylamino-N-phenylpyrazine-2-carboxamides. Bioorganic Med. Chem. 2015, 23, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Zitko, J.; Servusová, B.; Paterová, P.; Mandíková, J.; Kubíček, V.; Kučera, R.; Hrabcová, V.; Kuneš, J.; Soukup, O.; Doležal, M. Synthesis, Antimycobacterial Activity and In Vitro Cytotoxicity of 5-Chloro-N-phenylpyrazine-2-carboxamides. Molecules 2013, 18, 14807–14825. [Google Scholar] [CrossRef] [Green Version]
- Semelková, L.; Janošcová, P.; Fernandes, C.; Bouz, G.; Janďourek, O.; Konečná, K.; Paterová, P.; Navrátilová, L.; Kuneš, J.; Doležal, M.; et al. Design, Synthesis, Antimycobacterial Evaluation, and In Silico Studies of 3-(Phenylcarbamoyl)-pyrazine-2-carboxylic Acids. Molecules 2017, 22, 1491. [Google Scholar] [CrossRef] [Green Version]
- Franzblau, S.G.; Witzig, R.S.; McLaughlin, J.C.; Torres, P.; Madico, G.; Hernandez, A.; Degnan, M.T.; Cook, M.B.; Quenzer, V.K.; Ferguson, R.M.; et al. Rapid, low-technology MIC determination with clinical Mycobacterium tuberculosis isolates by using the microplate Alamar Blue assay. J. Clin. Microbiol. 1998, 36, 362–366. [Google Scholar] [CrossRef] [Green Version]
- Heinrichs, M.T.; May, R.J.; Heider, F.; Reimers, T.; Sy, S.K.B.; Peloquin, C.A.; Derendorf, H. Mycobacterium tuberculosis Strains H37ra and H37rv have equivalent minimum inhibitory concentrations to most antituberculosis drugs. Int. J. Mycobacteriol. 2018, 7, 156. [Google Scholar] [CrossRef] [PubMed]
- Sood, S.; Yadav, A.; Shrivastava, R. Mycobacterium aurum is Unable to Survive Mycobacterium tuberculosis Latency Associated Stress Conditions: Implications as Non-suitable Model Organism. Indian J. Microbiol. 2016, 56, 198–204. [Google Scholar] [CrossRef] [Green Version]
- Mahesh, R.; Devadoss, T.; Pandey, D.K.; Bhatt, S.; Yadav, S.K. Design, synthesis and structure–activity relationship of novel quinoxalin-2-carboxamides as 5-HT3 receptor antagonists for the management of depression. Bioorganic Med. Chem. Lett. 2010, 20, 6773–6776. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.R.; Dodd, P.G.; Gayton, S.; Greener, B.S.; Harbottle, G.W.; Mantell, S.J.; Maw, G.N.; Osborne, S.A.; Rees, H.; Ringer, T.J.; et al. Structure–activity relationships of novel non-competitive mGluR1 antagonists: A potential treatment for chronic pain. Bioorganic Med. Chem. Lett. 2007, 17, 486–490. [Google Scholar] [CrossRef]
- Juhás, M.; Kučerová, L.; Horáček, O.; Janďourek, O.; Kubíček, V.; Konečná, K.; Kučera, R.; Barta, P.; Janoušek, J.; Paterová, P.; et al. N-Pyrazinoyl Substituted Amino Acids as Potential Antimycobacterial Agents—the Synthesis and Biological Evaluation of Enantiomers. Molecules 2020, 25, 1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yew, W.W.; Leung, C.C. Antituberculosis drugs and hepatotoxicity. Respirology 2006, 11, 699–707. [Google Scholar] [CrossRef]
- Kaushal, T.; Srivastava, G.; Sharma, A.; Negi, A.S. An insight into medicinal chemistry of anticancer quinoxalines. Bioorganic Med. Chem. 2019, 27, 16–35. [Google Scholar] [CrossRef]
- Chen, J.; Chen, W.; Wang, F.; He, Z.; Dai, W.; Li, Q.; Liu, X.; Zhang, Z.; Zhai, D. The role of the vascular endothelial growth factor/vascular endothelial growth factor receptors axis mediated angiogenesis in curcumin-loaded nanostructured lipid carriers induced human HepG2 cells apoptosis. J. Cancer Res. Ther. 2015, 11, 597–605. [Google Scholar] [CrossRef]
- Yang, C.; Qin, S. Apatinib targets both tumor and endothelial cells in hepatocellular carcinoma. Cancer Med. 2018, 7, 4570–4583. [Google Scholar] [CrossRef] [Green Version]
- Novy, Z.; Janousek, J.; Barta, P.; Petrik, M.; Hajduch, M.; Trejtnar, F. Preclinical evaluation of anti-VEGFR2 monoclonal antibody ramucirumab labelled with zirconium-89 for tumour imaging. J. Label. Compd. Radiopharm. 2021, 64, 262–270. [Google Scholar] [CrossRef]
- Szabo, E.; Schneider, H.; Seystahl, K.; Rushing, E.J.; Herting, F.; Weidner, K.M.; Weller, M. Autocrine VEGFR1 and VEGFR2 signaling promotes survival in human glioblastoma models in vitro and in vivo. Neuro-Oncology 2016, 18, 1242–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.Y.; Hwang, J.; Lee, S.H.; Lee, H.J.; Jelinek, J.; Jeong, H.; Lim, J.S.; Kim, J.M.; Song, K.S.; Kim, B.H.; et al. Decreased efficacy of drugs targeting the vascular endothelial growth factor pathway by the epigenetic silencing of FLT1 in renal cancer cells. Clin. Epigenet. 2015, 7, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
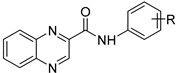 | |||||||
| Cmpd | R | LogP | MIC µg/mL (µM) | HepG2 | SI †† | ||
| Mtb H37Ra | M. smeg | M. aurum | IC50 (µM) | ||||
| 1 | H | 2.43 | ≥500 | ≥250 | ≥250 | >50 † | |
| 2 | 3-OH | 2.04 | 15.625 (58.9) | ≥125 | ≥125 | >250 † | >4.20 |
| 3 | 4-OH | 2.04 | 62.5 (235.6) | ≥500 | ≥500 | >250 † | >1.01 |
| 4 | 3,5-diOCH3 | 2.18 | 62.5 (202) | ≥125 | ≥125 | >25 † | >0.12 |
| 5 | 4-OCH3 | 2.31 | ≥125 | ≥125 | ≥125 | >50 † | |
| 6 | Cyclohexyl * | 2.32 | 31.25 (122.4) | 31.25 | ≥500 | >1000 | >8.17 |
| 7 | 2-F | 2.59 | ≥500 | 62.5 | ≥500 | >100 † | |
| 8 | 3-F | 2.59 | 250 | ≥500 | ≥500 | >100 † | |
| 9 | 4-F | 2.59 | 15.625 (58.5) | ≥500 | ≥500 | >100 † | >1.71 |
| 10 | 4-N(CH3)2 | 2.72 | ≥500 | ≥500 | ≥500 | >250 † | |
| 11 | 2,4-diF | 2.75 | ≥500 | ≥500 | ≥500 | >250 † | |
| 12 | 2-Cl | 2.99 | ≥125 | ≥125 | ≥125 | >50 † | |
| 13 | 3-Cl | 2.99 | ≥500 | ≥500 | ≥500 | >25 † | |
| 14 | 3-CF3 | 3.35 | ≥500 | ≥125 | ≥125 | >25 † | |
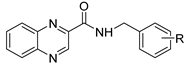 | |||||||
| Cmpd | R | LogP | MIC µg/mL (µM) | HepG2 | SI †† | ||
| MtbH37Ra | M. smeg | M. aurum | IC50 (µM) | ||||
| 15 | H | 2.50 | 15.625 (59.3) | ≥125 | ≥125 | >1000 | >17 |
| 16 | 3-OH | 2.11 | 31.25 (111.9) | 62.5 | ≥ 500 | 600.7 | 5.40 |
| 17 | 4-OH | 2.11 | 62.5 (223.8) | ≥500 | 500 | >250 † | >1.1 |
| 18 | 2,5-diOCH3 | 2.25 | 3.91 (12.1) | ≥500 | ≥500 | >1000 | 82.60 |
| 19 | 3-OCH3 | 2.38 | 15.625 (47) | ≥500 | ≥500 | >1000 | >21.3 |
| 20 | 4-OCH3 | 2.38 | 3.91 (11.8) | ≥250 | 125 | 209.4 | 16.1 |
| 21 | 3-F | 2.66 | 7.81 (27.8) | ≥500 | ≥500 | 248.0 | 8.92 |
| 22 | 2,4-diF | 2.75 | ≥500 | ≥500 | ≥500 | >25 † | |
| 23 | 2-CH3 | 2.99 | 7.81 (28.2) | ≥125 | ≥125 | 156.7 | 5.60 |
| 24 | 4-CH3 | 2.99 | 7.81 (28.2) | ≥250 | ≥250 | 87.7 | 3.11 |
| 25 | 2-Cl | 3.06 | 15.625 (52.5) | ≥500 | ≥500 | 527.5 | 22.45 |
| 26 | 3-Cl | 3.06 | 7.81 (26.2) | 31.25 | 31.25 | 140.1 | 5.34 |
| 27 | 4-Br | 3.33 | 7.81 (22.8) | ≥500 | ≥500 | >100 † | >4.4 |
| 28 | 3-CF3 | 3.42 | 3.91 (11.8) | 250 | ≥500 | 48.6 | 4.12 |
| 29 | Naphthyl ** | 3.50 | ≥250 | ≥125 | ≥125 | 37.3 | |
| 30 | 2,4-diCl | 3.62 | ≥500 | ≥125 | ≥125 | >100 † | |
| 31 | 3,4-diCl | 3.62 | 7.81 (23.5) | ≥500 | ≥500 | 66.9 | 2.80 |
 | |||||||
| Cmpd | R | LogP | MIC µg/mL (µM) | HepG2 | SI †† | ||
| MtbH37Ra | M. smeg | M. aurum | IC50 (µM) | ||||
| 32 | H | 2.78 | ≥500 | ≥500 | ≥500 | >1000 | |
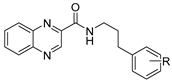 | |||||||
| Cmpd | R | LogP | MIC µg/mL (µM) | HepG2 | SI †† | ||
| MtbH37Ra | M. smeg | M. aurum | IC50 (µM) | ||||
| 33 | H | 3.20 | 15.625 (53.6) | ≥500 | ≥500 | 145.9 | 2.7 |
| Standards | LogP | MIC µg/mL (µM) | HepG2 | SI †† | |||
| MtbH37Ra | M. smeg | M. aurum | IC50 (µM) | ||||
| PZA | −1.31 | ≥500 (≥4061) ††† | ≥500 | ≥500 | >1000 | ||
| INH | −0.64 | 0.25 (1.82) | 15.625 | 3.91 | >1000 | >459.5 | |
| CPX | 1.32 | 0.25 (0.75) | 0.125 | 0.0156 | >500 † | >666.7 | |
| RIF | 4.24 | 0.003125 (0.0038) | 12.5 | 0.39 | >500 † | >131.579 | |
| Tamoxifen | 6.07 | na | na | na | 19.56 | na | |
* 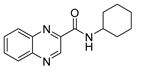 | ** 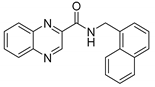 | ||||||
| Semelkova et al. [10] | This Paper | ||
|---|---|---|---|
| R | 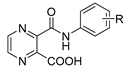 |  | 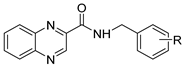 |
| H | ≥500 | ≥500 (1) | 15.625 (15) |
| 4-OCH3 | ≥500 | ≥125 (5) | 3.91 (20) |
| 3-CF3 | 250 | ≥500 (14) | 3.91 (28) |
| Activity against Mycobacterial Strains Expressed as MIC (µM) | |||||||
| Mtb H37Ra | M. smeg | M. aurum | |||||
| ≥798 | ≥399 | ≥399 | |||||
| Activity against Bacterial Strains Expressed as MIC (µM) | |||||||
| S. aureus | MRSA | E. faecalis | E. coli | P. aeruginosa | S. epidermidis | K. pneumoniae | S. marcescens |
| ≥500 | ≥500 | ≥500 | ≥500 | ≥500 | ≥500 | ≥500 | ≥500 |
| Activity against Fungal Strains Expressed as MIC (µM) | |||||||
| C. albicans | C. krusei | C. parapsilosis | C. tropicalis | A. flavus | L. corymbifera | T. interdigitale | A. fumigatus |
| ≥500 | ≥500 | ≥500 | ≥500 | ≥500 | ≥500 | ≥500 | ≥500 |
| Cytotoxicity against Human Cell Lines Expressed as IC50 (µM) | |||||||
| HepG2 | HK-2 | U-87 MG | A498 | PC-3 | SK-OV-3 | ||
| 37.3 | >250 | >250 | >250 | 68.5 | 49.6 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouz, G.; Bouz, S.; Janďourek, O.; Konečná, K.; Bárta, P.; Vinšová, J.; Doležal, M.; Zitko, J. Synthesis, Biological Evaluation, and In Silico Modeling of N-Substituted Quinoxaline-2-Carboxamides. Pharmaceuticals 2021, 14, 768. https://doi.org/10.3390/ph14080768
Bouz G, Bouz S, Janďourek O, Konečná K, Bárta P, Vinšová J, Doležal M, Zitko J. Synthesis, Biological Evaluation, and In Silico Modeling of N-Substituted Quinoxaline-2-Carboxamides. Pharmaceuticals. 2021; 14(8):768. https://doi.org/10.3390/ph14080768
Chicago/Turabian StyleBouz, Ghada, Sarah Bouz, Ondřej Janďourek, Klára Konečná, Pavel Bárta, Jarmila Vinšová, Martin Doležal, and Jan Zitko. 2021. "Synthesis, Biological Evaluation, and In Silico Modeling of N-Substituted Quinoxaline-2-Carboxamides" Pharmaceuticals 14, no. 8: 768. https://doi.org/10.3390/ph14080768
APA StyleBouz, G., Bouz, S., Janďourek, O., Konečná, K., Bárta, P., Vinšová, J., Doležal, M., & Zitko, J. (2021). Synthesis, Biological Evaluation, and In Silico Modeling of N-Substituted Quinoxaline-2-Carboxamides. Pharmaceuticals, 14(8), 768. https://doi.org/10.3390/ph14080768