
Synthesis and Biological Evaluation of a Radiolabeled PET (Positron Emission Tomography) Probe for Visualization of In Vivo α-Fucosidase Expression

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
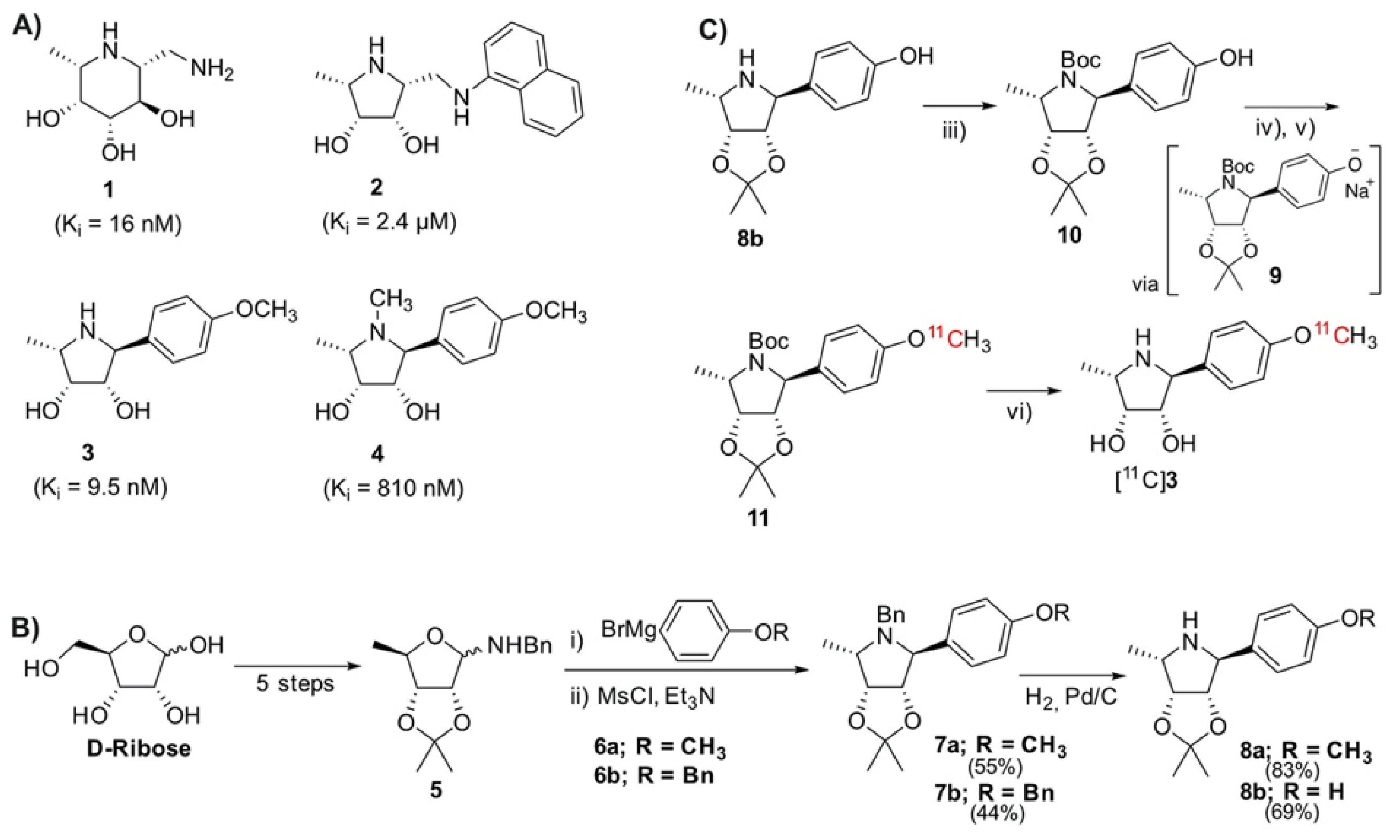
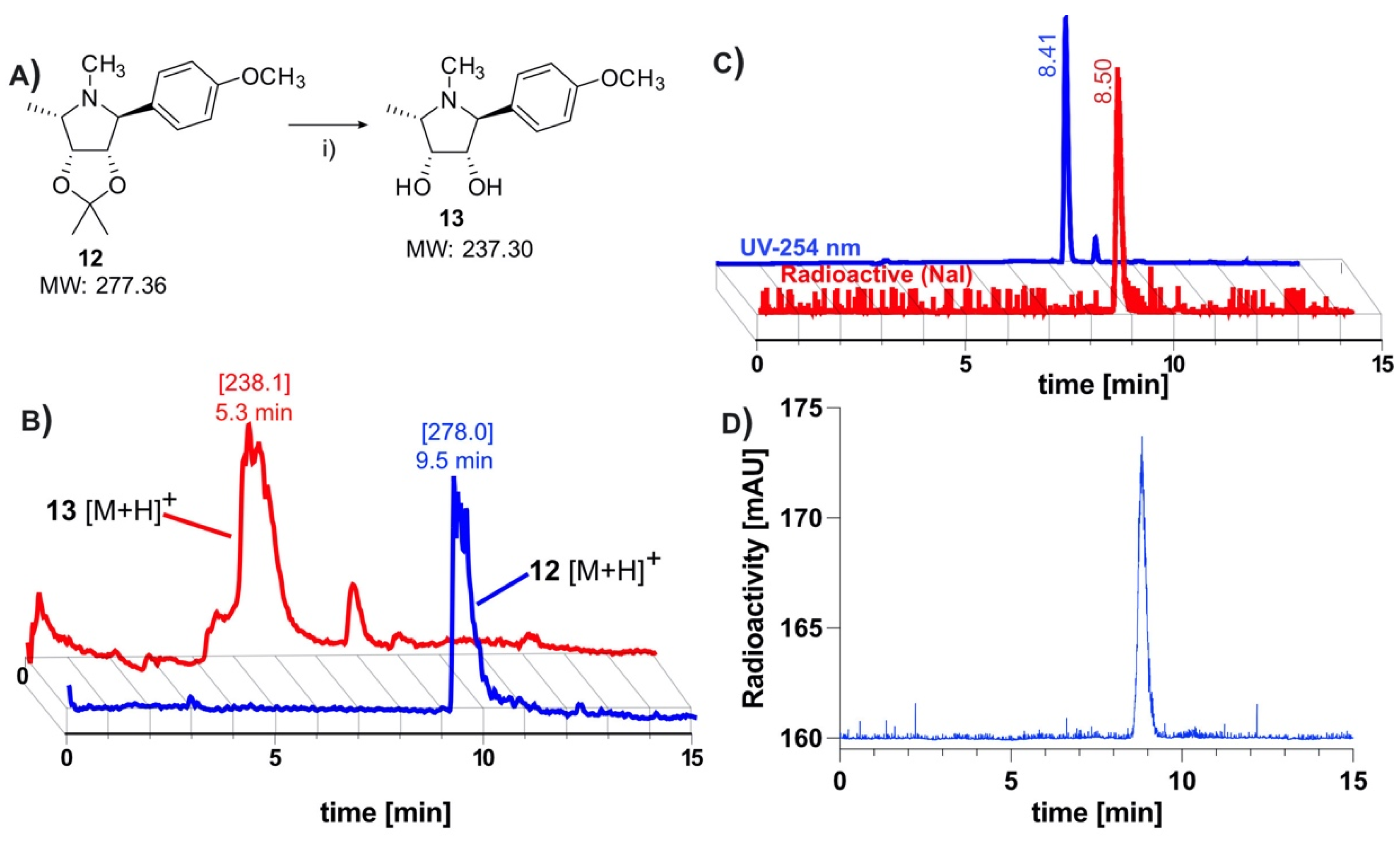
2.1. Chemical Synthesis of Precursor and Optimization of Deprotection
2.2. Radiolabeling and Automation of Tracer Synthesis
2.3. In Vitro Evaluation of Tracer
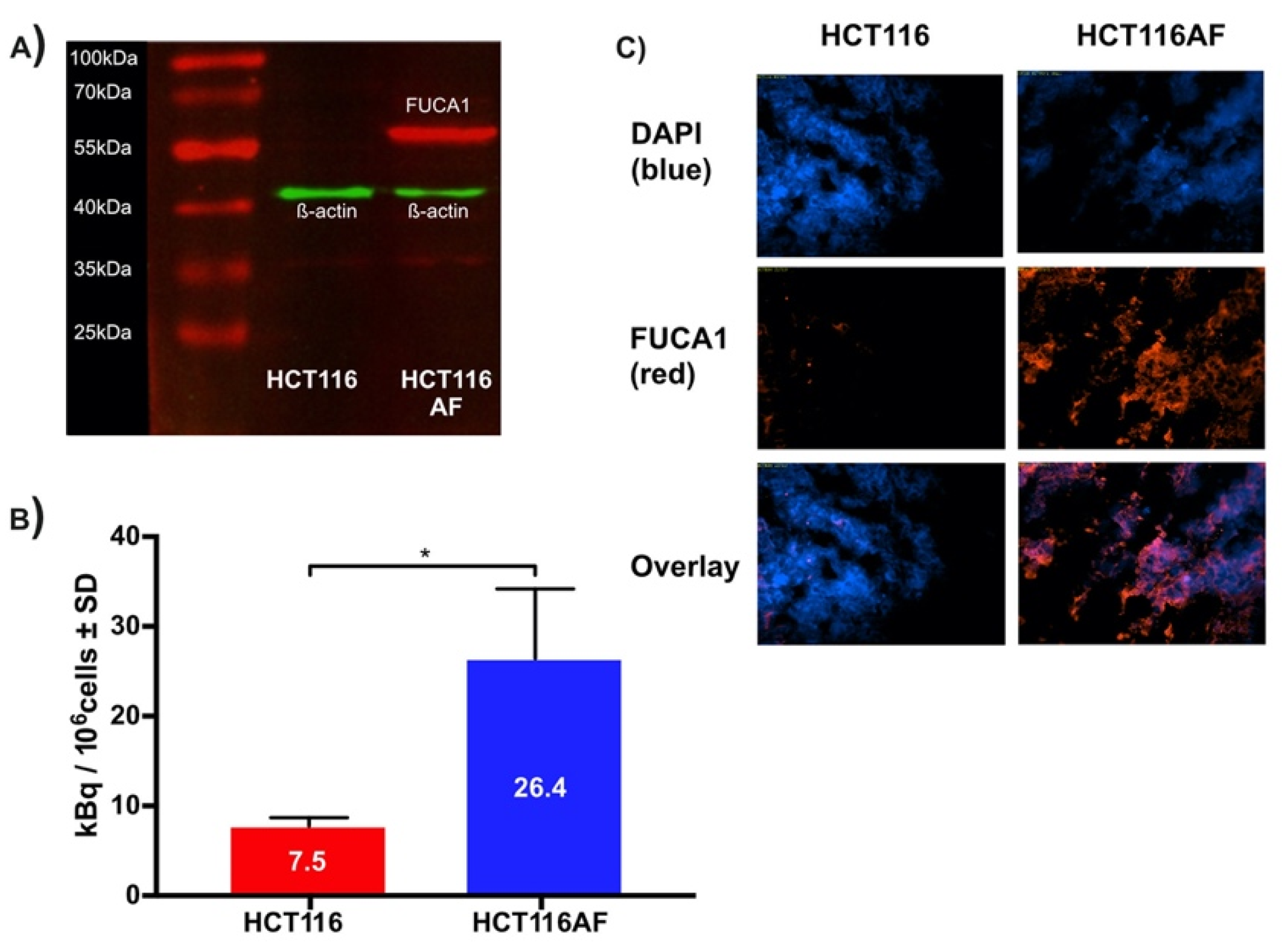
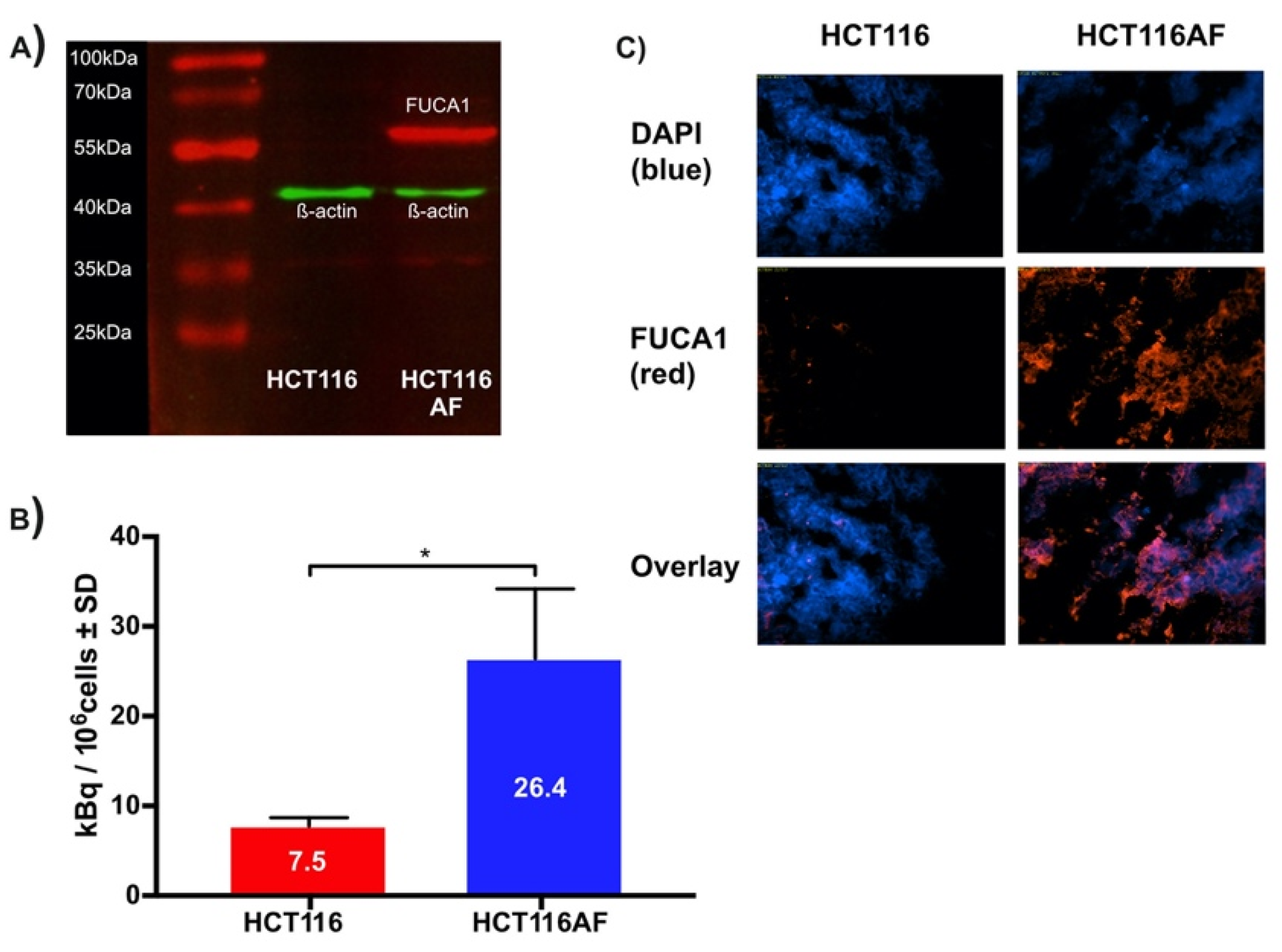
2.3.1. Producing AF Overexpressing Cell Line
2.3.2. In Vitro Evaluation of Tracer
2.3.3. Ex Vivo Evaluation of Tumors
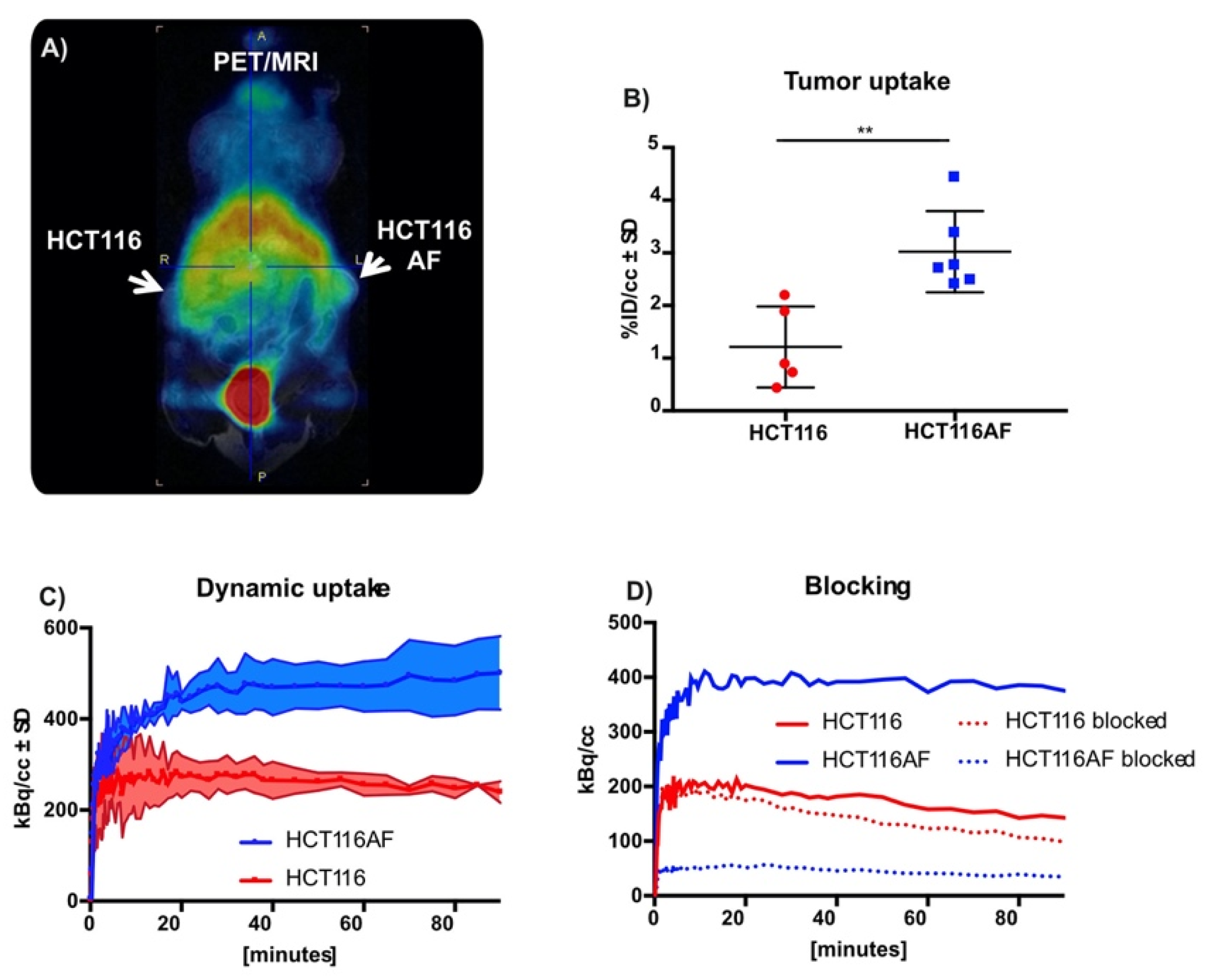
2.3.4. In Vivo Evaluation of Tracer
3. Materials and Methods
3.1. General
3.1.1. Chemistry
3.1.2. Radiochemistry and Tracer Validation
3.1.3. Cell Culture and Western Blot
3.1.4. In Vitro Evaluation
3.1.5. Ex Vivo Evaluation
3.1.6. PET Studies and Blocking
3.1.7. Statistics
4. Conclusions and Outlook
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sobkowicz, A.D.; Gallagher, M.E.; Reid, C.J.; Crean, D.; Carrington, S.D.; Irwin, J.A. Modulation of Expression in BEAS-2B Airway Epithelial Cells of α-l-Fucosidase A1 and A2 by Th1 and Th2 Cytokines, and Overexpression of α-l-Fucosidase 2. Mol. Cell Biochem. 2014, 390, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Scanlin, T.F.; Glick, M.C. Terminal Glycosylation in Cystic Fibrosis. Biochim. Biophys. Acta 1999, 1455, 241–253. [Google Scholar] [CrossRef] [Green Version]
- Cheng, T.; Tu, S.; Chen, L.-C.; Chen, M.-Y.; Chen, W.-Y.; Lin, Y.-K.; Ho, C.-T.; Lin, S.-Y.; Wu, C.-H.; Ho, Y.-S. Down-Regulation of α-L-Fucosidase 1 Expression Confers Inferior Survival for Triple-Negative Breast Cancer Patients by Modulating the Glycosylation Status of the Tumor Cell Surface. Oncotarget 2015, 6, 21283–21300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otero-Estévez, O.; Martínez-Fernández, M.; Vázquez-Iglesias, L.; Páez de la Cadena, M.; Rodríguez-Berrocal, F.J.; Martínez-Zorzano, V.S. Decreased Expression of Alpha-L-Fucosidase gene FUCA1 in Human Colorectal Tumors. Int. J. Mol. Sci. 2013, 14, 16986–16998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giardina, M.G.; Matarazzo, M.; Varriale, A.; Morante, R.; Napoli, A.; Martino, R. Serum Alpha-L-Fucosidase. A Useful Marker in the Diagnosis of Hepatocellular Carcinoma. Cancer 1992, 70, 1044–1048. [Google Scholar] [CrossRef]
- Mossad, N.A.; Mahmoud, E.H.; Osman, E.A.; Mahmoud, S.H.; Shousha, H.I. Evaluation of Squamous Cell Carcinoma Antigen-Immunoglobulin M Complex (SCCA-IGM) and Alpha-L-Fucosidase (AFU) as Novel Diagnostic Biomarkers for Hepatocellular Carcinoma. Tumor Biol. 2014, 35, 11559–11564. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.-W.; Ho, C.-W.; Huang, H.-H.; Chang, S.-M.; Popat, S.D.; Wang, Y.-T.; Wu, M.-S.; Chen, Y.-J.; Lin, C.-H. Role for Alpha-L-Fucosidase in the Control of Helicobacter Pylori-Infected Gastric Cancer Cells. Proc. Natl. Acad. Sci. USA 2009, 106, 14581–14586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, C.-W.; Lin, Y.-N.; Chang, C.-F.; Li, S.-T.; Wu, Y.-T.; Wu, C.-Y.; Chang, C.-F.; Liu, S.-W.; Li, Y.-K.; Lin, C.-H. Discovery of Different Types of Inhibition between the Human and Thermotoga maritima α-Fucosidases by Fuconojirimycin-Based Derivatives. Biochemistry 2006, 45, 5695–5702. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Clavijo, E.; Carmona, A.T.; Vera-Ayoso, Y.; Moreno-Vargas, A.J.; Bello, C.; Vogel, P.; Robina, I. Synthesis of Novel Pyrrolidine 3,4-Diol Derivatives as Inhibitors of Alpha-L-Fucosidases. Org. Biomol. Chem. 2009, 7, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Kotland, A.; Accadbled, F.; Robeyns, K.; Behr, J.B. Synthesis and Fucosidase Inhibitory Study of Unnatural Pyrrolidine Alkaloid 4-epi-(+)-Codonopsinine. J. Org. Chem. 2011, 76, 4094–4098. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-T.; Lin, T.-C.; Chen, Y.-H.; Lin, C.-H.; Fang, J.-M. Polyhydroxylated Pyrrolidine and 2-Oxapyrrolizidine as Glycosidase Inhibitors. Med. Chem. Comm. 2013, 4, 783. [Google Scholar] [CrossRef]
- McOmie, J.F.W.; Watts, M.L.; West, D.E. Demethylation of Aryl Methyl Ethers by Boron Tribromide. Tetrahedron 1968, 24, 2289–2292. [Google Scholar] [CrossRef]
- Bhatt, V.M.; Babu, J.R. New Reagents 3: Aluminium Iodide-a Highly Regioselective Ether-Cleaving Reagent with Novel Cleavage Pattern. Tetrahedron. Lett. 1984, 25, 3497–3500. [Google Scholar]
- Du, Z.; Lu, J.; Yu, H.; Xu, Y.; Li, A. A Facile Demethylation of Ortho Substituted Aryl Methyl Ethers Promoted by AlCl3. J. Chem. Res. 2010, 2010, 222–227. [Google Scholar] [CrossRef]
- Steiger, C.; Finnema, S.J.; Raus, L.; Schou, M.; Nakao, R.; Suzuki, K.; Pike, V.W.; Wikström, H.V.; Halldin, C. A Two-Step One-Pot Radiosynthesis of the Potent Dopamine D 2/D 3 Agonist PET Radioligand [11 C]MNPA. J. Label. Compd. Radiopharm. 2009, 52, 158–165. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cotton, J.; Goehring, C.M.; Kuehn, A.; Maurer, A.; Fuchs, K.; Pichler, B.J. Synthesis and Biological Evaluation of a Radiolabeled PET (Positron Emission Tomography) Probe for Visualization of In Vivo α-Fucosidase Expression. Pharmaceuticals 2021, 14, 745. https://doi.org/10.3390/ph14080745
Cotton J, Goehring CM, Kuehn A, Maurer A, Fuchs K, Pichler BJ. Synthesis and Biological Evaluation of a Radiolabeled PET (Positron Emission Tomography) Probe for Visualization of In Vivo α-Fucosidase Expression. Pharmaceuticals. 2021; 14(8):745. https://doi.org/10.3390/ph14080745
Chicago/Turabian StyleCotton, Jonathan, Chris Marc Goehring, Anna Kuehn, Andreas Maurer, Kerstin Fuchs, and Bernd J. Pichler. 2021. "Synthesis and Biological Evaluation of a Radiolabeled PET (Positron Emission Tomography) Probe for Visualization of In Vivo α-Fucosidase Expression" Pharmaceuticals 14, no. 8: 745. https://doi.org/10.3390/ph14080745
APA StyleCotton, J., Goehring, C. M., Kuehn, A., Maurer, A., Fuchs, K., & Pichler, B. J. (2021). Synthesis and Biological Evaluation of a Radiolabeled PET (Positron Emission Tomography) Probe for Visualization of In Vivo α-Fucosidase Expression. Pharmaceuticals, 14(8), 745. https://doi.org/10.3390/ph14080745