
Metabolic Host–Microbiota Interactions in Autophagy and the Pathogenesis of Inflammatory Bowel Disease (IBD)

Abstract
:1. Introduction
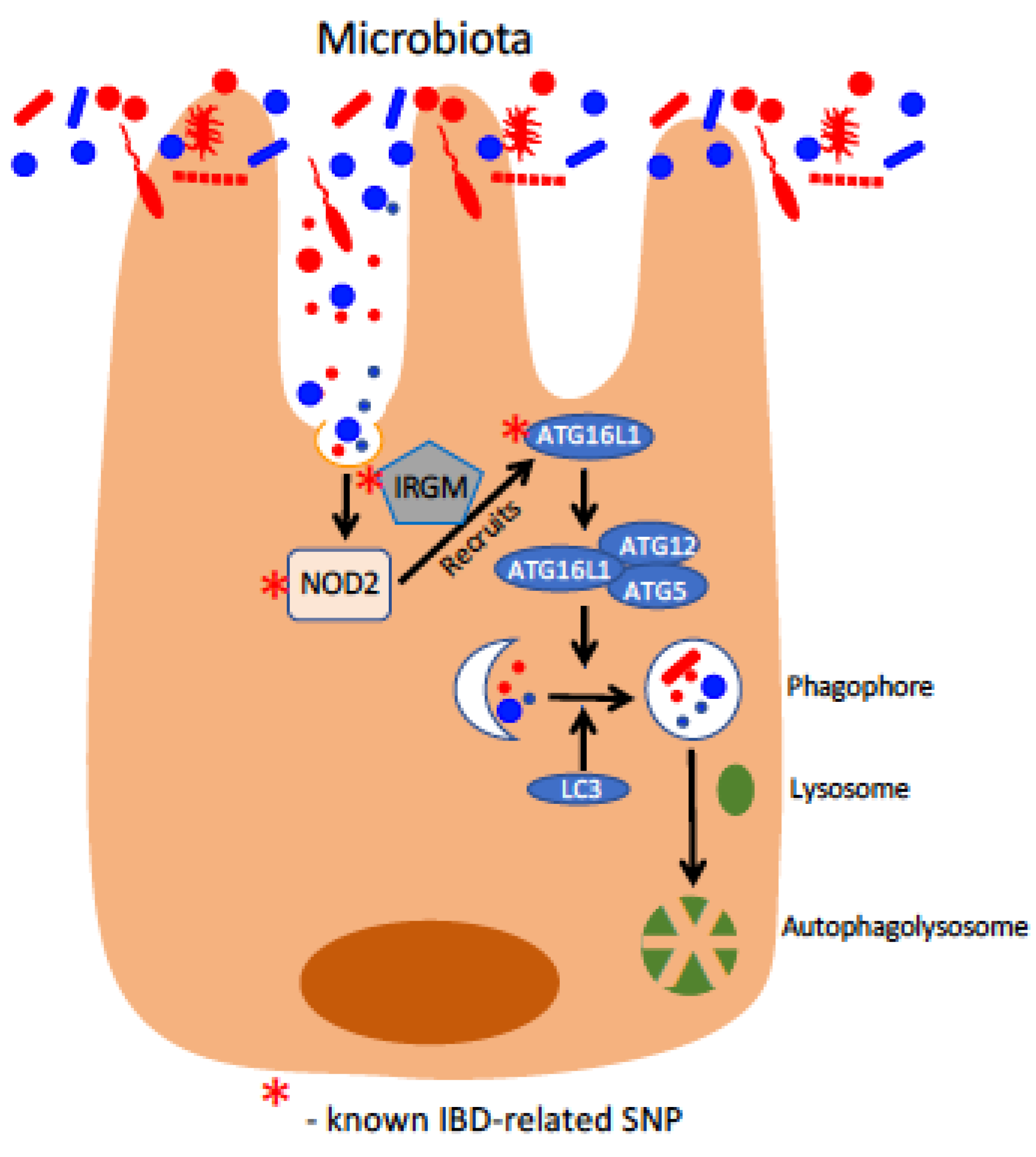
2. Genetic Basis for IBD: Loss of Homeostatic Intestine–Microbe Interactions
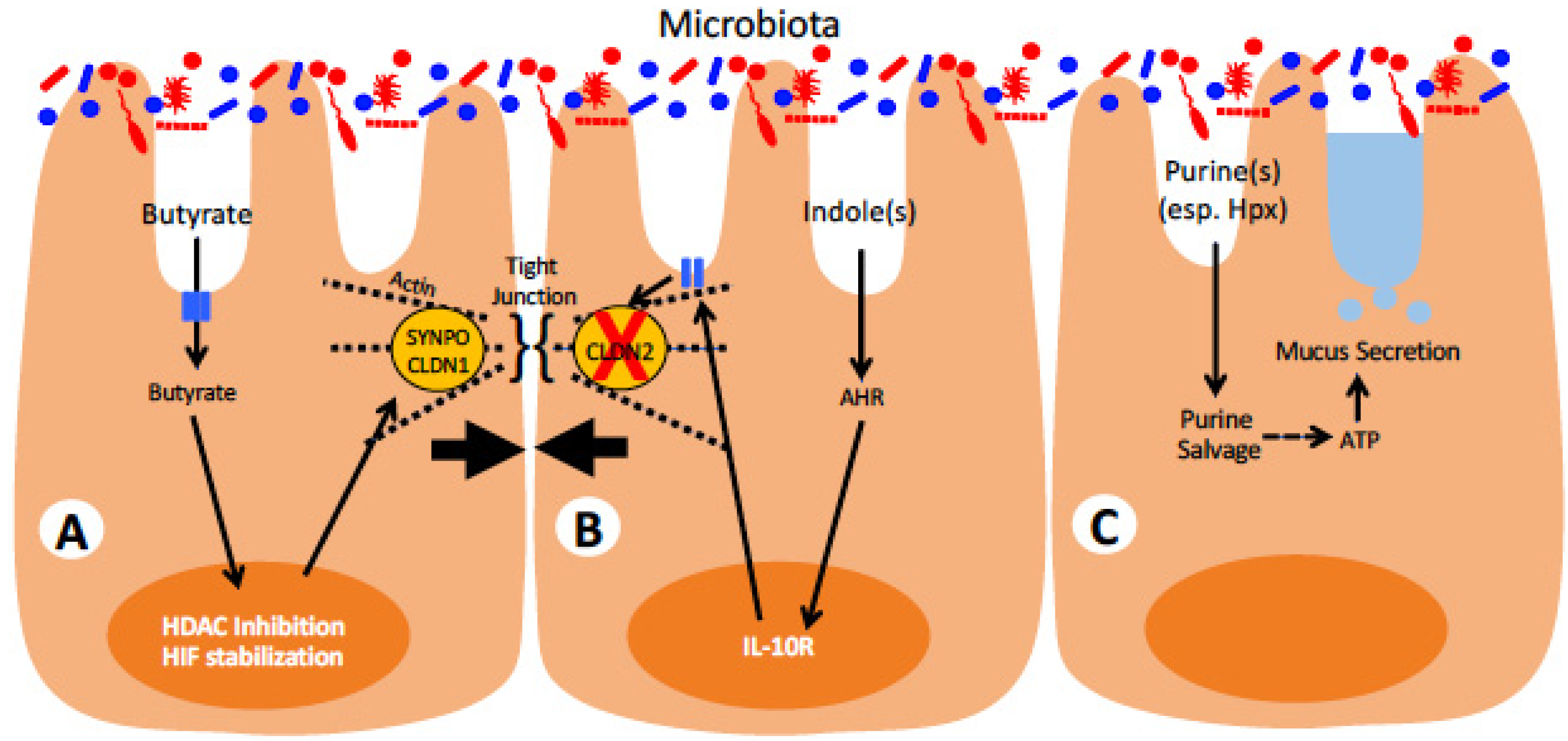
3. Role of Microbiota in Modulating Intestinal Barrier and Inflammation
4. Approaches to Intervene in Modulating Host–Microbe Interactions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sairenji, T.; Collins, K.L.; Evans, D.V. An Update on Inflammatory Bowel Disease. Prim. Care 2017, 44, 673–692. [Google Scholar] [CrossRef]
- Flynn, S.; Eisenstein, S. Inflammatory Bowel Disease Presentation and Diagnosis. Surg. Clin. N. Am. 2019, 99, 1051–1062. [Google Scholar] [CrossRef]
- Zhang, Y.Z.; Li, Y.Y. Inflammatory bowel disease: Pathogenesis. World J. Gastroenterol. 2014, 20, 91–99. [Google Scholar] [CrossRef]
- Windsor, J.W.; Kaplan, G.G. Evolving Epidemiology of IBD. Curr. Gastroenterol. Rep. 2019, 21, 40. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N. Epidemiology and risk factors for IBD. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 205–217. [Google Scholar] [CrossRef]
- Liu, J.Z.; van Sommeren, S.; Huang, H.; Ng, S.C.; Alberts, R.; Takahashi, A.; Ripke, S.; Lee, J.C.; Jostins, L.; Shah, T.; et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015, 47, 979–986. [Google Scholar] [CrossRef]
- Park, S.C.; Jeen, Y.T. Genetic Studies of Inflammatory Bowel Disease-Focusing on Asian Patients. Cells 2019, 8, 404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mentella, M.C.; Scaldaferri, F.; Pizzoferrato, M.; Gasbarrini, A.; Miggiano, G.A.D. Nutrition, IBD and Gut Microbiota: A Review. Nutrients 2020, 12, 944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ananthakrishnan, A.N.; Bernstein, C.N.; Iliopoulos, D.; Macpherson, A.; Neurath, M.F.; Ali, R.A.R.; Vavricka, S.R.; Fiocchi, C. Environmental triggers in IBD: A review of progress and evidence. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 39–49. [Google Scholar] [CrossRef]
- Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nature 2007, 447, 661–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libioulle, C.; Louis, E.; Hansoul, S.; Sandor, C.; Farnir, F.; Franchimont, D.; Vermeire, S.; Dewit, O.; de Vos, M.; Dixon, A.; et al. Novel Crohn disease locus identified by genome-wide association maps to a gene desert on 5p13.1 and modulates expression of PTGER4. PLoS Genet. 2007, 3, e58. [Google Scholar] [CrossRef] [PubMed]
- Lavelle, A.; Sokol, H. Gut microbiota-derived metabolites as key actors in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Parent, K.; Mitchell, P. Cell wall-defective variants of pseudomonas-like (group Va) bacteria in Crohn’s disease. Gastroenterology 1978, 75, 368–372. [Google Scholar] [CrossRef]
- Frank, D.N.; St Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, D.A.; Frank, D.N.; Pace, N.R.; Gordon, J.I. Metagenomic approaches for defining the pathogenesis of inflammatory bowel diseases. Cell Host Microbe 2008, 3, 417–427. [Google Scholar] [CrossRef] [Green Version]
- Walker, A.W.; Sanderson, J.D.; Churcher, C.; Parkes, G.C.; Hudspith, B.N.; Rayment, N.; Brostoff, J.; Parkhill, J.; Dougan, G.; Petrovska, L. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol. 2011, 11, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manichanh, C.; Rigottier-Gois, L.; Bonnaud, E.; Gloux, K.; Pelletier, E.; Frangeul, L.; Nalin, R.; Jarrin, C.; Chardon, P.; Marteau, P.; et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 2006, 55, 205–211. [Google Scholar] [CrossRef] [Green Version]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J.; et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019, 4, 293–305. [Google Scholar] [CrossRef]
- Jacobs, J.P.; Goudarzi, M.; Singh, N.; Tong, M.; McHardy, I.H.; Ruegger, P.; Asadourian, M.; Moon, B.H.; Ayson, A.; Borneman, J.; et al. A Disease-Associated Microbial and Metabolomics State in Relatives of Pediatric Inflammatory Bowel Disease Patients. Cell Mol. Gastroenterol. Hepatol. 2016, 2, 750–766. [Google Scholar] [CrossRef] [Green Version]
- Mosli, M.H.; Zou, G.; Garg, S.K.; Feagan, S.G.; MacDonald, J.K.; Chande, N.; Sandborn, W.J.; Feagan, B.G. C-Reactive Protein, Fecal Calprotectin, and Stool Lactoferrin for Detection of Endoscopic Activity in Symptomatic Inflammatory Bowel Disease Patients: A Systematic Review and Meta-Analysis. Am. J. Gastroenterol. 2015, 110, 802–819. [Google Scholar] [CrossRef] [PubMed]
- Vermeire, S.; Peeters, M.; Vlietinck, R.; Joossens, S.; Den Hond, E.; Bulteel, V.; Bossuyt, X.; Geypens, B.; Rutgeerts, P. Anti-Saccharomyces cerevisiae antibodies (ASCA), phenotypes of IBD, and intestinal permeability: A study in IBD families. Inflamm. Bowel. Dis. 2001, 7, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Irvine, E.J.; Marshall, J.K. Increased intestinal permeability precedes the onset of Crohn’s disease in a subject with familial risk. Gastroenterology 2000, 119, 1740–1744. [Google Scholar] [CrossRef] [PubMed]
- Orholm, M.; Munkholm, P.; Langholz, E.; Nielsen, O.H.; Sorensen, T.I.; Binder, V. Familial occurrence of inflammatory bowel disease. N. Engl. J. Med. 1991, 324, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Hugot, J.P.; Chamaillard, M.; Zouali, H.; Lesage, S.; Cezard, J.P.; Belaiche, J.; Almer, S.; Tysk, C.; O’Morain, C.A.; Gassull, M.; et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 2001, 411, 599–603. [Google Scholar] [CrossRef]
- Ogura, Y.; Bonen, D.K.; Inohara, N.; Nicolae, D.L.; Chen, F.F.; Ramos, R.; Britton, H.; Moran, T.; Karaliuskas, R.; Duerr, R.H.; et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 2001, 411, 603–606. [Google Scholar] [CrossRef]
- Hugot, J.P.; Laurent-Puig, P.; Gower-Rousseau, C.; Olson, J.M.; Lee, J.C.; Beaugerie, L.; Naom, I.; Dupas, J.L.; Van Gossum, A.; Orholm, M.; et al. Mapping of a susceptibility locus for Crohn’s disease on chromosome 16. Nature 1996, 379, 821–823. [Google Scholar] [CrossRef] [PubMed]
- Inohara, N.; Ogura, Y.; Fontalba, A.; Gutierrez, O.; Pons, F.; Crespo, J.; Fukase, K.; Inamura, S.; Kusumoto, S.; Hashimoto, M.; et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J. Biol. Chem. 2003, 278, 5509–5512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabeen, M.T.; Jacobs-Wagner, C. Bacterial cell shape. Nat. Rev. Microbiol. 2005, 3, 601–610. [Google Scholar] [CrossRef]
- Caruso, R.; Warner, N.; Inohara, N.; Nunez, G. NOD1 and NOD2: Signaling, host defense, and inflammatory disease. Immunity 2014, 41, 898–908. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S.; Ma, X. Role of Nod2 in the development of Crohn’s disease. Microbes Infect. 2009, 11, 912–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonen, D.K.; Ogura, Y.; Nicolae, D.L.; Inohara, N.; Saab, L.; Tanabe, T.; Chen, F.F.; Foster, S.J.; Duerr, R.H.; Brant, S.R.; et al. Crohn’s disease-associated NOD2 variants share a signaling defect in response to lipopolysaccharide and peptidoglycan. Gastroenterology 2003, 124, 140–146. [Google Scholar] [CrossRef]
- Sidiq, T.; Yoshihama, S.; Downs, I.; Kobayashi, K.S. Nod2: A Critical Regulator of Ileal Microbiota and Crohn’s Disease. Front. Immunol. 2016, 7, 367. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.H.; Lam, W.; Ma, D.L.; Gullen, E.A.; Cheng, Y.C. Butyrate mediates nucleotide-binding and oligomerisation domain (NOD) 2-dependent mucosal immune responses against peptidoglycan. Eur. J. Immunol. 2009, 39, 3529–3537. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, O.; Pipaon, C.; Inohara, N.; Fontalba, A.; Ogura, Y.; Prosper, F.; Nunez, G.; Fernandez-Luna, J.L. Induction of Nod2 in myelomonocytic and intestinal epithelial cells via nuclear factor-kappa B activation. J. Biol. Chem. 2002, 277, 41701–41705. [Google Scholar] [CrossRef] [Green Version]
- Wehkamp, J.; Harder, J.; Weichenthal, M.; Schwab, M.; Schaffeler, E.; Schlee, M.; Herrlinger, K.R.; Stallmach, A.; Noack, F.; Fritz, P.; et al. NOD2 (CARD15) mutations in Crohn’s disease are associated with diminished mucosal alpha-defensin expression. Gut 2004, 53, 1658–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petnicki-Ocwieja, T.; Hrncir, T.; Liu, Y.J.; Biswas, A.; Hudcovic, T.; Tlaskalova-Hogenova, H.; Kobayashi, K.S. Nod2 is required for the regulation of commensal microbiota in the intestine. Proc. Natl. Acad. Sci. USA 2009, 106, 15813–15818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramanan, D.; Tang, M.S.; Bowcutt, R.; Loke, P.; Cadwell, K. Bacterial sensor Nod2 prevents inflammation of the small intestine by restricting the expansion of the commensal Bacteroides vulgatus. Immunity 2014, 41, 311–324. [Google Scholar] [CrossRef] [Green Version]
- Cooney, R.; Baker, J.; Brain, O.; Danis, B.; Pichulik, T.; Allan, P.; Ferguson, D.J.; Campbell, B.J.; Jewell, D.; Simmons, A. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat. Med. 2010, 16, 90–97. [Google Scholar] [CrossRef]
- Rahman, M.K.; Midtling, E.H.; Svingen, P.A.; Xiong, Y.; Bell, M.P.; Tung, J.; Smyrk, T.; Egan, L.J.; Faubion, W.A., Jr. The pathogen recognition receptor NOD2 regulates human FOXP3+ T cell survival. J. Immunol. 2010, 184, 7247–7256. [Google Scholar] [CrossRef] [Green Version]
- Umiker, B.; Lee, H.H.; Cope, J.; Ajami, N.J.; Laine, J.P.; Fregeau, C.; Ferguson, H.; Alves, S.E.; Sciammetta, N.; Kleinschek, M.; et al. The NLRP3 inflammasome mediates DSS-induced intestinal inflammation in Nod2 knockout mice. Innate. Immun. 2019, 25, 132–143. [Google Scholar] [CrossRef] [Green Version]
- Barreau, F.; Meinzer, U.; Chareyre, F.; Berrebi, D.; Niwa-Kawakita, M.; Dussaillant, M.; Foligne, B.; Ollendorff, V.; Heyman, M.; Bonacorsi, S.; et al. CARD15/NOD2 is required for Peyer’s patches homeostasis in mice. PLoS ONE 2007, 2, e523. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.G.; Kamada, N.; Shaw, M.H.; Warner, N.; Chen, G.Y.; Franchi, L.; Nunez, G. The Nod2 sensor promotes intestinal pathogen eradication via the chemokine CCL2-dependent recruitment of inflammatory monocytes. Immunity 2011, 34, 769–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampe, J.; Franke, A.; Rosenstiel, P.; Till, A.; Teuber, M.; Huse, K.; Albrecht, M.; Mayr, G.; De La Vega, F.M.; Briggs, J.; et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 2007, 39, 207–211. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B. Autophagy in Human Diseases. N. Engl. J. Med. 2020, 383, 1564–1576. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Levine, B. Autosis and autophagic cell death: The dark side of autophagy. Cell Death Differ. 2015, 22, 367–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Noda, T.; Ohsumi, Y. Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway. EMBO J. 1999, 18, 3888–3896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lystad, A.H.; Carlsson, S.R.; Simonsen, A. Toward the function of mammalian ATG12-ATG5-ATG16L1 complex in autophagy and related processes. Autophagy 2019, 15, 1485–1486. [Google Scholar] [CrossRef] [PubMed]
- Rioux, J.D.; Xavier, R.J.; Taylor, K.D.; Silverberg, M.S.; Goyette, P.; Huett, A.; Green, T.; Kuballa, P.; Barmada, M.M.; Datta, L.W.; et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 2007, 39, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Cadwell, K.; Liu, J.Y.; Brown, S.L.; Miyoshi, H.; Loh, J.; Lennerz, J.K.; Kishi, C.; Kc, W.; Carrero, J.A.; Hunt, S.; et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008, 456, 259–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lassen, K.G.; Kuballa, P.; Conway, K.L.; Patel, K.K.; Becker, C.E.; Peloquin, J.M.; Villablanca, E.J.; Norman, J.M.; Liu, T.C.; Heath, R.J.; et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc. Natl. Acad. Sci. USA 2014, 111, 7741–7746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuballa, P.; Huett, A.; Rioux, J.D.; Daly, M.J.; Xavier, R.J. Impaired autophagy of an intracellular pathogen induced by a Crohn’s disease associated ATG16L1 variant. PLoS ONE 2008, 3, e3391. [Google Scholar] [CrossRef] [PubMed]
- Murthy, A.; Li, Y.; Peng, I.; Reichelt, M.; Katakam, A.K.; Noubade, R.; Roose-Girma, M.; DeVoss, J.; Diehl, L.; Graham, R.R.; et al. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature 2014, 506, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Homer, C.R.; Richmond, A.L.; Rebert, N.A.; Achkar, J.P.; McDonald, C. ATG16L1 and NOD2 interact in an autophagy-dependent antibacterial pathway implicated in Crohn’s disease pathogenesis. Gastroenterology 2010, 139, 1630–1641.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travassos, L.H.; Carneiro, L.A.; Ramjeet, M.; Hussey, S.; Kim, Y.G.; Magalhaes, J.G.; Yuan, L.; Soares, F.; Chea, E.; Le Bourhis, L.; et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 2010, 11, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Plantinga, T.S.; Crisan, T.O.; Oosting, M.; van de Veerdonk, F.L.; de Jong, D.J.; Philpott, D.J.; van der Meer, J.W.; Girardin, S.E.; Joosten, L.A.; Netea, M.G. Crohn’s disease-associated ATG16L1 polymorphism modulates pro-inflammatory cytokine responses selectively upon activation of NOD2. Gut 2011, 60, 1229–1235. [Google Scholar] [CrossRef]
- Sorbara, M.T.; Ellison, L.K.; Ramjeet, M.; Travassos, L.H.; Jones, N.L.; Girardin, S.E.; Philpott, D.J. The protein ATG16L1 suppresses inflammatory cytokines induced by the intracellular sensors Nod1 and Nod2 in an autophagy-independent manner. Immunity 2013, 39, 858–873. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Green, D.R. Autophagy-Independent Functions of the Autophagy Machinery. Cell 2019, 177, 1682–1699. [Google Scholar] [CrossRef]
- Kabat, A.M.; Harrison, O.J.; Riffelmacher, T.; Moghaddam, A.E.; Pearson, C.F.; Laing, A.; Abeler-Dorner, L.; Forman, S.P.; Grencis, R.K.; Sattentau, Q.; et al. The autophagy gene Atg16l1 differentially regulates Treg and TH2 cells to control intestinal inflammation. Elife 2016, 5, e12444. [Google Scholar] [CrossRef]
- Parkes, M.; Barrett, J.C.; Prescott, N.J.; Tremelling, M.; Anderson, C.A.; Fisher, S.A.; Roberts, R.G.; Nimmo, E.R.; Cummings, F.R.; Soars, D.; et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat. Genet. 2007, 39, 830–832. [Google Scholar] [CrossRef]
- Bekpen, C.; Hunn, J.P.; Rohde, C.; Parvanova, I.; Guethlein, L.; Dunn, D.M.; Glowalla, E.; Leptin, M.; Howard, J.C. The interferon-inducible p47 (IRG) GTPases in vertebrates: Loss of the cell autonomous resistance mechanism in the human lineage. Genome Biol. 2005, 6, R92. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.B.; Davis, A.S.; Taylor, G.A.; Deretic, V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 2006, 313, 1438–1441. [Google Scholar] [CrossRef]
- Collazo, C.M.; Yap, G.S.; Sempowski, G.D.; Lusby, K.C.; Tessarollo, L.; Vande Woude, G.F.; Sher, A.; Taylor, G.A. Inactivation of LRG-47 and IRG-47 reveals a family of interferon gamma-inducible genes with essential, pathogen-specific roles in resistance to infection. J. Exp. Med. 2001, 194, 181–188. [Google Scholar] [CrossRef] [Green Version]
- Brest, P.; Lapaquette, P.; Souidi, M.; Lebrigand, K.; Cesaro, A.; Vouret-Craviari, V.; Mari, B.; Barbry, P.; Mosnier, J.F.; Hebuterne, X.; et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease. Nat. Genet. 2011, 43, 242–245. [Google Scholar] [CrossRef]
- McCarroll, S.A.; Huett, A.; Kuballa, P.; Chilewski, S.D.; Landry, A.; Goyette, P.; Zody, M.C.; Hall, J.L.; Brant, S.R.; Cho, J.H.; et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease. Nat. Genet. 2008, 40, 1107–1112. [Google Scholar] [CrossRef]
- Gregoire, I.P.; Richetta, C.; Meyniel-Schicklin, L.; Borel, S.; Pradezynski, F.; Diaz, O.; Deloire, A.; Azocar, O.; Baguet, J.; Le Breton, M.; et al. IRGM is a common target of RNA viruses that subvert the autophagy network. PLoS Pathog. 2011, 7, e1002422. [Google Scholar] [CrossRef] [PubMed]
- Mehto, S.; Jena, K.K.; Nath, P.; Chauhan, S.; Kolapalli, S.P.; Das, S.K.; Sahoo, P.K.; Jain, A.; Taylor, G.A.; Chauhan, S. The Crohn’s Disease Risk Factor IRGM Limits NLRP3 Inflammasome Activation by Impeding Its Assembly and by Mediating Its Selective Autophagy. Mol. Cell 2019, 73, 429–445.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, S.; Mandell, M.A.; Deretic, V. IRGM governs the core autophagy machinery to conduct antimicrobial defense. Mol. Cell 2015, 58, 507–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.B.; Ornatowski, W.; Vergne, I.; Naylor, J.; Delgado, M.; Roberts, E.; Ponpuak, M.; Master, S.; Pilli, M.; White, E.; et al. Human IRGM regulates autophagy and cell-autonomous immunity functions through mitochondria. Nat. Cell Biol. 2010, 12, 1154–1165. [Google Scholar] [CrossRef] [Green Version]
- Lapaquette, P.; Glasser, A.L.; Huett, A.; Xavier, R.J.; Darfeuille-Michaud, A. Crohn’s disease-associated adherent-invasive E. coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell Microbiol. 2010, 12, 99–113. [Google Scholar] [CrossRef] [Green Version]
- Darfeuille-Michaud, A.; Boudeau, J.; Bulois, P.; Neut, C.; Glasser, A.L.; Barnich, N.; Bringer, M.A.; Swidsinski, A.; Beaugerie, L.; Colombel, J.F. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology 2004, 127, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Heim, V.J.; Dagley, L.F.; Stafford, C.A.; Hansen, F.M.; Clayer, E.; Bankovacki, A.; Webb, A.I.; Lucet, I.S.; Silke, J.; Nachbur, U. A regulatory region on RIPK2 is required for XIAP binding and NOD signaling activity. EMBO Rep. 2020, 21, e50400. [Google Scholar] [CrossRef]
- Kobayashi, K.; Inohara, N.; Hernandez, L.D.; Galan, J.E.; Nunez, G.; Janeway, C.A.; Medzhitov, R.; Flavell, R.A. RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature 2002, 416, 194–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eng, V.V.; Wemyss, M.A.; Pearson, J.S. The diverse roles of RIP kinases in host-pathogen interactions. Semin. Cell Dev. Biol. 2021, 109, 125–143. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.K.; Tait, S.W.; Lamkanfi, M.; Amer, A.O.; Nunez, G.; Pages, G.; Pouyssegur, J.; McGargill, M.A.; Green, D.R.; Kanneganti, T.D. TLR2 and RIP2 pathways mediate autophagy of Listeria monocytogenes via extracellular signal-regulated kinase (ERK) activation. J. Biol. Chem. 2011, 286, 42981–42991. [Google Scholar] [CrossRef] [Green Version]
- Topal, Y.; Gyrd-Hansen, M. RIPK2 NODs to XIAP and IBD. Semin. Cell Dev. Biol. 2021, 109, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Zeissig, Y.; Petersen, B.S.; Milutinovic, S.; Bosse, E.; Mayr, G.; Peuker, K.; Hartwig, J.; Keller, A.; Kohl, M.; Laass, M.W.; et al. XIAP variants in male Crohn’s disease. Gut 2015, 64, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Lekbua, A.; Ouahed, J.; O’Connell, A.E.; Kahn, S.A.; Goldsmith, J.D.; Imamura, T.; Duncan, C.N.; Kelsen, J.R.; Worthey, E.; Snapper, S.B.; et al. Risk-factors Associated with Poor Outcomes in VEO-IBD Secondary to XIAP Deficiency: A Case Report and Literature Review. J. Pediatr. Gastroenterol. Nutr. 2019, 69, e13–e18. [Google Scholar] [CrossRef]
- Graham, D.B.; Xavier, R.J. Pathway paradigms revealed from the genetics of inflammatory bowel disease. Nature 2020, 578, 527–539. [Google Scholar] [CrossRef]
- Marcil, V.; Sinnett, D.; Seidman, E.; Boudreau, F.; Gendron, F.P.; Beaulieu, J.F.; Menard, D.; Lambert, M.; Bitton, A.; Sanchez, R.; et al. Association between genetic variants in the HNF4A gene and childhood-onset Crohn’s disease. Genes Immun. 2012, 13, 556–565. [Google Scholar] [CrossRef] [Green Version]
- Prager, M.; Buttner, J.; Buning, C. PTGER4 modulating variants in Crohn’s disease. Int. J. Colorectal. Dis. 2014, 29, 909–915. [Google Scholar] [CrossRef]
- Glas, J.; Seiderer, J.; Wagner, J.; Olszak, T.; Fries, C.; Tillack, C.; Friedrich, M.; Beigel, F.; Stallhofer, J.; Steib, C.; et al. Analysis of IL12B gene variants in inflammatory bowel disease. PLoS ONE 2012, 7, e34349. [Google Scholar] [CrossRef]
- Diaz-Gallo, L.M.; Palomino-Morales, R.J.; Gomez-Garcia, M.; Cardena, C.; Rodrigo, L.; Nieto, A.; Alcain, G.; Cueto, I.; Lopez-Nevot, M.A.; Martin, J. STAT4 gene influences genetic predisposition to ulcerative colitis but not Crohn’s disease in the Spanish population: A replication study. Hum. Immunol. 2010, 71, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Akyuz, F.; Besisik, F.; Ustek, D.; Ekmekci, C.; Uyar, A.; Pinarbasi, B.; Demir, K.; Ozdil, S.; Kaymakoglu, S.; Boztas, G.; et al. Association of the MEFV gene variations with inflammatory bowel disease in Turkey. J. Clin. Gastroenterol. 2013, 47, e23–e27. [Google Scholar] [CrossRef] [PubMed]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Perencevich, M.; Burakoff, R. Use of antibiotics in the treatment of inflammatory bowel disease. Inflamm. Bowel Dis. 2006, 12, 651–664. [Google Scholar] [CrossRef] [PubMed]
- Guinane, C.M.; Cotter, P.D. Role of the gut microbiota in health and chronic gastrointestinal disease: Understanding a hidden metabolic organ. Therap. Adv. Gastroenterol. 2013, 6, 295–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sellon, R.K.; Tonkonogy, S.; Schultz, M.; Dieleman, L.A.; Grenther, W.; Balish, E.; Rennick, D.M.; Sartor, R.B. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect. Immun. 1998, 66, 5224–5231. [Google Scholar] [CrossRef] [Green Version]
- Aranda, R.; Sydora, B.C.; McAllister, P.L.; Binder, S.W.; Yang, H.Y.; Targan, S.R.; Kronenberg, M. Analysis of intestinal lymphocytes in mouse colitis mediated by transfer of CD4+, CD45RBhigh T cells to SCID recipients. J. Immunol. 1997, 158, 3464–3473. [Google Scholar] [PubMed]
- Pastor Rojo, O.; Lopez San Roman, A.; Albeniz Arbizu, E.; de la Hera Martinez, A.; Ripoll Sevillano, E.; Albillos Martinez, A. Serum lipopolysaccharide-binding protein in endotoxemic patients with inflammatory bowel disease. Inflamm. Bowel Dis. 2007, 13, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Lakatos, P.L.; Kiss, L.S.; Palatka, K.; Altorjay, I.; Antal-Szalmas, P.; Palyu, E.; Udvardy, M.; Molnar, T.; Farkas, K.; Veres, G.; et al. Serum lipopolysaccharide-binding protein and soluble CD14 are markers of disease activity in patients with Crohn’s disease. Inflamm. Bowel Dis. 2011, 17, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.; Frances, R.; Amoros, A.; Zapater, P.; Garmendia, M.; Ndongo, M.; Cano, R.; Jover, R.; Such, J.; Perez-Mateo, M. Cytokine association with bacterial DNA in serum of patients with inflammatory bowel disease. Inflamm. Bowel Dis. 2009, 15, 508–514. [Google Scholar] [CrossRef]
- Caruso, R.; Lo, B.C.; Nunez, G. Host-microbiota interactions in inflammatory bowel disease. Nat. Rev. Immunol. 2020, 20, 411–426. [Google Scholar] [CrossRef] [PubMed]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef] [Green Version]
- Dalal, S.R.; Chang, E.B. The microbial basis of inflammatory bowel diseases. J. Clin. Investig. 2014, 124, 4190–4196. [Google Scholar] [CrossRef]
- Axelrad, J.E.; Cadwell, K.H.; Colombel, J.F.; Shah, S.C. Systematic review: Gastrointestinal infection and incident inflammatory bowel disease. Aliment. Pharmacol. Ther. 2020, 51, 1222–1232. [Google Scholar] [CrossRef]
- Yu, Y.; Zhu, S.; Li, P.; Min, L.; Zhang, S. Helicobacter pylori infection and inflammatory bowel disease: A crosstalk between upper and lower digestive tract. Cell Death Dis. 2018, 9, 961. [Google Scholar] [CrossRef] [PubMed]
- Wroblewski, L.E.; Peek, R.M., Jr.; Wilson, K.T. Helicobacter pylori and gastric cancer: Factors that modulate disease risk. Clin. Microbiol. Rev. 2010, 23, 713–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duvallet, C.; Gibbons, S.M.; Gurry, T.; Irizarry, R.A.; Alm, E.J. Meta-analysis of gut microbiome studies identifies disease-specific and shared responses. Nat. Commun. 2017, 8, 1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostic, A.D.; Xavier, R.J.; Gevers, D. The microbiome in inflammatory bowel disease: Current status and the future ahead. Gastroenterology 2014, 146, 1489–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fangmann, D.; Theismann, E.M.; Turk, K.; Schulte, D.M.; Relling, I.; Hartmann, K.; Keppler, J.K.; Knipp, J.R.; Rehman, A.; Heinsen, F.A.; et al. Targeted Microbiome Intervention by Microencapsulated Delayed-Release Niacin Beneficially Affects Insulin Sensitivity in Humans. Diabetes Care 2018, 41, 398–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalile, B.; Van Oudenhove, L.; Vervliet, B.; Verbeke, K. The role of short-chain fatty acids in microbiota-gut-brain communication. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 461–478. [Google Scholar] [CrossRef]
- Wang, R.X.; Lee, J.S.; Campbell, E.L.; Colgan, S.P. Microbiota-derived butyrate dynamically regulates intestinal homeostasis through regulation of actin-associated protein synaptopodin. Proc. Natl. Acad. Sci. USA 2020, 117, 11648–11657. [Google Scholar] [CrossRef]
- Roediger, W.E. Role of anaerobic bacteria in the metabolic welfare of the colonic mucosa in man. Gut 1980, 21, 793–798. [Google Scholar] [CrossRef] [Green Version]
- Roediger, W.E. Utilization of nutrients by isolated epithelial cells of the rat colon. Gastroenterology 1982, 83, 424–429. [Google Scholar] [CrossRef]
- Donohoe, D.R.; Garge, N.; Zhang, X.; Sun, W.; O’Connell, T.M.; Bunger, M.K.; Bultman, S.J. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011, 13, 517–526. [Google Scholar] [CrossRef] [Green Version]
- Roediger, W.E. The colonic epithelium in ulcerative colitis: An energy-deficiency disease? Lancet 1980, 2, 712–715. [Google Scholar] [CrossRef]
- Cummins, E.P.; Seeballuck, F.; Keely, S.J.; Mangan, N.E.; Callanan, J.J.; Fallon, P.G.; Taylor, C.T. The hydroxylase inhibitor dimethyloxalylglycine is protective in a murine model of colitis. Gastroenterology 2008, 134, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.J.; Zheng, L.; Campbell, E.L.; Saeedi, B.; Scholz, C.C.; Bayless, A.J.; Wilson, K.E.; Glover, L.E.; Kominsky, D.J.; Magnuson, A.; et al. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe 2015, 17, 662–671. [Google Scholar] [CrossRef] [Green Version]
- Robinson, A.; Keely, S.; Karhausen, J.; Gerich, M.E.; Furuta, G.T.; Colgan, S.P. Mucosal protection by hypoxia-inducible factor prolyl hydroxylase inhibition. Gastroenterology 2008, 134, 145–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saeedi, B.J.; Kao, D.J.; Kitzenberg, D.A.; Dobrinskikh, E.; Schwisow, K.D.; Masterson, J.C.; Kendrick, A.A.; Kelly, C.J.; Bayless, A.J.; Kominsky, D.J.; et al. HIF-dependent regulation of claudin-1 is central to intestinal epithelial tight junction integrity. Mol. Biol. Cell. 2015, 26, 2252–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.X.; Henen, M.A.; Lee, J.S.; Vogeli, B.; Colgan, S.P. Microbiota-derived butyrate is an endogenous HIF prolyl hydroxylase inhibitor. Gut Microbes 2021, in press. [Google Scholar] [CrossRef]
- Zheng, L.; Kelly, C.J.; Battista, K.D.; Schaefer, R.; Lanis, J.M.; Alexeev, E.E.; Wang, R.X.; Onyiah, J.C.; Kominsky, D.J.; Colgan, S.P. Microbial-Derived Butyrate Promotes Epithelial Barrier Function through IL-10 Receptor-Dependent Repression of Claudin-2. J. Immunol. 2017, 199, 2976–2984. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly, Y.M.; Glickman, J.N.; Garrett, W.S. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Correa-Oliveira, R.; Fachi, J.L.; Vieira, A.; Sato, F.T.; Vinolo, M.A. Regulation of immune cell function by short-chain fatty acids. Clin. Transl. Immunol. 2016, 5, e73. [Google Scholar] [CrossRef] [PubMed]
- Gantois, I.; Ducatelle, R.; Pasmans, F.; Haesebrouck, F.; Hautefort, I.; Thompson, A.; Hinton, J.C.; Van Immerseel, F. Butyrate specifically down-regulates salmonella pathogenicity island 1 gene expression. Appl. Environ. Microbiol. 2006, 72, 946–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byndloss, M.X.; Olsan, E.E.; Rivera-Chavez, F.; Tiffany, C.R.; Cevallos, S.A.; Lokken, K.L.; Torres, T.P.; Byndloss, A.J.; Faber, F.; Gao, Y.; et al. Microbiota-activated PPAR-gamma signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 2017, 357, 570–575. [Google Scholar] [CrossRef]
- Rigottier-Gois, L. Dysbiosis in inflammatory bowel diseases: The oxygen hypothesis. ISME J. 2013, 7, 1256–1261. [Google Scholar] [CrossRef]
- Fellows, R.; Denizot, J.; Stellato, C.; Cuomo, A.; Jain, P.; Stoyanova, E.; Balazsi, S.; Hajnady, Z.; Liebert, A.; Kazakevych, J.; et al. Microbiota derived short chain fatty acids promote histone crotonylation in the colon through histone deacetylases. Nat. Commun. 2018, 9, 105. [Google Scholar] [CrossRef]
- Sivaprakasam, S.; Bhutia, Y.D.; Yang, S.; Ganapathy, V. Short-Chain Fatty Acid Transporters: Role in Colonic Homeostasis. Compr. Physiol. 2017, 8, 299–314. [Google Scholar] [CrossRef]
- Priyadarshini, M.; Kotlo, K.U.; Dudeja, P.K.; Layden, B.T. Role of Short Chain Fatty Acid Receptors in Intestinal Physiology and Pathophysiology. Compr. Physiol. 2018, 8, 1091–1115. [Google Scholar] [CrossRef]
- Melhem, H.; Kaya, B.; Ayata, C.K.; Hruz, P.; Niess, J.H. Metabolite-Sensing G Protein-Coupled Receptors Connect the Diet-Microbiota-Metabolites Axis to Inflammatory Bowel Disease. Cells 2019, 8, 450. [Google Scholar] [CrossRef] [Green Version]
- Husted, A.S.; Trauelsen, M.; Rudenko, O.; Hjorth, S.A.; Schwartz, T.W. GPCR-Mediated Signaling of Metabolites. Cell Metab. 2017, 25, 777–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundmann, M.; Bender, E.; Schamberger, J.; Eitner, F. Pharmacology of Free Fatty Acid Receptors and Their Allosteric Modulators. Int. J. Mol. Sci. 2021, 22, 1763. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Wang, R.X.; Alexeev, E.E.; Lanis, J.M.; Battista, K.D.; Glover, L.E.; Colgan, S.P. Hypoxanthine is a checkpoint stress metabolite in colonic epithelial energy modulation and barrier function. J. Biol. Chem. 2018, 293, 6039–6051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glover, L.E.; Bowers, B.E.; Saeedi, B.; Ehrentraut, S.F.; Campbell, E.L.; Bayless, A.J.; Dobrinskikh, E.; Kendrick, A.A.; Kelly, C.J.; Burgess, A.; et al. Control of creatine metabolism by HIF is an endogenous mechanism of barrier regulation in colitis. Proc. Natl. Acad. Sci. USA 2013, 110, 19820–19825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, C.H.T.; Lee, J.S.; Murphy, E.M.; Gerich, M.E.; Dran, R.; Glover, L.E.; Abdulla, Z.I.; Skelton, M.R.; Colgan, S.P. Creatine Transporter, Reduced in Colon Tissues from Patients with Inflammatory Bowel Diseases, Regulates Energy Balance in Intestinal Epithelial Cells, Epithelial Integrity, and Barrier Function. Gastroenterology 2020, 159, 984–998.e1. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Wang, R.X.; Goldberg, M.S.; Clifford, G.P.; Kao, D.J.; Colgan, S.P. Microbiota-Sourced Purines Support Wound Healing and Mucous Barrier Function. iScience 2020, 23, 101226. [Google Scholar] [CrossRef]
- Lee, J.S.; Wang, R.X.; Alexeev, E.E.; Colgan, S.P. Intestinal Inflammation as a Dysbiosis of Energy Procurement: New Insights into an Old Topic. Gut Microbes 2021, 13, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef] [Green Version]
- Roager, H.M.; Licht, T.R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 2018, 9, 3294. [Google Scholar] [CrossRef] [Green Version]
- Stockinger, B.; Shah, K.; Wincent, E. AHR in the intestinal microenvironment: Safeguarding barrier function. Nat. Rev. Gastroenterol. Hepatol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Lanis, J.M.; Alexeev, E.E.; Curtis, V.F.; Kitzenberg, D.A.; Kao, D.J.; Battista, K.D.; Gerich, M.E.; Glover, L.E.; Kominsky, D.J.; Colgan, S.P. Tryptophan metabolite activation of the aryl hydrocarbon receptor regulates IL-10 receptor expression on intestinal epithelia. Mucosal. Immunol. 2017, 10, 1133–1144. [Google Scholar] [CrossRef]
- Alexeev, E.E.; Lanis, J.M.; Kao, D.J.; Campbell, E.L.; Kelly, C.J.; Battista, K.D.; Gerich, M.E.; Jenkins, B.R.; Walk, S.T.; Kominsky, D.J.; et al. Microbiota-Derived Indole Metabolites Promote Human and Murine Intestinal Homeostasis through Regulation of Interleukin-10 Receptor. Am. J. Pathol. 2018, 188, 1183–1194. [Google Scholar] [CrossRef] [Green Version]
- Alexeev, E.E.; Dowdell, A.S.; Henen, M.A.; Lanis, J.M.; Lee, J.S.; Cartwright, I.M.; Schaefer, R.E.M.; Ornelas, A.; Onyiah, J.C.; Vogeli, B.; et al. Microbial-derived indoles inhibit neutrophil myeloperoxidase to diminish bystander tissue damage. FASEB J. 2021, 35, e21552. [Google Scholar] [CrossRef]
- Lamb, C.A.; Kennedy, N.A.; Raine, T.; Hendy, P.A.; Smith, P.J.; Limdi, J.K.; Hayee, B.; Lomer, M.C.E.; Parkes, G.C.; Selinger, C.; et al. British Society of Gastroenterology consensus guidelines on the management of inflammatory bowel disease in adults. Gut 2019, 68, s1–s106. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, C.N. Treatment of IBD: Where we are and where we are going. Am. J. Gastroenterol. 2015, 110, 114–126. [Google Scholar] [CrossRef]
- Gupta, A.; Khanna, S. Fecal Microbiota Transplantation. JAMA 2017, 318, 102. [Google Scholar] [CrossRef] [PubMed]
- Ott, S.J.; Waetzig, G.H.; Rehman, A.; Moltzau-Anderson, J.; Bharti, R.; Grasis, J.A.; Cassidy, L.; Tholey, A.; Fickenscher, H.; Seegert, D.; et al. Efficacy of Sterile Fecal Filtrate Transfer for Treating Patients With Clostridium difficile Infection. Gastroenterology 2017, 152, 799–811. [Google Scholar] [CrossRef] [Green Version]
- Kelly, C.R.; Yen, E.F.; Grinspan, A.M.; Kahn, S.A.; Atreja, A.; Lewis, J.D.; Moore, T.A.; Rubin, D.T.; Kim, A.M.; Serra, S.; et al. Fecal Microbiota Transplantation Is Highly Effective in Real-World Practice: Initial Results from the FMT National Registry. Gastroenterology 2021, 160, 183–192.e3. [Google Scholar] [CrossRef]
- Weingarden, A.R.; Vaughn, B.P. Intestinal microbiota, fecal microbiota transplantation, and inflammatory bowel disease. Gut Microbes 2017, 8, 238–252. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.G.; Liu, S.P.; Ma, T.H.; Yan, W.; Zhou, J.F.; Shi, Y.T.; Shen, P.; Yang, X.Z.; Wu, S.N. Fecal microbiota transplantation treatment for refractory ulcerative colitis with allergy to 5-aminosalicylic acid: A case report. Medicine 2018, 97, e0675. [Google Scholar] [CrossRef] [PubMed]
- Nakase, H.; Uchino, M.; Shinzaki, S.; Matsuura, M.; Matsuoka, K.; Kobayashi, T.; Saruta, M.; Hirai, F.; Hata, K.; Hiraoka, S.; et al. Evidence-based clinical practice guidelines for inflammatory bowel disease 2020. J. Gastroenterol. 2021. [Google Scholar] [CrossRef]
- Sood, A.; Mahajan, R.; Singh, A.; Midha, V.; Mehta, V.; Narang, V.; Singh, T.; Singh Pannu, A. Role of Faecal Microbiota Transplantation for Maintenance of Remission in Patients with Ulcerative Colitis: A Pilot Study. J. Crohn’s Colitis 2019, 13, 1311–1317. [Google Scholar] [CrossRef]
- Fehily, S.R.; Basnayake, C.; Wright, E.K.; Kamm, M.A. Fecal microbiota transplantation therapy in Crohn’s disease: Systematic review. J. Gastroenterol. Hepatol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Huang, Z.; Wei, W.; Li, Z. Fecal microbiota transplantation for Crohn’s disease: A systematic review and meta-analysis. Tech. Coloproctol. 2021, 25, 495–504. [Google Scholar] [CrossRef]
- Sokol, H.; Landman, C.; Seksik, P.; Berard, L.; Montil, M.; Nion-Larmurier, I.; Bourrier, A.; Le Gall, G.; Lalande, V.; De Rougemont, A.; et al. Fecal microbiota transplantation to maintain remission in Crohn’s disease: A pilot randomized controlled study. Microbiome 2020, 8, 12. [Google Scholar] [CrossRef]
- Duncan, S.H.; Louis, P.; Flint, H.J. Cultivable bacterial diversity from the human colon. Lett. Appl. Microbiol. 2007, 44, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Chu, N.D.; Smith, M.B.; Perrotta, A.R.; Kassam, Z.; Alm, E.J. Profiling Living Bacteria Informs Preparation of Fecal Microbiota Transplantations. PLoS ONE 2017, 12, e0170922. [Google Scholar] [CrossRef]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermudez-Humaran, L.G.; Gratadoux, J.J.; Blugeon, S.; Bridonneau, C.; Furet, J.P.; Corthier, G.; et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef] [Green Version]
- Costello, S.P.; Hughes, P.A.; Waters, O.; Bryant, R.V.; Vincent, A.D.; Blatchford, P.; Katsikeros, R.; Makanyanga, J.; Campaniello, M.A.; Mavrangelos, C.; et al. Effect of Fecal Microbiota Transplantation on 8-Week Remission in Patients With Ulcerative Colitis: A Randomized Clinical Trial. JAMA 2019, 321, 156–164. [Google Scholar] [CrossRef] [Green Version]
- Jakubczyk, D.; Leszczynska, K.; Gorska, S. The Effectiveness of Probiotics in the Treatment of Inflammatory Bowel Disease (IBD)-A Critical Review. Nutrients 2020, 12, 1973. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, H.; Nakase, H.; Inoue, S.; Kawanami, C.; Itani, T.; Ohana, M.; Kusaka, T.; Uose, S.; Hisatsune, H.; Tojo, M.; et al. Efficacy of probiotic treatment with Bifidobacterium longum 536 for induction of remission in active ulcerative colitis: A randomized, double-blinded, placebo-controlled multicenter trial. Dig. Endosc. 2016, 28, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Kruis, W.; Fric, P.; Pokrotnieks, J.; Lukas, M.; Fixa, B.; Kascak, M.; Kamm, M.A.; Weismueller, J.; Beglinger, C.; Stolte, M.; et al. Maintaining remission of ulcerative colitis with the probiotic Escherichia coli Nissle 1917 is as effective as with standard mesalazine. Gut 2004, 53, 1617–1623. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, J.G.; Chain, F.; Martin, R.; Bermudez-Humaran, L.G.; Courau, S.; Langella, P. Beneficial effects on host energy metabolism of short-chain fatty acids and vitamins produced by commensal and probiotic bacteria. Microb Cell Fact 2017, 16, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagpal, R.; Wang, S.; Ahmadi, S.; Hayes, J.; Gagliano, J.; Subashchandrabose, S.; Kitzman, D.W.; Becton, T.; Read, R.; Yadav, H. Human-origin probiotic cocktail increases short-chain fatty acid production via modulation of mice and human gut microbiome. Sci. Rep. 2018, 8, 12649. [Google Scholar] [CrossRef] [Green Version]
- Deriu, E.; Liu, J.Z.; Pezeshki, M.; Edwards, R.A.; Ochoa, R.J.; Contreras, H.; Libby, S.J.; Fang, F.C.; Raffatellu, M. Probiotic bacteria reduce salmonella typhimurium intestinal colonization by competing for iron. Cell Host Microbe 2013, 14, 26–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deutsch, S.M.; Mariadassou, M.; Nicolas, P.; Parayre, S.; Le Guellec, R.; Chuat, V.; Peton, V.; Le Marechal, C.; Burati, J.; Loux, V.; et al. Identification of proteins involved in the anti-inflammatory properties of Propionibacterium freudenreichii by means of a multi-strain study. Sci. Rep. 2017, 7, 46409. [Google Scholar] [CrossRef] [PubMed]
- Steidler, L.; Hans, W.; Schotte, L.; Neirynck, S.; Obermeier, F.; Falk, W.; Fiers, W.; Remaut, E. Treatment of murine colitis by Lactococcus lactis secreting interleukin-10. Science 2000, 289, 1352–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, D.P.; Gysemans, C.; Mathieu, C. Lactococcus lactis As a Versatile Vehicle for Tolerogenic Immunotherapy. Front. Immunol. 2017, 8, 1961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braat, H.; Rottiers, P.; Hommes, D.W.; Huyghebaert, N.; Remaut, E.; Remon, J.P.; van Deventer, S.J.; Neirynck, S.; Peppelenbosch, M.P.; Steidler, L. A phase I trial with transgenic bacteria expressing interleukin-10 in Crohn’s disease. Clin. Gastroenterol. Hepatol. 2006, 4, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Vallance, B.A.; Boyer, L.; Bergstrom, K.S.; Walker, J.; Madsen, K.; O’Kusky, J.R.; Buchan, A.M.; Jacobson, K. Saccharomyces boulardii ameliorates Citrobacter rodentium-induced colitis through actions on bacterial virulence factors. Am. J. Physiol. Gastrointest. Liver. Physiol. 2008, 294, G295–G306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalmasso, G.; Cottrez, F.; Imbert, V.; Lagadec, P.; Peyron, J.F.; Rampal, P.; Czerucka, D.; Groux, H.; Foussat, A.; Brun, V. Saccharomyces boulardii inhibits inflammatory bowel disease by trapping T cells in mesenteric lymph nodes. Gastroenterology 2006, 131, 1812–1825. [Google Scholar] [CrossRef] [PubMed]
- Jawhara, S.; Poulain, D. Saccharomyces boulardii decreases inflammation and intestinal colonization by Candida albicans in a mouse model of chemically-induced colitis. Med. Mycol. 2007, 45, 691–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Nogales, A.; Algieri, F.; Garrido-Mesa, J.; Vezza, T.; Utrilla, M.P.; Chueca, N.; Garcia, F.; Rodriguez-Cabezas, M.E.; Galvez, J. Intestinal anti-inflammatory effect of the probiotic Saccharomyces boulardii in DSS-induced colitis in mice: Impact on microRNAs expression and gut microbiota composition. J. Nutr. Biochem. 2018, 61, 129–139. [Google Scholar] [CrossRef]
- Garcia Vilela, E.; De Lourdes De Abreu Ferrari, M.; Oswaldo Da Gama Torres, H.; Guerra Pinto, A.; Carolina Carneiro Aguirre, A.; Paiva Martins, F.; Marcos Andrade Goulart, E.; Sales Da Cunha, A. Influence of Saccharomyces boulardii on the intestinal permeability of patients with Crohn’s disease in remission. Scand. J. Gastroenterol. 2008, 43, 842–848. [Google Scholar] [CrossRef]
- Everard, A.; Matamoros, S.; Geurts, L.; Delzenne, N.M.; Cani, P.D. Saccharomyces boulardii administration changes gut microbiota and reduces hepatic steatosis, low-grade inflammation, and fat mass in obese and type 2 diabetic db/db mice. MBio 2014, 5, e01011-14. [Google Scholar] [CrossRef] [Green Version]
- Pothoulakis, C. Review article: Anti-inflammatory mechanisms of action of Saccharomyces boulardii. Aliment. Pharmacol. Ther. 2009, 30, 826–833. [Google Scholar] [CrossRef]
- Scott, B.M.; Gutierrez-Vazquez, C.; Sanmarco, L.M.; da Silva Pereira, J.A.; Li, Z.; Plasencia, A.; Hewson, P.; Cox, L.M.; O’Brien, M.; Chen, S.K.; et al. Self-tunable engineered yeast probiotics for the treatment of inflammatory bowel disease. Nat. Med. 2021, 27, 1212–1222. [Google Scholar] [CrossRef]
- Aherne, C.M.; Saeedi, B.; Collins, C.B.; Masterson, J.C.; McNamee, E.N.; Perrenoud, L.; Rapp, C.R.; Curtis, V.F.; Bayless, A.; Fletcher, A.; et al. Epithelial-specific A2B adenosine receptor signaling protects the colonic epithelial barrier during acute colitis. Mucosal. Immunol. 2015, 8, 1324–1338. [Google Scholar] [CrossRef]
- Curtis, V.F.; Cartwright, I.M.; Lee, J.S.; Wang, R.X.; Kao, D.J.; Lanis, J.M.; Burney, K.M.; Welch, N.; Hall, C.H.T.; Goldberg, M.S.; et al. Neutrophils as sources of dinucleotide polyphosphates and metabolism by epithelial ENPP1 to influence barrier function via adenosine signaling. Mol. Biol. Cell 2018, 29, 2687–2699. [Google Scholar] [CrossRef]
- Cartwright, I.M.; Curtis, V.F.; Lanis, J.M.; Alexeev, E.E.; Welch, N.; Goldberg, M.S.; Schaefer, R.E.M.; Gao, R.Y.; Chun, C.; Fennimore, B.; et al. Adaptation to inflammatory acidity through neutrophil-derived adenosine regulation of SLC26A3. Mucosal. Immunol. 2020, 13, 230–244. [Google Scholar] [CrossRef]
- Meini, S.; Laureano, R.; Fani, L.; Tascini, C.; Galano, A.; Antonelli, A.; Rossolini, G.M. Breakthrough Lactobacillus rhamnosus GG bacteremia associated with probiotic use in an adult patient with severe active ulcerative colitis: Case report and review of the literature. Infection 2015, 43, 777–781. [Google Scholar] [CrossRef]
- Limketkai, B.N.; Akobeng, A.K.; Gordon, M.; Adepoju, A.A. Probiotics for induction of remission in Crohn’s disease. Cochrane Database Syst. Rev. 2020, 7, CD006634. [Google Scholar] [CrossRef]
- Rolfe, V.E.; Fortun, P.J.; Hawkey, C.J.; Bath-Hextall, F. Probiotics for maintenance of remission in Crohn’s disease. Cochrane Database Syst. Rev. 2006. [Google Scholar] [CrossRef] [PubMed]
- Bauset, C.; Gisbert-Ferrandiz, L.; Cosin-Roger, J. Metabolomics as a Promising Resource Identifying Potential Biomarkers for Inflammatory Bowel Disease. J. Clin. Med. 2021, 10, 622. [Google Scholar] [CrossRef]
- Mishiro, T.; Kusunoki, R.; Otani, A.; Ansary, M.M.; Tongu, M.; Harashima, N.; Yamada, T.; Sato, S.; Amano, Y.; Itoh, K.; et al. Butyric acid attenuates intestinal inflammation in murine DSS-induced colitis model via milk fat globule-EGF factor 8. Lab. Investig. 2013, 93, 834–843. [Google Scholar] [CrossRef]
- Scheppach, W.; Sommer, H.; Kirchner, T.; Paganelli, G.M.; Bartram, P.; Christl, S.; Richter, F.; Dusel, G.; Kasper, H. Effect of butyrate enemas on the colonic mucosa in distal ulcerative colitis. Gastroenterology 1992, 103, 51–56. [Google Scholar] [CrossRef]
- Steinhart, A.H.; Brzezinski, A.; Baker, J.P. Treatment of refractory ulcerative proctosigmoiditis with butyrate enemas. Am. J. Gastroenterol. 1994, 89, 179–183. [Google Scholar]
- Vernia, P.; Annese, V.; Bresci, G.; d’Albasio, G.; D’Inca, R.; Giaccari, S.; Ingrosso, M.; Mansi, C.; Riegler, G.; Valpiani, D.; et al. Topical butyrate improves efficacy of 5-ASA in refractory distal ulcerative colitis: Results of a multicentre trial. Eur. J. Clin. Investg. 2003, 33, 244–248. [Google Scholar] [CrossRef]
- Jamka, M.; Kokot, M.; Kaczmarek, N.; Bermagambetova, S.; Nowak, J.K.; Walkowiak, J. The Effect of Sodium Butyrate Enemas Compared with Placebo on Disease Activity, Endoscopic Scores, and Histological and Inflammatory Parameters in Inflammatory Bowel Diseases: A Systematic Review of Randomised Controlled Trials. Complement. Med. Res. 2020, 1–13. [Google Scholar] [CrossRef]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsden, S.R.; Hilton, M.G.; Waller, J.M. The end products of the metabolism of aromatic amino acids by Clostridia. Arch. Microbiol. 1976, 107, 283–288. [Google Scholar] [CrossRef]
- Dodd, D.; Spitzer, M.H.; Van Treuren, W.; Merrill, B.D.; Hryckowian, A.J.; Higginbottom, S.K.; Le, A.; Cowan, T.M.; Nolan, G.P.; Fischbach, M.A.; et al. A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature 2017, 551, 648–652. [Google Scholar] [CrossRef]
- Venkatesh, M.; Mukherjee, S.; Wang, H.; Li, H.; Sun, K.; Benechet, A.P.; Qiu, Z.; Maher, L.; Redinbo, M.R.; Phillips, R.S.; et al. Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity 2014, 41, 296–310. [Google Scholar] [CrossRef] [Green Version]
- Chyan, Y.J.; Poeggeler, B.; Omar, R.A.; Chain, D.G.; Frangione, B.; Ghiso, J.; Pappolla, M.A. Potent neuroprotective properties against the Alzheimer beta-amyloid by an endogenous melatonin-related indole structure, indole-3-propionic acid. J. Biol. Chem. 1999, 274, 21937–21942. [Google Scholar] [CrossRef] [Green Version]
- Hwang, I.K.; Yoo, K.Y.; Li, H.; Park, O.K.; Lee, C.H.; Choi, J.H.; Jeong, Y.G.; Lee, Y.L.; Kim, Y.M.; Kwon, Y.G.; et al. Indole-3-propionic acid attenuates neuronal damage and oxidative stress in the ischemic hippocampus. J. Neurosci. Res. 2009, 87, 2126–2137. [Google Scholar] [CrossRef]
- Naganuma, M.; Sugimoto, S.; Mitsuyama, K.; Kobayashi, T.; Yoshimura, N.; Ohi, H.; Tanaka, S.; Andoh, A.; Ohmiya, N.; Saigusa, K.; et al. Efficacy of Indigo Naturalis in a Multicenter Randomized Controlled Trial of Patients With Ulcerative Colitis. Gastroenterology 2018, 154, 935–947. [Google Scholar] [CrossRef] [Green Version]
- Maronek, M.; Link, R.; Ambro, L.; Gardlik, R. Phages and Their Role in Gastrointestinal Disease: Focus on Inflammatory Bowel Disease. Cells 2020, 9, 1013. [Google Scholar] [CrossRef] [Green Version]
- Breitbart, M.; Hewson, I.; Felts, B.; Mahaffy, J.M.; Nulton, J.; Salamon, P.; Rohwer, F. Metagenomic analyses of an uncultured viral community from human feces. J. Bacteriol. 2003, 185, 6220–6223. [Google Scholar] [CrossRef] [Green Version]
- Lusiak-Szelachowska, M.; Weber-Dabrowska, B.; Jonczyk-Matysiak, E.; Wojciechowska, R.; Gorski, A. Bacteriophages in the gastrointestinal tract and their implications. Gut Pathog. 2017, 9, 44. [Google Scholar] [CrossRef] [Green Version]
- Duerkop, B.A.; Kleiner, M.; Paez-Espino, D.; Zhu, W.; Bushnell, B.; Hassell, B.; Winter, S.E.; Kyrpides, N.C.; Hooper, L.V. Murine colitis reveals a disease-associated bacteriophage community. Nat. Microbiol. 2018, 3, 1023–1031. [Google Scholar] [CrossRef]
- Gogokhia, L.; Buhrke, K.; Bell, R.; Hoffman, B.; Brown, D.G.; Hanke-Gogokhia, C.; Ajami, N.J.; Wong, M.C.; Ghazaryan, A.; Valentine, J.F.; et al. Expansion of Bacteriophages Is Linked to Aggravated Intestinal Inflammation and Colitis. Cell Host Microbe 2019, 25, 285–299.e8. [Google Scholar] [CrossRef] [Green Version]
- Gordillo Altamirano, F.L.; Barr, J.J. Phage Therapy in the Postantibiotic Era. Clin. Microbiol. Rev. 2019, 32. [Google Scholar] [CrossRef] [Green Version]
- Galtier, M.; De Sordi, L.; Sivignon, A.; de Vallee, A.; Maura, D.; Neut, C.; Rahmouni, O.; Wannerberger, K.; Darfeuille-Michaud, A.; Desreumaux, P.; et al. Bacteriophages Targeting Adherent Invasive Escherichia coli Strains as a Promising New Treatment for Crohn’s Disease. J. Crohn’s Colitis 2017, 11, 840–847. [Google Scholar] [CrossRef] [Green Version]
- Nale, J.Y.; Spencer, J.; Hargreaves, K.R.; Buckley, A.M.; Trzepinski, P.; Douce, G.R.; Clokie, M.R. Bacteriophage Combinations Significantly Reduce Clostridium difficile Growth In Vitro and Proliferation In Vivo. Antimicrob. Agents Chemother. 2016, 60, 968–981. [Google Scholar] [CrossRef] [Green Version]
- Selle, K.; Fletcher, J.R.; Tuson, H.; Schmitt, D.S.; McMillan, L.; Vridhambal, G.S.; Rivera, A.J.; Montgomery, S.A.; Fortier, L.C.; Barrangou, R.; et al. In Vivo Targeting of Clostridioides difficile Using Phage-Delivered CRISPR-Cas3 Antimicrobials. MBio 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Cassini, A.; Hogberg, L.D.; Plachouras, D.; Quattrocchi, A.; Hoxha, A.; Simonsen, G.S.; Colomb-Cotinat, M.; Kretzschmar, M.E.; Devleesschauwer, B.; Cecchini, M.; et al. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: A population-level modelling analysis. Lancet Infect. Dis. 2019, 19, 56–66. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Treatment | Model | Outcome | Reference |
|---|---|---|---|
| Fecal microbiota transplantation (FMT) | Case report, patient with steroid-refractory UC | Induction/maintenance of remission | [144] |
| Randomized controlled trial, UC patients | Maintenance of steroid-free remission | [146] | |
| Randomized controlled trial, CD patients | Decrease in the endoscopic index of severity, prevention of increase in serum CRP | [149] | |
| Faecalibacterium prausnitzii | TNBS mouse model of colitis | Attenuation of colitis, reduction in colitis-driven gut dysbiosis | [152] |
| Bifidobacterium longum 536 | Randomized controlled trial, UC patients | Significant decreases in the disease activity index, the Rachmilewitz endoscopic index, and the Mayo subscore | [155] |
| Escherichia coli Nissle 1917 | Clinical trial, UC patients | Equivalent to mesalazine in preventing relapse | [156] |
| IL-10-secreting Lactococcus lactis | DSS, Il-10−/− mouse models of colitis | Reduction in intestinal histopathology | [162] |
| Clinical trial, CD patients | Decrease in disease activity, serum CRP | [164] | |
| Saccharomyces boulardii | Randomized controlled trial, CD patients | Decrease in intestinal permeability | [157] |
| C. rodentium mouse model of colitis | Decreases in body weight loss, histopathology, tissue MPO, and intestinal permeability observed | [165] | |
| CD4+CD45RBhi T cell adoptive transfer mouse model of colitis | Decreases in body weight loss, histopathology, intestinal pro-inflammatory cytokines, and NF-κB activation observed | [166] | |
| DSS mouse model of colitis | Reduction in clinical score, histopathology, and colonization by colitis-associated Candida albicans | [167] | |
| DSS mouse model of colitis | Reduction in the disease activity index, improved weight recovery, amelioration of colitis-driven gut dysbiosis | [168] | |
| Extracellular ATP-degrading S. cerevisiae | TNBS, DSS, and anti-CD3 mouse models of intestinal inflammation | Decreases in colon length shortening, histopathology, weight loss, and intestinal pro-inflammatory cytokine expression | [171] |
| Hypoxanthine | DSS mouse model of colitis; mice pre-treated with streptomycin | Decreases in ER stress, intestinal epithelial apoptosis, body weight loss, and colon shortening; increases in mucus secretion, energy homeostasis, and cellular proliferation | [130] |
| Indole-3-propionic acid (IPA) | DSS mouse model of colitis | Decreases in histopathology, intestinal pro-inflammatory cytokines, and colon shortening | [136] |
| Indomethacin mouse model of intestinal inflammation | Reduction in intestinal permeability | [187] | |
| Indigo naturalis | Randomized controlled trial, active UC patients | Increases in clinical remission and mucosal healing | [190] |
| Butyrate | DSS mouse model of colitis | Decreases in histopathology, colon shortening, pro-inflammatory cytokines in colon tissue | [179] |
| Clinical trial, treatment-refractory UC patients | Decreases in endoscopic score, histologic degree of inflammation, stool frequency, and blood discharge | [180] | |
| Clinical trial, treatment-refractory UC patients | 60% response rate based on positive change in activity score | [181] | |
| Clinical trial, UC patients | Significantly greater improvement in the disease activity index when butyrate combined with 5-ASA vs. 5-ASA alone | [182] | |
| Phage therapy | DSS mouse model of colitis; mice pre-colonized with AIEC | Anti-AIEC bacteriophages reduced the disease activity index and gut AIEC burdens | [197] |
| C. difficile Syrian Golden hamster model of colitis | Reduced bacterial colonization and delay in symptom onset | [198] | |
| Cefoperazone-pretreatment/C. difficile mouse model of colitis | Reduction in intestinal C. difficile burdens | [199] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dowdell, A.S.; Colgan, S.P. Metabolic Host–Microbiota Interactions in Autophagy and the Pathogenesis of Inflammatory Bowel Disease (IBD). Pharmaceuticals 2021, 14, 708. https://doi.org/10.3390/ph14080708
Dowdell AS, Colgan SP. Metabolic Host–Microbiota Interactions in Autophagy and the Pathogenesis of Inflammatory Bowel Disease (IBD). Pharmaceuticals. 2021; 14(8):708. https://doi.org/10.3390/ph14080708
Chicago/Turabian StyleDowdell, Alexander S., and Sean P. Colgan. 2021. "Metabolic Host–Microbiota Interactions in Autophagy and the Pathogenesis of Inflammatory Bowel Disease (IBD)" Pharmaceuticals 14, no. 8: 708. https://doi.org/10.3390/ph14080708
APA StyleDowdell, A. S., & Colgan, S. P. (2021). Metabolic Host–Microbiota Interactions in Autophagy and the Pathogenesis of Inflammatory Bowel Disease (IBD). Pharmaceuticals, 14(8), 708. https://doi.org/10.3390/ph14080708