
Interaction between DNA, Albumin and Apo-Transferrin and Iridium(III) Complexes with Phosphines Derived from Fluoroquinolones as a Potent Anticancer Drug

 , ,
, ,
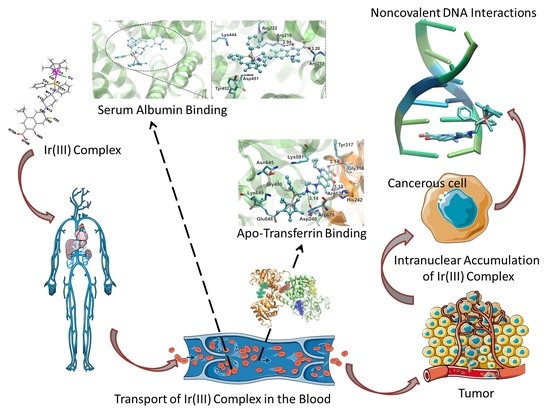
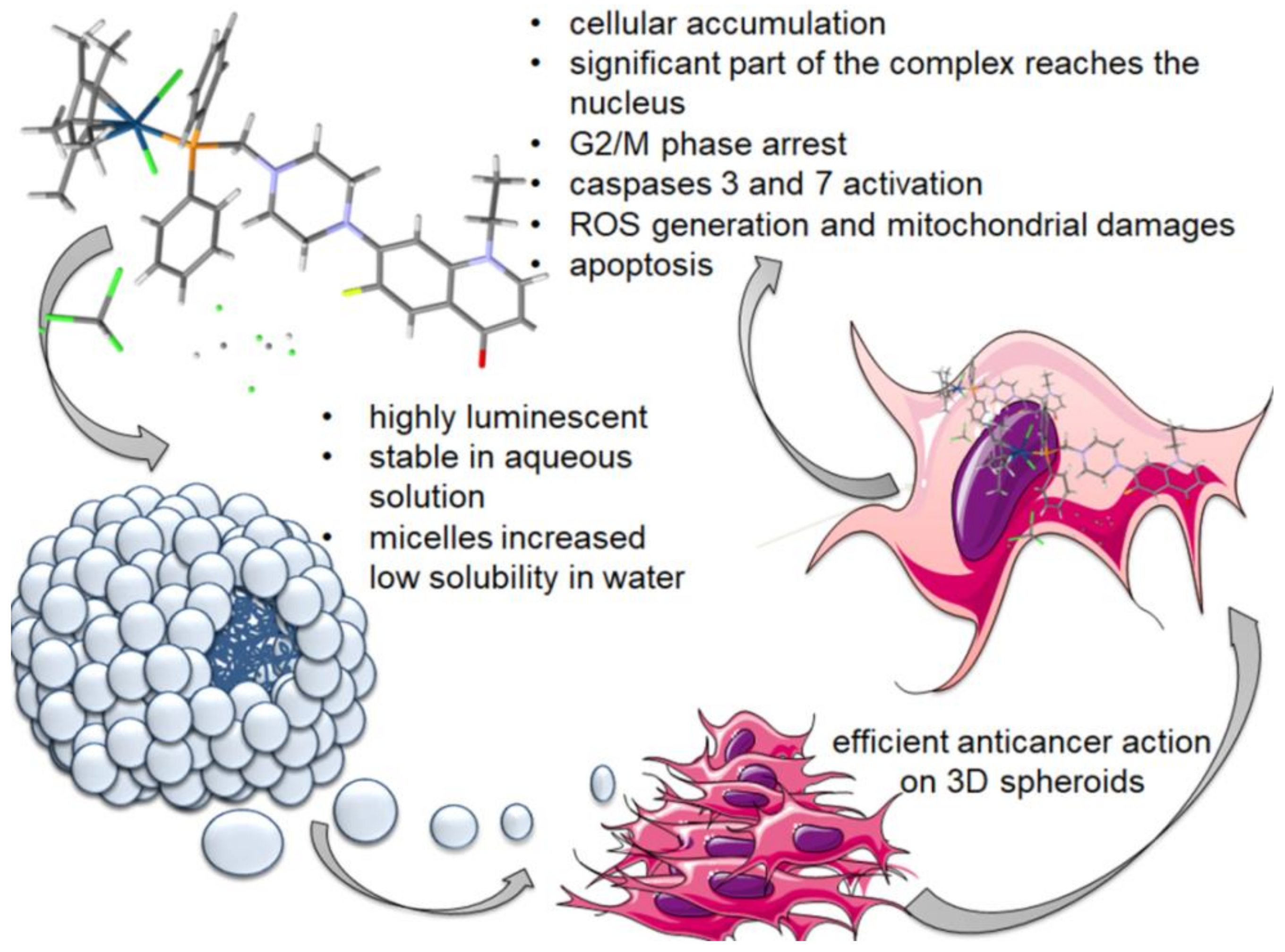
Abstract
:
1. Introduction
2. Results and Discussion
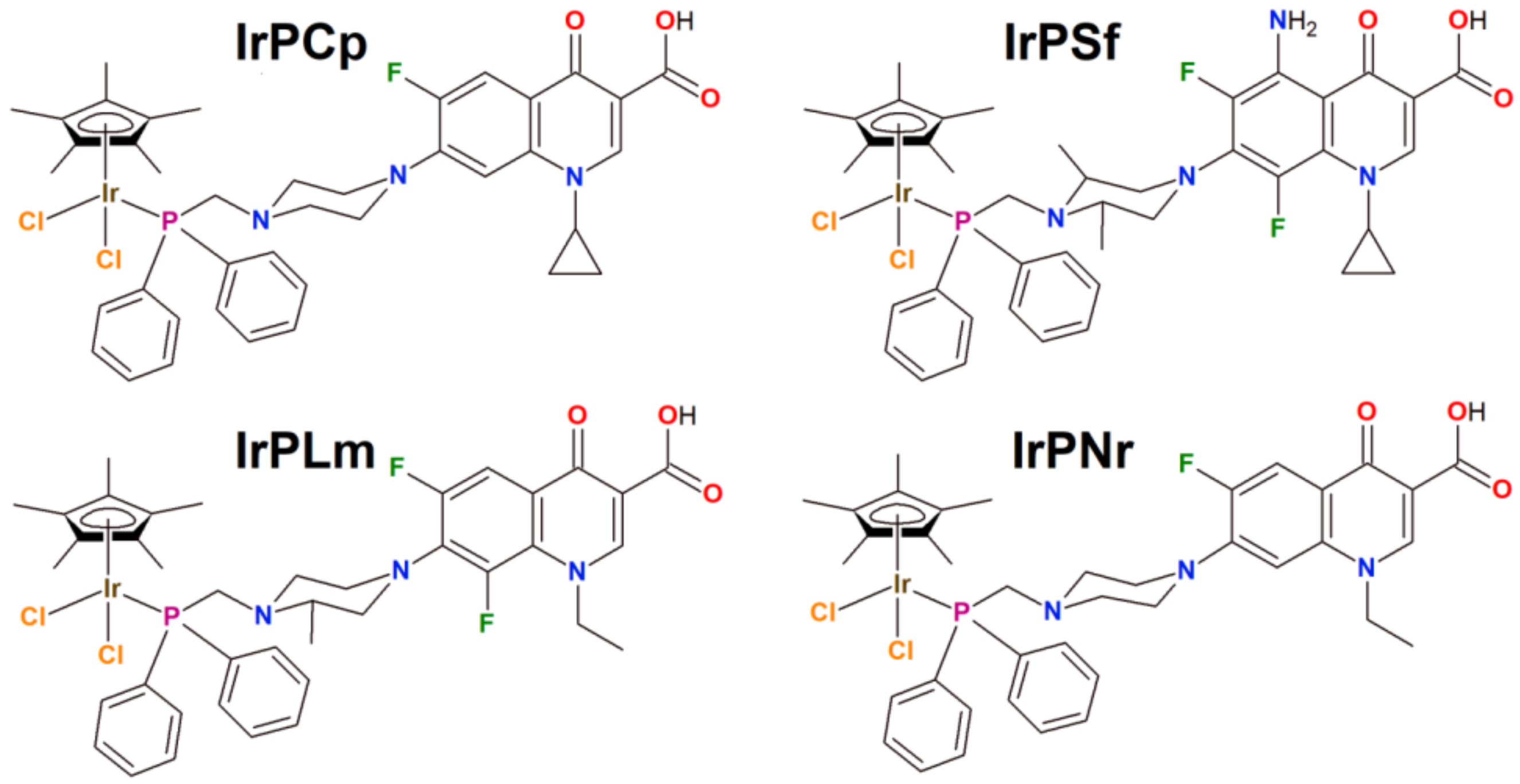
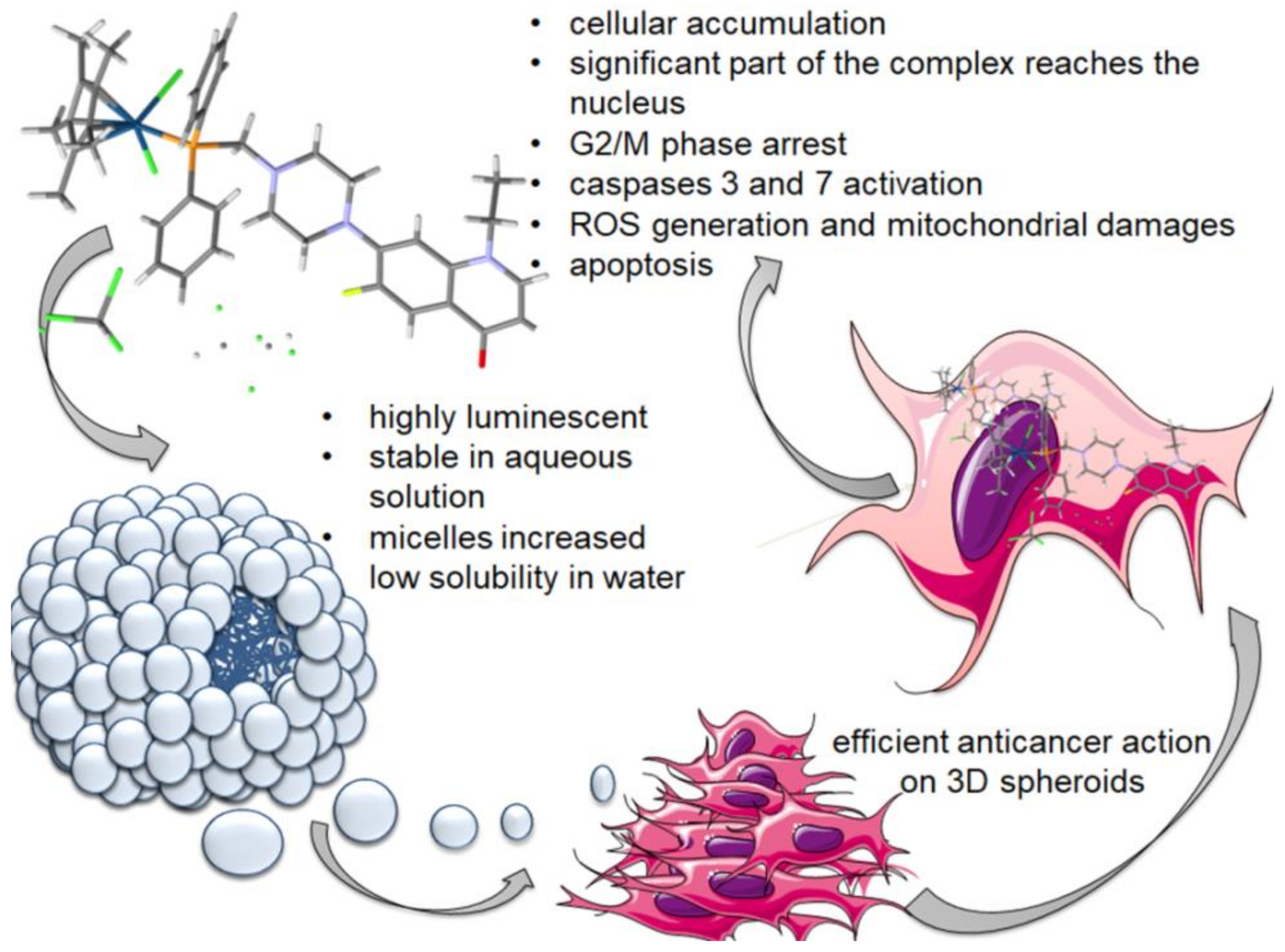
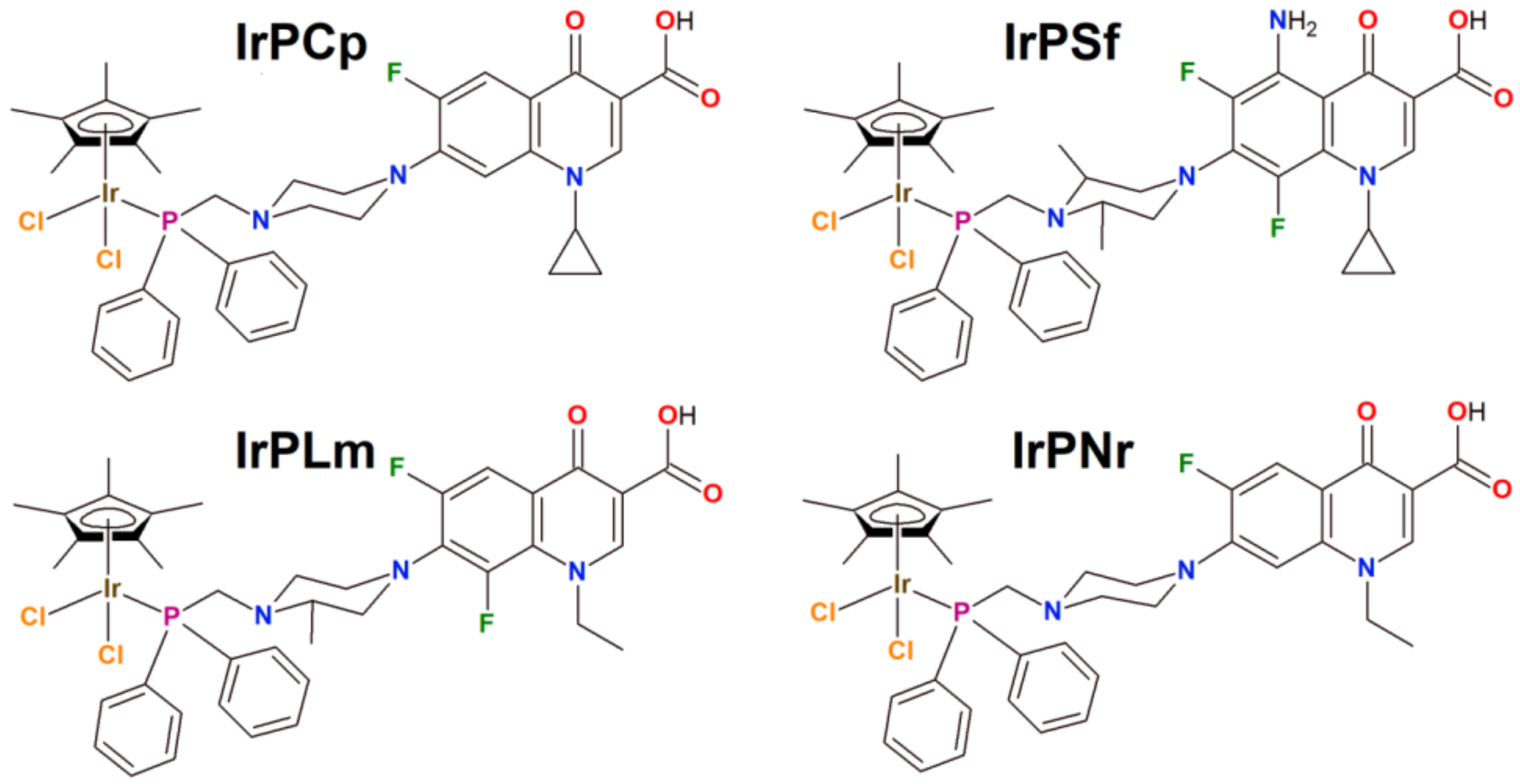
2.1. Synthesis, Physicochemical and Biological Characterization
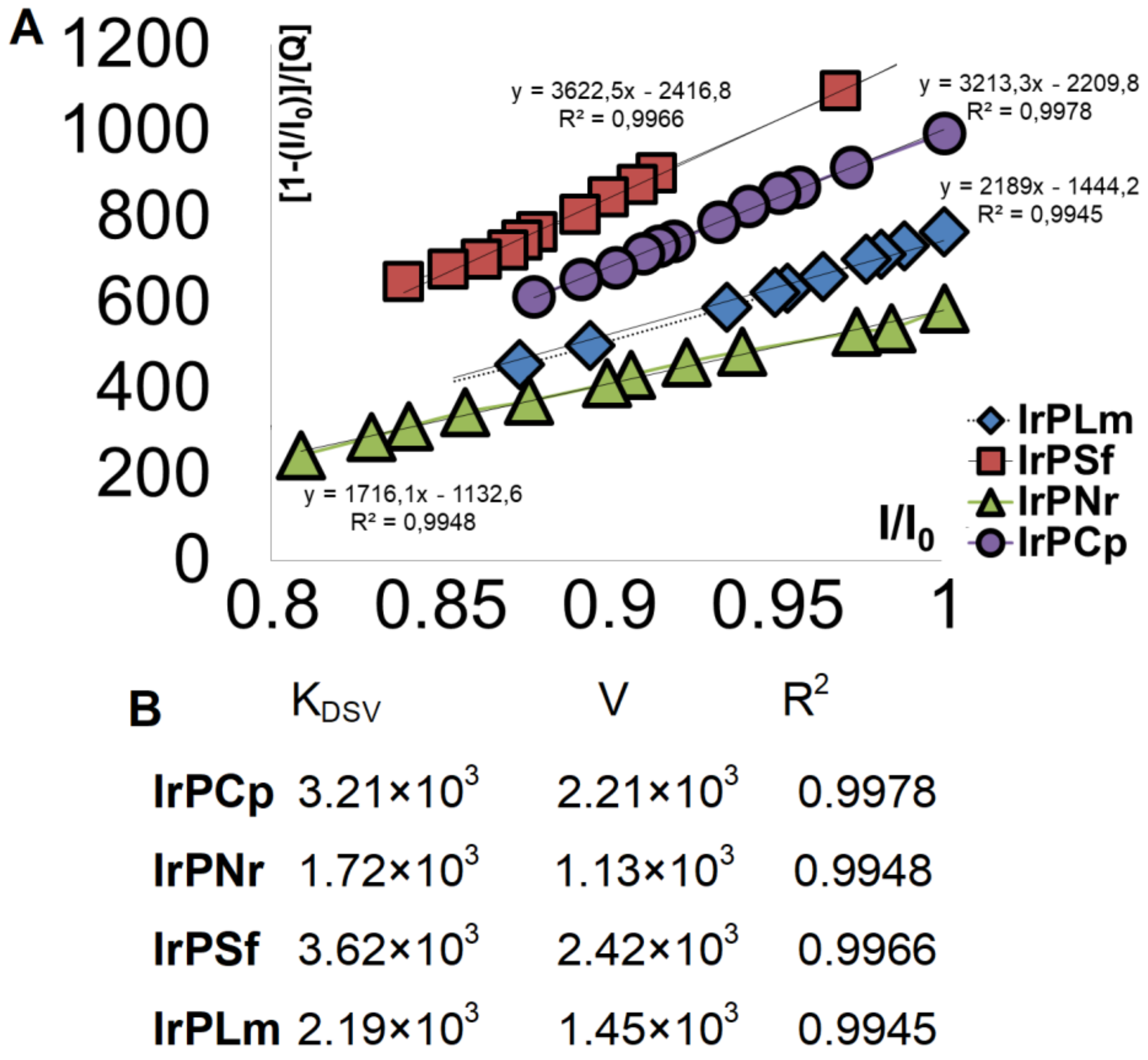
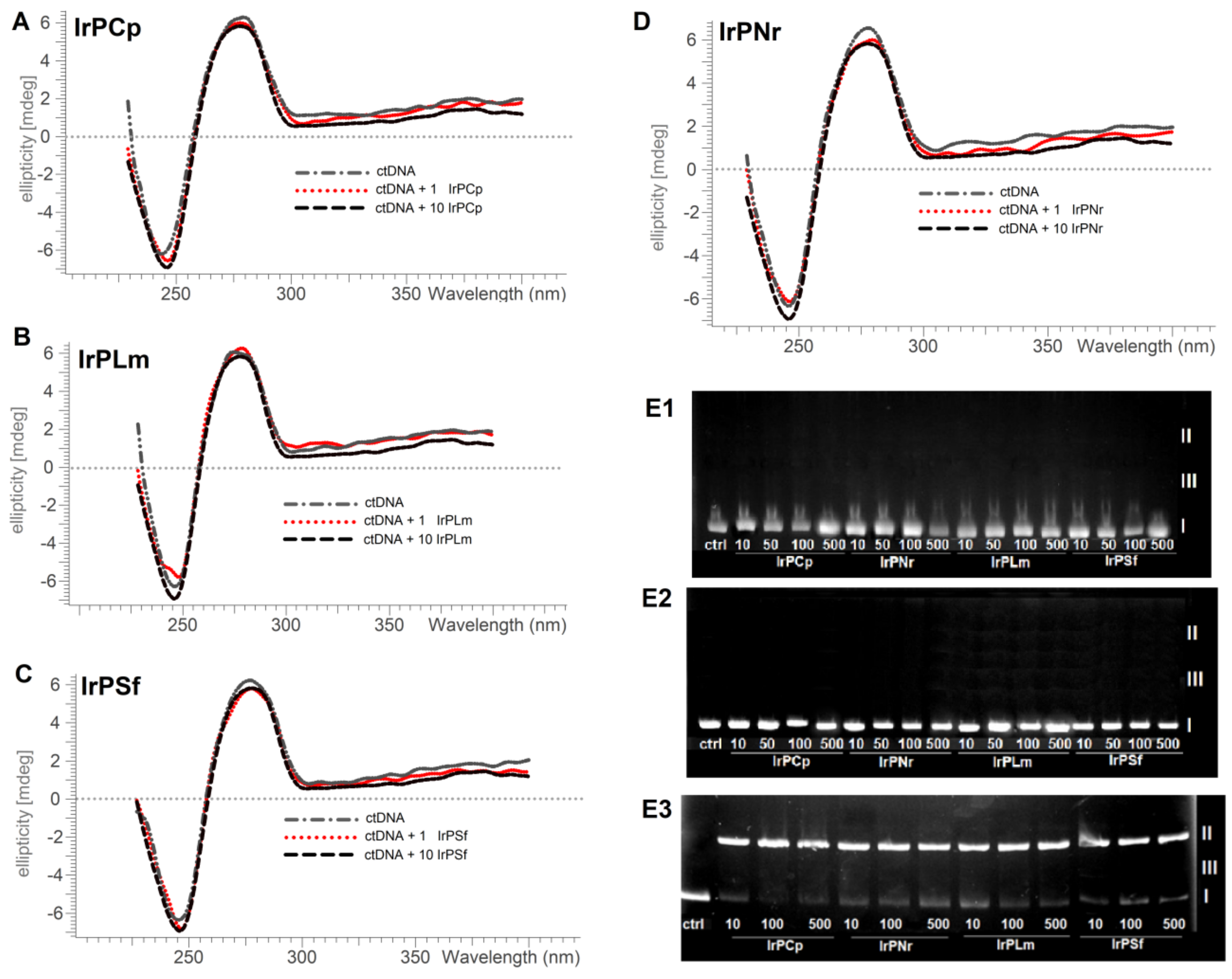
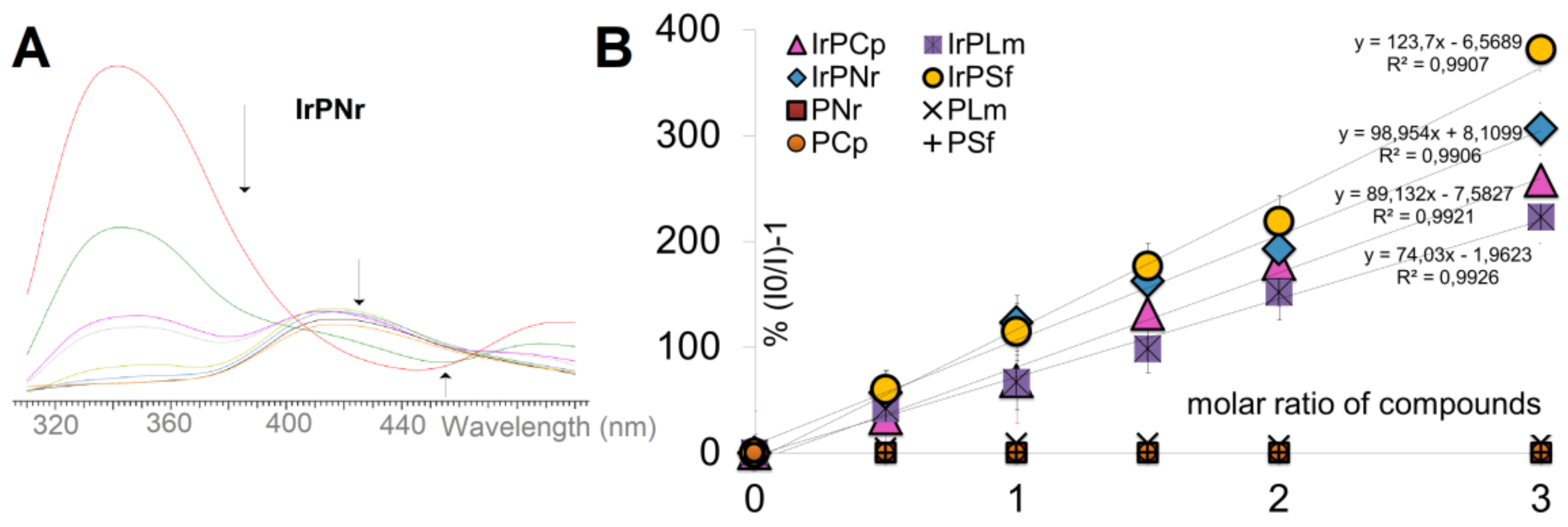
2.2. Interaction with DNA
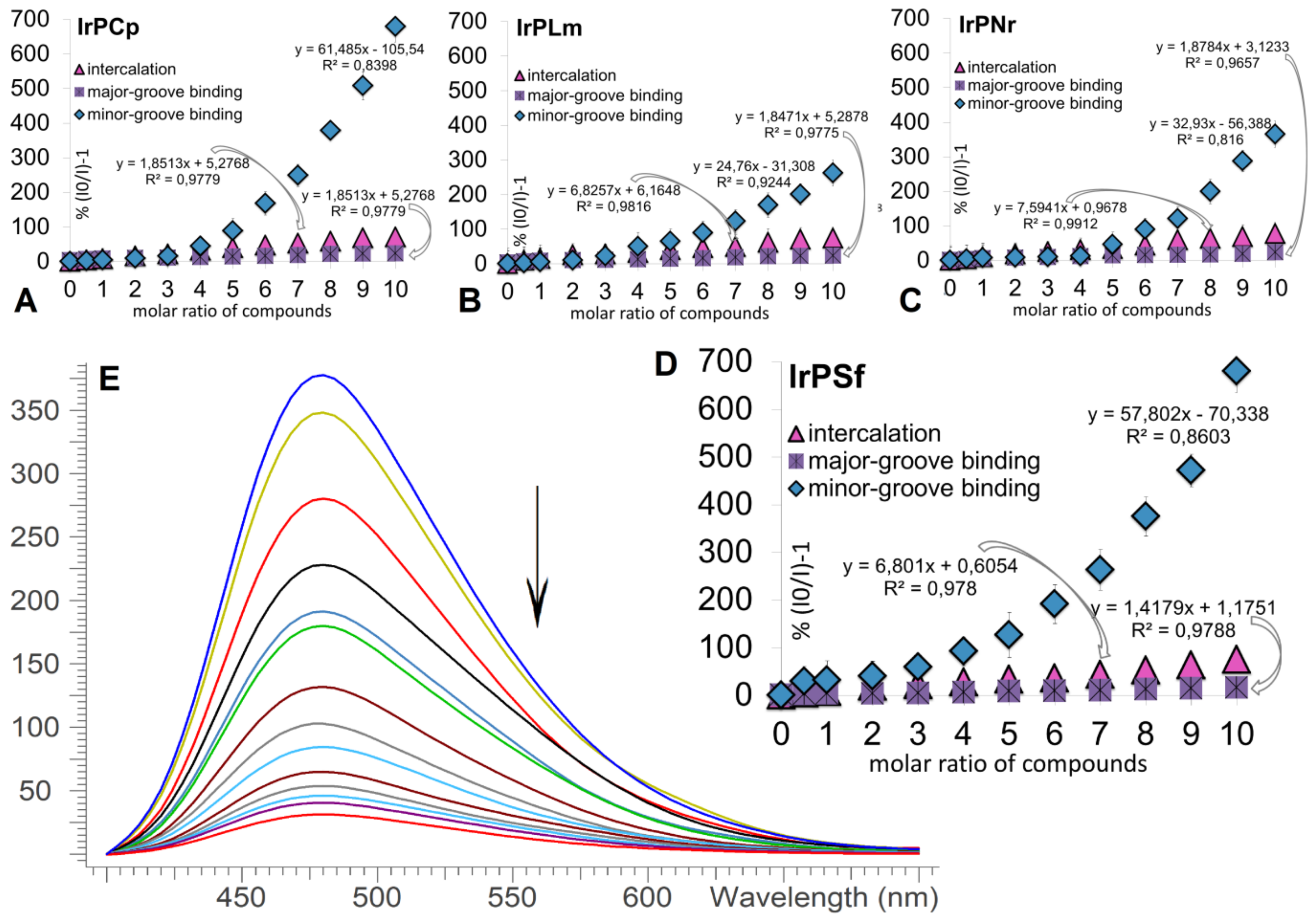
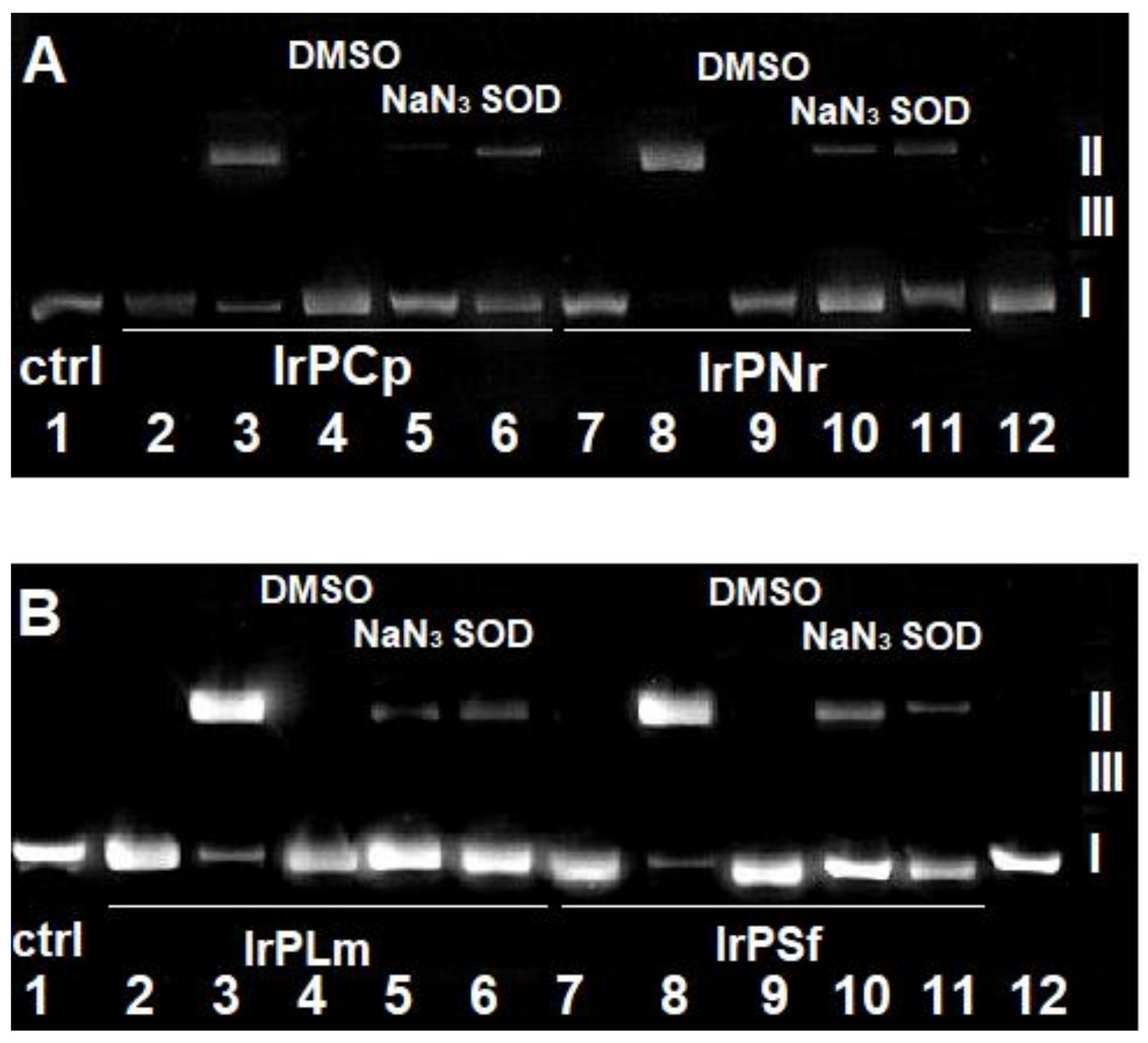
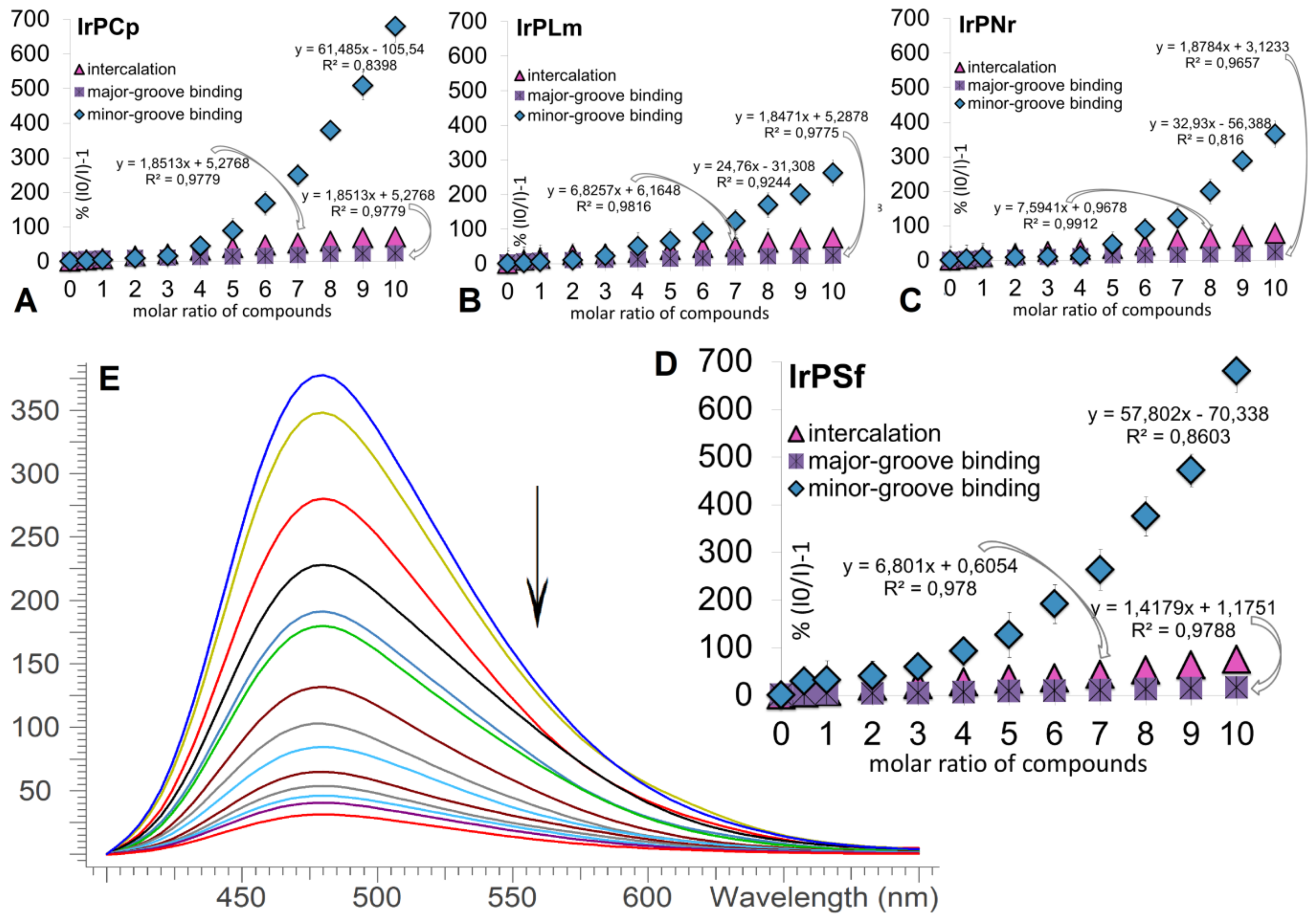
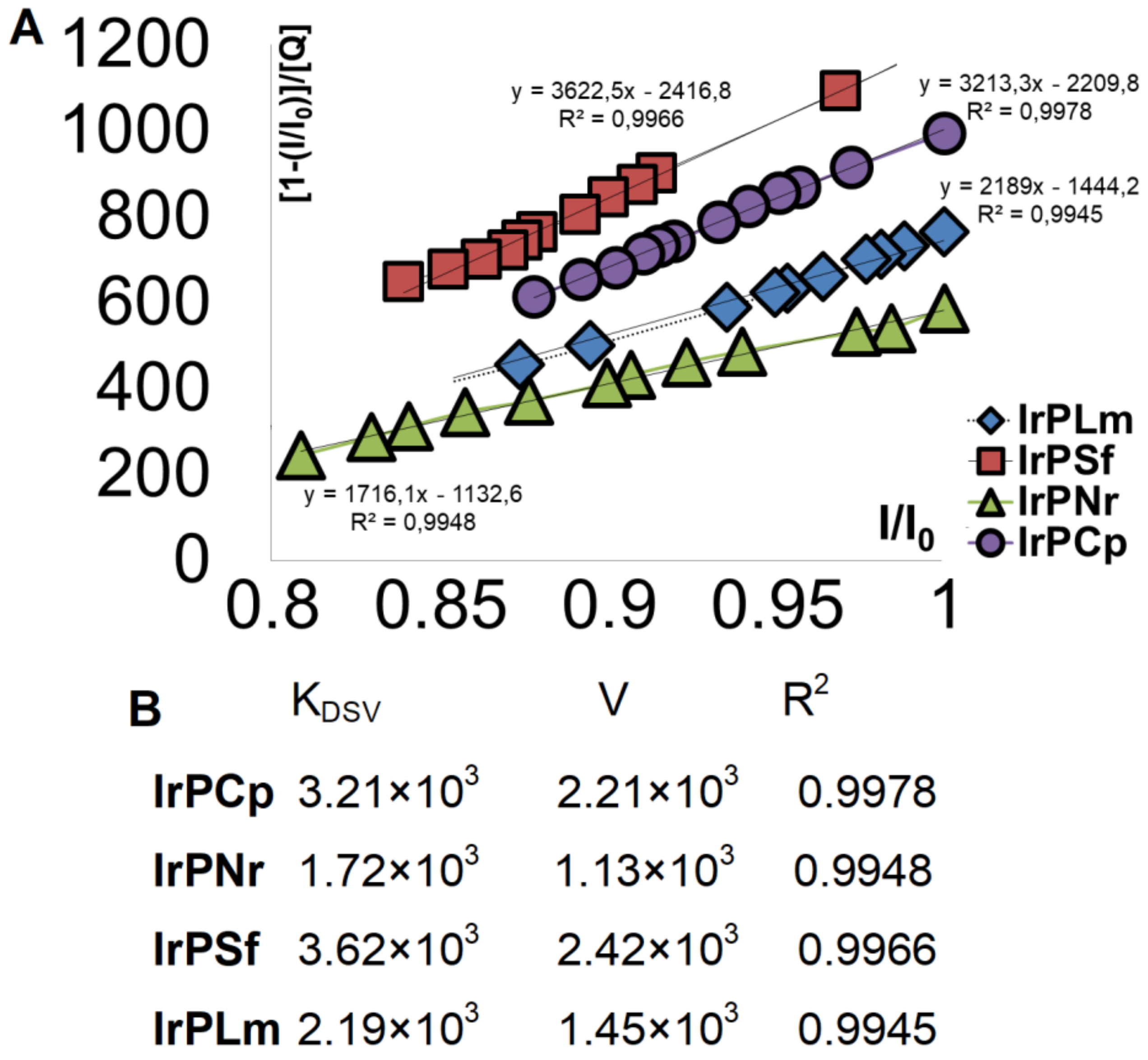
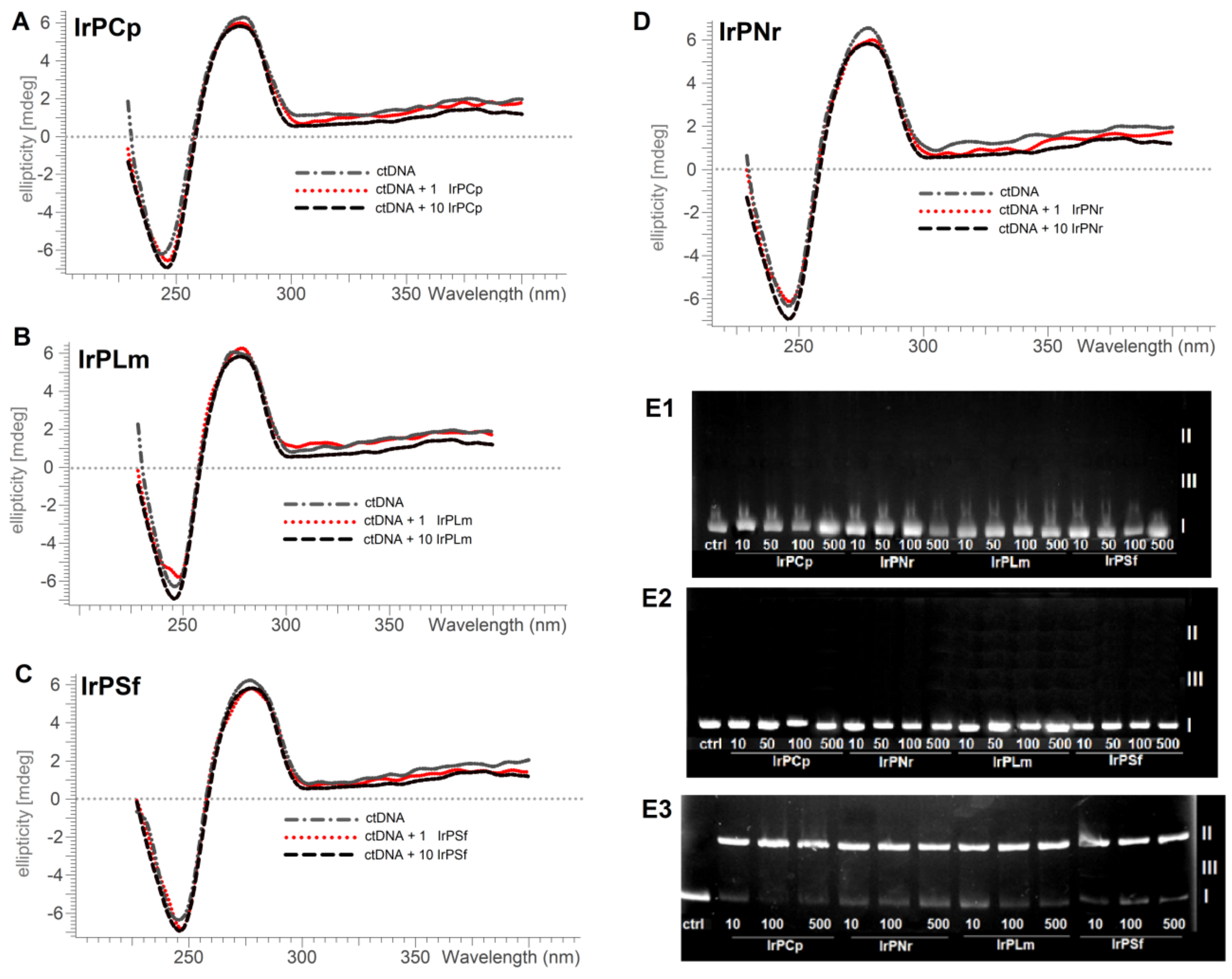
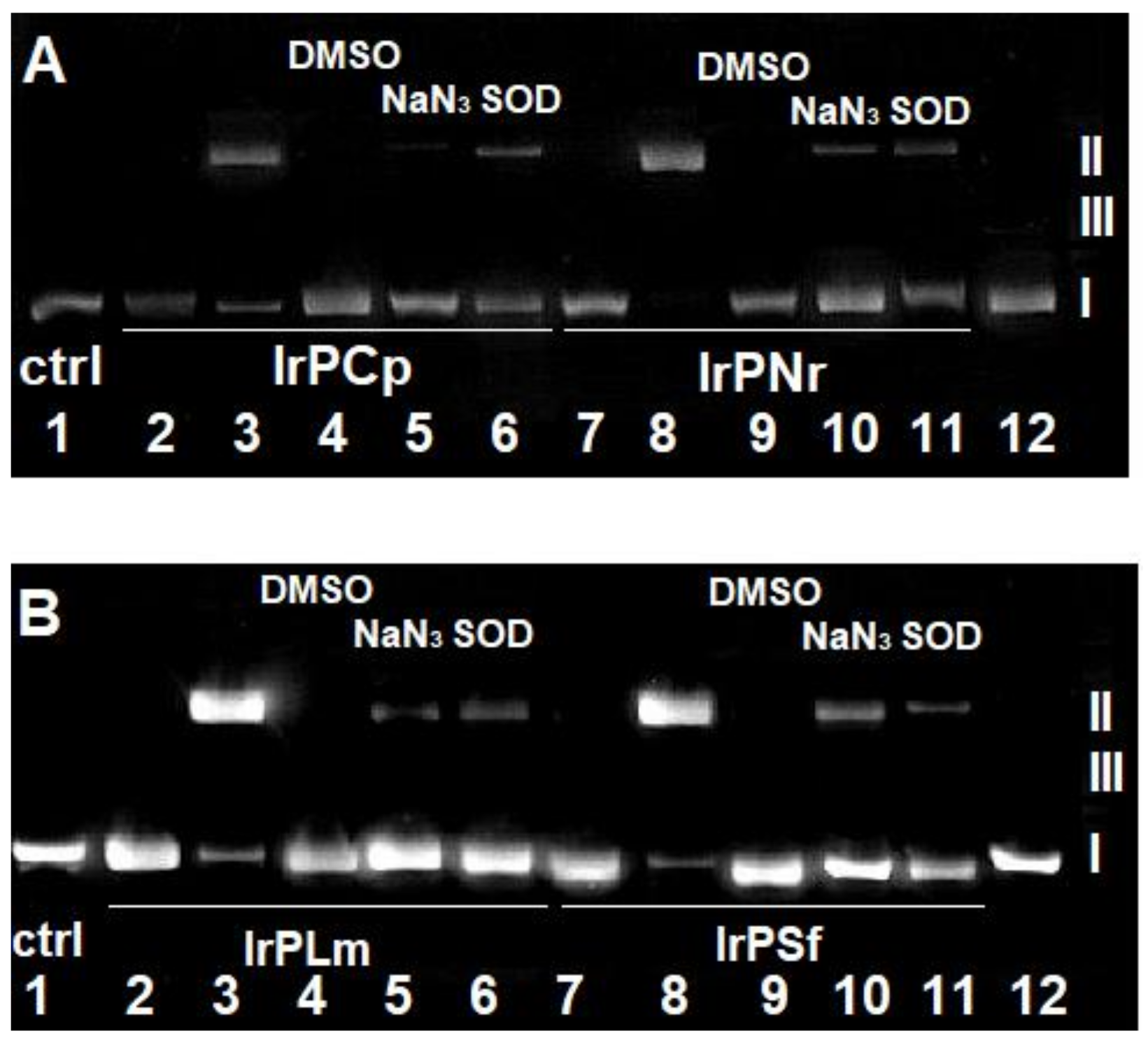
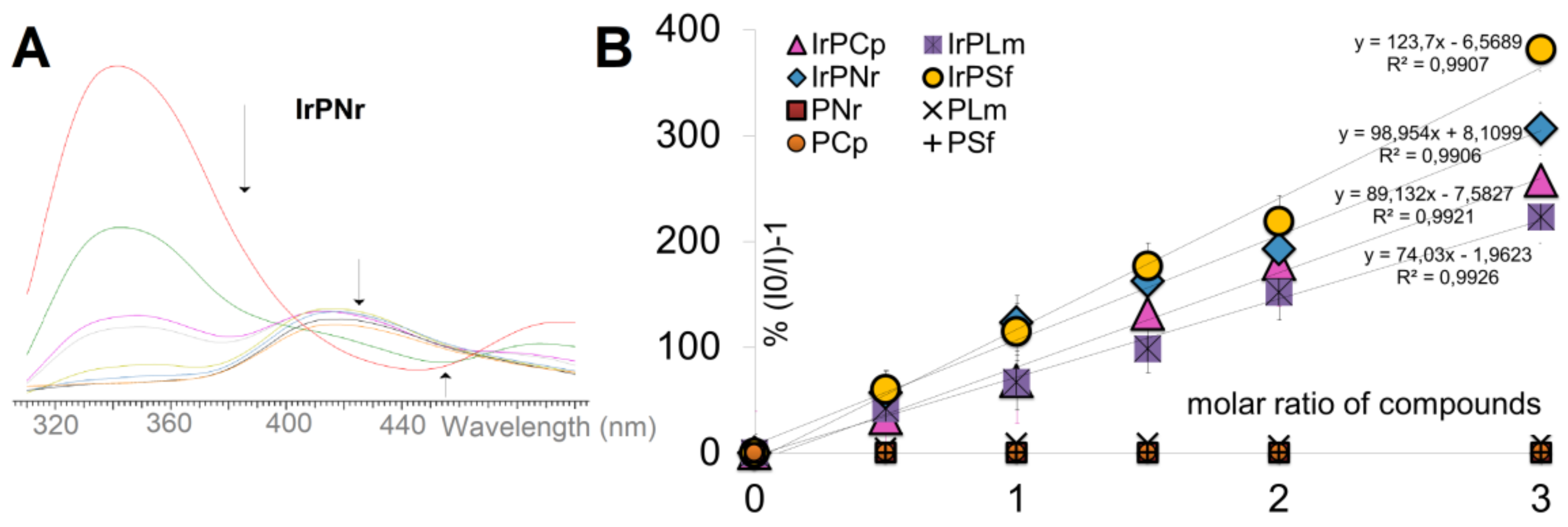
2.2.1. Competitive Fluorescence Studies and DNA Degeneration
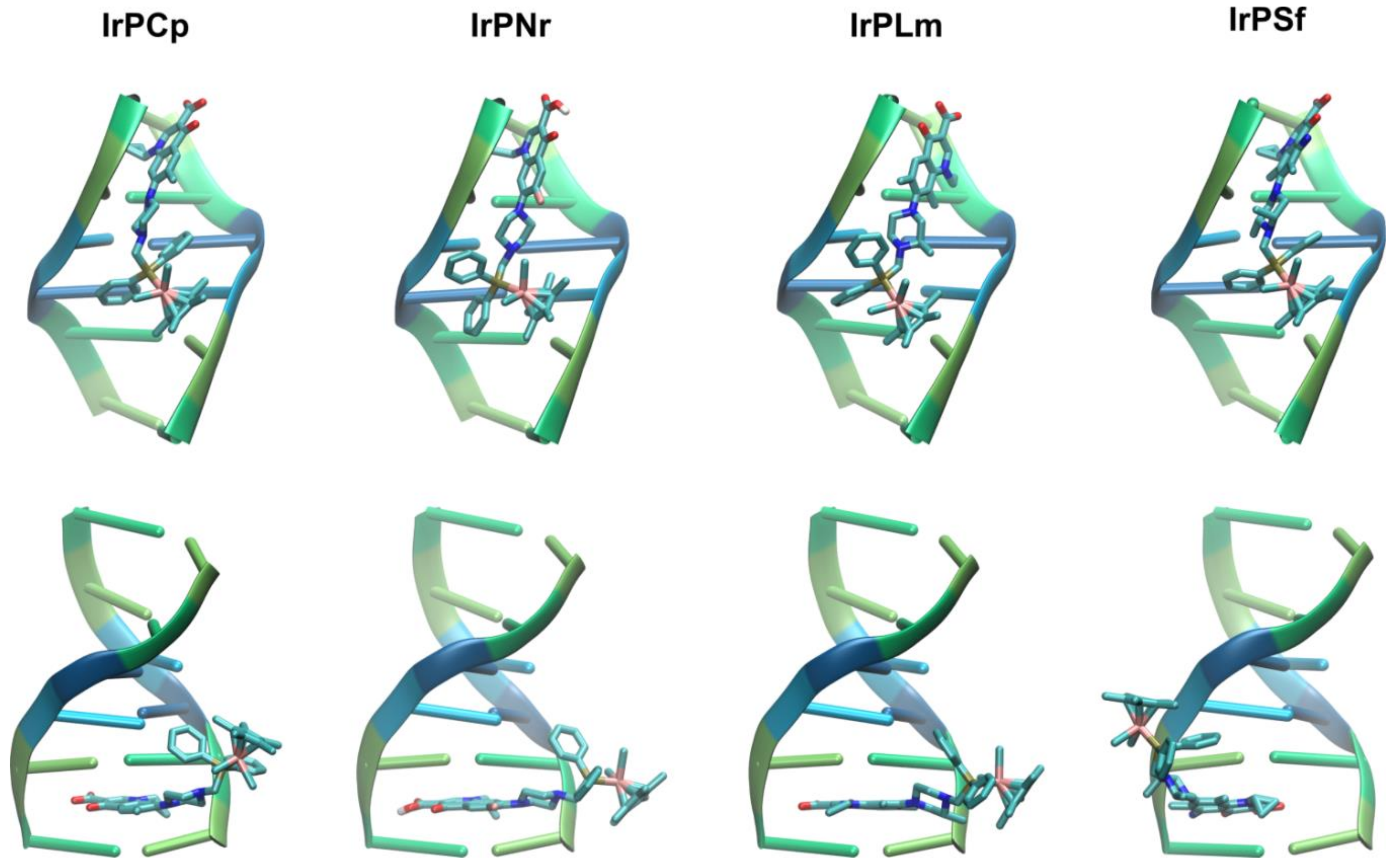
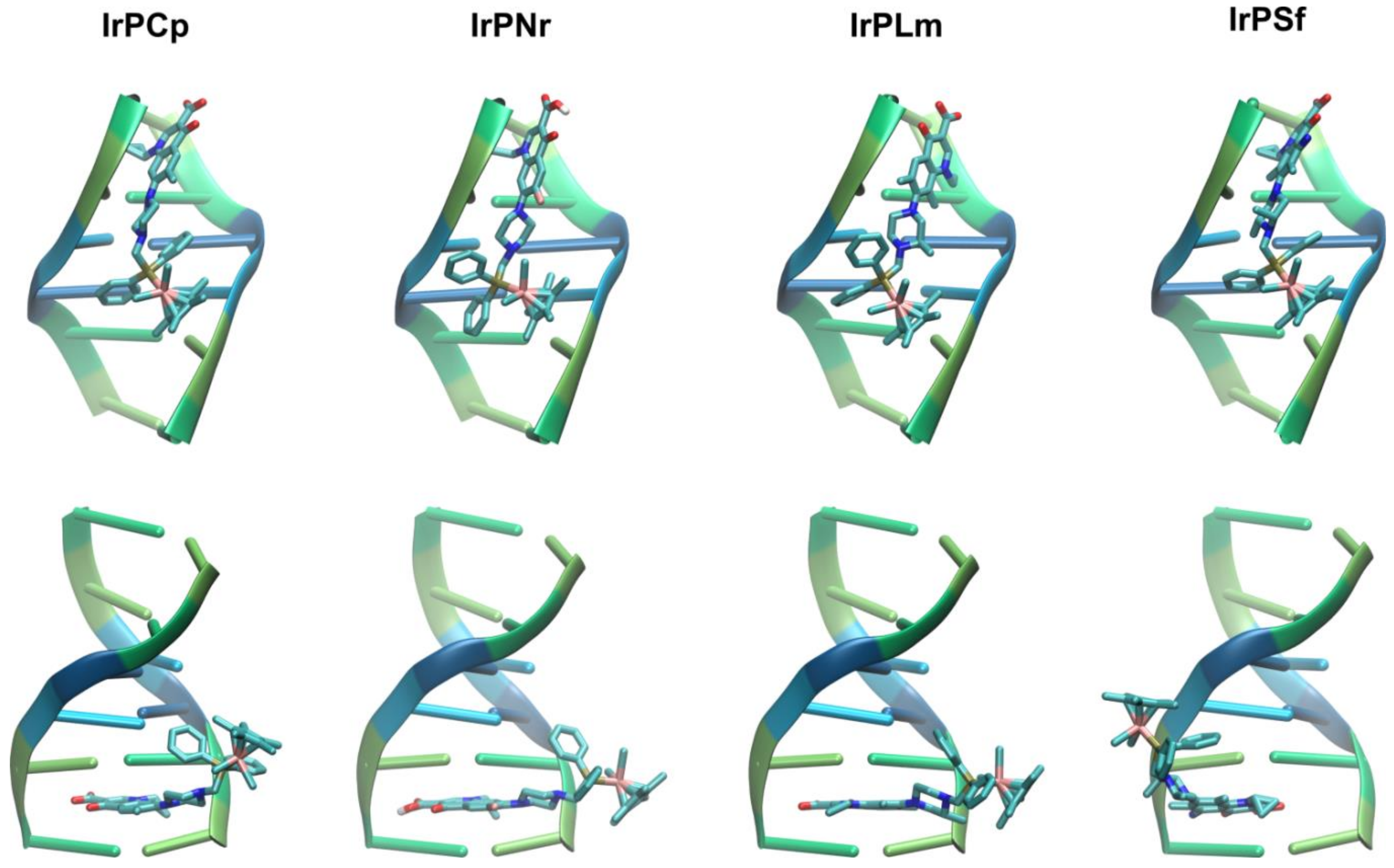
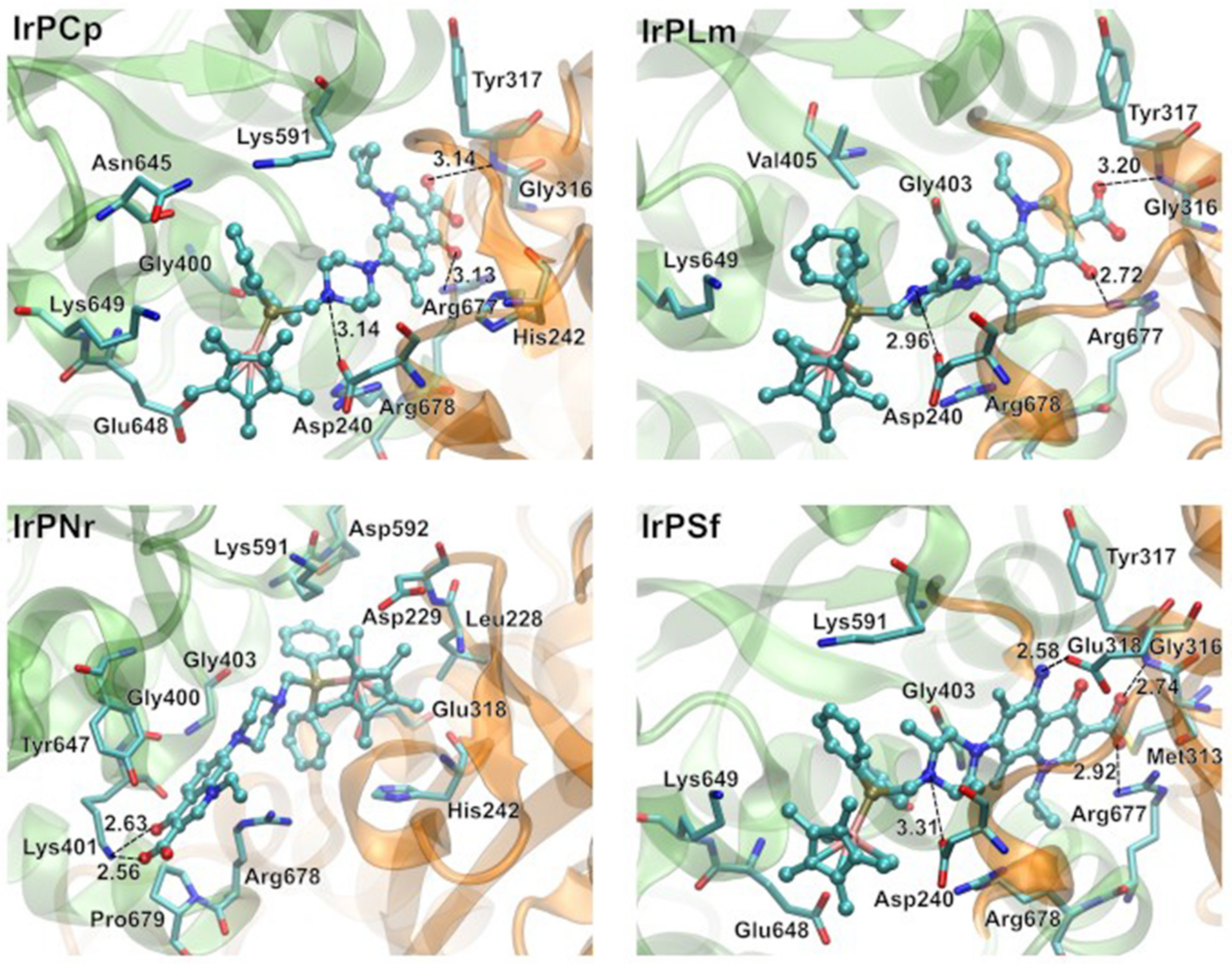
2.2.2. Molecular Docking Study of the Interactions between DNA and Ir(III) Complexes
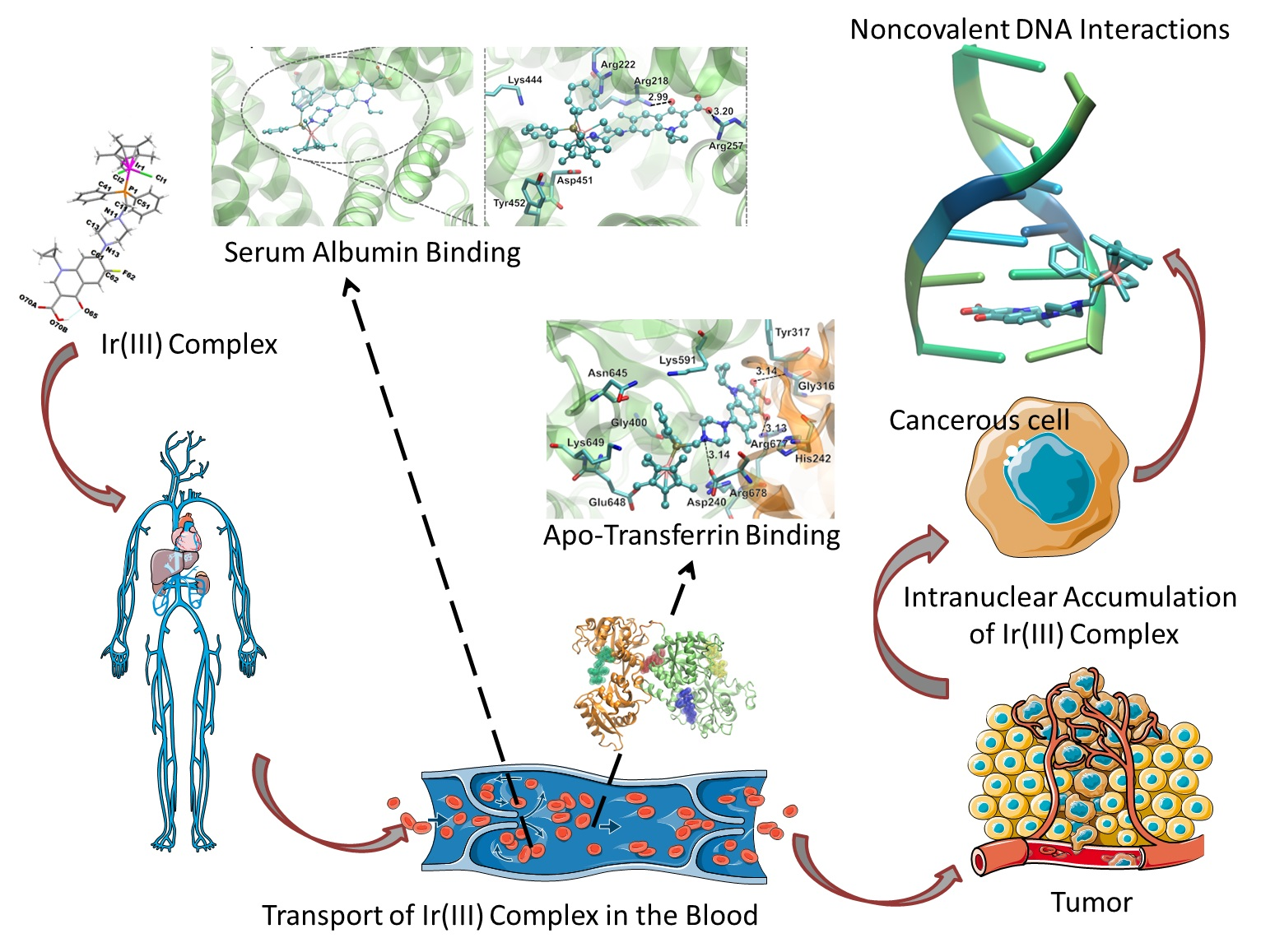
2.3. Possible Reaction with Proteins
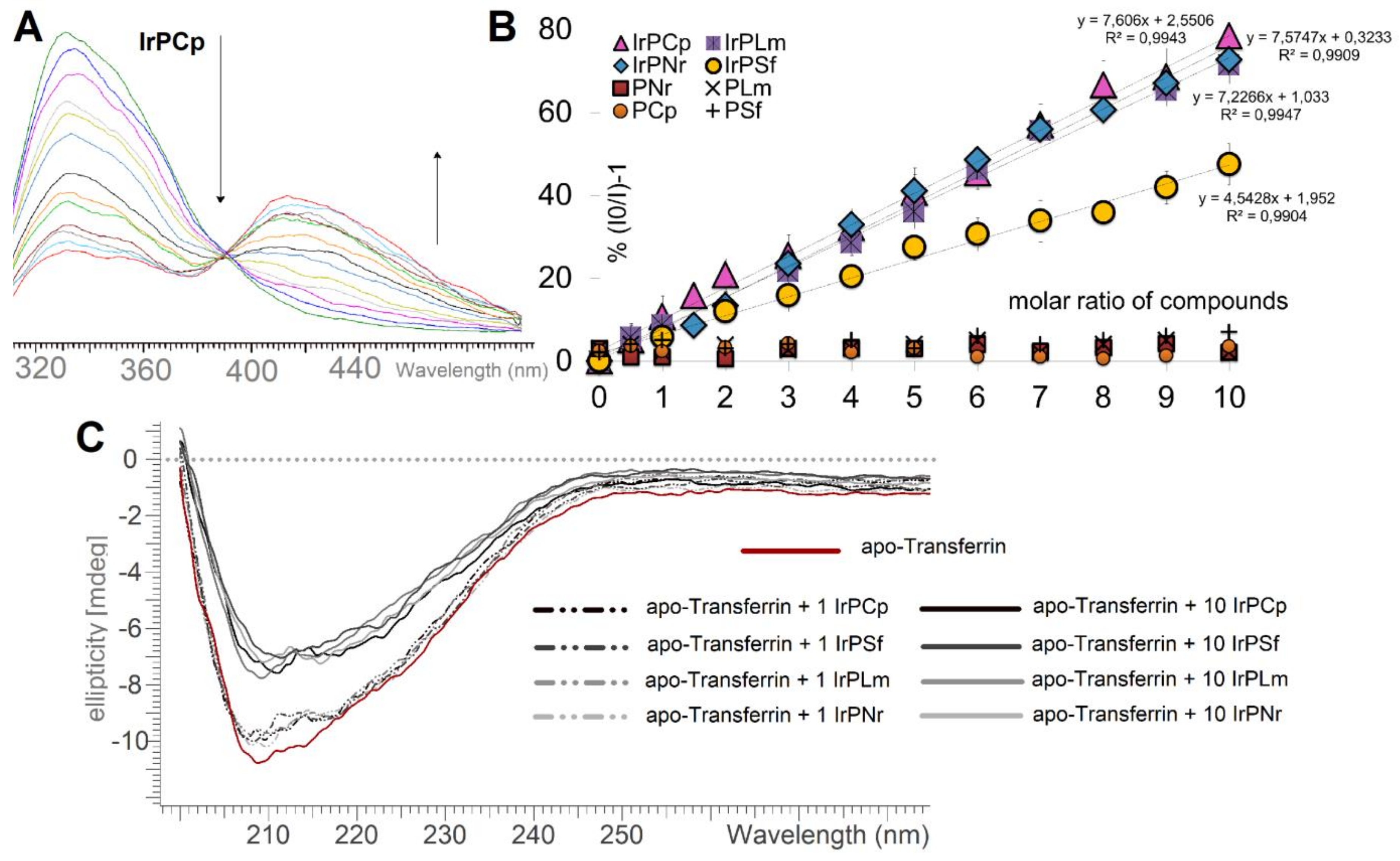
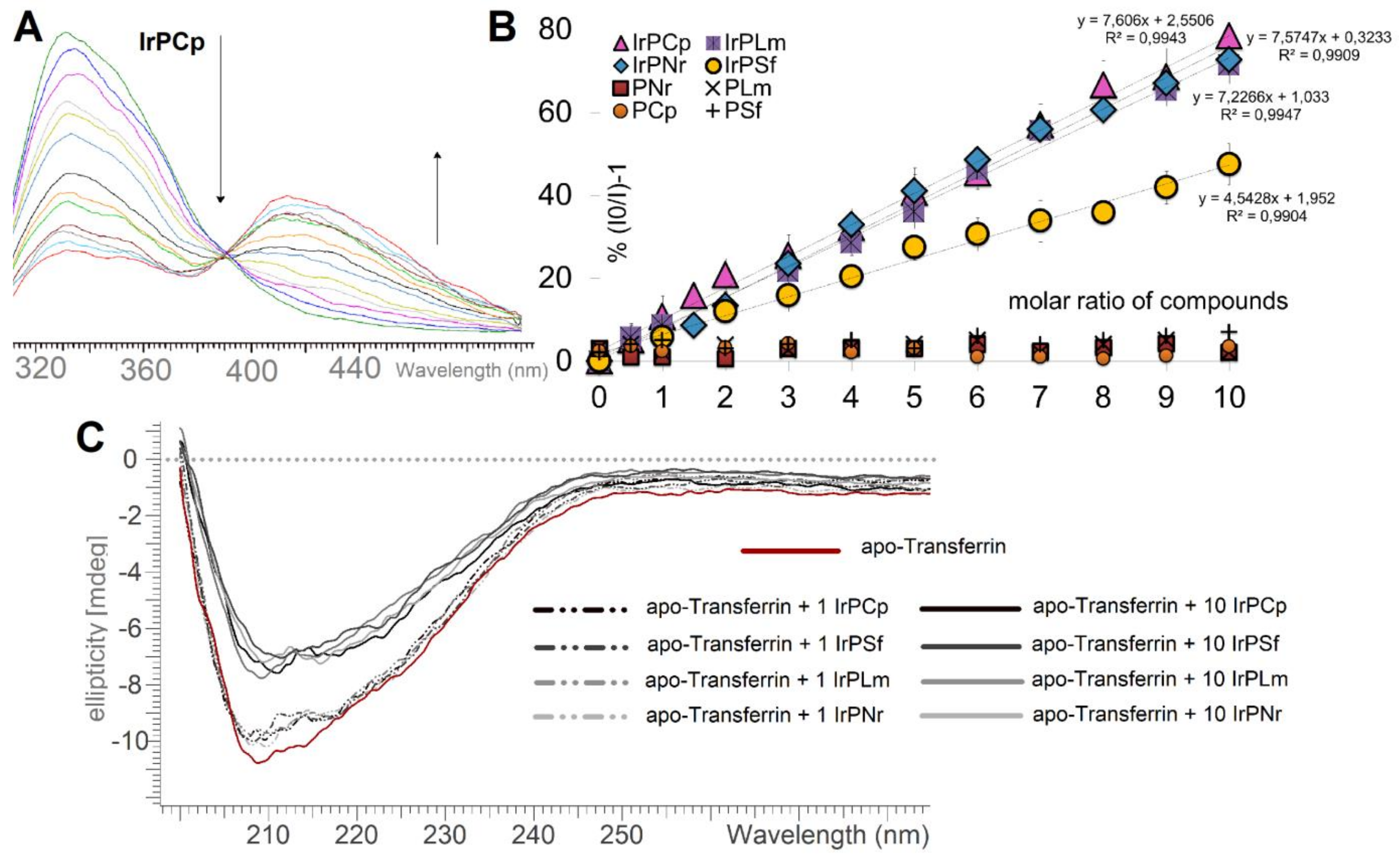
2.3.1. Human Serum Albumin Interaction Study
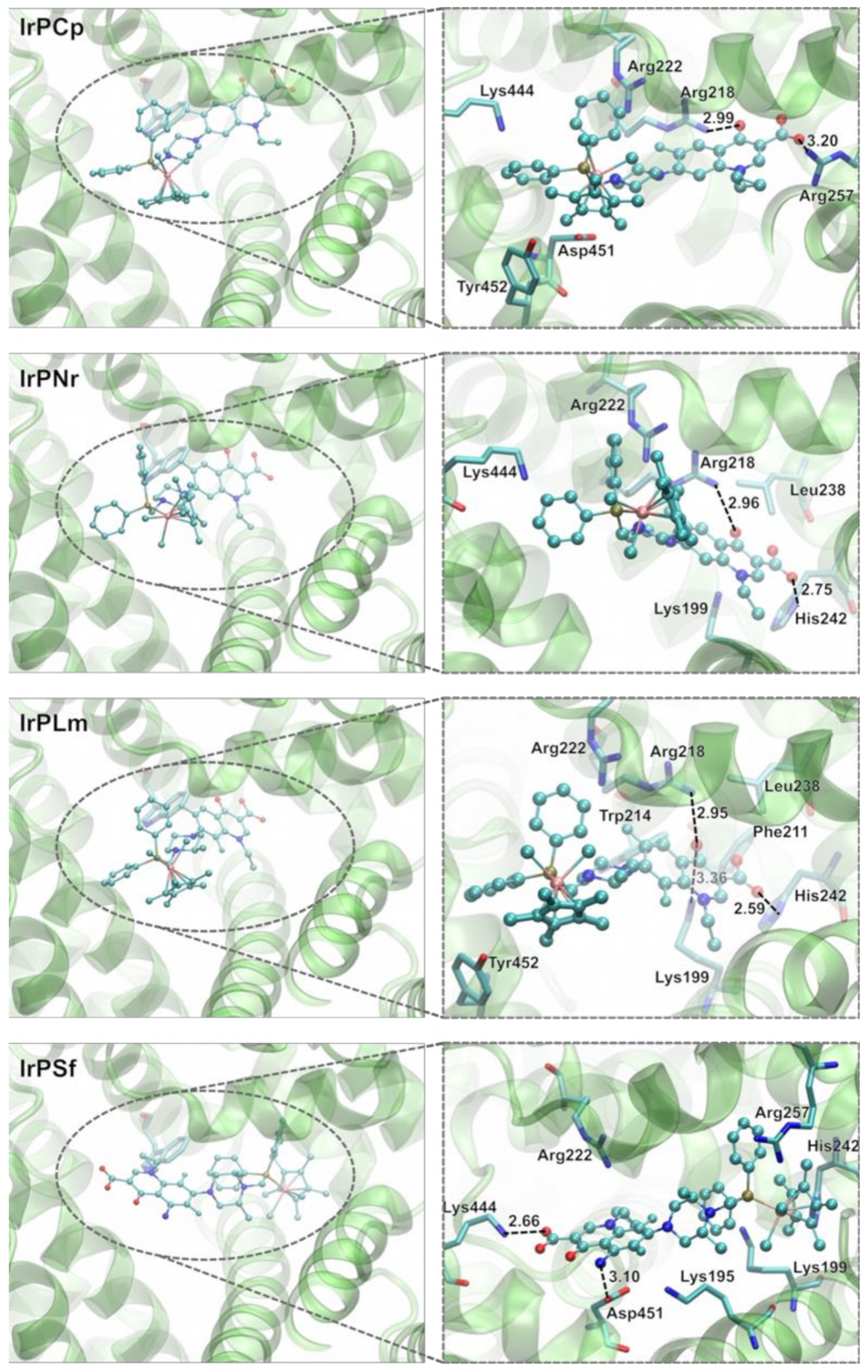
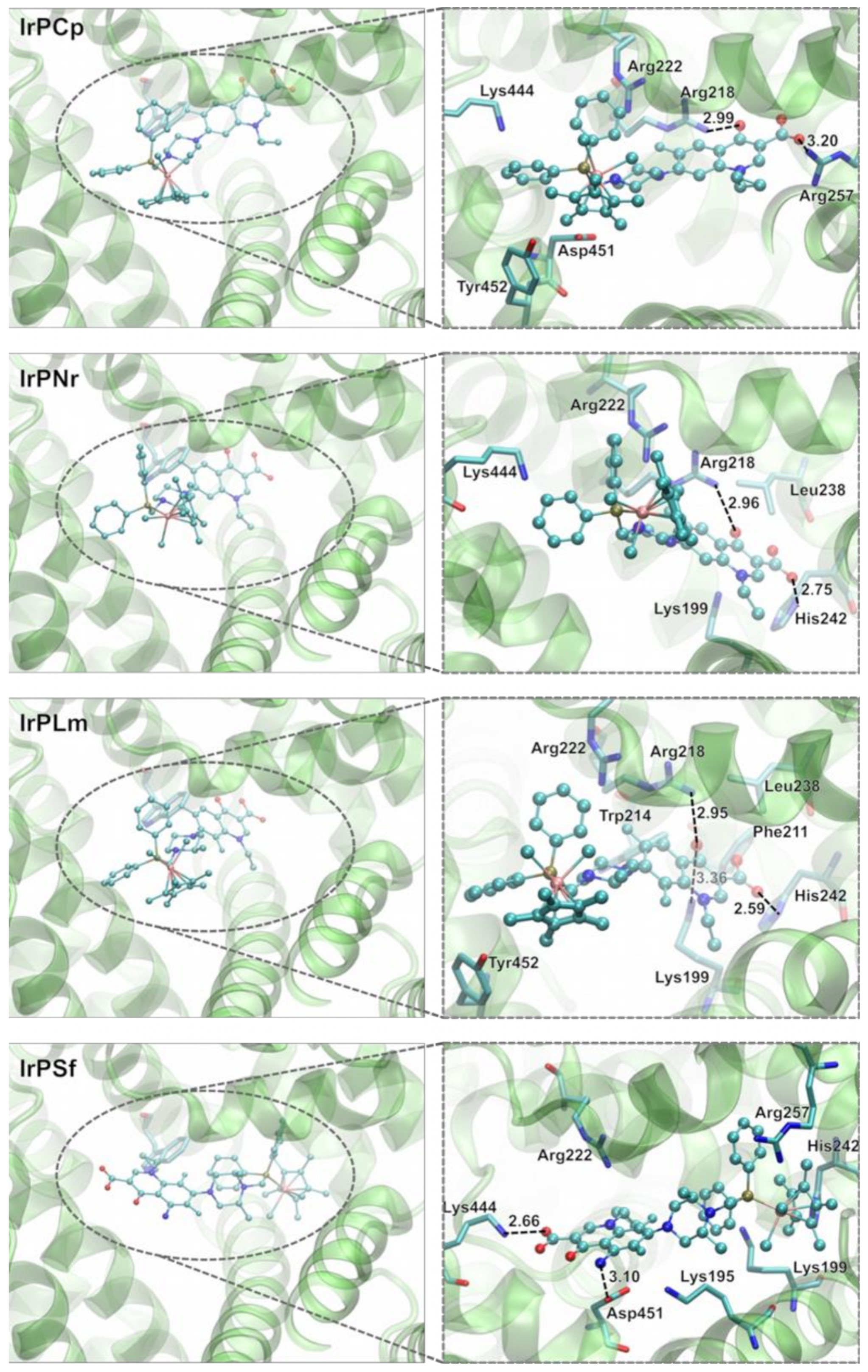
2.3.2. Molecular Docking Study of the Interactions between Human Albumin and Ir(III) Complexes
2.3.3. Apo-Tranferrin Interaction Study
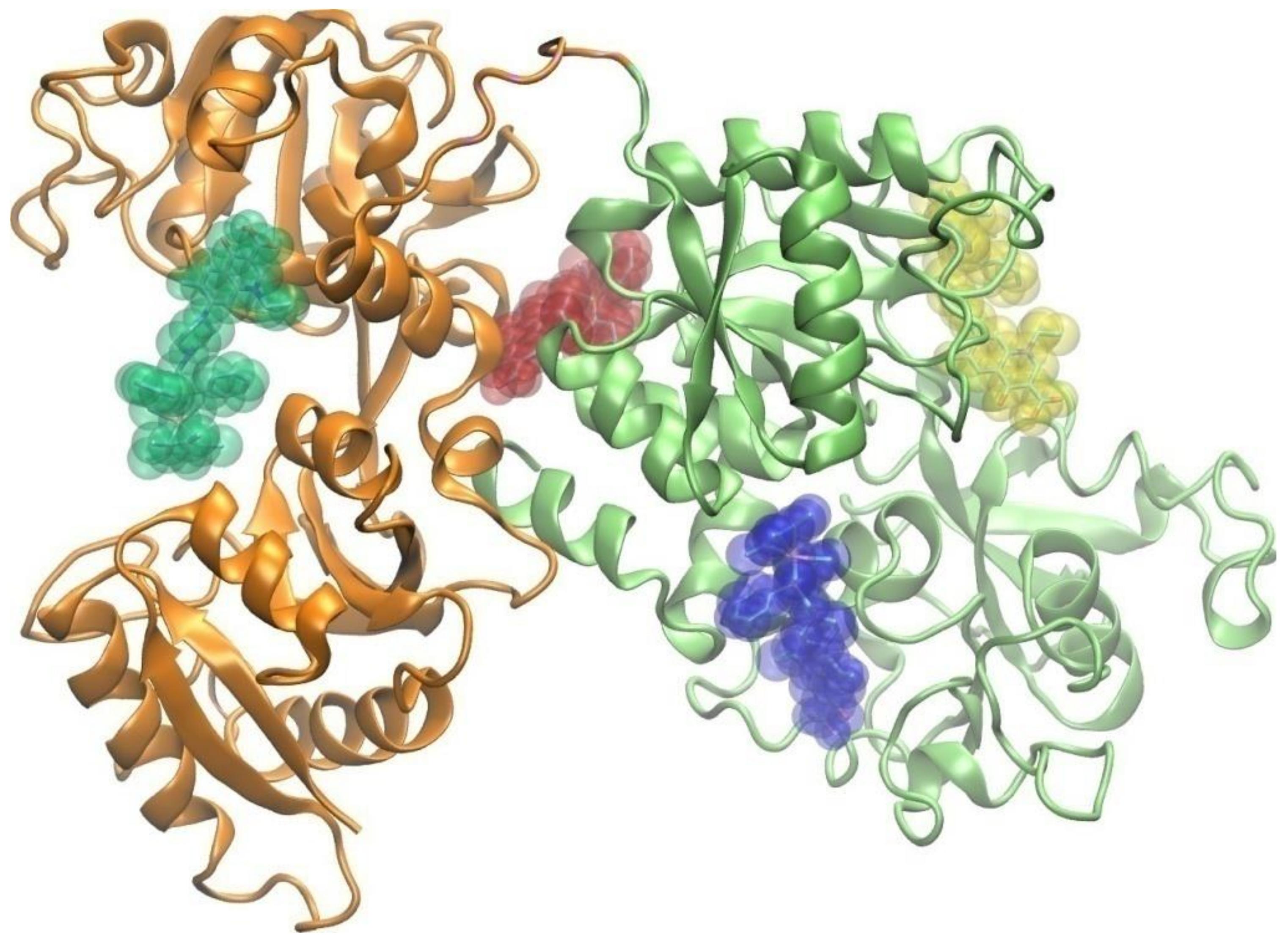
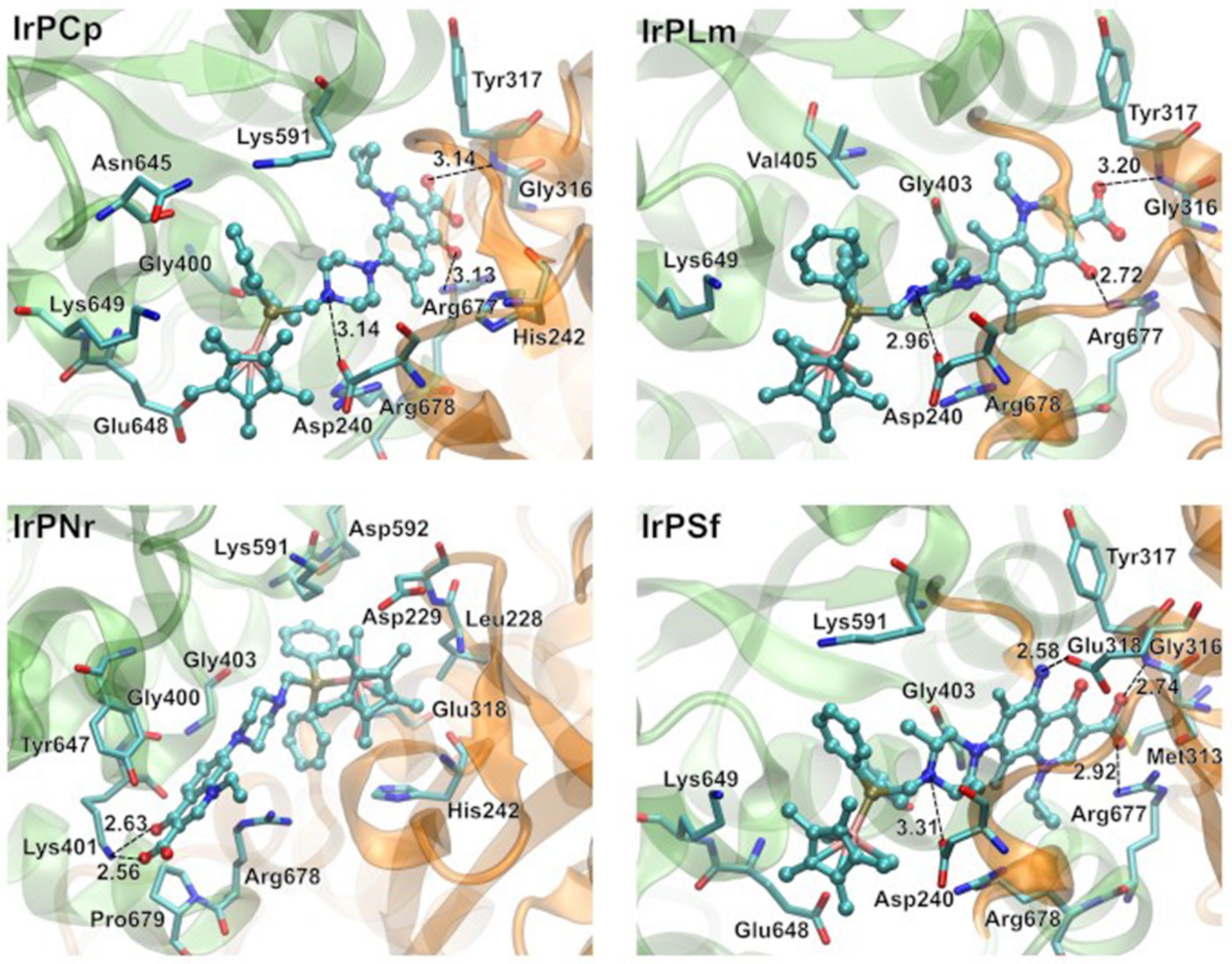
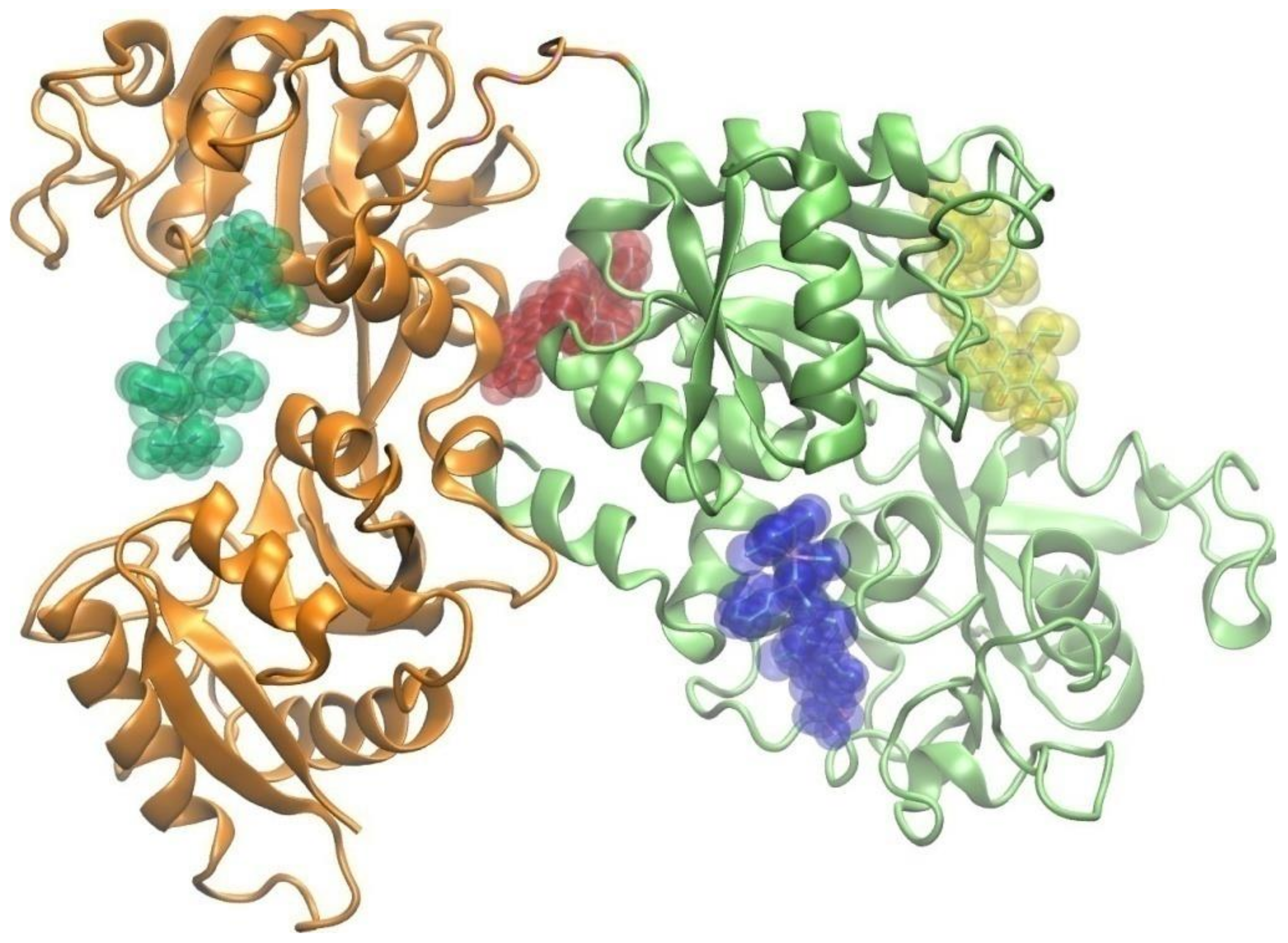
2.3.4. Molecular Docking Study of the Interactions between Apo-Transferrin and Ir(III) Complexes
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. Physical Measurements
3.2.2. Characterization of Organometallic Iridium(III) Complexes
- Data for [Ir(η5-Cp*)Cl2PCp] (IrPCp)
- Data for [Ir(η5-Cp*)Cl2PSf] (IrPSf)
- Data for [Ir(η5-Cp*)Cl2PLm] (IrPLm)
- Data for [Ir(η5-Cp*)Cl2PNr] (IrPNr)
3.2.3. Interaction with Calf Thymus DNA
3.2.4. DNA Strand Break Analysis
3.2.5. Interaction with Human Serum Albumin
3.2.6. Interaction with Transferring
3.2.7. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fuertes, M.A.; Alonso, A.C.; Pérez, J.M. Biochemical Modulation of Cisplatin Mechanisms of Action: Enhancement of Antitumor Activity and Circumvention of Drug Resistance. Chem. Rev. 2003, 103, 645–662. [Google Scholar] [CrossRef]
- Uversky, V.N.; Kretsinger, R.H.; Permyakov, E.A. Encyclopedia of Metalloproteins. Springer: New York, NY, USA, 2013; Volume 1, pp. 1–89. [Google Scholar]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz, J.; Rodriguez, V.; Cutillas, N.; Samper, K.G.; Capdevila, M.; Palacios, Ò.; Espinosa, A. Novel C,N-chelate rhodium(iii) and iridium(iii) antitumor complexes incorporating a lipophilic steroidal conjugate and their interaction with DNA. Dalton Trans. 2012, 41, 12847–12856. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Li, Q.; Zhang, X.; Shi, C.; Li, G.; Wang, M.; Li, K.; Yuan, A. Tuning the Photophysical and Excited State Properties of Phosphorescent Iridium(III) Complexes by Polycyclic Unit Substitution. Chem. Open 2019, 8, 339–343. [Google Scholar] [CrossRef] [Green Version]
- Štarha, P.; Trávníček, Z. Non-platinum complexes containing releasable biologically active ligands. Coord. Chem. Rev. 2019, 395, 130–145. [Google Scholar] [CrossRef]
- Millett, A.J.; Habtemariam, A.; Romero-Canelon, I.; Clarkson, G.J.; Sadler, P.J. Contrasting Anticancer Activity of Half-Sandwich Iridium(III) Complexes Bearing Functionally Diverse 2-Phenylpyridine Ligands. Organometallics 2015, 34, 2683–2694. [Google Scholar] [CrossRef]
- Lapasam, A.; Hussain, O.; Phillips, R.M.; Kaminsky, W.; Kollipara, M.R. Synthesis, characterization and chemosensitivity studies of half-sandwich ruthenium, rhodium and iridium complexes containing κ1(S) and κ2(N,S) aroylthiourea ligands. J. Organomet. Chem. 2019, 880, 272–280. [Google Scholar] [CrossRef] [Green Version]
- Ma, D.-L.; Wu, C.; Wu, K.-J.; Leung, C.-H. Iridium(III) Complexes Targeting Apoptotic Cell Death in Cancer Cells. Molecules 2019, 24, 2739. [Google Scholar] [CrossRef] [Green Version]
- Yadav, V.; Talwar, P. Repositioning of fluoroquinolones from antibiotic to anti-cancer agents: An underestimated truth. Biomed. Pharmacother. 2019, 111, 934–946. [Google Scholar] [CrossRef]
- Bykowska, A.; Starosta, R.; Komarnicka, U.; Ciunik, L.; Kyzioł, A.; Guz-Regner, K.; Bugla-Płoskońska, G.; Jeżowska-Bojczuk, M. Phosphine derivatives of ciprofloxacin and norfloxacin, a new class of potential therapeutic agents. New J. Chem. 2014, 38, 1062. [Google Scholar] [CrossRef]
- Komarnicka, U.; Starosta, R.; Kyzioł, A.; Jeżowska-Bojczuk, M. Copper(i) complexes with phosphine derived from sparfloxacin. Part I—Structures, spectroscopic properties and cytotoxicity. Dalton Trans. 2015, 44, 12688–12699. [Google Scholar] [CrossRef] [PubMed]
- Komarnicka, U.; Starosta, R.; Kyzioł, A.; Płotek, M.; Puchalska, M.; Jeżowska-Bojczuk, M. New copper(I) complexes bearing lomefloxacin motif: Spectroscopic properties, in vitro cytotoxicity and interactions with DNA and human serum albumin. J. Inorg. Biochem. 2016, 165, 25–35. [Google Scholar] [CrossRef]
- Kozieł, S.; Komarnicka, U.K.; Ziółkowska, A.; Skórska-Stania, A.; Pucelik, B.; Płotek, M.; Sebastian, V.; Bieńko, A.; Stochel, G.; Kyzioł, A. Anticancer potency of novel organometallic Ir(iii) complexes with phosphine derivatives of fluoroquinolones encapsulated in polymeric micelles. Inorg. Chem. Front. 2020, 7, 3386–3401. [Google Scholar] [CrossRef]
- Van Holde, K.E.; Zlatanova, J. The Evolution of Molecular Biology; Elsevier: Amsterdam, The Netherlands, 2018; pp. 57–63. [Google Scholar]
- Płotek, M.; Starosta, R.; Komarnicka, U.; Skórska-Stania, A.; Kołoczek, P.; Kyzioł, A. Ruthenium(II) piano stool coordination compounds with aminomethylphosphanes: Synthesis, characterisation and preliminary biological study in vitro. J. Inorg. Biochem. 2017, 170, 178–187. [Google Scholar] [CrossRef]
- Kumar, M.; Kumar, G.; Mogha, N.K.; Jain, R.; Hussain, F.; Masram, D.T. Structure, DNA/proteins binding, docking and cytotoxicity studies of copper(II) complexes with the first quinolone drug nalidixic acid and 2,2′-dipyridylamine. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019, 212, 94–104. [Google Scholar] [CrossRef]
- Manna, S.C.; Mistri, S.; Patra, A.; Mahish, M.K.; Saren, D.; Manne, R.K.; Santra, M.K.; Zangrando, E.; Puschmann, H. Synthesis, structure, DNA/protein binding, molecular docking and in vitro anticancer activity of two Schiff base coordinated copper(II) complexes. Polyhedron 2019, 171, 77–85. [Google Scholar] [CrossRef]
- Kratz, F.; Hartmann, M.; Keppler, B.; Messori, L. The binding properties of two antitumor ruthenium(III) complexes to apotransferrin. J. Biol. Chem. 1994, 269, 2581–2588. [Google Scholar] [CrossRef]
- Pessoa, J.C.; Tomaz, A.I. Transport of Therapeutic Vanadium and Ruthenium Complexes by Blood Plasma Components. Curr. Med. Chem. 2010, 17, 3701–3738. [Google Scholar] [CrossRef]
- Yousefi, R.; Kafrani, A.T.; Nabavizadeh, S.M.; Pouryasin, Z.; Shahsavani, M.B.; Khoshaman, K.; Rashidi, M. The binding assessment with human serum albumin of novel six-coordinate Pt(IV) complexes, containing bidentate nitrogen donor/methyl ligands. Mol. Boil. Res. Commun. 2015, 4, 167–179. [Google Scholar]
- Shahraki, S.; Shiri, F.; Mansouri-Torshizi, H.; Shahraki, J. Characterization of the interaction between a platinum(II) complex and human serum albumin: Spectroscopic analysis and molecular docking. J. Iran. Chem. Soc. 2016, 13, 723–731. [Google Scholar] [CrossRef]
- Bergamo, A.; Messori, L.; Piccioli, F.; Cocchietto, M.; Sava, G. Biological role of adduct formation of the ruthenium(III) complex NAMI-A with serum albumin and serum transferrin. Investig. New Drugs 2003, 21, 401–411. [Google Scholar] [CrossRef]
- Pongratz, M.; Schluga, P.; Jakupec, M.; Arion, V.B.; Hartinger, C.; Keppler, B.K. Transferrin binding and transferrin-mediated cellular uptake of the ruthenium coordination compound KP1019, studied by means of AAS, ESI-MS and CD spectroscopy. J. Anal. At. Spectrom. 2003, 19, 46–51. [Google Scholar] [CrossRef]
- Gibaldi, M.; Koup, J.R. Pharmacokinetic concepts ? Drug binding, apparent volume of distribution and clearance. Eur. J. Clin. Pharmacol. 1981, 20, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Moghadam, N.H.; Salehzadeh, S.; Tanzadehpanah, H.; Saidijam, M.; Karimi, J.; Khazalpour, S. In vitro cytotoxicity and DNA/HSA interaction study of triamterene using molecular modelling and multi-spectroscopic methods. J. Biomol. Struct. Dyn. 2018, 37, 2242–2253. [Google Scholar] [CrossRef]
- Kudarha, R.R.; Sawant, K.K. Albumin based versatile multifunctional nanocarriers for cancer therapy: Fabrication, surface modification, multimodal therapeutics and imaging approaches. Mater. Sci. Eng. C 2017, 81, 607–626. [Google Scholar] [CrossRef]
- Zhao, P.; Wang, Y.; Wu, A.; Rao, Y.; Huang, Y. Roles of Albumin-Binding Proteins in Cancer Progression and Biomimetic Targeted Drug Delivery. ChemBioChem 2018, 19, 1796–1805. [Google Scholar] [CrossRef]
- Hoogenboezem, E.N.; Duvall, C.L. Harnessing albumin as a carrier for cancer therapies. Adv. Drug Deliv. Rev. 2018, 130, 73–89. [Google Scholar] [CrossRef]
- Li, C.; Wang, X.; Song, H.; Deng, S.; Li, W.; Li, J.; Sun, J. Current multifunctional albumin-based nanoplatforms for cancer multi-mode therapy. Asian J. Pharm. Sci. 2020, 15, 1–12. [Google Scholar] [CrossRef]
- Lambert, L.A.; Perri, H.; Halbrooks, P.J.; Mason, A.B. Evolution of the transferrin family: Conservation of residues associated with iron and anion binding. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2005, 142, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Thorstensen, K.; Romslo, I. The role of transferrin in the mechanism of cellular iron uptake. Biochem. J. 1990, 271, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Guy, J.; Drabek, D.; Antoniou, M. Delivery of drugs, proteins and genes into cells using transferrin as a ligand for receptor-mediated endocytosis. Mol. Biotechnol. 1995, 3, 237–248. [Google Scholar] [CrossRef]
- Sun, H.; Li, H.; Sadler, P.J. Transferrin as a Metal Ion Mediator. Chem. Rev. 1999, 99, 2817–2842. [Google Scholar] [CrossRef]
- Wang, J.; Tian, S.; Petros, R.A.; Napier, M.E.; DeSimone, J.M. The Complex Role of Multivalency in Nanoparticles Targeting the Transferrin Receptor for Cancer Therapies. J. Am. Chem. Soc. 2010, 132, 11306–11313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Qian, Z.M. Transferrin/transferrin receptor-mediated drug delivery. Med. Res. Rev. 2002, 22, 225–250. [Google Scholar] [CrossRef] [PubMed]
- Luck, A.N.; Mason, A.B. Structure and dynamics of drug carriers and their interaction with cellular receptors: Focus on serum transferrin. Adv. Drug Deliv. Rev. 2013, 65, 1012–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Li, Y.; Tian, Z.; Cao, R.; Yang, B. Transferrin-conjugated nanodiamond as an intracellular transporter of chemotherapeutic drug and targeting therapy for cancer cells. Ther. Deliv. 2014, 5, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Daniels, T.R.; Bernabeu, E.; Rodríguez, J.A.; Patel, S.; Kozman, M.; Chiappetta, D.A.; Holler, E.; Ljubimova, J.Y.; Helguera, G.; Penichet, M.L. The transferrin receptor and the targeted delivery of therapeutic agents against cancer. Biochim. et Biophys. Acta (BBA) Gen. Subj. 2012, 1820, 291–317. [Google Scholar] [CrossRef] [Green Version]
- Sparreboom, A.; Verweij, J. Advances in Cancer Therapeutics. Clin. Pharmacol. Ther. 2009, 85, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Komarnicka, U.K.; Starosta, R.; Płotek, M.; De Almeida, R.F.M.; Jeżowska-Bojczuk, M.; Kyzioł, A. Copper(i) complexes with phosphine derived from sparfloxacin. Part II: A first insight into the cytotoxic action mode. Dalton Trans. 2016, 45, 5052–5063. [Google Scholar] [CrossRef]
- Bykowska, A.; Starosta, R.; Brzuszkiewicz, A.; Bażanów, B.; Florek, M.; Jackulak, N.; Król, J.; Grzesiak, J.; Kaliński, K.; Jeżowska-Bojczuk, M. Synthesis, properties and biological activity of a novel phosphines ligand derived from ciprofloxacin. Polyhedron 2013, 60, 23–29. [Google Scholar] [CrossRef]
- Komarnicka, U.K.; Starosta, R.; Guz-Regner, K.; Bugla-Płoskońska, G.; Kyzioł, A.; Jeżowska-Bojczuk, M. Phosphine derivatives of sparfloxacin—Synthesis, structures and in vitro activity. J. Mol. Struct. 2015, 1096, 55–63. [Google Scholar] [CrossRef]
- Kołoczek, P.; Skórska-Stania, A.; Cierniak, A.; Sebastian, V.; Komarnicka, U.K.; Płotek, M.; Kyzioł, A. Polymeric micelle-mediated delivery of half-sandwich ruthenium(II) complexes with phosphanes derived from fluoroloquinolones for lung adenocarcinoma treatment. Eur. J. Pharm. Biopharm. 2018, 128, 69–81. [Google Scholar] [CrossRef] [Green Version]
- Malik, M.; Bieńko, D.C.; Komarnicka, U.K.; Kyzioł, A.; Dryś, M.; Świtlicka, A.; Dyguda-Kazimierowicz, E.; Jedwabny, W. Synthesis, structural characterization, docking simulation and in vitro antiproliferative activity of the new gold(III) complex with 2-pyridineethanol. J. Inorg. Biochem. 2021, 215, 111311. [Google Scholar] [CrossRef]
- Subastri, A.; Ramamurthy, C.; Suyavaran, A.; Mareeswaran, R.; Rao, P.L.; Krishna, K.H.; Kumar, M.S.; Sujatha, V.; Thirunavukkarasu, C. Spectroscopic and molecular docking studies on the interaction of troxerutin with DNA. Int. J. Biol. Macromol. 2015, 78, 122–129. [Google Scholar] [CrossRef]
- Varlan, A.; Ionescu, S.; Hillebrand, M. Study of the interaction between ofloxacin and human serum albumin by spectroscopic methods. Luminescence 2011, 26, 710–715. [Google Scholar] [CrossRef]
- Komarnicka, U.K.; Kozieł, S.; Starosta, R.; Kyzioł, A. Selective Cu(I) complex with phosphine-peptide (SarGly) conjugate contra breast cancer: Synthesis, spectroscopic characterization and insight into cytotoxic action. J. Inorg. Biochem. 2018, 186, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Kyzioł, A.; Cierniak, A.; Gubernator, J.; Markowski, A.; Jezowska-Bojczuk, M.; Komarnicka, U.K. Copper(i) complexes with phosphine derived from sparfloxacin. Part III: Multifaceted cell death and preliminary study of liposomal formulation of selected copper(i) complexes. Dalton Trans. 2017, 47, 1981–1992. [Google Scholar] [CrossRef] [PubMed]
- Santini, C.; Pellei, M.; Gandin, V.; Porchia, M.; Tisato, F.; Marzano, C. Advances in Copper Complexes as Anticancer Agents. Chem. Rev. 2014, 114, 815–862. [Google Scholar] [CrossRef] [PubMed]
- Bancirova, M. Sodium azide as a specific quencher of singlet oxygen during chemiluminescent detection by luminol and Cypridina luciferin analogues. Luminescence 2011, 26, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Huey, R.; Morris, G.; Olson, A.J.; Goodsell, D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Živec, P.; Perdih, F.; Turel, I.; Giester, G.; Psomas, G. Different types of copper complexes with the quinolone antimicrobial drugs ofloxacin and norfloxacin: Structure, DNA- and albumin-binding. J. Inorg. Biochem. 2012, 117, 35–47. [Google Scholar] [CrossRef]
- Sudlow, G.; Birkett, D.J.; Wade, D.N. The characterization of two specific drug binding sites on human serum albumin. Mol. Pharmacol. 1975, 11, 824–832. [Google Scholar]
- Sudlow, G.; Birkett, D.J.; Wade, D.N. Further characterization of specific drug binding sites on human serum albumin. Mol. Pharmacol. 1976, 12, 1052–1061. [Google Scholar]
- Ahmad, B.; Parveen, S.; Khan, R.H. Effect of Albumin Conformation on the Binding of Ciprofloxacin to Human Serum Albumin: A Novel Approach Directly Assigning Binding Site. Biomacromolecules 2006, 7, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.B.; Love, S. The binding and transport of alternative metals by transferrin. Biochim. et Biophys. Acta (BBA) Gen. Subj. 2012, 1820, 362–378. [Google Scholar] [CrossRef]
- Martínez, A.; Suárez, J.; Shand, T.; Magliozzo, R.S.; Sánchez-Delgado, R.A. Interactions of arene–Ru(II)–chloroquine complexes of known antimalarial and antitumor activity with human serum albumin (HSA) and transferrin. J. Inorg. Biochem. 2011, 105, 39–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saboury, A.A. Application of a new method for data analysis of isothermal titration calorimetry in the interaction between human serum albumin and Ni2+. J. Chem. Thermodyn. 2003, 35, 1975–1981. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, J.; Jin, L.; Chang, Y.; Duan, J.; Lu, Y. A new conjugated poly(pyridinium salt) derived from phenanthridine diamine: Its synthesis, optical properties and interaction with calf thymus DNA. Polym. J. 2015, 47, 753–759. [Google Scholar] [CrossRef]
- Kumar, H.M.S.; Kunabenchi, R.S.; Nishti, S.V.; Biradar, J.S.; Kadadevarmath, J.S. Effect of Solvent Polarity on Fluorescence Quenching of New Indole Derivatives by CCl4. Spectrosc. Lett. 2009, 42, 226–234. [Google Scholar] [CrossRef]
- Hanagodimath, S.M.; Manohara, S.R.; Biradar, D.S.; Hadimani, S.K.B. Fluorescence Quenching of 2,2″-dimethyl-p-terphenyl by Carbon Tetrachloride in Binary Mixtures. Spectrosc. Lett. 2008, 41, 242–250. [Google Scholar] [CrossRef]
- Baslak, C.; Kuş, M.; Cengeloglu, Y.; Ersoz, M. A comparative study on fluorescence quenching of CdTe nanocrystals with a serial of polycyclic aromatic hydrocarbons. J. Lumin. 2014, 153, 177–181. [Google Scholar] [CrossRef]
- Lesiów, M.K.; Komarnicka, U.K.; Kyzioł, A.; Bieńko, A.; Pietrzyk, P. ROS-mediated lipid peroxidation as a result of Cu(ii) interaction with FomA protein fragments of F. nucleatum: Relevance to colorectal carcinogenesis. Metallomics 2019, 11, 2066–2077. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Nordén, B. Methyl green. A DNA major-groove binding drug. FEBS Lett. 1993, 315, 61–64. [Google Scholar] [CrossRef] [Green Version]
- De Castro, L.F.P.; Zacharias, M. DAPI binding to the DNA minor groove: A continuum solvent analysis. J. Mol. Recognit. 2002, 15, 209–220. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Autodock. Available online: http://autodock.1369657.n2.nabble.com/ADL-Parameters-for-docking-with-metal-ions-in-receptor-td2505649.html (accessed on 30 March 2021).
- Canals, A.; Purciolas, M.; Aymamí, J.; Coll, M. The anticancer agent ellipticine unwinds DNA by intercalative binding in an orientation parallel to base pairs. Acta Crystallogr. Sect. D Biol. Crystallogr. 2005, 61, 1009–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wardell, M.; Wang, Z.; Ho, J.X.; Robert, J.; Rueker, F.; Ruble, J.; Carter, D.C. The Atomic Structure of Human Methemalbumin at 1.9 Å. Biochem. Biophys. Res. Commun. 2002, 291, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, J.; Doerr, S.; Martínez-Rosell, G.; Rose, A.; De Fabritiis, G. DeepSite: Protein-binding site predictor using 3D-convolutional neural networks. Bioinformatics 2017, 33, 3036–3042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capra, J.A.; Laskowski, R.; Thornton, J.; Singh, M.; Funkhouser, T.A. Predicting Protein Ligand Binding Sites by Combining Evolutionary Sequence Conservation and 3D Structure. PLoS Comput. Biol. 2009, 5, e1000585. [Google Scholar] [CrossRef] [Green Version]
- Jendele, L.; Krivak, R.; Skoda, P.; Novotny, M.; Hoksza, D. PrankWeb: A web server for ligand binding site prediction and visualization. Nucleic Acids Res. 2019, 47, W345–W349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krivák, R.; Hoksza, D. P2Rank: Machine learning based tool for rapid and accurate prediction of ligand binding sites from protein structure. J. Chemin. 2018, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- Wally, J.; Halbrooks, P.J.; Vonrhein, C.; Rould, M.A.; Everse, S.J.; Mason, A.B.; Buchanan, S.K. The Crystal Structure of Iron-free Human Serum Transferrin Provides Insight into Inter-lobe Communication and Receptor Binding. J. Biol. Chem. 2006, 281, 24934–24944. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ksv [M−1] | |||
|---|---|---|---|
| Intercalation | Minor Groove Binding | Major Groove Binding | |
| Ligands | |||
| PCp | 7.62 × 101 | 7.43 × 103 | 0.03 × 101 |
| PNr | 5.27 × 102 | 1.57 × 103 | 0.25 × 101 |
| PLm | 2.59 × 102 | 1.91 × 102 | 0.14 × 101 |
| PSf | 2.05 × 102 | 7.39 × 103 | 0.13 × 101 |
| Complexes | |||
| IrPCp | 1.42 × 103 | 1.23 × 104 | 3.25 × 101 |
| IrPNr | 1.54 × 103 | 8.25 × 103 | 0.46 × 101 |
| IrPLm | 1.48 × 103 | 6.25 × 103 | 2.13 × 101 |
| IrPSf | 1.51 × 103 | 1.36 × 104 | 0.41 × 101 |
| IrPCp | IrPNr | IrPLm | IrPSf | |||||
|---|---|---|---|---|---|---|---|---|
| All Clusters | Clusters with at Least 6 Members | All Clusters | Clusters with at Least 6 Members | All Clusters | Clusters with at Least 6 Members | All Clusters | Clusters with at Least 6 Members | |
| Minor groove binding | 47% | 59% | 48% | 42% | 60% | 89% | 47% | 81% |
| Intercalat ion and major groove binding | 41% | 41% | 44% | 58% | 34% | 11% | 36% | 19% |
| Intercalat ion and minor groove binding | 12% | - | 8% | - | 6% | - | 17% | - |
| Number of binding poses | 100 | 61 | 100 | 53 | 100 | 53 | 100 | 37 |
| Ksv [M−1] | |||
|---|---|---|---|
| PCp | 0.21 × 101 | IrPCp | 2.08 × 106 |
| PSf | 0.11 × 101 | IrPSf | 5.67 × 106 |
| PLm | 0.53 × 101 | IrPLm | 5.06 × 106 |
| PNr | 0.19 × 101 | IrPNr | 6.98 × 105 |
| Ksv [M−1] | |||
|---|---|---|---|
| PCp | 0.45 × 101 | IrPCp | 2.18 × 104 |
| PSf | 1.10 × 104 | IrPSf | 1.32 × 104 |
| PLm | 1.52 × 102 | IrPLm | 1.99 × 104 |
| PNr | 4.56 × 101 | IrPNr | 2.02 × 104 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kozieł, S.A.; Lesiów, M.K.; Wojtala, D.; Dyguda-Kazimierowicz, E.; Bieńko, D.; Komarnicka, U.K. Interaction between DNA, Albumin and Apo-Transferrin and Iridium(III) Complexes with Phosphines Derived from Fluoroquinolones as a Potent Anticancer Drug. Pharmaceuticals 2021, 14, 685. https://doi.org/10.3390/ph14070685
Kozieł SA, Lesiów MK, Wojtala D, Dyguda-Kazimierowicz E, Bieńko D, Komarnicka UK. Interaction between DNA, Albumin and Apo-Transferrin and Iridium(III) Complexes with Phosphines Derived from Fluoroquinolones as a Potent Anticancer Drug. Pharmaceuticals. 2021; 14(7):685. https://doi.org/10.3390/ph14070685
Chicago/Turabian StyleKozieł, Sandra Amanda, Monika Katarzyna Lesiów, Daria Wojtala, Edyta Dyguda-Kazimierowicz, Dariusz Bieńko, and Urszula Katarzyna Komarnicka. 2021. "Interaction between DNA, Albumin and Apo-Transferrin and Iridium(III) Complexes with Phosphines Derived from Fluoroquinolones as a Potent Anticancer Drug" Pharmaceuticals 14, no. 7: 685. https://doi.org/10.3390/ph14070685
APA StyleKozieł, S. A., Lesiów, M. K., Wojtala, D., Dyguda-Kazimierowicz, E., Bieńko, D., & Komarnicka, U. K. (2021). Interaction between DNA, Albumin and Apo-Transferrin and Iridium(III) Complexes with Phosphines Derived from Fluoroquinolones as a Potent Anticancer Drug. Pharmaceuticals, 14(7), 685. https://doi.org/10.3390/ph14070685