
Design and Synthesis of Novel Thiazolo[5,4-d]pyrimidine Derivatives with High Affinity for Both the Adenosine A1 and A2A Receptors, and Efficacy in Animal Models of Depression


 ,
, 
 ,
,  , ,
, ,  and
and 
Abstract
:
1. Introduction
2. Results and Discussion
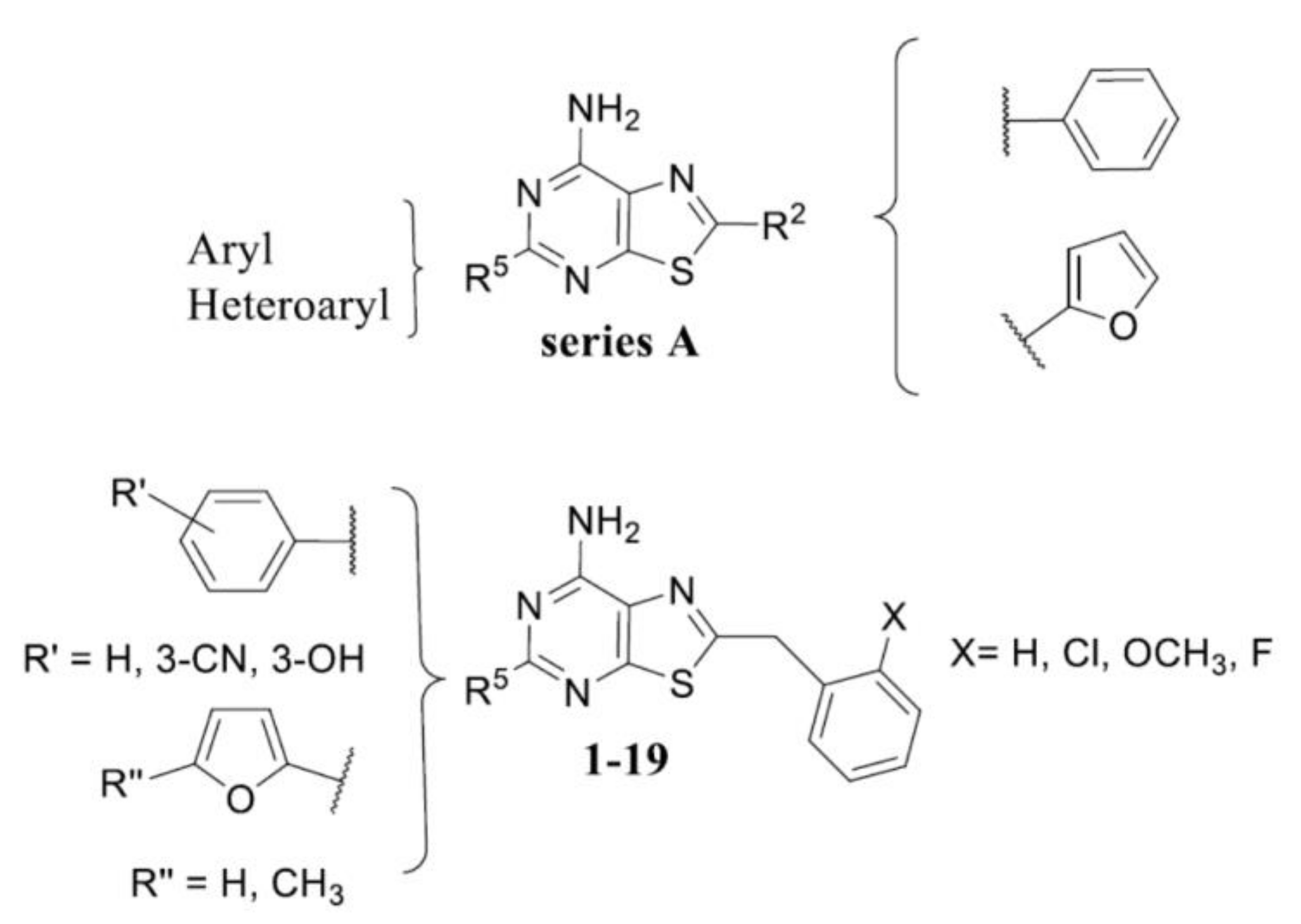
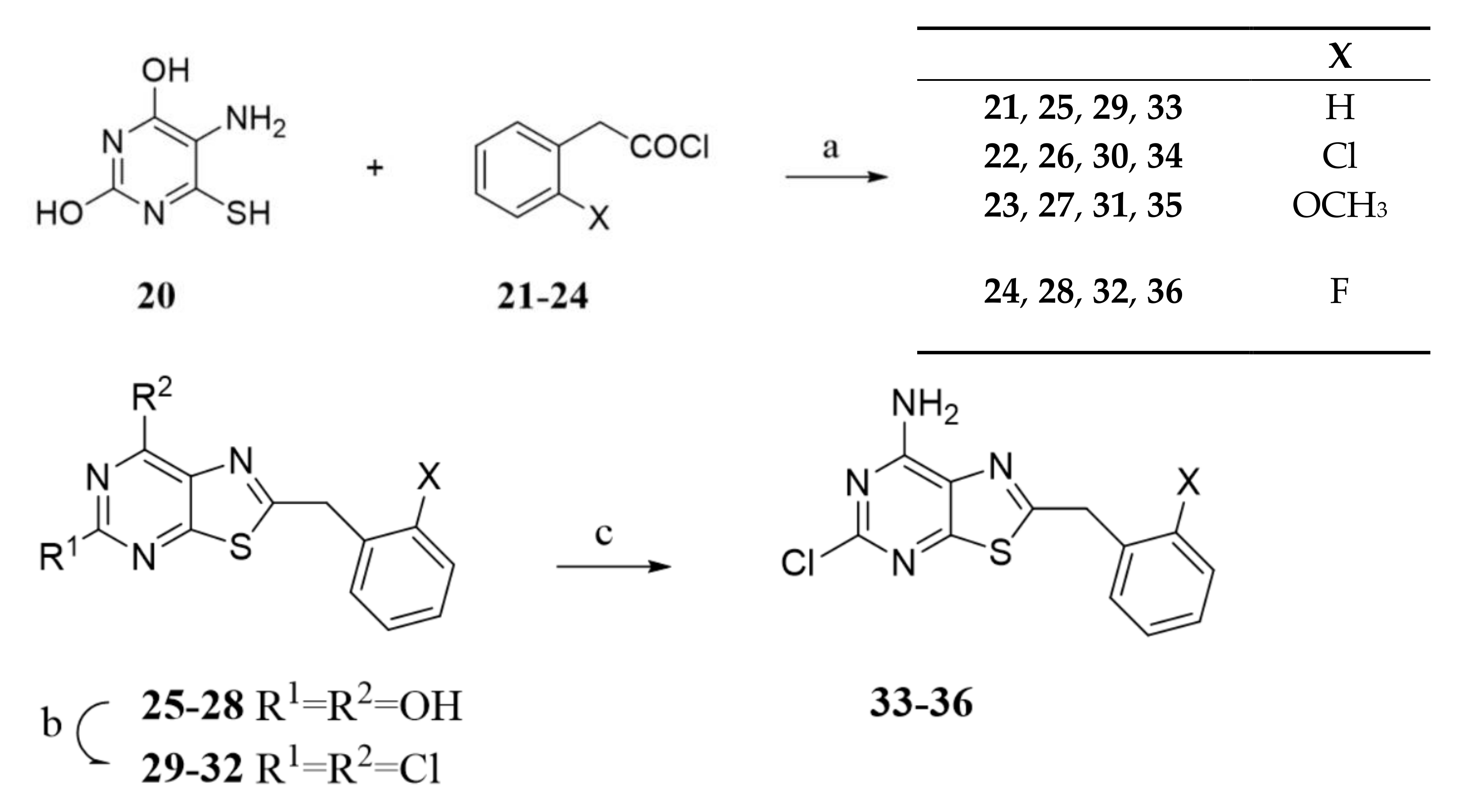
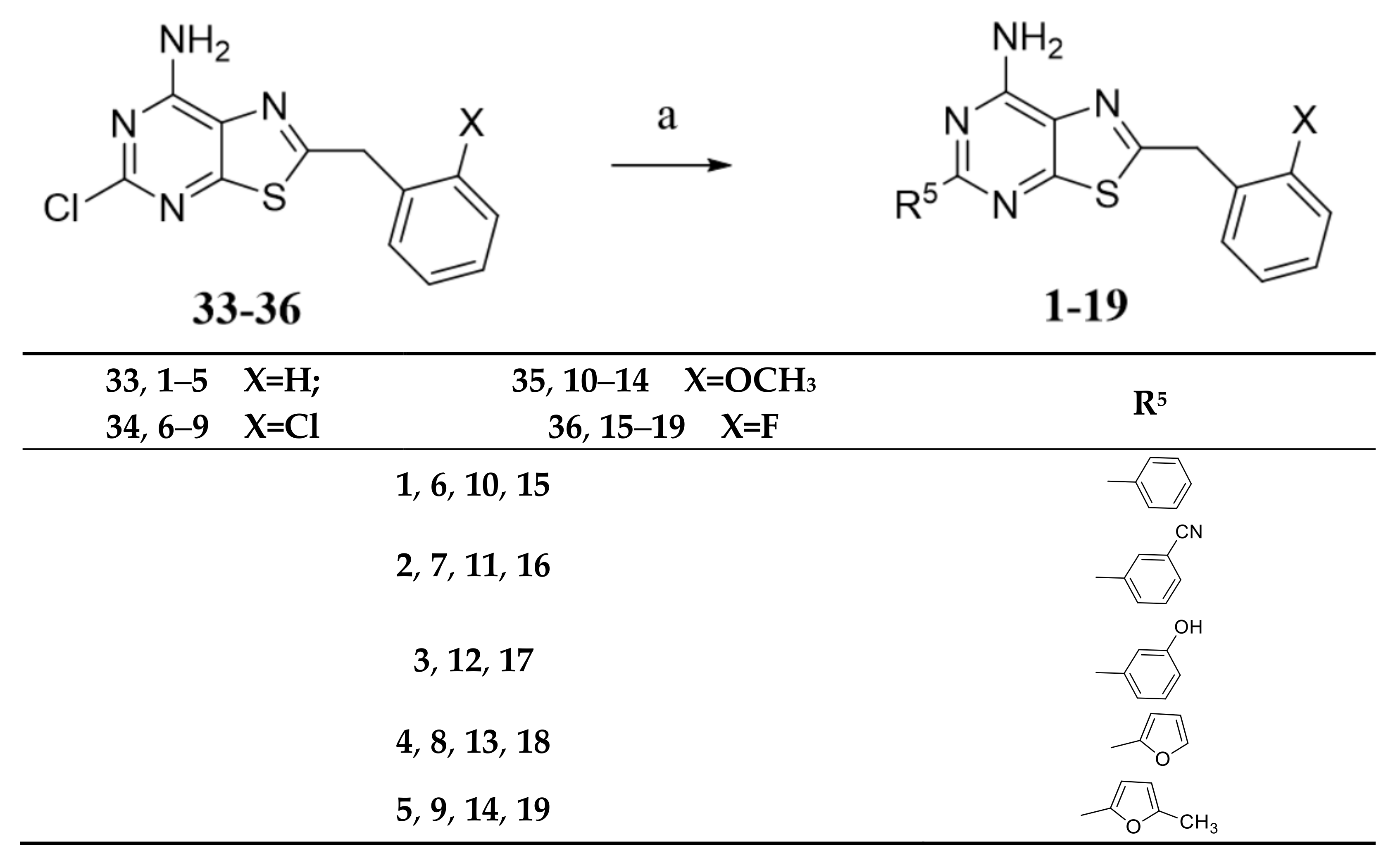
2.1. Chemistry
2.2. Binding and cAMP Assays
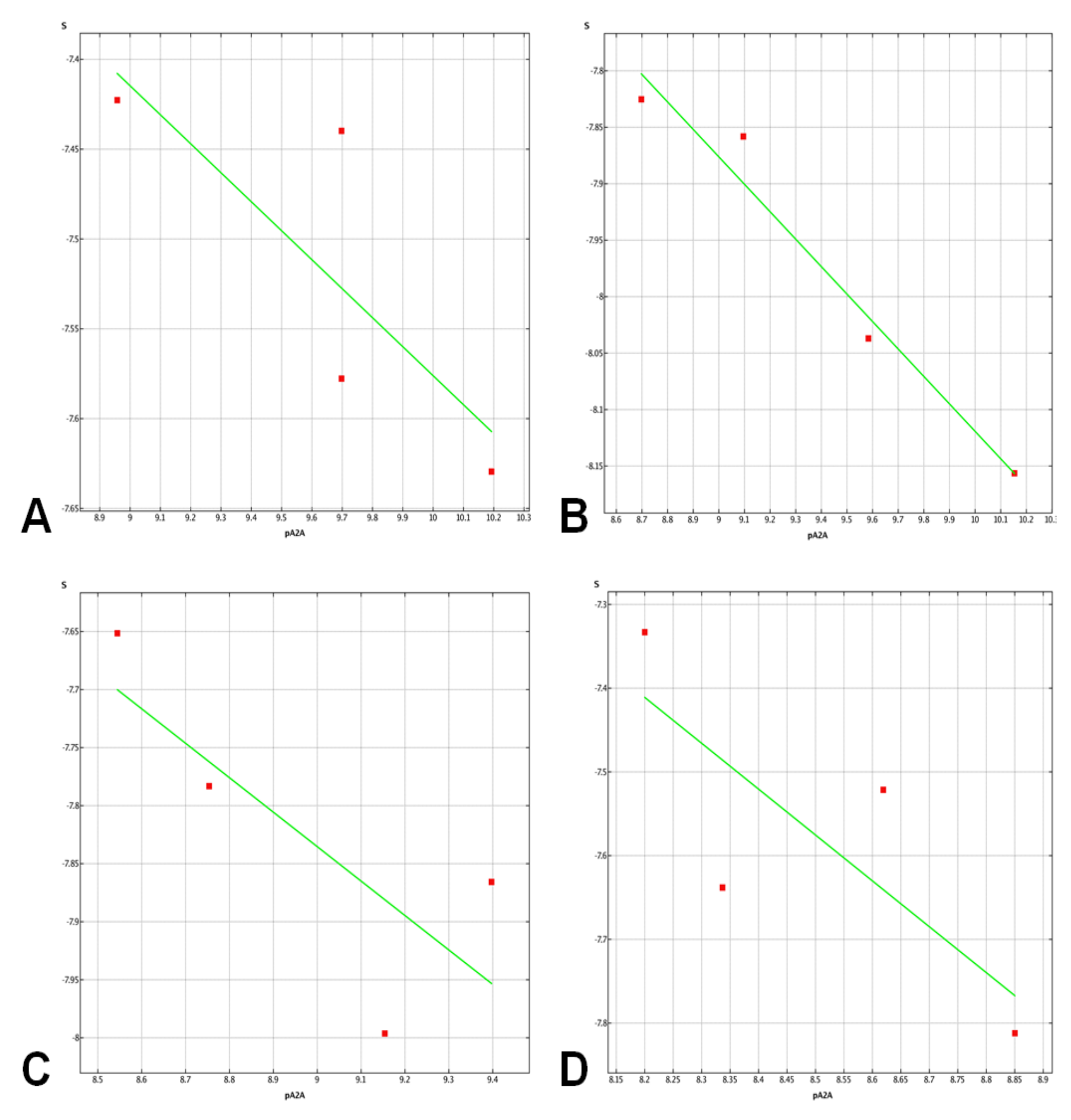
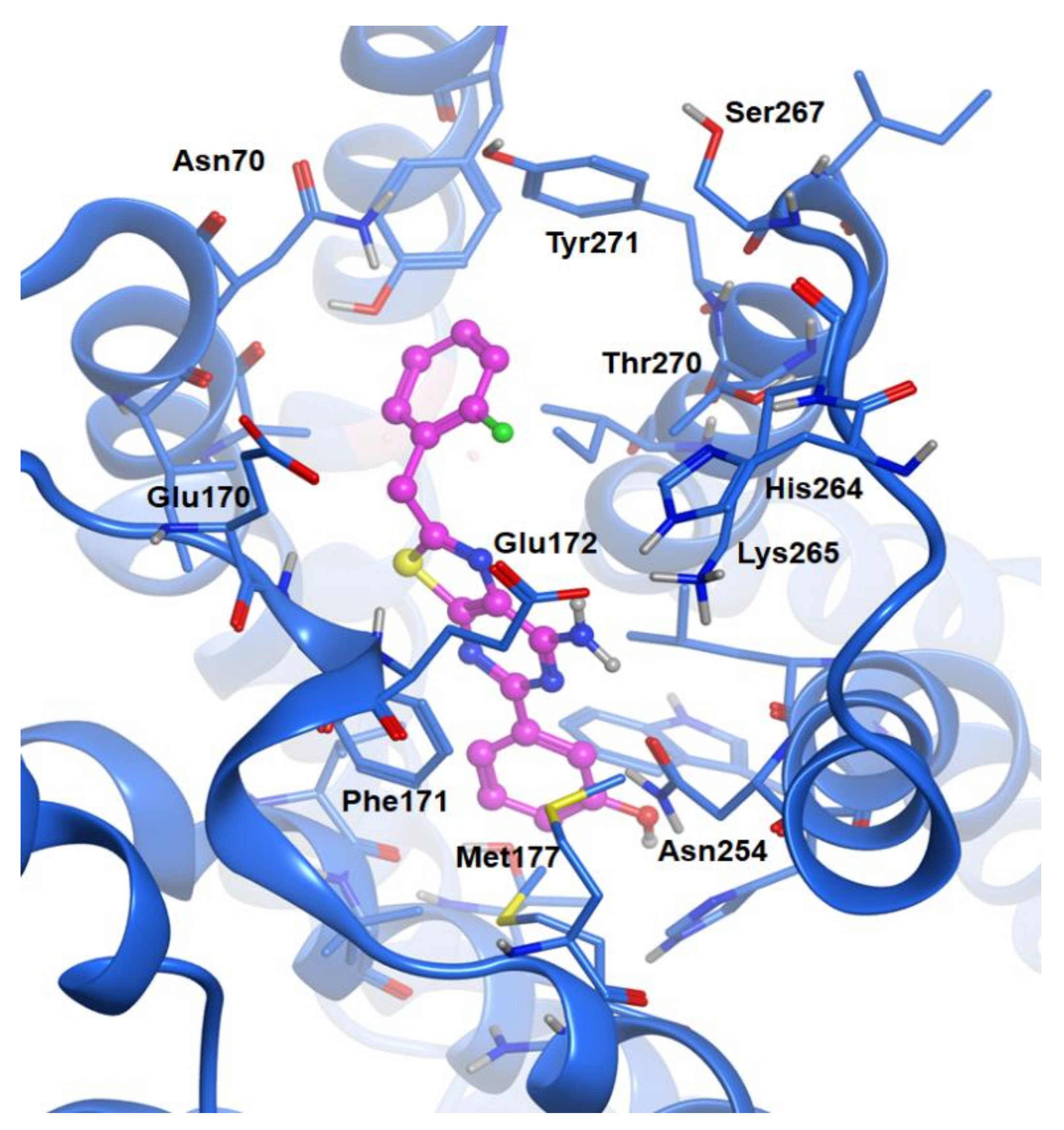
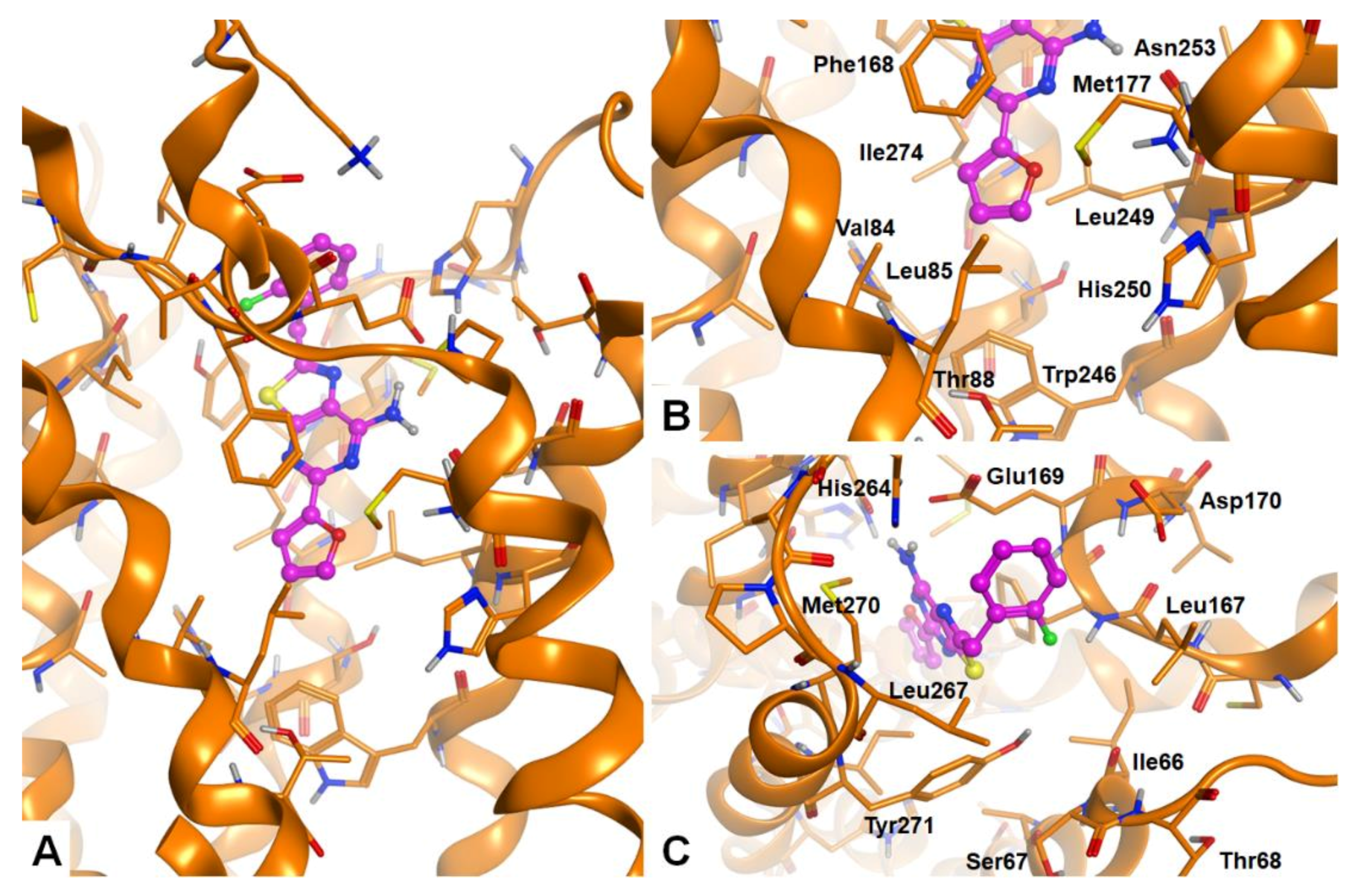
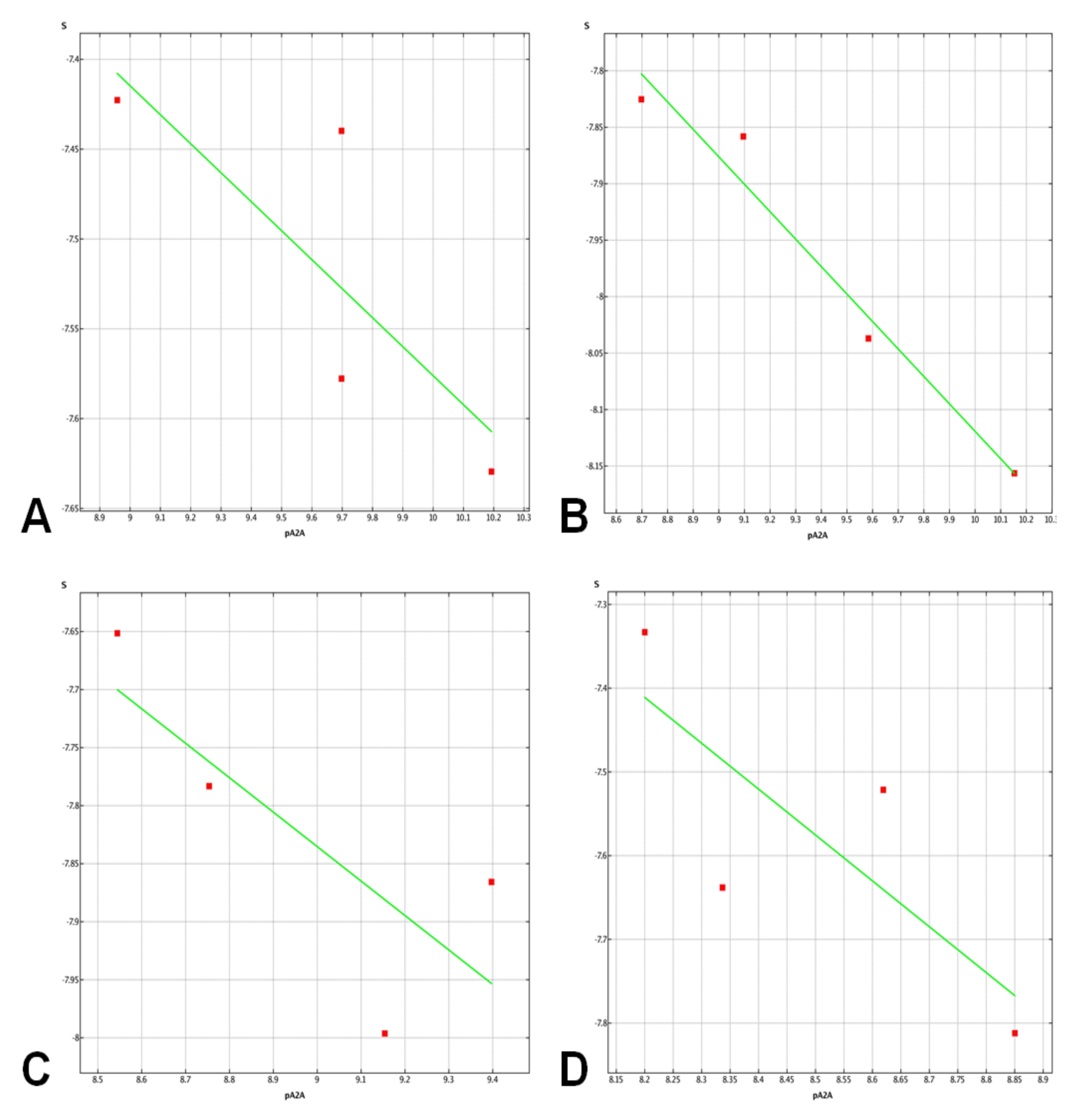
2.3. Molecular Docking Studies
2.4. Behavioral In Vivo Tests
2.4.1. Forced Swimming Test and Tail Suspension Test
2.4.2. Sucrose Preference Test
3. Materials and Methods
3.1. Chemistry
3.1.1. General Methods
3.1.2. General Procedure for the Synthesis of 25–28
3.1.3. General Procedure for the Synthesis of 29–32
3.1.4. General Procedure for the Synthesis of 33–36
3.1.5. General Procedure for the Synthesis of 1–19
3.2. Pharmacological Assays
3.2.1. Membrane Preparation
3.2.2. Radioligand Binding
3.2.3. GloSensor cAMP Assay
3.2.4. Statistical Analysis
3.3. In Vivo Assays
3.3.1. Animals
3.3.2. Forced Swimming Test
3.3.3. Tail Suspension Test
3.3.4. LPS-Induced Anhedonia
3.3.5. Sucrose Preference Test
3.3.6. Statistical Analysis
3.4. Molecular Modeling Studies
3.4.1. Refinement of the Human A2A AR and A1 AR Structures
3.4.2. Molecular Docking Analysis
3.4.3. Post Docking Analysis. Residue Interaction Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pan American Health Organization. The Burden of Mental Disorders in the Region of the Americas, 2018; PAHO: Washington, DC, USA, 2018. [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Arlington, VA, USA, 2013. [Google Scholar]
- Currier, M.B.; Nemeroff, C.B. Depression as a risk factor for cancer: From pathophysiological advances to treatment implications. Annu. Rev. Med. 2014, 65, 203–221. [Google Scholar] [CrossRef] [PubMed]
- Knol, M.J.; Twisk, J.W.R.; Beekman, A.T.F.; Heine, R.J.; Snoek, F.J.; Pouwer, F. Depression as a risk factor for the onset of type 2 diabetes mellitus. A meta-analysis. Diabetologia 2006, 49, 837–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiedorowicz, J.G. Depression and cardiovascular disease: An update on how course of illness may influence risk. Curr. Psychiatry Rep. 2014, 16, 492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santiago, J.A.; Potashkin, J.A. The impact of disease comorbidities in Alzheimer’s disease. Front. Aging Neurosci. 2021, 13, 631770. [Google Scholar] [CrossRef] [PubMed]
- Timmer, M.H.M.; van Beek, M.H.C.T.; Bloem, B.R.; Esselink, R.A.J. What a neurologist should know about depression in Parkinson’s disease. Pract. Neurol. 2017, 17, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Robinson, R.G.; Jorge, R.E. Post-stroke depression: A review. Am. J. Psychiatry 2016, 173, 221–231. [Google Scholar] [CrossRef] [Green Version]
- Doan, L.; Manders, T.; Wang, J. Neuroplasticity underlying the comorbidity of pain and depression. Neuronal Plast. 2015, 2015, 504691. [Google Scholar] [CrossRef]
- Maffioletti, E.; Minelli, A.; Tardito, D.; Gennarelli, M. Blues in the brain and beyond: Molecular bases of major depressive disorder and relative pharmacological and non-pharmacological treatments. Genes 2020, 11, 1089. [Google Scholar] [CrossRef]
- Berton, O.; Nestler, E.J. New approaches to antidepressant drug discovery: Beyond monoamines. Nat. Rev. Neurosci. 2006, 7, 137–151. [Google Scholar] [CrossRef]
- Szopa, A.; Socala, K.; Serefko, A.; Doboszewska, U.; Wrobel, A.; Poleszak, E.; Wlaz, P. Purinergic transmission in depressive disorders. Pharm. Ther. 2021, 224, 107821. [Google Scholar] [CrossRef]
- Bartoli, F.; Burnstock, G.; Crocamo, C.; Carrà, G. Purinergic signaling and related biomarkers in depression. Brain Sci. 2020, 10, 160. [Google Scholar] [CrossRef] [Green Version]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of adenosine receptors: The state of the art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef]
- Lopez-Cruz, L.; Salamone, J.D.; Correa, M. Caffeine and selective adenosine receptor antagonists as new therapeutic tools for the motivational symptoms of depression. Front. Pharmacol. 2018, 9, 526. [Google Scholar] [CrossRef]
- Szopa, A.; Poleszak, E.; Bogatko, K.; Wyska, E.; Wośko, S.; Doboszewska, U.; Świąder, K.; Wlaź, A.; Dudka, J.; Wróbel, A.; et al. DPCPX, a selective adenosine A1 receptor antagonist, enhances the antidepressant-like effects of imipramine, escitalopram, and reboxetine in mice behavioral tests. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2018, 391, 1361–1371. [Google Scholar] [CrossRef] [Green Version]
- El Yacoubi, M.; Ledent, C.; Parmentier, M.; Bertorelli, R.; Ongini, E.; Costentin, J.; Vaugeois, J.M. Adenosine A2A receptor antagonists are potential antidepressant: Evidence based on pharmacology and A2A receptor knockout mice. Br. J. Pharmacol. 2001, 134, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K.; Kobayashi, M.; Mori, A.; Jenner, P.; Kanda, T. Antidepressant-like activity of the adenosine A2A receptor antagonist, istradefylline (KW-6002), in the forced swim test and the tail suspension test in rodents. Pharmacol. Biochem. Behav. 2013, 114–115, 23–30. [Google Scholar] [CrossRef]
- Szopa, A.; Poleszak, E.; Wyska, E.; Serefko, A.; Wośko, S.; Wlaź, A.; Pieróg, M.; Wróbel, A.; Wlaź, P. Caffeine enhances the antidepressant-like activity of common antidepressant drugs in the forced swim test in mice. Naunyn Schmiedebergs Arch. Pharmacol. 2016, 389, 211–221. [Google Scholar] [CrossRef] [Green Version]
- Bogatko, K.; Poleszak, E.; Szopa, A.; Wyska, E.; Wlaź, P.; Świąder, K.; Wlaź, A.; Doboszewska, U.; Rojek, K.; Serefko, A. The influence of selective A1 and A2A receptor antagonists on the antidepressant-like activity of moclobemide, venlafaxine and bupropion in mice. J. Pharm. Pharmacol. 2018, 70, 1200–1208. [Google Scholar] [CrossRef]
- Zheng, J.; Zhang, X.; Zhen, X. Development of adenosine A2A receptor antagonists for the treatment of Parkinson’s disease: A recent update and challenge. ACS Chem. Neurosci. 2019, 10, 783–791. [Google Scholar] [CrossRef]
- Yamada, K.; Kobayashi, M.; Shiozaki, S.; Ohta, T.; Mori, A.; Jenner, P.; Kanda, T. Antidepressant activity of the adenosine A2A receptor antagonist, istradefylline (KW-6002) on learned helplessness in rats. Psychopharmacology 2014, 231, 2839–2849. [Google Scholar] [CrossRef]
- Basu, S.; Barawkar, D.A.; Ramdas, V.; Naykodi, M.; Shejul, Y.D.; Patel, M.; Thorat, S.; Panmand, A.; Kashinath, K.; Bonagiri, R.; et al. Discovery of potent and selective A2A antagonists with efficacy in animal models of Parkinson’s disease and depression. ACS Med. Chem. Lett. 2017, 8, 835–840. [Google Scholar]
- Mihara, T.; Mihara, K.; Yarimizu, J.; Mitani, Y.; Matsuda, R.; Yamamoto, H.; Aoki, S.; Akahane, A.; Iwashita, A.; Matsuoka, N. Pharmacological characterization of a novel, potent adenosine A1 and A2A receptor dual antagonist, 5-[5-amino-3-(4-fluorophenyl)pyrazine-2-yl]-1-isopropylpyridine-2(1H)-one (ASP5854), in models of Parkinson’s disease and cognition. J. Pharmacol. Exp. Ther. 2007, 323, 708–719. [Google Scholar] [CrossRef] [Green Version]
- Atack, J.R.; Shook, B.C.; Rassnick, S.; Jackson, P.F.; Rhodes, K.; Drinkenburg, W.H.; Ahnaou, A.; Te Riele, P.; Langlois, X.; Hrupka, B.; et al. JNJ-40255293, a novel adenosine A2A/A1 antagonist with efficacy in preclinical models of Parkinson’s disease. ACS Chem. Neurosci. 2014, 5, 1005–1019. [Google Scholar] [CrossRef]
- Squarcialupi, L.; Falsini, M.; Catarzi, D.; Varano, F.; Betti, M.; Varani, K.; Vincenzi, F.; Dal Ben, D.; Lambertucci, C.; Volpini, R.; et al. Exploring the 2- and 5-positions of the pyrazolo[4,3-d]pyrimidin-7-amino scaffold to target human A1 and A2A adenosine receptors. Bioorg. Med. Chem. 2016, 24, 2794–2808. [Google Scholar] [CrossRef]
- Poli, D.; Falsini, M.; Varano, F.; Betti, M.; Varani, K.; Vincenzi, F.; Pugliese, A.M.; Pedata, F.; Dal Ben, D.; Thomas, A.; et al. Imidazo[1,2-a]pyrazin-8-amine core for the design of new adenosine receptor antagonists: Structural exploration to target the A3 and A2A subtypes. Eur. J. Med. Chem. 2017, 125, 611–628. [Google Scholar] [CrossRef]
- Falsini, M.; Squarcialupi, L.; Catarzi, D.; Varano, F.; Betti, M.; Dal Ben, D.; Marucci, G.; Buccioni, M.; Volpini, R.; De Vita, T.; et al. The 1,2,4-triazolo[4,3-a]pyrazin-3-one as a versatile scaffold for the design of potent adenosine human receptor antagonists. Structural investigations to target the A2A receptor subtype. J. Med. Chem. 2017, 60, 5772–5790. [Google Scholar] [CrossRef]
- Falsini, M.; Catarzi, D.; Varano, F.; Dal Ben, D.; Marucci, G.; Buccioni, M.; Volpini, R.; Di Cesare Mannelli, L.; Ghelardini, C.; Colotta, V. Novel 8-amino-1,2,4-triazolo[4,3-a]pyrazin-3-one derivatives as potent human adenosine A1 and A2A receptor antagonists. Evaluation of their protective effect against β-amyloidinduced neurotoxicity in SH-SY5Y cells. Bioorg. Chem. 2019, 87, 380–394. [Google Scholar] [CrossRef]
- Betti, M.; Catarzi, D.; Varano, F.; Falsini, M.; Varani, K.; Vincenzi, F.; Pasquini, S.; Di Cesare Mannelli, L.; Ghelardini, C.; Lucarini, E.; et al. Modifications on the amino-3,5-dicyanopyridine core to obtain multifaceted adenosine receptor ligands with antineuropathic activity. J. Med. Chem. 2019, 62, 6894–6912. [Google Scholar] [CrossRef]
- Varano, F.; Catarzi, D.; Vincenzi, F.; Betti, M.; Falsini, M.; Ravani, A.; Borea, P.A.; Colotta, V.; Varani, K. Design, Synthesis and pharmacological characterization of 2-(2-furanyl)thiazolo[5,4-d]pyrimidine-5,7-diamine derivatives: New highly potent A2A adenosine receptor inverse agonists with antinociceptive activity. J. Med. Chem. 2016, 59, 10564–10576. [Google Scholar] [CrossRef]
- Varano, F.; Catarzi, D.; Falsini, M.; Vincenzi, F.; Pasquini, S.; Varani, K.; Colotta, V. Identification of novel thiazolo[5,4-d]pyrimidine derivatives as human A1 and A2A adenosine receptor antagonists/inverse agonists. Bioorg. Med. Chem. 2018, 26, 3688–3695. [Google Scholar] [CrossRef]
- Varano, F.; Catarzi, D.; Vincenzi, F.; Falsini, M.; Pasquini, S.; Borea, P.A.; Colotta, V.; Varani, K. Structure-activity relationship studies and pharmacological characterization of N5-heteroarylalkyl-substituted-2-(2-furanyl)thiazolo[5,4-d]pyrimidine-5,7-diamine-based derivatives as inverse agonists at human A2A adenosine receptor. Eur. J. Med. Chem. 2018, 155, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Varano, F.; Catarzi, D.; Falsini, M.; Dal Ben, D.; Buccioni, M.; Marucci, G.; Volpini, R.; Colotta, V.; Novel human adenosine receptor antagonists based on the 7-amino-thiazolo[5,4-d]pyrimidine scaffold. Structural investigations at the 2-, 5- and 7-positions to enhance affinity and tune selectivity. Bioorg. Med. Chem. Lett. 2019, 29, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Varano, F.; Catarzi, D.; Vigiani, E.; Vincenzi, F.; Pasquini, S.; Varani, K.; Colotta, V. Piperazine- and piperidine-containing thiazolo[5,4-d]pyrimidine derivatives as new potent and selective adenosine A2A receptor inverse agonists. Pharmaceuticals 2020, 13, 161. [Google Scholar] [CrossRef] [PubMed]
- Hager, G.P.; Kaiser, C. Oxazolopyrimidine and Thiazolopyrimidine Derivatives Related to Xantines. J. Am. Pharm. Assoc. 1955, 44, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.R.; Grande, F.; Chouhan, B.S.; Naik, P.P.; Giridhar, R.; Garofalo, A.; Neamati, N. Cytotoxic potential of novel 6,7-dimethoxyquinazolines. Eur. J. Med. Chem. 2012, 48, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Salado, I.G.; Redondo, M.; Bello, M.L.; Perez, C.; Liachko, N.F.; Kraemer, B.C.; Miguel, L.; Lecourtois, M.; Gil, C.; Martinez, A.; et al. Protein kinase CK-1 inhibitors ass new potential drugs for amyotrophic lateral sclerosis. J. Med. Chem. 2014, 57, 2755–2772. [Google Scholar] [CrossRef] [PubMed]
- Li, W.Y.; Ni, W.W.; Ye, Y.X.; Fang, H.L.; Pan, X.M.; He, J.L.; Zhou, T.L.; Yi, J.; Liu, S.S.; Zhou, M.; et al. N-monoarylacetothioureas as potent urease inhibitors: Synthesis, SAR, and biological evaluation. J. Enzyme Inhib. Med. Chem. 2020, 35, 404–413. [Google Scholar] [CrossRef] [Green Version]
- Weinert, T.; Olieric, N.; Cheng, R.; Brunle, S.; James, D.; Ozerov, D.; Gashi, D.; Vera, L.; Marsh, M.; Jaeger, K.; et al. Serial millisecond crystallography for routine room-temperature structure determination at synchrotrons. Nat. Commun. 2017, 8, 542. [Google Scholar] [CrossRef]
- Molecular Operating Environment, C.C.G., Inc., 1255 University St., Suite 1600, Montreal, QC, Canada, H3B 3X3. Available online: https://www.chemcomp.com/ (accessed on 15 November 2020).
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Flare Version 2.0; Cresset BioMolecular Discovery Ltd.: Cambridgeshire, UK. Available online: http://www.cresset-group.com/flare/ (accessed on 5 December 2020).
- Cheng, R.K.Y.; Segala, E.; Robertson, N.; Deflorian, F.; Dore, A.S.; Errey, J.C.; Fiez-Vandal, C.; Marshall, F.H.; Cooke, R.M. Structures of human A1 and A2A adenosine receptors with xanthines reveal determinants of selectivity. Structure 2017, 25, 1275–1285. [Google Scholar] [CrossRef]
- Shadnia, H.; Wright, J.S.; Anderson, J.M. Interaction force diagrams: New insight into ligand-receptor binding. J. Comput. Aided Mol. Des. 2009, 23, 185–194. [Google Scholar] [CrossRef]
- Dal Ben, D.; Buccioni, M.; Lambertucci, C.; Marucci, G.; Thomas, A.; Volpini, R.; Cristalli, G. Molecular modeling study on potent and selective adenosine A3 receptor agonists. Bioorg. Med. Chem. 2010, 18, 7923–7930. [Google Scholar] [CrossRef]
- Varano, F.; Catarzi, D.; Squarcialupi, L.; Betti, M.; Vincenzi, F.; Ravani, A.; Varani, K.; Dal Ben, D.; Thomas, A.; Volpini, R.; et al. Exploring the 7-oxo-thiazolo[5,4-d]pyrimidine core for the design of new human adenosine A3 receptor antagonists. Synthesis, molecular modeling studies and pharmacological evaluation. Eur. J. Med. Chem. 2015, 96, 105–121. [Google Scholar] [CrossRef]
- Pallanti, S.; Tofani, T.; Zanardelli, M.; Di Cesare Mannelli, L.; Ghelardini, C. BDNF and Artemin are increased in drug-naïve non-depressed GAD patients: Preliminary data. Int. J. Psychiatry Clin. Pract. 2014, 18, 255–260. [Google Scholar] [CrossRef]
- Micheli, L.; Spitoni, S.; Di Cesare Mannelli, L.; Bilia, A.R.; Ghelardini, C.; Pallanti, S. Bacopa monnieri as augmentation therapy in the treatment of anhedonia, preclinical and clinical evaluation. Phytother. Res. 2020, 34, 2331–2340. [Google Scholar] [CrossRef]
- Buccioni, M.; Marucci, G.; Dal Ben, D.; Giacobbe, D.; Lambertucci, C.; Soverchia, L.; Thomas, A.; Volpini, R.; Cristalli, G. Innovative functional cAMP assay for studying G protein-coupled receptors: Application to the pharmacological characterization of GPR17. Purinergic Signal. 2011, 7, 463–468. [Google Scholar] [CrossRef] [Green Version]
- Thomas, A.; Buccioni, M.; Dal Ben, D.; Lambertucci, C.; Marucci, G.; Santinelli, C.; Spinaci, A.; Kachler, S.; Klotz, K.-N.; Volpini, R. The length and flexibility of the 2-substituent of 9-ethyladenine derivatives modulate affinity and selectivity for the human A2A adenosine receptor. ChemMedChem 2016, 11, 1829–1839. [Google Scholar] [CrossRef]
- Porsolt, R.D.; Le Pichon, M.; Jalfre, M. Depression: A new animal model sensitive to antidepressant treatments. Nature 1977, 266, 730–732. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| X | R5 | hA1Ki (nM) a | hA2AKi (nM) b | hA3Ki (nM) c | hA2BIC50 (nM) d | |
|---|---|---|---|---|---|---|
| 1 | H | C6H5 | 20 ± 4.3 | 2.9 ± 0.04 | 26.6 ± 6.0 | >30,000 |
| 2 | H | C6H4-3-CN | 51.7 ± 11.7 | 4.6 ± 0.9 | 76 ± 0.9 | >30,000 |
| 3 | H | C6H4-3-OH | 15.7 ± 3.4 | 7.3 ± 1.0 | 103.6 ± 21.1 | >30,000 |
| 4 | H | furan-2yl | 15.5 ± 3.6 | 1.1 ± 0.2 | 65.4 ± 12 | 794 ± 196 |
| 5 | H | furan-2yl-5-CH3 | 6.8 ± 1.2 | 0.8 ± 0.04 | 18.8 ± 1.9 | 1096 ± 228 |
| 6 | Cl | C6H5 | 2.8 ± 0.05 | 0.4 ± 0.04 | 143 ± 0.2 | >30,000 |
| 7 | Cl | C6H4-3-CN | 72.8 ± 15 | 6.3 ± 0.18 | 295.1 ± 0.6 | 6785 ± 62 |
| 8 | Cl | furan-2yl | 3.8 ± 0.6 | 0.2 ± 0.03 | 4786 ± 5.1 | 1887 ± 175 |
| 9 | Cl | furan-2yl-5-CH3 | 0.5 ± 0.1 | 0.07 ± 0.006 | 8.5 ± 1.6 | 8847 ± 1445 |
| 10 | OCH3 | C6H5 | 4.5 ± 0.8 | 0.7 ± 0.1 | 37.4 ± 1 | >30,000 |
| 11 | OCH3 | C6H4-3-CN | 28.4 ± 4 | 2.4 ± 0.09 | 54.2 ± 1.2 | 1536 ± 35 |
| 12 | OCH3 | C6H4-3-OH | 10.2 ± 0.3 | 1.9 ± 0.4 | 79.6 ± 2.3 | 7424 ± 876 |
| 13 | OCH3 | furan-2yl | 2.4 ± 0.1 | 0.2 ± 0.03 | 48.6 ± 2.4 | 1536 ± 35 |
| 14 | OCH3 | furan-2yl-5-CH3 | 11.5 ± 2.8 | 2.0 ± 0.6 | 18.2 ± 5 | 2416 ± 545 |
| 15 | F | C6H5 | 1.9 ± 0.05 | 1.8 ± 0.6 | 72.1 ± 5.3 | >30,000 |
| 16 | F | C6H4-3-CN | 21.5 ± 5.1 | 1.4 ± 0.3 | 95.5 ± 1.5 | 1514 ± 121 |
| 17 | F | C6H4-3-OH | 0.5 ± 0.1 | 13.8 ± 0.06 | 455.9 ± 15 | 3237 ± 136 |
| 18 | F | furan-2yl | 1.9 ± 0.08 | 0.06 ± 0.02 | 93.1 ± 2.8 | 384 ± 55 |
| 19 | F | furan-2yl-5-CH3 | 5.7 ± 0.2 | 0.26 ± 0.01 | 50.2 ± 9.2 | 1695 ± 228 |
| 20e | 148 ± 16 | 19 ± 6.2 | 84 ± 13 | >30,000 |
| hA2A IC50 (nM) a | hA2A IC50 (nM) a | ||
|---|---|---|---|
| 5 | 88 ± 21 | 13 | 35 ± 8.5 |
| 8 | 39±9 | 19 | 51 ± 12 |
| 9 | 7.7 ± 1.3 | 18 | 14 ± 3.4 (360 ± 115) b |
| 10 | 125 ± 33 |
| 8 | 9 | 6 | 18 | 19 | 15 | 4 | 5 | 1 | |
| pKi A2A | 9.70 | 10.15 | 9.40 | 10.19 | 9.59 | 8.75 | 8.96 | 9.10 | 8.55 |
| R5 | 2-furyl | 2-furyl-5-CH3 | C6H5 | 2-furyl | 2-furyl-5-CH3 | C6H5 | 2-furyl | 2-furyl-5-CH3 | C6H5 |
| Val84 | −0.0 | −0.2 | −0.1 | −0.1 | −0.3 | −0.1 | −0.0 | −0.4 | −0.2 |
| Leu85 | −0.8 | −1.1 | −0.5 | −0.8 | −1.1 | −0.5 | −0.8 | −1.1 | −0.6 |
| Thr88 | −0.0 | −0.2 | −0.2 | −0.0 | −0.2 | −0.2 | −0.0 | −0.3 | −0.3 |
| Phe168 | −5.0 | −4.9 | −5.3 | −5.7 | −5.4 | −5.8 | −3.9 | −4.0 | −2.9 |
| Met177 | −2.0 | −2.3 | −0.1 | −2.0 | −2.3 | −0.1 | −2.1 | −2.3 | −0.1 |
| Trp246 | −0.6 | −1.3 | −0.5 | −0.6 | −1.3 | −0.5 | −0.6 | −1.3 | −0.6 |
| Leu249 | −2.5 | −2.7 | −2.1 | −2.4 | −2.7 | −2.0 | −2.5 | −2.8 | −2.0 |
| His250 | 0.1 | −107 | −0.6 | 0.1 | −0.3 | −0.7 | 0.1 | −0.3 | −0.7 |
| Asn253 | −7.0 | −6.9 | −4.9 | −6.9 | −6.9 | −4.9 | −7.3 | −7.1 | −4.4 |
| Ile274 | 0.6 | 0.3 | −0.2 | 0.5 | 0.3 | −0.2 | 0.6 | 0.3 | 0.1 |
| tot | −17.2 | −21.0 | −14.5 | −17.9 | −20.2 | −15.0 | −16.5 | −19.3 | −11.7 |
| Treatment | Immobility Time (s) a |
|---|---|
| vehicle | 193.7 ± 8.9 |
| 18 10 mg kg−1 p.o. | 184.7 ± 5.6 |
| 18 30 mg kg−1 p.o. | 143.5 ± 9.1 ** |
| amitriptyline 15 mg kg−1 s.c. | 149.4 ± 4.6 ** |
| Treatment | Immobility Time (s) a |
|---|---|
| vehicle | 99.0 ± 6.8 |
| 18 10 mg kg−1 p.o. | 88.9 ± 10.16 |
| 18 30 mg kg−1 p.o. | 56.5 ± 6.1 ** |
| amitriptyline 15 mg kg−1 s.c. | 61.6 ± 8.4 ** |
| Treatment | Sucrose Preference (%), 6 h a | Sucrose Preference (%), 24 h a |
|---|---|---|
| vehicle | 78.1 ± 2.0 | 75.3 ± 1.8 |
| Vehicle + LPS | 42.3 ± 3.1 ^^ | 38.0 ± 5.6 ^^ |
| LPS + 18 10 mg kg−1 p.o. | 48.9 ± 6.7 | 39.7 ± 3.7 |
| LPS + 18 30 mg kg−1 p.o. | 67.3 ± 1.6 ** | 64.9 ± 2.6 ** |
| LPS + amitriptyline 15 mg kg−1 s.c. | 69.1 ± 4.8 ** | 70.3 ± 6.4 ** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varano, F.; Catarzi, D.; Vigiani, E.; Dal Ben, D.; Buccioni, M.; Marucci, G.; Di Cesare Mannelli, L.; Lucarini, E.; Ghelardini, C.; Volpini, R.; et al. Design and Synthesis of Novel Thiazolo[5,4-d]pyrimidine Derivatives with High Affinity for Both the Adenosine A1 and A2A Receptors, and Efficacy in Animal Models of Depression. Pharmaceuticals 2021, 14, 657. https://doi.org/10.3390/ph14070657
Varano F, Catarzi D, Vigiani E, Dal Ben D, Buccioni M, Marucci G, Di Cesare Mannelli L, Lucarini E, Ghelardini C, Volpini R, et al. Design and Synthesis of Novel Thiazolo[5,4-d]pyrimidine Derivatives with High Affinity for Both the Adenosine A1 and A2A Receptors, and Efficacy in Animal Models of Depression. Pharmaceuticals. 2021; 14(7):657. https://doi.org/10.3390/ph14070657
Chicago/Turabian StyleVarano, Flavia, Daniela Catarzi, Erica Vigiani, Diego Dal Ben, Michela Buccioni, Gabriella Marucci, Lorenzo Di Cesare Mannelli, Elena Lucarini, Carla Ghelardini, Rosaria Volpini, and et al. 2021. "Design and Synthesis of Novel Thiazolo[5,4-d]pyrimidine Derivatives with High Affinity for Both the Adenosine A1 and A2A Receptors, and Efficacy in Animal Models of Depression" Pharmaceuticals 14, no. 7: 657. https://doi.org/10.3390/ph14070657
APA StyleVarano, F., Catarzi, D., Vigiani, E., Dal Ben, D., Buccioni, M., Marucci, G., Di Cesare Mannelli, L., Lucarini, E., Ghelardini, C., Volpini, R., & Colotta, V. (2021). Design and Synthesis of Novel Thiazolo[5,4-d]pyrimidine Derivatives with High Affinity for Both the Adenosine A1 and A2A Receptors, and Efficacy in Animal Models of Depression. Pharmaceuticals, 14(7), 657. https://doi.org/10.3390/ph14070657